

Abstract
Background
Pine moths (Lepidoptera; Bombycoidea; Lasiocampidae: Dendrolimus spp.) are among the most serious insect pests of forests, especially in southern China. Although COI barcodes (a standardized portion of the mitochondrial cytochrome c oxidase subunit I gene) can distinguish some members of this genus, the evolutionary relationships of the three morphospecies Dendrolimus punctatus, D. tabulaeformis and D. spectabilis have remained largely unresolved. We sequenced whole mitochondrial genomes of eight specimens, including D. punctatuswenshanensis. This is an unambiguous subspecies of D. punctatus, and was used as a reference for inferring the relationships of the other two morphospecies of the D. punctatus complex. We constructed phylogenetic trees from this data, including twelve published mitochondrial genomes of other Bombycoidea species, and examined the relationships of the Dendrolimus taxa using these trees and the genomic features of the mitochondrial genome.
Results
The eight fully sequenced mitochondrial genomes from the three morphospecies displayed similar genome structures as other Bombycoidea species in terms of gene content, base composition, level of overall AT-bias and codon usage. However, the Dendrolimus genomes possess a unique feature in the large ribosomal 16S RNA subunits (rrnL), which are more than 60 bp longer than other members of the superfamily and have a higher AC proportion. The eight mitochondrial genomes of Dendrolimus were highly conservative in many aspects, for example with identical stop codons and overlapping regions. But there were many differences in start codons, intergenic spacers, and numbers of mismatched base pairs of tRNA (transfer RNA genes).
Our results, based on phylogenetic trees, genetic distances, species delimitation and genomic features (such as intergenic spacers) of the mitochondrial genome, indicated that D. tabulaeformis is as close to D. punctatus as is D. punctatus wenshanensis, whereas D. spectabilis evolved independently from D. tabulaeformis and D. punctatus. Whole mitochondrial DNA phylogenies showed that D. spectabilis formed a well-supported monophyletic clade, with a clear species boundary separating it from the other congeners examined here. However, D. tabulaeformis often clustered with D. punctatus and with the subspecies D. punctatus wenshanensis. Genetic distance analyses showed that the distance between D. tabulaeformis and D. punctatus is generally less than the intraspecific distance of D. punctatus and its subspecies D. punctatus wenshanensis. In the species delimitation analysis of Poisson Tree Processes (PTP), D. tabulaeformis, D. punctatus and D. punctatus wenshanensis clustered into a putative species separated from D. spectabilis. In comparison with D. spectabilis, D. tabulaeformis and D. punctatus also exhibit a similar structure in intergenic spacer characterization. These different types of evidence suggest that D. tabulaeformis is very close to D. punctatus and its subspecies D. punctatus wenshanensis, and is likely to be another subspecies of D. punctatus.
Conclusions
Whole mitochondrial genomes possess relatively rich genetic information compared with the traditional use of single or multiple genes for phylogenetic purposes. They can be used to better infer phylogenetic relationships and degrees of relatedness of taxonomic groups, at least from the aspect of maternal lineage: caution should be taken due to the maternal-only inheritance of this genome. Our results indicate that D. spectabilis is an independent lineage, while D. tabulaeformis shows an extremely close relationship to D. punctatus.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-1566-5) contains supplementary material, which is available to authorized users.
Keywords: Mitochondrial genome, Dendrolimus punctatus, D. tabulaeformis, D. spectabilis, Subspecies
Background
Insect mitochondrial genomes are usually a closed-circular molecule 14-20 kilo base pairs (kbp) in size [1]. They contain 13 protein-coding genes (PCGs), 22 transfer RNAs (tRNAs), two ribosomal RNAs (rRNAs), and a large non-coding region called the control region (CR, or the AT-rich region) which includes the essential regulatory elements for transcription and replication [1,2]. Due to their unique characters, which include small genome size, easily accessible nature, faster nucleotide substitution rates and the presence of strictly orthologous genes, mitochondrial genomes have been widely used as molecular markers for phylogenetic analyses and the investigation of questions concerning comparative and evolutionary genomics [3-10]. Whole mitochondrial genomes provide not only more general genetic information than shorter sequences of individual genes such as the COI gene, but also sets of genome-level characters, such as the relative position of different genes, structural genomic features and compositional features [1,2,11,12]. Whole mitochondrial genomes are widely used to infer phylogenetic relationships at different taxonomic levels [7,13-18]. Furthermore, mitochondrial genomes often evolve faster through higher mutation rates than nuclear genomes, especially in intergenic regions [19,20], and can be powerful markers for the inference of phylogenetic relationships among closely related taxa [9,21-24].
