

Abstract
Familial British dementia (FBD) is an early-onset non-amyloid-β (Aβ) cerebral amyloidosis that presents with severe cognitive decline and strikingly similar neuropathological features to those present in Alzheimer’s disease (AD). FBD is associated with a T to A single nucleotide transition in the stop codon of a gene encoding BRI2, leading to the production of an elongated precursor protein. Furin-like proteolytic processing at its C-terminus releases a longer-than-normal 34 amino acid peptide, ABri, exhibiting amyloidogenic properties not seen in its 23 amino acid physiologic counterpart Bri1-23. Deposited ABri exhibits abundant post-translational pyroglutamate (pE) formation at the N-terminus, a feature seen in truncated forms of Aβ found in AD deposits, and co-exists with neurofibrillary tangles almost identical to those found in AD. We tested the impact of the FBD mutation alone and in conjunction with the pE post-translational modification on the structural properties and associated neurotoxicity of the ABri peptide. The presence of pE conferred to the ABri molecule enhanced hydrophobicity and accelerated aggregation/fibrillization properties. ABri pE was capable of triggering oxidative stress, loss of mitochondrial membrane potential and activation of caspase-mediated apoptotic mechanisms in neuronal cells, whereas homologous peptides lacking the elongated C-terminus and/or the N-terminal pE were unable to induce similar detrimental cellular pathways. The data indicate that the presence of N-terminal pE is not in itself sufficient to induce pathogenic changes in the physiologic Bri1-23 peptide but that its combination with the ABri mutation is critical for the molecular pathogenesis of FBD.
Keywords: familial British dementia, cerebral amyloidosis, oligomeric amyloid assemblies, apoptosis, cytochrome c, oxidative stress
1. Introduction
The aberrant aggregation and/or folding of proteins is a recurrent problem of many proteopathies, including amyloidosis. Alzheimer’s disease (AD), the most common form of amyloidosis and dementia in humans, is characterized by the deposition of amyloid-β (Aβ) in the brain parenchyma and cerebral vasculature as well as by the presence of hyperphosphorylated tau in intraneuronal neurofibrillary tangles [1, 2]. Although it is unclear what primarily triggers and drives the progression of AD, strong evidence supports a pathogenic role of Aβ oligomeric conformations [3–6]. According to the current amyloid cascade hypothesis, changes in brain Aβ homeostasis lead to the accumulation of oligomers capable of inducing oxidative injury, inflammation, synaptic dysfunction, tau hyperphosphorylation, and ultimately neurodegenerative cell death. In support of the relevance of Aβ for disease pathogenesis, individuals with Down’s syndrome – who feature an overexpression of the Aβ precursor protein (APP) due to the presence of the extra chromosome 21– develop AD pathology by middle age [7, 8]. In this line, familial APP mutations exhibiting elevated Aβ production translate into an earlier onset of the disease [9–11] and transgenic animal models of such familial mutations develop amyloid pathology and cognitive deficits in the absence of neurofibrillary tangles [12].
Perhaps the strongest support for the amyloid cascade hypothesis comes from an autosomal dominant form of non-Aβ cerebral amyloidosis – Familial British Dementia (FBD) – which shares many clinical and pathological features with AD. This fatal disease is characterized by progressive dementia, cerebellar ataxia, and spastic tetraparesis with an age of onset in the fourth to fifth decade of life and a duration of about 7 to 9 years [13]. Neuropathologically, affected individuals present with severe widespread amyloid deposition in the vasculature of the brain and spinal cord, perivascular plaques, periventricular white matter changes, amyloid plaques predominantly in the limbic areas, and hippocampal neurofibrillary tangles morphologically and biochemically identical to those found in AD [14–16]. FBD is associated with a single nucleotide transition (T to A) at the stop codon of the BRI2 gene located on the long arm of chromosome 13 encoding a 266 amino acid long transmembrane protein BRI2 with a presently undefined physiological function. This stop-to-arginine mutation produces an elongated 277 amino acid precursor protein, ABriPP, which is cleaved at peptide bond 243–244 by furin-like normal proteolytic processing to release a 34 amino acid amyloidogenic peptide, ABri. In non-carrier individuals, BRI2 is also cleaved in the same position to produce a 23 amino acid circulating peptide, Bri1-23 or WT Bri [16–20].
A soluble form of ABri is also present in the circulation of affected individuals [14, 17, 18]. Interestingly, while this circulating form bears a regular N-terminal glutamate residue, biochemical analysis of deposited ABri reveals the presence of a post-translationally modified N-terminus in which the free amino group of the glutamate moiety is cyclized to form pyroglutamate (pE), a reaction that proceeds through the action of glutaminyl cyclase [21, 22]. N-terminal pE formation is a common finding in the heavy and light chains of immunoglobulin molecules (http://www.ncbi.nlm.nih.gov/protein) and has been reported for several truncated forms of Alzheimer’s Aβ peptide, specifically AβpE3 and AβpE11. The loss of a negative charge occurring as a result of the posttranslational modification increases the β-sheet content, aggregation propensity, hydrophobicity, and resistance to degradation by peptidases in the Aβ molecule [23, 24]. These modified forms of Aβ have been shown to be more neurotoxic than full-length, unmodified Aβ [25–28]. Accordingly, pE-containing Aβ is more abundant in the AD brain compared to healthy age-matched controls, suggesting an important role of this modification in the pathogenesis of this disease [29–33].
Previous studies indicate that ABri pE exhibits rapid fibrillogenesis and neurotoxicity [34–39] while the normal circulating Bri1-23 lacks β-sheet secondary structure and is not prone to aggregation [34, 38]. The formation of the N-terminal pyroglutamate moiety has been shown to influence the aggregation propensity of ABri as well as of the other BRI2-related amyloid ADan, and in the latter, enhance oligomer formation and impair synaptic potentiation. [25]. The present work includes a comprehensive analysis of the influence of the FBD mutation as well as the posttranslational pE formation on the aggregation/fibrillization and associated neurotoxicity of ABri. A detailed analysis of the cellular mechanisms elicited by the oligomeric amyloid assemblies revealed the engagement of mitochondrial pathways involving the release of cytochrome c (cyt c), loss of the organelle membrane potential, and oxidative stress that in turn lead to the induction of caspase 9-mediated apoptotic pathways.
2. Materials and methods
2.1. Peptide synthesis
Synthetic homologues of ABri and Bri1-23, both with either an N-terminal pE or an N-terminal glutamate, as well as Aβ42 were synthesized using N-tert-butyloxycarbonyl chemistry by James I. Elliott at Yale University (New Haven, CT) and purified by reverse phase-high performance liquid chromatography on a Vydac C4 column (Western Analytical, Murrieta, CA). Molecular masses were corroborated by matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry, and concentrations were assessed by amino acid analysis, as previously reported [40].
2.2. Circular dichroism spectroscopy
To evaluate peptide solubilization and disruption of pre-existing conformations with potential to seed and enhance subsequent aggregation, ABri and Bri1-23 (either N-terminal pE or E) synthetic homologues were incubated at a concentration of 1 mg/ml in hexafluoroisopropanol (HFIP; Sigma Chemical Co., St. Louis, MO), a pretreatment that breaks down β-sheet structures and disrupts hydrophobic forces leading to monodisperse amyloid subunit preparations [41]. Formation of stable α-helix structures was monitored over time until superimposed circular dichroism (CD) scans were obtained in subsequent days. To measure changes in peptide secondary structure over time at physiological salt concentrations the lyophilized HFIP-pretreated peptides were reconstituted to 1 mM in 1% ammonium hydroxide followed by further dilution in a 10 mM phosphate buffer, pH 7.4, containing 150 mM sodium fluoride to a final concentration of 50 μM. Under both conditions, spectra in the far-ultraviolet light (wavelength range: 190–260 nm; band-width 1 nm; intervals 1 nm; scan rate 60 nm/min) yielded by the different peptides at various time points of aggregation (up to 2 days) were recorded at 24°C with a Jasco J-720 spectropolarimeter (Jasco Inc., Easton, MD). A 0.2 mm path quartz cell was used for the samples in HFIP, while a 1 mm path quartz cell was used for the samples in salt-containing buffer. For each sample, 15 consecutive spectra were obtained and averaged, and the baseline reading was subtracted. Results are expressed in terms of molar ellipticity (deg·cm2·dmol −1), as previously described [40].