Dendrolimus species (Lepidoptera, Lasiocampidae) are the most serious phytophagous pests worldwide. Dendrolimus pini causes destructive damage of the Scots pine (Pinus sylvestris) in Europe [25], and Dendrolimus moths cause extensive forest damage in China [26-31]. The occurrence of different species of these pine moths and the existence of natural hybrids leads to heterosis and strong tolerance to pesticides, giving high survival rates and making them difficult to eradicate [27]. The need for improved biological pest control means that it is very important to elucidate the genetic relationships of pine moth species. However, three (D. punctatus [32], D. tabulaeformis [33], D. spectabilis [34]) of the six commonly occurring Dendrolimus species in China cannot be readily discriminated, and their taxonomic status is uncertain. Previous studies revealed that these three can hybridize, and it has been suggested that D. tabulaeformis and D. spectabilis are subspecies of D. punctatus [26,27], although other studies treat all three as different species [29-31]. A recent extensive molecular systematic study, looking at mitochondrial COI and several nuclear genes, used different phylogenetic and DNA barcoding methods to assess this complex. The phylogenetic relationships of D. punctatus, D. tabulaeformis, D. spectabilis could not be fully resolved, although their close relationship was confirmed [35]. This species group thus provides a good model for investigating the utility of the mitochondrial genome in exploring relationships of closely related species groups.
To clarify the taxonomic and phylogenetic relationships of these species, we report here eight complete mitochondrial genomes of the three morphospecies Dendrolimus punctatus, D. tabulaeformis and D. spectabilis. We compare their genome structures in detail and analyze the relationships between them.
Methods
Specimen collection, DNA extraction, PCR amplification, cloning and sequencing
Eight adults of three morphospecies (D. punctatus (2), D. punctatus wenshanensis (2), D. tabulaeformis (2) and D. spectabilis (2)) were collected from five locations (Additional file 1). A taxonomic expert (Professor Chun-sheng Wu, Institute of Zoology, Chinese Academy of Sciences) used traditional morphological approaches to identify these specimens. All specimens were preserved in 95% ethanol and maintained at 4°C for long term storage. DNA was isolated from thoracic muscle tissue using the DNeasy Blood and Tissue kit (QIAGEN) following the manufacturer’s protocol.
Mitochondrial genomes were PCR-amplified and sequenced by Sanger sequencing. Whole genomes of the eight specimens were mainly assembled from 14 overlapping PCR fragments (Additional file 2). Eight pairs of universal primers were selected and used for initial amplifications [3,4]. From these sequences, six species-specific primer sets were designed to amplify remaining sections. A specific fragment of DNA was amplified using universal primers by the following conditions: an initial denaturation at 94°C for 4 min, followed by 40 cycles of denaturation at 94°C for 30 s, annealing at 45–55°C for 30 s, elongation at 72°C for 45 s, and a final extension step of 72°C for 10 min. Species-specific primers were amplified with long PCR reaction conditions: an initial denaturation at 92°C for 2 min, followed by 40 cycles of denaturation at 92°C for 30 s, annealing at 45–55°C for 30 s, elongation at 60°C for 12 min, and a final extension step of 60°C for 20 min. All reactions were performed using Takara LA taq (TaKaRa Co., Dalian, China). PCR fragments containing the control region could not be sequenced directly; they were cloned into the pEASY-T3 Cloning Vector (Beijing TransGen Biotech Co., Ltd., Beijing, China) and then sequenced by M13-F (CGCCAGGGTTTTCCCAGTCACGAC) and M13-R (GAGCGGATAACAATTTCACACAGG) primers.