2.3. Peptide solubilization and aggregation
Synthetic ABri and Bri1-23 homologues (with or without N-terminal pE) were pretreated in HFIP at a concentration of 1 mg/ml for 4–7 days, depending on the inherent characteristics of each molecule. Following lyophilization, peptides were thoroughly dissolved to 10 mM in dimethyl sulfoxide (DMSO, Sigma) then to 1 mM in deionized water followed by further dilution in 1X phosphate-buffered saline (PBS) to reach the desired concentration. Reconstituted peptides were incubated at 37°C for up to 24 hours for the aggregation studies. Structural properties of the synthetic homologues at different time points were assessed by thioflavin T binding, 8-anilinonapthalene-1-sulfonic acid (ANS; Sigma) binding, and Western blot (WB) analysis under denaturing and non-denaturing conditions, as described below. For cell culture experiments, peptides were dissolved to 10 mM in DMSO followed by the addition of deionized water to 1 mM, and diluted into the pertinent culture medium at the required concentration.
2.4. Thioflavin T binding assay
Binding of the ABri and Bri1-23 homologues (with or without N-terminal pE) to thioflavin T was monitored by fluorescence evaluation as described previously [40, 42]. Briefly, 6 μl aliquots from each 50 μM peptide aggregation time point were added to 184 μl of 50 mM tris-HCl buffer, pH 8.5, and 10 μl of 0.1 mM thioflavin T (Sigma). Fluorescence was recorded for 300 s in an LS-50B spectrometer (Perkin Elmer, Waltham, MA) with a slit width of 10 nm and excitation and emission wavelengths of 435 and 490 nm, respectively [43].
2.5. ANS binding assay
Exposure of hydrophobic residues in the ABri and Bri1-23 peptides (with or without N-terminal pE) was evaluated by their ability to bind the fluorescent dye ANS. Fifty μl from each of the 50 μM peptide aggregation time points were added to 200 μl of PBS, pH 7.4, containing 2 μl of 7.5 mg/ml ANS dissolved in dimethyl formamide (Sigma). Spectra were recorded from 450 to 570 nm in a Perkin Elmer LS-50B spectrometer with excitation at 370 nm, slit width of 10 nm, and scan speed of 500 nm/min. For each sample, 7 consecutive scans were obtained and the maximum fluorescence values from each scan were averaged and plotted as ANS binding in arbitrary units of fluorescence [44].
2.6. Gel electrophoresis and Western blotting
Peptide aggregation was assessed using the Blue Native PAGE (BN-PAGE) technique [45]. Briefly, samples were electrophoresed in a 4–13% gradient polyacrylamide gel, in the absence of SDS, with a running buffer containing 50 mM tricine, 7.5 mM imidazole, and 0.02% Coomassie blue G-250 (Sigma) and an anode buffer containing 25 mM imidazole. Molecular mass was determined using the Amersham High Molecular Weight Calibration Kit for Native Electrophoresis (GE Healthcare Life Sciences, Piscataway, NJ) in combination with the low molecular weight proteins insulin and soybean trypsin inhibitor. Aggregation was also assessed under denaturing conditions using 16.5% tris-tricine SDS gels with a running buffer composed of 100 mM Tris-HCl, 100 mM Tricine, and 0.1% SDS with 200 mM Tris-HCl used as the anode buffer. Following electrophoretic separation, proteins were electrotransferred to polyvinylidene fluoride (PVDF) membranes (0.45 μm pore size; Immobilon, Millipore, Billerica, MA) at 400 mA for 2.5 h using 10 mM 3-cyclohexamino-1-propanesulfonic acid (CAPS, Sigma) buffer, pH 11.0, containing 10% (v/v) methanol. The membranes were blocked with 5% nonfat milk in Tris-buffered saline containing 0.1% Tween 20 (TBST) and incubated overnight at 4°C with a custom-made rabbit polyclonal antibody specifically recognizing the C-terminal portion of the ABri peptide (Ab 338, 1:5,000) [14, 46, 47]. Membranes were immunoreacted with horseradish peroxidase (HRP)-labeled F(ab′)2 anti-rabbit IgG (1:5,000; GE) and developed by enhanced chemiluminescence (ECL) using the SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, Waltham, MA, USA).
2.7. Cell culture
Human neuroblastoma cells (SH-SY5Y) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in DMEM medium (Mediatech, Manassas, VA) with 10% fetal bovine serum (FBS).
2.8. Cell death assays
The extent of apoptosis induced by the ABri and Bri1-23 synthetic derivatives was evaluated by quantitation of DNA-histone complex formation resulting from DNA fragmentation using the Cell Death ELISAplus kit (Roche Applied Science, Indianapolis, IN) as previously described [40, 43, 48]. SH-SY5Y cells were seeded at a density of 2 x 104 cells/well on 24-well plates and allowed to attach for 1 day prior to the addition of 50 μM peptide in DMEM, 0% FBS. Following incubation for 4 to 24 hours, plates were centrifuged for 10 minutes at 1,000 RPM (Beckman J-6B, Beckman Instruments, Fullerton, CA) to collect detached cells. Supernatants were saved for the analyses of lactate dehydrogenase (LDH) release (see below), and cells were lysed for evaluation of fragmented DNA-histone complexes (mono- and oligo-nucleosomes) following the manufacturer’s instructions. For LDH quantitation, the supernatants from the peptide-treated cultures were further centrifuged at 14,000 RPM for 5 minutes to pellet any remaining cell debris, and assayed with the Cytotoxicity Detection Kit (Roche Applied Science) per the manufacturer’s instructions.
2.9. Immunocytochemical evaluation of cytochrome c subcellular localization and mitochondrial membrane potential
SH-SY5Y cells were seeded on 12 mm poly-D-lysine coated glass coverslips (BD Biosciences, Franklin Lakes, NJ) at a density of 7 x 104 cells/coverslip and allowed to attach for 1 day prior to treatment with 50 μM ABri pE/E, Bri1-23 pE/E, or Aβ42 for 4–16 hours. After washing once with cold PBS, cells were fixed with 4% paraformaldehyde and blocked with 20 mg/ml BSA in PBS containing 0.3% Triton X-100. Cells were subsequently incubated with mouse monoclonal anti-cyt c antibody (BD Biosciences; 1:200 in PBS containing 5 mg/ml BSA, 2h at RT) followed by Alexa Fluor 488-conjugated anti-mouse IgG (Life Technologies, Carlsbad, CA; 1:200 in PBS with 5 mg/ml BSA, 1h at RT), and nuclei were counterstained TO-PRO-3 iodide (Life Technologies; 1:1,000 in PBS, 10 minutes at RT) as previously described. To examine mitochondrial localization in conjunction with changes in the membrane potential of the organelle, cells–after peptide treatment–were washed once with warm PBS and incubated for 30 minutes with 1.5 μM Mitrotracker Red CM-H2X Ros (Life Technologies), followed by cyt c immunostaining as above. All images were acquired using a Zeiss LSM 510 confocal microscope and analyzed using Image J (NIH, Bethesda, MD; http://rsbweb.nih.gov).
2.10. Detection of reactive oxygen species
SH-SY5Y cells were seeded at a density of 104 cells/well on 96-well plates and challenged with 50 μM ABri pE/E for 4 hours. The cells were subsequently incubated at 37°C with 5μM CellROX Deep Red (Life Technologies) and 0.2 μg/ml Hoechst Stain (Immunochemistry Technologies, Bloomington, MN) followed by fixation in 4% paraformaldehyde. Images were acquired using a Nikon Eclipse Ti microscope and analyzed using Image J (NIH).
2.11. Caspase-9 activation and inhibition assays
Caspase-9 activation was measured using the Caspase-Glo 9 luminescent assay (Promega, Madison, WI). Cells were plated at a density of 104 cells/well in white 96-well plates with clear bottoms followed by incubation with freshly solubilized ABri pE at a concentration of 50 μM in DMEM with no FBS for 0–16 h. Caspase-Glo reagent was added to the cells to promote cell lysis, followed by active caspase cleavage of the substrate and generation of a luminescent signal produced by a luciferase reaction. After 40 minute incubation, the signal, proportional to the amount of caspase activity present, was evaluated on a plate-reading luminometer (Tecan Freedom Evo 150, Tecan, Männedorf, Switzerland). To inhibit nonspecific background activity, the proteasome inhibitor MG-13 was added to the Caspase-Glo reagent prior to the experiment as indicated by the manufacturer.