Sequence assembly and annotation
Raw sequences were checked manually by eye using BioEdit [36] and assembled using the SeqMan program from the laser gene package DNAStar (Madison, USA). The tRNA genes were identified by tRNAscan-SE Search Server v.1.21 [37], with source set for Mito/Chloroplast and Genetic Code for tRNA Isotype Prediction set for Invertebrate Mito. To detect overall tRNA genes, the cove cutoff score could be set lower (≥1). The locations of protein-coding and rRNA genes were determined by comparing homologous sequences with other published Lepidoptera mitochondrial sequences downloaded from GenBank.
Evolutionary relationships based on phylogenetic analysis
There has been much controversy concerning the evolutionary relationships of closely related species of Dendrolimus [26-31]. The three morphospecies D. spectabilis, D. punctatus and D. tabulaeformis can hybridize with each other. Furthermore, D. punctatus and D. tabulaeformis have highly similar diets and morphological features. Different geographical environments and host plants can result in varied morphology, and D. spectabilis and D. tabulaeformis have been treated as subspecies of D. punctatus [26-28]. We assessed their genetic relationships using whole mitochondrial genome data (including genomic features), with D. punctatus wenshanensis (an unambiguous subspecies of D. punctatus) as a baseline reference to infer relationships between the two ambiguous morphospecies (D. tabulaeformis and D. spectabilis) and D. punctatus.
Phylogenetic analyses were performed on 20 complete mitochondrial genomes of Bombycoidea. Eight mitochondrial genomes were newly sequenced in this study (Table 1) and 12 others downloaded from GenBank. Two Geometroidea species were used as outgroups: Biston panterinaria (NC_020004) [38] and Phthonandria atrilineata (NC_010522) [39]. Nucleotide sequences of the 13 protein-coding genes were aligned using a perl script on the basis of protein alignments. The basic algorithm firstly translated DNA sequences into proteins, the protein sequences were then aligned, and finally the DNA sequences were aligned on the protein sequences. This method generated a robust DNA sequence alignment of both distantly and closely related species, as direct DNA sequence alignment may produce unreliable alignments by introducing many indels (gaps). The pipeline can be provided on request (at email zhangab2008@gmail.com or zhangab2008@mail.cnu.edu.cn). The tRNA and rRNA genes were aligned using MUSCLE [40]. Individual genes and aligned partitions were concatenated with SequenceMatrix [41]. Two datasets were generated to infer phylogenetic relationships. One included 13 protein-coding genes (13PCGs), the other 37 genes, including 13 protein coding genes, 22 transfer RNA genes and two ribosomal RNA genes (37gene). In order to improve the reliability of our phylogenetic analyses, several partitions were set in the two datasets. The first dataset was divided into 13 partitions for each protein coding gene, and the second dataset was divided into 15 partitions: each protein-coding gene, the concatenated 22 tRNA genes, and the concatenated rRNA genes. PartitionFinder software was subsequently used to select the optimal scheme and the best-fitting substitution model for each partition set under the Bayesian Information Criterion [42]. Two datasets were analyzed by both Maximum likelihood (ML) and Bayesian Inference (BI) methods [43,44].
Table 1.
List of taxa analyzed in this study
| Family | Species | Acc.number | Reference |
|---|---|---|---|
| Bombycidae | Rondotia menciana | NC_021962 | Kong,W.Q.unpublished |
| Bombyx mori | NC_002355 | Lee et al., unpulished | |
| Bombyx mandarina | NC_003395 | [41] | |
| Sphingidae | Sphinx morio | NC_020780 | [42] |
| Manduca sexta | NC_010266 | [43] | |
| Saturniidae | Samia cynthia ricini | NC_017869 | [44] |
| Eriogyna pyretorum | NC_012727 | [45] | |
| Antheraea pernyi | NC_004622 | [46] | |
| Attacus atlas | NC_021770 | Liu,Y.-Q., unpulished | |
| Antheraea yamamai | NC_012739 | [47] | |
| Saturnia boisduvalii | NC_010613 | [48] | |
| Actias selene | NC_018133 | [49] | |
| Lasiocampidae | Dendrolimus spectabilis02 | KJ_913815 | In this study |
| Dendrolimus spectabilis13 | KJ_913816 | In this study | |
| Dendrolimus punctatus04 | KJ_913813 | In this study | |
| Dendrolimus punctatus05 | KJ_913814 | In this study | |
| Dendrolimus tabulaeformis06 | KJ_913817 | In this study | |
| Dendrolimus tabulaeformis38 | KJ_913818 | In this study | |
| Dendrolimus punctatus wenshanensis03 | KJ_913811 | In this study | |
| Dendrolimus punctatus wenshanensis06 | KJ_913812 | In this study |
The ML analyses were conducted using RaxML [43] with the selected partition scheme (Additional file 3). Because different partition models cannot be set in RaxML, GTRGAMMAI was selected for both the 13PCGs dataset and the 37genes dataset. In RaxML, the rapid hill-climbing algorithm starting from 100 randomized maximum-parsimony trees was used for ML searches. Confidence values of the ML trees were evaluated via bootstrap tests with 1000 iterations.