Caspase participation was confirmed through the use of specific caspase-9 inhibitor as well as pan-caspase inhibitors, as described previously [49]. SH-SY5Y cells were seeded at a density of 2 x 104 cells/well on 24-well plates and allowed to attach for 1 day. Cells were then challenged with ABri pE (50 μM) or the well-studied apoptosis inducer staurosporine (Enzo Life Sciences, Farmingdale, NY; 1 μM) in DMEM, 0% FBS for 8 or 24 hours in the presence or absence of 100 μM caspase-9 inhibitor Z-LEHD-FMK (R&D Systems, Minneapolis, MN) or pan-caspase inhibitor Z-VAD(OMe)-FMK (Enzo Life Sciences). In all cases, cells were then subjected to Cell Death ELISA as above.
2.12. Statistical analysis
ANOVA for comparison of multiple groups with Tukey post hoc tests was performed using GraphPad Prism (GraphPad, La Jolla, CA). Values of P ≤ 0.05 were considered significant.
3. Results
3.1 Structural analysis of ABri and Bri1-23 derivatives
HFIP pre-treatment is known to improve peptide solubilization while disrupting pre-existing β-sheet-rich structures with the potential to seed and enhance the process of aggregation. Both parameters were evaluated by CD spectroscopy to test the influence of the stop-to-arginine mutation and of the existence of pE post-translational modification in the furin-generated BRI2 C-terminal fragments ABri and Bri1-23. As indicated in Figure 1A, ABri pE had a different behavior from the rest of the peptides, showing poor solubility (low CD signal) and resistance to adopt a stable α-helical structure even after the standard 3 days incubation in HFIP, requiring a minimum of 7 days pretreatment (note the increase of negative signals at 208 and 222 nm as the incubation time progressed in the left panel). Conversely, ABri E, Bri1-23 pE and Bri1-23 E all exhibited similar behavior; after 3 days in HFIP, they generated strong CD signals and reached α-helical structures that remained unchanged with further incubation time (right panel).
Figure 1. Circular dichroism spectroscopy of ABri pE/E and Bri1-23 pE/E.

Changes in secondary structure were monitored recording the CD spectra after incubation in HFIP for 3–8 days (room temperature; peptide concentrations of 1 mg/ml) (A) and incubation for up to 48 hours in 10 mM PO4 buffer containing 150 mM NaF (37°C; peptide concentrations of 50 μM) (B). In all cases, data represent the mean of 15 scans after subtraction of background readings of the respective buffer blanks.
Structural rearrangements in the pretreated peptides generated by their solubilization in aqueous buffer containing physiologic salt concentration were monitored for 2 days while recording changes in the CD spectra. As illustrated in Figure 1B, ABri pE and ABri E scans (top panels) were compatible with β-sheet conformations containing some random structures (minimum centered at ~210 nm instead of the classic 218 nm of pure β-structures). In addition, ABri pE was less stable than the ABri E homologue; the CD signal faded with time, indicative of the peptide falling out of solution (top left panel). Notably, Bri1-23 pE and Bri1-23 E exhibited a primarily random coil conformation (minima at 198 nm) that remained unchanged throughout the length of the experiments.
The fibrillization kinetics of the ABri and Bri1-23 with and without N-terminal pE were analyzed using thioflavin T, a dye that displays enhanced fluorescence upon binding fibrillar and protofibrillar amyloid conformations [40, 42]. In accordance with their high β-sheet content in physiologic salt concentrations, both ABri pE and ABri E showed thioflavin T fluorescence values that increased rapidly and reached a plateau after only 2 hours (Figure 2A). ABri pE thioflavin T fluorescence remained significantly higher than that of ABri E, a trend that continued for the 24 hour duration of the experiment. In agreement with their unstructured conformations shown by the CD data, both Bri1-23 pE and Bri1-23 E displayed negligible binding to Thioflavin T during the first 24 hours of incubation, a trend that remained unaltered when followed up to 5 days (data not shown). To further evaluate the structural differences between ABri and Bri1-23 containing either pE or E at their N-terminus, we examined their binding to the fluorescent molecular probe ANS (Figure 2B) [50]. When in the presence of hydrophobic regions, ANS produces an increase in fluorescence intensity and a blue shift in the emission maximum from ~520nm to ~490nm. As indicated in Figure 2B (left panel), ABri pE displayed a rapid and robust increase in ANS binding reaching a fluorescence intensity plateau in about 2 hours, with kinetics comparable to that observed with thioflavin T. In comparison, ABri E bound ANS to a lesser degree, whereas Bri1-23 pE and E did not show significant increase in fluorescence or blue shift to the emission maximum upon ANS binding for up to 24 hours, a trend that persisted with longer incubation times (data not shown). Figure 2B (right panel) illustrates a snap shot of the comparative blue shift in the fluorescence maximum observed after 24 hour peptide aggregation for all the peptides tested.
Figure 2. Aggregation/fibrillization of ABri pE/E and Bri1-23 pE/E.

(A) Oligomerization / fibrillization of ABri pE/E and Bri 1–23 pE/E was assessed by fluorescence evaluation of thioflavin T binding to 50 μM peptide in 1X PBS over 24 hours. (B) Exposure of hydrophobic regions of the molecule was evaluated by assessing the binding of the peptides to ANS. Left panel: Fluorescence evaluation in arbitrary units (A.U); graph illustrates mean ± SEM of three independent experiments after subtraction of blank levels. Right panel: representative scan illustrating ANS binding curve for ABri pE, ABri E, and Bri 1–23 pE/E at 24 hours and highlighting the blue shift of the maximum fluorescence values for ABri pE/E homologues. (C) Blue Native-PAGE. Peptides were incubated at 20 μM in 1X HBSS at 37°C for up to 24 hours and aliquots at different time-points were separated in a 4–13% Blue Native gel followed by Western blot analysis. (D) SDS-PAGE. Peptides were incubated at 20 μM in 1X HBSS at 37°C for up to 24 hours and samples at different time-points were separated in a 5–20% Tris-tricine SDS gel followed by Western blot analysis. All blots were probed with the anti-ABri 338 antibody (1:5,000).
The rapid and distinct aggregation/fibrillization of ABri pE and ABri E was further visualized by Western blot (WB) as illustrated in Figure 2C. Following 0–24 hour aggregation in aqueous-based buffer containing physiologic salt concentrations, ABri pE and ABri E were subjected to non-denaturing WB using BN-PAGE [45, 51, 52]. ABri pE monomers polymerized rapidly with the appearance of high molecular weight oligomers (~ 200 kD–700 kD) after only 30 minutes. These oligomers continued to increase in size over time until 24 hours, the longest incubation time studied, at which point very little peptide entered the gel. ABri E monomers aggregated less aggressively to form intermediate molecular weight oligomers after 30 minutes, followed by further polymerization to form a mixture of intermediate and high molecular weight oligomers that persisted for the 24 hours tested. In contrast to ABri pE monomers, which aggregated extremely fast and were largely nonexistent after the initial time point, monomers of ABri E remained evident throughout the 24 hours of the experiment. Electrophoresis under denaturing conditions (Figure 2D) confirmed that both ABri pE and ABri E aggregated rapidly, albeit SDS-resistant ABri pE oligomers reached a higher molecular mass faster than those of ABri E. It is also important to note that the oligomeric species present under native conditions had much higher molecular masses than those seen under denaturing conditions, indicating that most of these aggregates were not SDS-resistant. Taken together, the enhanced binding of thioflavin T and ANS by ABri peptides compared to Bri1-23 demonstrates that the structural changes induced by the stop to arginine mutation originate a peptide more prone to aggregation/fibrillization and amplify the β-sheet content and hydrophobicity of the molecule. Also of note, the presence of an N-terminal pyroglutamate on ABri exacerbates these pro-amyloidogenic structural properties.
3.2 ABri pE-induced mitochondrial dysfunction, oxidative stress, and apoptosis in neuronal cells
DNA fragmentation, an event indicative of apoptosis, was evaluated by Cell Death ELISA in SH-SY5Y neuronal cells following incubation with the peptide homologues at 50 μM concentration, a dose in the lower range of those typically employed (25–300 μM) for the assessment of amyloid toxicity [38, 39, 53–57]. Although 20 μM was the lowest concentration of ABri pE that exhibited a measurable – albeit modest– effect in Cell Death ELISA in our dose-response experimental paradigm (not shown), a 50 μM concentration was selected for all the studies described below based on the well-defined 2.5 fold-change observed over untreated controls. It is important to emphasize that the actual levels of ABri present in vivo in parenchymal and CAA FBD lesions still remain undefined– as is also the case for the widely studied Aβ in AD–although it is clear that the local tissue concentrations are by far much higher than the physiological levels of their soluble counterparts in biological fluids. Discrepancies based on the different areas of affected brains, genetic variability among patients, years of disease progression, and differences in the extraction procedures as well as in the biochemical analysis of the different deposited species contribute to obscure the problem. Among the few quantitative data available, the amount of Aβ retrieved from AD brain deposits could reach an impressive ratio of 140 μg per gram of tissue depending on the brain area selected for analysis [58]. The complexity of the problem is amplified by the uncertainty about the in vivo ratio among the broad spectrum of oligomeric species identified to date and their actual relevance to human disease [11], an issue poorly defined in AD and basically unexplored in FBD.
Figure 3A illustrates that ABri pE induced nucleosome formation after only 4 hours of peptide challenge, while ABri E was not neurotoxic in the same time frame. The level of ABri pE-induced apoptosis increased steadily over time and after 8 and 24 hours of peptide challenge the level of DNA fragmentation rose to about 2-fold and 2.5-fold compared to control cells that received no peptide treatment, respectively. Cells treated with the same concentration of Aβ42, the primary component of AD parenchymal plaques, exhibited apoptotic levels slightly higher than those of ABri pE-treated cells. Both forms of Bri1-23 did not induce apoptosis even after 24 hours of peptide challenge (Figure 3A, right panel). LDH is a cytosolic enzyme that is released into culture medium following plasma membrane damage, such as during necrotic cell death. None of the ABri or Bri1-23 peptide variants caused an increase in LDH release from the neuronal cells, indicating that the plasma membrane of treated cells remained intact during the time frame of the experiments (Figure 3B) and that neurotoxicity proceeded through an apoptotic mechanism. Cells treated with Aβ42 showed a slight, albeit not statistically significant, increase in LDH release, most likely connected to early stage of secondary necrosis subsequent to the initiation of apoptosis. Overall, these results indicate that the stop-to-Arg mutation in BRI2 associated with FBD, coupled with the N-terminal pE posttranslational modification, renders the ABri peptide neurotoxic via an apoptotic mechanism.
Figure 3. Apoptosis induction by ABri pE.

SH-SY5Y cells were challenged with 50 μM ABri pE/E, Bri 1–23 pE/E, or Aβ42 for 4, 8, or 24 hours. (A) Apoptosis was evaluated by Cell Death ELISA. Results are expressed as a fold of change of nucleosome formation compared to no-peptide controls at the respective time points. (B) The release of lactate dehydrogenate (LDH) from the cells was used as a measure of necrotic cell death. Results are expressed as a fold of change of LDH release compared to no-peptide controls at the respective time points; data represent the mean ± SEM of three independent experiments. ** = p<.01, *** = p<.001.
To further delineate the cell death mechanisms engaged by the peptide variants, cyt c subcellular localization was evaluated by confocal microscopy. Cyt c is a heme protein that is normally present in the mitochondrial intermembrane space; however during apoptosis cyt c is released to the cytosol where it plays a key role in the caspase activation cascade [59]. Figure 4 demonstrates the punctate localization of cyt c immunofluorescence to the mitochondrial chains in untreated control SH-SY5Y cells that is maintained for the 16 hours tested. In accordance with the above Cell Death ELISA results, cells challenged with ABri pE for only 4 hours exhibited diffuse cyt c staining, indicative of its release from the mitochondria to the cytoplasm. This diffuse staining was also apparent in cells treated with ABri pE for longer time periods (8 and 16 hours). In contrast, cells incubated with ABri E maintained almost entirely the mitochondrial cyt c localization consistent with the lack of neurotoxicity associated with this peptide. Similarly, in cells challenged with Bri1-23 pE or Bri1-23 E, cyt c remained confined to the mitochondria as in the control cells. Cells challenged with Aβ42 exhibited cyt c release at all time-points studied, in agreement with previous reports [60–62].
Figure 4. Mitochondrial cytochrome c release.

SH-SY5Y cells were challenged with 50 μM ABri pE/E, Bri 1–23 pE/E, or Aβ42 for 4, 8, or 16 hours. Green fluorescence indicates cyt c immunostaining; blue fluorescence represents nuclear DNA counterstained with To-Pro. Magnification = X63 in all cases.
Changes in the mitochondrial membrane potential induced by the synthetic peptide homologues were illustrated using the mitochondrial marker Mitotracker in conjunction with cyt c immunofluorescence (Figure 5). In untreated control SH-SY5Y cells, cyt c maintained a punctate localization that significantly overlapped with the Mitotracker signal, as illustrated by the merged images. This was also the case following ABri E treatment in agreement with the non-proapoptotic behavior of the peptide. Challenge with ABri pE resulted not only in the release of cyt c but also in poor mitochondrial localization of the MitoTracker stain, indicative of a loss of membrane potential of the organelle. Cells treated with Aβ42 produced a similar diffuse staining pattern for both cyt c and MitoTracker. Loss of mitochondrial membrane integrity is often associated with the generation of reactive oxygen species (ROS) that are key players in neurodegenerative cellular demise [63–66]. In Figure 6, cells treated with ABri pE for only 4 hours showed a high level of oxidative stress, as highlighted by the CellROX fluorogenic probe. Conversely, cells challenged with ABri E did not show evidence of ROS production, as with the untreated cells. The fast generation of ROS, visualized much earlier than other apoptotic markers suggests that this mechanism could be an initiating event in the cell death cascade elicited by ABri. Whether the different elements in this cascade are differentially sensitive to peptide dosages remain to be determined.
Figure 5. Mitochondrial dysfunction induced by ABri pE.

SH-SY5Y cells were challenged with ABri pE/E or Aβ42 for 16 hours followed by staining with MitoTracker Red CM-H2XRos and cyt c immunocytochemistry. Top panel: cyt c fluorescence shown in green; Central panel: oxidized Mitotracker highlighted in red; Bottom panel: merged images. Magnification = X63 in all cases.
Figure 6. Oxidative stress in cells challenged with ABri pE.

SH-SY5Y cells were treated with 50 μM ABri pE/E for 4 hours followed by staining with the CellROX Deep Red fluorogenic probe. Top panel: Hoechst nuclear stain shown in blue; Central panel: oxidized CellROX reagent highlighted in red; Bottom panel: merged images. Magnification = X20 in all cases.
Mitochondrial dysfunction, such as failure to maintain the appropriate membrane potential, is often associated with the intrinsic apoptotic pathway. Cyt c release and caspase-9 activation are important steps in the activation of this cell death program [66–69]. In accordance with the loss of mitochondrial membrane potential and cytochrome c release seen above, SH-SY5Y cells challenged with ABri pE exhibited caspase-9 activation that as early as 2 hours after peptide treatment (Figure 7A), providing additional evidence for the rapid induction of neurotoxicity by this peptide. Conversely, ABri E did not induce caspase-9 activation consistent with the inability of the peptide to induce an apoptotic response in this time-frame. Blocking caspase activation with the specific caspase-9 inhibitor Z-LEHD-FMK or the pan-caspase inhibitor Z-VAD-FMK protected the cells from apoptosis following incubation with ABri pE for 8 or 24 hours (Figure 7B). Staurosporine, a known inducer of neuronal apoptosis and activator of caspase-9, was used as a control [70–72].
Figure 7. Activation of caspase-mediated apoptotic pathways by ABri pE.