The BI analyses were conducted using MrBayes ver.3.1 [44] with the best partition scheme (Additional file 3). All BI analyses were conducted with two sets of four Markov chains (one cold and three hot chains). Three criteria were used to verify convergence of BI analyses in order to be confident about the reliability of the results: (1) the average standard deviation of split frequencies was less than 0.01, (2) the potential scale reduction factors (PSRF) [45] were close to 1.0 for all parameters, and (3) the effective sample size value in tracer software exceeded 200 [46]. Each set was sampled with a burnin of 25%. Bayesian posterior probabilities were estimated as the confidence values of the BI tree (BBP).
Evolutionary relationships based on genome analysis and genetic distance calculations
Genome analysis was performed with the 20 mitochondrial sequences of Bombycoidea (Table 1). Nucleotide compositions, codon usage (excluding stop codons) and Relative Synonymous Codon Usage (RSCU) were calculated with MEGA 4.0 [47]. Composition skew analysis used the formulae AT skew = [A-T]/[A + T] and GC skew = [G-C]/[G + C] [48].
Genetic distance is generally considered to be an important aspect of species discrimination. A clear gap in genetic distance between intraspecific and interspecific variation usually indicates a species boundary [49,50,35]. To explore the relationships of D. punctatus, D. tabulaeformis and D. spectabilis, we calculated Kimura's two parameter (K2P) genetic distances among different genes and regions using MEGA 5.0[51] (Figure 1). We calculated pairwise distances among all eight individuals of D. punctatus, D. spectabilis, D. tabulaeformis and D. punctatus wenshanensis, for different genes and regions (Additional file 4). Then average genetic distances (with range) were calculated. Genetic distances of D. punctatus to D. tabulaeformis, and D. punctatus to D. spectabilis, were then compared with the intraspecific distance of D. punctatus and with the genetic distance of D. punctatus and its recognized subspecies D. punctatus wenshanensis.
Figure 1.
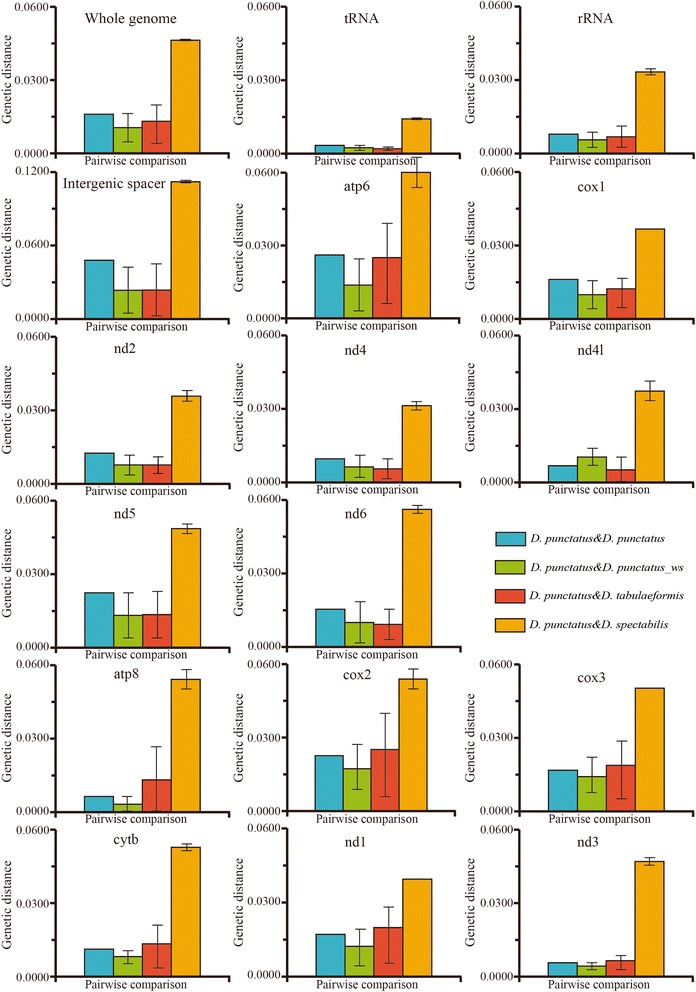
Average K2P genetic distances for different genes and regions of: intraspecific D. punctatus (blue histogram), and genetic distances of D. tabulaeformis to D. punctatus (red), D. punctatus wenshanensis to D. punctatus (green), and D. spectabilis to D. punctatus (yellow) for different genes and regions. Bars show range of values.