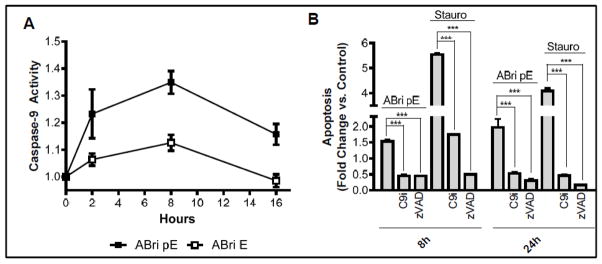
(A) SH-SY5Y cells were treated with 50 μM ABri pE/E and activation of caspase-9 monitored for up to 16 hours using a luminescent assay. Results are expressed as a fold of change compared to no-peptide controls; data represent the mean ± SEM of three independent experiments. (B) SH-SY5Y cells were treated with 50 μM ABri pE or 1 μM of the apoptosis-inducer staurosporine. Apoptosis in the presence or absence of caspase-9 inhibitor Z-LEHD-FMK or the pan-caspase inhibitor Z-VAD-FMK was evaluated by Cell Death ELISA; results are expressed as fold of change compared to no peptide controls in the absence of inhibitors and represent the mean ± SEM of three independent experiments. *** = p<.001.
4. Discussion
The most frequent forms of amyloidosis in humans are localized to the central nervous system (CNS) and typically result in parenchymal/vascular deposits causing cognitive deficits, dementia, stroke, cerebellar and extrapyramidal signs, or a combination of these clinical phenotypes [73–75]. An important argument supporting the importance of amyloid in the pathogenesis of these disorders is provided by rare familial diseases in which the presence of mutated amyloid subunits is consistently associated with early onset of the disease. In general terms, familial mutations often result in highly aggregation-prone species exhibiting increased hydrophobicity, propensity to form β-sheet structures, and alterations in peptide charge [76]. This is also the case with FBD, a disorder that shares many neuropathological and clinical features with AD in spite of the completely different primary structure of the respective amyloid subunits. The results presented herein show that, as a result of the FBD mutation, elongation of the BRI2 cleavage product by 11 amino acids resulted in the exceptionally amyloidogenic ABri molecule bearing high β-sheet content and showing rapid oligomerization/fibrillization to form high molecular mass oligomers with increased hydrophobicity when compared to the wild type Bri1-23 counterpart.
Amyloid subunits are known to sustain a number of post-translational modifications which in turn largely contribute to the heterogeneity of the amyloid deposits. Among the many post translational modifications identified in systemic and cerebral amyloidosis, phosphorylation, isomerization, racemization, oxidation and cyclization occurring in conjunction with proteolytic fragmentation are the most relevant [75]. Perhaps the most frequent post-translational modification is the formation of pE, a form of blocked N-terminus that precludes amino acid sequencing using Edman degradation, and which is believed to confer resistance against N-terminal peptidases delaying and/or preventing the protein removal from the lesions by normal clearance mechanisms thereby contributing to the accumulation of pathogenic species in affected tissues [77–79]. The formation of the pyroglutamyl moiety may result from the post-translational modification of either glutamine or glutamic acid residues. The former is the most common mechanism and involves the nucleophilic attack of the α-amino group on the amidated carboxyl group, a reaction catalyzed at neutral pH by glutaminyl cyclase [21, 22]. Although cyclization of N-terminal glutamate by glutaminyl cyclase is not frequent, it interestingly takes place both in truncated Aβ species, as well as in the ABri and ADan peptides associated with mutations in the BRI2 gene. The formation of pE has been reported to alter the physicochemical properties of amyloid proteins favoring aggregation. In the case of ABri, our results demonstrate that the presence of pE exacerbates the pro-amyloidogenic properties conferred by the FBD mutation, further enhancing the β-sheet content, aggregability, and hydrophobicity of the molecule, consistent with the effect of the posttranslational modification in truncated Aβ species [23, 24, 32].
Enhanced oligomerization/fibrillization typically correlates with amyloid-mediated cytotoxicity. In the case of Alzheimer’s Aβ, numerous reports illustrate that species containing N-terminal pE are more toxic than the unmodified counterparts in cell culture and animal model paradigms [25, 27, 29, 31]. Further supporting the importance of posttranslational modifications for amyloid toxicity, the data presented herein clearly indicate that when coupled with the FBD mutation, posttranslational pE formation produced high molecular mass oligomers that induced neuronal cell death within very short time frames. In addition, our present findings also demonstrate the lack of aggregation potential and consequent inability to induce toxicity of the pE-modified ABri1-23, arguing against the notion that this posttranslational modification might constitute in itself a general pathological mechanism triggering amyloid deposition and toxicity [37].
In contrast to ABri pE, the molecule exhibiting glutamate at the N-terminus, ABri E, aggregated less aggressively, with slower kinetics, and did not exhibit signs of toxicity in our experimental paradigm. Whether this relates to the inability of the peptide to form the specific high molecular mass oligomeric assemblies reached by the pE counterpart remains to be determined. The intermediate hydrophobicity/fibrillization potential of peptide demonstrated in our studies also suggests that ABri E could contribute to the pathogenesis of the deposits in alternative ways. In view of the current evidence suggesting that formation of pE takes place at the site of deposition, both in the brain and systemic organs [46], it is likely that ABri E is the precursor of the more pathogenic pE species. Moreover, it is conceivable that the intermediate conformational behavior of ABri E could be tilted to more amyloidogenic conformations by existing fibrillar structures of ABri pE acting as seeding elements capable of accelerating and/or enhancing the formation of more pathogenic elements, a mechanism resembling the infectivity of prion diseases, which is currently considered as a significant contributor to the mechanisms of AD pathogenesis [80]. In this light, in vitro and in vivo evidence supporting the seeding capabilities of pE-modified peptides as well as consequential propagation via prion-like mechanisms has been presented using truncated forms of Aβ [28, 81] and substantiated by complementary biophysical studies [82].
Although the exact mechanisms of cellular demise in neurodegenerative disorders remain elusive, in the case of AD, a great deal of evidence points to Aβ-mediated activation of intrinsic and extrinsic apoptotic pathways [40, 83] as key elements for the pathogenesis of the disease. Intrinsic apoptosis is typically triggered through intracellular stressors that activate one or more members of the Bcl-2 family of proteins, leading to pore formation in the outer mitochondrial membrane. These pores disrupt mitochondrial membrane potential and cause the release of cyt c, which in turn leads to the formation of the apoptosome with subsequent activation of caspase-9 and further propogation of the caspase activation cascade. It is thought that reactive oxygen species play an important role in the execution of intrinsic apoptosis, either by providing the intracellular stress required for the initiation of the cascade or through direct modification of the protein components that facilitate formation of the apoptosome [66]. The data presented herein demonstrate that ABri pE-mediated toxicity proceeds through an apoptotic mechanism, with levels of DNA fragmentation comparable to those produced by Aβ42. The process was accompanied by mitochondrial compromise with release of cytochrome c, changes in membrane potential, and induction of oxidative stress mechanisms. The presence of downstream caspase-9 activation, in addition to the abrogation of cell death in the presence of specific caspase-9 inhibitors, confirmed the involvement of the intrinsic apoptotic pathway. Whether mitochondrial engagement results from a direct association of ABri with the organelle leading to its dysfunction, as shown for Aβ, remains to be determined [84]. Alternatively, it is conceivable that the initiation of the intrinsic apoptotic path could originate in a direct interaction of ABri-pE with the plasma membrane and the formation of ion-channel like structures, as we previously reported [85], a common feature exhibited also by Aβ and other amyloid subunits. Nevertheless, irrespective of the nature of the initiating events, the similarities in cell death pathways triggered by ABri and Aβ support the concept that different amyloids, despite entirely dissimilar origin and primary sequence, can cause similar pathological processes that ultimately lead to neurodegeneration and dementia.
Overall, our data link the induction of amyloid-mediated neuronal cell apoptosis to the structural stability of the respective subunits. Through the accelerated and enhanced formation of oligomeric/pre-fibrillar assemblies, genetic variants and post-translational modifications exacerbate neuronal dysfunction and accelerate the initiation of cell death mechanisms engaging mitochondrial pathways. The data convey the notion that comparable oligomeric amyloid assemblies, regardless of the primary structure of the amyloid subunit, have the capacity to elicit common pathological mechanisms that ultimately result in cell death. Genetic mutations, although rare, through their accelerated effect and enhanced response constitute unique alternative models to unveil the mechanisms of amyloid-mediated disease pathogenesis.
Highlights.
The FBD mutation originates the highly amyloidogenic molecule ABri
pE posttranslational modification amplifies the amyloidogenic properties of ABri
ABri pE induces neuronal apoptosis while ABri E and Bri1-23 pE/E are not toxic
ABri pE elicits oxidative stress, mitochondrial dysfunction and caspase activation
The mutation and the pE modification are both critical for ABri neurotoxicity
Acknowledgments
This work was supported in part by National Institute of Health [grant numbers NS051715, AG030539, and AG044817] and the Alzheimer’s Association
Abbreviations
- AD
Alzheimer’s disease
- Aβ
amyloid-β
- ANS
8-anilinonapthalene-1-sulfonic acid
- BSA
bovine serum albumin
- BN-PAGE
Blue Native polyacrilamide gel electrophoresis
- CAA
cerebral amyloid angiopathy
- CAPS
3-cyclohexamino-1-propanesulfonic acid
- cyt c
cytochrome c
- CD
circular dichroism
- DMSO
di-methylsulfoxide
- ECL
enhanced chemiluminescence
- FBD
familial British dementia
- FBS
fetal bovine serum
- HFIP
hexafluoroisopropanol
- HRP
horseradish peroxidase
- LDH
lactate dehydrogenase
- PBS
phosphate buffered saline
- pE
pyroglulatmate
- PVDF
polyvinylidene fluoride
- RT
room temperature
- TBST
Tris-buffered saline containing 0.1% Tween 20
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could a3ect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiological Reviews. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 2.Ghiso J, Frangione B. Cerebral amyloidosis, amyloid angiopathy, and their relationship to stroke and dementia. J Alzheimers Dis. 2001;3:65–73. doi: 10.3233/jad-2001-3110. [DOI] [PubMed] [Google Scholar]
- 3.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Querfurth H, LaFerla F. Alzheimer’s disease. The New England journal of medicine. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 5.Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Science translational medicine. 2011;3:77sr71. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 7.Busciglio J, Pelsman A, Wong C, Pigino G, Yuan M, Mori H, Yankner BA. Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down’s syndrome. Neuron. 2002;33:677–688. doi: 10.1016/s0896-6273(02)00604-9. [DOI] [PubMed] [Google Scholar]
- 8.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morawe T, Hiebel C, Kern A, Behl C. Protein homeostasis, aging and Alzheimer’s disease. Molecular neurobiology. 2012;46:41–54. doi: 10.1007/s12035-012-8246-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nature reviews Molecular cell biology. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 11.Benilova I, Karran E, De Strooper B. The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 12.Laurijssens B, Aujard F, Rahman A. Animal models of Alzheimer’s disease and drug development. Drug discovery today Technologies. 2013;10:e319–327. doi: 10.1016/j.ddtec.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 13.Plant GT, Revesz T, Barnard RO, Harding AE, Gautier-Smith PC. Familial cerebral amyloid angiopathy with nonneuritic amyloid plaque formation. Brain : a journal of neurology. 1990;113(Pt 3):721–747. doi: 10.1093/brain/113.3.721. [DOI] [PubMed] [Google Scholar]
- 14.Ghiso J, Vidal R, Rostagno A, Miravalle L, Holton JL, Mead S, Rvsz T, Plant G, Frangione B. Amyloidogenesis in familial British dementia is associated with a genetic defect on chromosome 13. Annals of the New York Academy of Sciences. 2000;920:84. doi: 10.1111/j.1749-6632.2000.tb06908.x. [DOI] [PubMed] [Google Scholar]
- 15.Revesz T, Holton JL, Doshi B, Anderton BH, Scaravilli F, Plant GT. Cytoskeletal pathology in familial cerebral amyloid angiopathy (British type) with non-neuritic amyloid plaque formation. Acta Neuropathologica. 1999;97:170–176. doi: 10.1007/s004010050970. [DOI] [PubMed] [Google Scholar]
- 16.Ghiso J, Rostagno A, Tomidokoro Y, Lashley T, Bojsen-Mller M, Braendgaard H, Plant G, Holton J, Lal R, Revesz T, Frangione B. Genetic alterations of the BRI2 gene: familial British and Danish dementias. Brain pathology. 2006;16:71. doi: 10.1111/j.1750-3639.2006.tb00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vidal R, Frangione B, Rostagno A, Mead S, Rvsz T, Plant G, Ghiso J. A stop-codon mutation in the BRI gene associated with familial British dementia. Nature. 1999;399:776. doi: 10.1038/21637. [DOI] [PubMed] [Google Scholar]
- 18.Rostagno A, Tomidokoro Y, Lashley T, Ng D, Plant G, Holton J, Frangione B, Revesz T, Ghiso J. Chromosome 13 dementias. Cellular and molecular life sciences. 2005;62:1814. doi: 10.1007/s00018-005-5092-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim SH, Wang R, Gordon DJ, Bass J, Steiner DF, Lynn DG, Thinakaran G, Meredith SC, Sisodia SS. Furin mediates enhanced production of fibrillogenic ABri peptides in familial British dementia. Nature neuroscience. 1999;2:984–988. doi: 10.1038/14783. [DOI] [PubMed] [Google Scholar]
- 20.Kim SH, Creemers JW, Chu S, Thinakaran G, Sisodia SS. Proteolytic processing of familial British dementia-associated BRI variants: evidence for enhanced intracellular accumulation of amyloidogenic peptides. The Journal of biological chemistry. 2002;277:1872–1877. doi: 10.1074/jbc.M108739200. [DOI] [PubMed] [Google Scholar]
- 21.Schilling S, Hoffmann T, Manhart S, Hoffmann M, Demuth HU. Glutaminyl cyclases unfold glutamyl cyclase activity under mild acid conditions. FEBS letters. 2004;563:191–196. doi: 10.1016/S0014-5793(04)00300-X. [DOI] [PubMed] [Google Scholar]
- 22.Schilling S, Wasternack C, Demuth HU. Glutaminyl cyclases from animals and plants: a case of functionally convergent protein evolution. Biological chemistry. 2008;389:983–991. doi: 10.1515/BC.2008.111. [DOI] [PubMed] [Google Scholar]
- 23.Saido TC, Yamao-Harigaya W, Iwatsubo T, Kawashima S. Amino- and carboxyl-terminal heterogeneity of beta-amyloid peptides deposited in human brain. Neuroscience letters. 1996;215:173–176. doi: 10.1016/0304-3940(96)12970-0. [DOI] [PubMed] [Google Scholar]
- 24.He W, Barrow CJ. The A beta 3-pyroglutamyl and 11-pyroglutamyl peptides found in senile plaque have greater beta-sheet forming and aggregation propensities in vitro than full-length A beta. Biochemistry. 1999;38:10871–10877. doi: 10.1021/bi990563r. [DOI] [PubMed] [Google Scholar]
- 25.Schlenzig D, Ronicke R, Cynis H, Ludwig HH, Scheel E, Reymann K, Saido T, Hause G, Schilling S, Demuth HU. N-Terminal pyroglutamate formation of Abeta38 and Abeta40 enforces oligomer formation and potency to disrupt hippocampal long-term potentiation. J Neurochem. 2012;121:774–784. doi: 10.1111/j.1471-4159.2012.07707.x. [DOI] [PubMed] [Google Scholar]
- 26.Wirths O, Breyhan H, Cynis H, Schilling S, Demuth HU, Bayer TA. Intraneuronal pyroglutamate-Abeta 3–42 triggers neurodegeneration and lethal neurological deficits in a transgenic mouse model. Acta Neuropathologica. 2009;118:487–496. doi: 10.1007/s00401-009-0557-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Kimpe L, van Haastert ES, Kaminari A, Zwart R, Rutjes H, Hoozemans JJ, Scheper W. Intracellular accumulation of aggregated pyroglutamate amyloid beta: convergence of aging and Abeta pathology at the lysosome. Age (Dordr) 2013;35:673–687. doi: 10.1007/s11357-012-9403-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, Tayler K, Wiltgen B, Hatami A, Ronicke R, Reymann K, Hutter-Paier B, Alexandru A, Jagla W, Graubner S, Glabe CG, Demuth HU, Bloom GS. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-beta. Nature. 2012;485:651–655. doi: 10.1038/nature11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu G, Miller RA, Connolly B, Marcus J, Renger J, Savage MJ. Pyroglutamate-Modified Amyloid-beta Protein Demonstrates Similar Properties in an Alzheimer’s Disease Familial Mutant Knock-In Mouse and Alzheimer’s Disease Brain. Neurodegener Dis. 2013 doi: 10.1159/000353634. [DOI] [PubMed] [Google Scholar]
- 30.Wirths O, Hillmann A, Pradier L, Hartig W, Bayer TA. Oligomeric pyroglutamate amyloid-beta is present in microglia and a subfraction of vessels in patients with Alzheimer’s disease: implications for immunotherapy. J Alzheimers Dis. 2013;35:741–749. doi: 10.3233/JAD-121945. [DOI] [PubMed] [Google Scholar]
- 31.Wirths O, Bethge T, Marcello A, Harmeier A, Jawhar S, Lucassen PJ, Multhaup G, Brody DL, Esparza T, Ingelsson M, Kalimo H, Lannfelt L, Bayer TA. Pyroglutamate Abeta pathology in APP/PS1KI mice, sporadic and familial Alzheimer’s disease cases. Journal of neural transmission (Vienna, Austria : 1996) 2010;117:85–96. doi: 10.1007/s00702-009-0314-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piccini A, Russo C, Gliozzi A, Relini A, Vitali A, Borghi R, Giliberto L, Armirotti A, D’Arrigo C, Bachi A, Cattaneo A, Canale C, Torrassa S, Saido TC, Markesbery W, Gambetti P, Tabaton M. beta-amyloid is different in normal aging and in Alzheimer disease. The Journal of biological chemistry. 2005;280:34186–34192. doi: 10.1074/jbc.M501694200. [DOI] [PubMed] [Google Scholar]
- 33.Frost JL, Le KX, Cynis H, Ekpo E, Kleinschmidt M, Palmour RM, Ervin FR, Snigdha S, Cotman CW, Saido TC, Vassar RJ, St George-Hyslop P, Ikezu T, Schilling S, Demuth HU, Lemere CA. Pyroglutamate-3 amyloid-beta deposition in the brains of humans, non-human primates, canines, and Alzheimer disease-like transgenic mouse models. Am J Pathol. 2013;183:369–381. doi: 10.1016/j.ajpath.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.El Agnaf OM, Sheridan JM, Sidera C, Siligardi G, Hussain R, Haris PI, Austen BM. Effect of the disulfide bridge and the C-terminal extension on the oligomerization of the amyloid peptide ABri implicated in familial British dementia. Biochemistry. 2001;40:3449. doi: 10.1021/bi002287i. [DOI] [PubMed] [Google Scholar]
- 35.Schlenzig D, Manhart S, Cinar Y, Kleinschmidt M, Hause G, Willbold D, Funke S, Schilling S, Demuth H-U. Pyroglutamate formation influences solubility and amyloidogenicity of amyloid peptides. Biochemistry. 2009;48:7072. doi: 10.1021/bi900818a. [DOI] [PubMed] [Google Scholar]
- 36.Srinivasan R, Jones E, Liu K, Ghiso J, Marchant R, Zagorski M. pH-dependent amyloid and protofibril formation by the ABri peptide of familial British dementia. Journal of Molecular Biology. 2003;333:1003. doi: 10.1016/j.jmb.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 37.Saul A, Lashley T, Revesz T, Holton J, Ghiso JA, Coomaraswamy J, Wirths O. Abundant pyroglutamate-modified ABri and ADan peptides in extracellular and vascular amyloid deposits in familial British and Danish dementias. Neurobiol Aging. 2013;34:1416–1425. doi: 10.1016/j.neurobiolaging.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Austen B, el Agnaf O, Nagala S, Patel B, Gunasekera N, Lee M, Lelyveld V. Properties of neurotoxic peptides related to the BRI gene. Biochemical Society transactions. 2002;30:557. doi: 10.1042/bst0300557. [DOI] [PubMed] [Google Scholar]
- 39.El Agnaf OM, Nagala S, Patel BP, Austen BM. Non-fibrillar oligomeric species of the amyloid ABri peptide, implicated in familial British dementia, are more potent at inducing apoptotic cell death than protofibrils or mature fibrils. Journal of Molecular Biology. 2001;310:157. doi: 10.1006/jmbi.2001.4743. [DOI] [PubMed] [Google Scholar]
- 40.Fossati S, Cam J, Meyerson J, Mezhericher E, Romero IA, Couraud PO, Weksler BB, Ghiso J, Rostagno A. Differential activation of mitochondrial apoptotic pathways by vasculotropic amyloid-beta variants in cells composing the cerebral vessel walls. The FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24:229–241. doi: 10.1096/fj.09-139584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stine WB, Jr, Dahlgren KN, Krafft GA, LaDu MJ. In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem. 2003;278:11612–11622. doi: 10.1074/jbc.M210207200. [DOI] [PubMed] [Google Scholar]
- 42.Walsh DM, Hartley DM, Kusumoto Y, Fezoui Y, Condron MM, Lomakin A, Benedek GB, Selkoe DJ, Teplow DB. Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. The Journal of biological chemistry. 1999;274:25945–25952. doi: 10.1074/jbc.274.36.25945. [DOI] [PubMed] [Google Scholar]
- 43.Viana RJ, Nunes AF, Castro RE, Ramalho RM, Meyerson J, Fossati S, Ghiso J, Rostagno A, Rodrigues CM. Tauroursodeoxycholic acid prevents E22Q Alzheimer’s Abeta toxicity in human cerebral endothelial cells. Cellular and molecular life sciences : CMLS. 2009;66:1094–1104. doi: 10.1007/s00018-009-8746-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bolognesi B, Kumita JR, Barros TP, Esbjorner EK, Luheshi LM, Crowther DC, Wilson MR, Dobson CM, Favrin G, Yerbury JJ. ANS binding reveals common features of cytotoxic amyloid species. ACS chemical biology. 2010;5:735–740. doi: 10.1021/cb1001203. [DOI] [PubMed] [Google Scholar]
- 45.Wittig I, Braun HP, Schagger H. Blue native PAGE. Nat Protoc. 2006;1:418–428. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
- 46.Ghiso JA, Holton J, Miravalle L, Calero M, Lashley T, Vidal R, Houlden H, Wood N, Neubert TA, Rostagno A, Plant G, Revesz T, Frangione B. Systemic amyloid deposits in familial British dementia. Journal of Biological Chemistry. 2001;276:43909. doi: 10.1074/jbc.M105956200. [DOI] [PubMed] [Google Scholar]
- 47.Ghiso J, Revesz T, Holton J, Rostagno A, Lashley T, Houlden H, Gibb G, Anderton B, Bek T, Bojsen-Moller M, Wood N, Vidal R, Braendgaard H, Plant G, Frangione B. Chromosome 13 dementia syndromes as models of neurodegeneration. Amyloid : the international journal of experimental and clinical investigation : the official journal of the International Society of Amyloidosis. 2001;8:277–284. doi: 10.3109/13506120108993826. [DOI] [PubMed] [Google Scholar]
- 48.Fossati S, Todd K, Sotolongo K, Ghiso J, Rostagno A. Differential contribution of isoaspartate post-translational modifications to the fibrillization and toxic properties of amyloid beta and the Asn23 Iowa mutation. The Biochemical journal. 2013;456:347–360. doi: 10.1042/BJ20130652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fossati S, Ghiso J, Rostagno A. TRAIL death receptors DR4 and DR5 mediate cerebral microvascular endothelial cell apoptosis induced by oligomeric Alzheimer’s Abeta. Cell death & disease. 2012;3:e321. doi: 10.1038/cddis.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hawe A, Sutter M, Jiskoot W. Extrinsic fluorescent dyes as tools for protein characterization. Pharmaceutical research. 2008;25:1487–1499. doi: 10.1007/s11095-007-9516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wittig I, Beckhaus T, Wumaier Z, Karas M, Schagger H. Mass estimation of native proteins by blue native electrophoresis: principles and practical hints. Molecular & cellular proteomics : MCP. 2010;9:2149–2161. doi: 10.1074/mcp.M900526-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wittig I, Schagger H. Features and applications of blue-native and clear-native electrophoresis. Proteomics. 2008;8:3974–3990. doi: 10.1002/pmic.200800017. [DOI] [PubMed] [Google Scholar]
- 53.Nicholson AM, Wold LA, Walsh DM, Ferreira A. beta-Amyloid carrying the Dutch mutation has diverse effects on calpain-mediated toxicity in hippocampal neurons. Mol Med. 2012;18:178–185. doi: 10.2119/molmed.2011.00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang MC, Lung FW. Neuroprotection of paliperidone on SH-SY5Y cells against beta-amyloid peptide(25–35), N-methyl-4-phenylpyridinium ion, and hydrogen peroxide-induced cell death. Psychopharmacology. 2011;217:397–410. doi: 10.1007/s00213-011-2291-7. [DOI] [PubMed] [Google Scholar]
- 55.Davis J, Van Nostrand WE. Enhanced pathologic properties of Dutch-type mutant amyloid beta-protein. Proc Natl Acad Sci U S A. 1996;93:2996–3000. doi: 10.1073/pnas.93.7.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gibson G, Gunasekera N, Lee M, Lelyveld V, El-Agnaf OMA, Wright A, Austen B. Oligomerization and neurotoxicity of the amyloid ADan peptide implicated in familial Danish dementia. Journal of neurochemistry. 2004;88:281. doi: 10.1046/j.1471-4159.2003.02134.x. [DOI] [PubMed] [Google Scholar]
- 57.Surolia I, Sarkar D, Sinha S. Form and dimensions of aggregates dictate cytotoxicities of Danish dementia peptides. Biochemical and biophysical research communications. 2008;372:62. doi: 10.1016/j.bbrc.2008.04.169. [DOI] [PubMed] [Google Scholar]
- 58.Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 60.Camilleri A, Zarb C, Caruana M, Ostermeier U, Ghio S, Hogen T, Schmidt F, Giese A, Vassallo N. Mitochondrial membrane permeabilisation by amyloid aggregates and protection by polyphenols. Biochim Biophys Acta. 2013;1828:2532–2543. doi: 10.1016/j.bbamem.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 61.Cozzolino M, Ferraro E, Ferri A, Rigamonti D, Quondamatteo F, Ding H, Xu ZS, Ferrari F, Angelini DF, Rotilio G, Cattaneo E, Carri MT, Cecconi F. Apoptosome inactivation rescues proneural and neural cells from neurodegeneration. Cell Death Differ. 2004;11:1179–1191. doi: 10.1038/sj.cdd.4401476. [DOI] [PubMed] [Google Scholar]
- 62.Tamagno E, Parola M, Guglielmotto M, Santoro G, Bardini P, Marra L, Tabaton M, Danni O. Multiple signaling events in amyloid beta-induced, oxidative stress-dependent neuronal apoptosis. Free Radic Biol Med. 2003;35:45–58. doi: 10.1016/s0891-5849(03)00244-2. [DOI] [PubMed] [Google Scholar]
- 63.Chaturvedi RK, Flint Beal M. Mitochondrial diseases of the brain. Free Radic Biol Med. 2013;63:1–29. doi: 10.1016/j.freeradbiomed.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 64.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry Biokhimiia. 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 65.Butterfield DA. Amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer’s disease brain. A review. Free radical research. 2002;36:1307–1313. doi: 10.1080/1071576021000049890. [DOI] [PubMed] [Google Scholar]
- 66.Wu CC, Bratton SB. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid Redox Signal. 2013;19:546–558. doi: 10.1089/ars.2012.4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Caroppi P, Sinibaldi F, Fiorucci L, Santucci R. Apoptosis and human diseases: mitochondrion damage and lethal role of released cytochrome C as proapoptotic protein. Current medicinal chemistry. 2009;16:4058–4065. doi: 10.2174/092986709789378206. [DOI] [PubMed] [Google Scholar]
- 68.Wurstle ML, Laussmann MA, Rehm M. The central role of initiator caspase-9 in apoptosis signal transduction and the regulation of its activation and activity on the apoptosome. Experimental cell research. 2012;318:1213–1220. doi: 10.1016/j.yexcr.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 69.Yuan S, Akey CW. Apoptosome structure, assembly, and procaspase activation. Structure. 2013;21:501–515. doi: 10.1016/j.str.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sabirzhanov B, Stoica BA, Hanscom M, Piao CS, Faden AI. Over-expression of HSP70 attenuates caspase-dependent and caspase-independent pathways and inhibits neuronal apoptosis. J Neurochem. 2012;123:542–554. doi: 10.1111/j.1471-4159.2012.07927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barberan S, McNair K, Iqbal K, Smith NC, Prendergast GC, Stone TW, Cobb SR, Morris BJ. Altered apoptotic responses in neurons lacking RhoB GTPase. Eur J Neurosci. 2011;34:1737–1746. doi: 10.1111/j.1460-9568.2011.07891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Higgins GC, Devenish RJ, Beart PM, Nagley P. Autophagic activity in cortical neurons under acute oxidative stress directly contributes to cell death. Cell Mol Life Sci. 2011;68:3725–3740. doi: 10.1007/s00018-011-0667-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, Masters CL, Merlini G, Saraiva MJ, Sipe JD. Amyloid fibril protein nomenclature. Amyloid: Intl J Exp Clin Invest. 2002;9:197–200. doi: 10.3109/13506120209114823. [DOI] [PubMed] [Google Scholar]
- 74.Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, Masters CL, Merlini G, Saraiva MJ, Sipe JD. Amyloid: toward terminology clarification. Report from the Nomenclature Committee of the International Society of Amyloidosis. Amyloid: Intl J Exp Clin Invest. 2005;12:1–4. doi: 10.1080/13506120500032196. [DOI] [PubMed] [Google Scholar]
- 75.Ghiso J, Frangione B. Amyloidosis and Alzheimer’s disease. Adv Drug Delivery Rev. 2002;54:1539–1551. doi: 10.1016/s0169-409x(02)00149-7. [DOI] [PubMed] [Google Scholar]
- 76.Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–808. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- 77.LeVine Hr. The amyloid hypothesis and the clearance and degradation of Alzheimer’s beta-peptide. J Alzheimers Dis. 2004;6:303–314. doi: 10.3233/jad-2004-6311. [DOI] [PubMed] [Google Scholar]
- 78.Moro ML, Collins MJ, Cappellini E. Alzheimer’s disease and amyloid beta-peptide deposition in the brain: a matter of ‘aging’? 2010;38:539–544. doi: 10.1042/BST0380539. [DOI] [PubMed] [Google Scholar]
- 79.Kuo YM, Webster S, Emmerling MR, De Lima N, Roher AE. Irreversible dimerization/tetramerization and post-translational modifications inhibit proteolytic degradation of A beta peptides of Alzheimer’s disease. Biochim Biophys Acts. 1998;1406:291–298. doi: 10.1016/s0925-4439(98)00014-3. [DOI] [PubMed] [Google Scholar]
- 80.Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 81.Schilling S, Zeitschel U, Hoffmann T, Heiser U, Francke M, Kehlen A, Holzer M, Hutter Paier B, Prokesch M, Windisch M, Jagla W, Schlenzig D, Lindner C, Rudolph T, Reuter G, Cynis H, Montag D, Demuth H-U, Rossner S. Glutaminyl cyclase inhibition attenuates pyroglutamate Abeta and Alzheimer’s disease-like pathology. Nature medicine. 2008;14:1106. doi: 10.1038/nm.1872. [DOI] [PubMed] [Google Scholar]
- 82.Matos JO, Goldblatt G, Jeon J, Chen B, Tatulian SA. Pyroglutamylated amyloid-beta peptide reverses cross beta-sheets by a prion-like mechanism. The journal of physical chemistry B. 2014;118:5637–5643. doi: 10.1021/jp412743s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Knowles J, Rajadas J, Nguyen T-V, Yang T, LeMieux M, Vander Griend L, Ishikawa C, Massa S, Wyss Coray T, Longo F. The p75 neurotrophin receptor promotes amyloid-beta(1–42)-induced neuritic dystrophy in vitro and in vivo. The Journal of neuroscience. 2009;29:10627–10637. doi: 10.1523/JNEUROSCI.0620-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta. 2014;1842:1219–1231. doi: 10.1016/j.bbadis.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Quist A, Doudevski I, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc Natl Acad Sci U S A. 2005;102:10427–10432. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]