A second investigation used the COI gene alone, but from much larger sample sizes ((D. punctatus (195), D. tabulaeformis (6), D. spectabilis (14), D. punctatus wenshanensis (35)) and different geographical populations. K2P genetic distances were calculated with a Perl script [11]. Confidence intervals were calculated using R with t.test function in a stats package ( http://cran.r-project.org/).
Evolutionary relationships based on species delimitation
We applied the Poisson Tree Processes (PTP) model to delimit species on a rooted phylogenetic tree for our two datasets (13PCGs and 37gene) [52]. The PTP program models speciation or coalescent events relative to number of substitutions represented by branch lengths, and uses heuristic algorithms to estimate the most likely classification of branches into population and species-level processes. It does not require ultrametrization of trees and outperforms the commonly used General Mixed Yule Coalescent (GMYC) model when species are closely related and evolutionary distances small [52,53]. The BI phylogenetic trees obtained with MrBayes 3.1 [44], described previously, were used as input trees. PTP analyses were run from Python using the ETE (Python Environment for Tree Exploration) package [54] for tree manipulation and visualization.
Results
Mitochondrial genome organization
Eight complete mitochondrial genomes from Dendrolimus were sequenced, annotated and deposited in GenBank (Table 1). Genome organizations are presented in an additional file (Additional file 5 a-h). All genomes contained the same set of 37 genes (13 protein-coding genes, 22 tRNA genes, and two rRNA genes) and a putative control region [1]. Genome sizes ranged from 15,407 bp to 15,419 bp, and all exhibited similar sequence characteristics. The gene order of these eight pine moths was identical to other ditrysian lepidopterans with the trnM gene location type (trnM-trnI-trnQ) [55-57]. All 13 PCGs start with typical ATN codons, except for cox1 which used CGA. Nine genes (nad2, atp8, atp6, cox3, nad5, nad4l, nad6, cob, nad1) share the same complete termination codon TAA, and four genes use incomplete stop codons (a single T for cox1, cox2 and nad4, TA for nad3) (Additional file 6). Incomplete stop codons are common in lepidopteran mitochondrial genomes and are presumed to be completed via post-transcriptional polyadenylation [58,59].
As for other Lepidoptera, 22 tRNA genes were detected in Dendrolimus mitochondrial genomes, and ranged in size from 64 to 71 bp. Fourteen tRNA genes were coded on the majority strand (J-strand) with eight coded on the minority strand (N-strand). All the tRNA genes have classic cloverleaf secondary structures except for the trnS1 (AGN) gene, where the dihydrouridine (DHU) arm is replaced by unpaired nucleotides. This feature is common to most Lepidopteran mitochondrial genomes except the tortricid Adoxophyes honmai, which has all tRNA genes with complete cloverleaf structures [60]. These aberrant tRNA genes may be modified via RNA-editing mechanisms [61,62]. Many mismatches were found in each of the newly sequenced genomes resulting in some small differences (Additional file 7). There were 16 identical mismatched base pairs and G-U wobble pairs in tRNA structures for all eight samples.
Base composition, AT-skew and codon usage were calculated for the eight newly sequenced genomes and the 12 published Bombycidae mitochondrial genomes (Table 1). The base composition of the J-strand of the Dendrolinus species fell within the range of other Bombycoidea species, and their nucleotide compositions were significantly biased toward A and T (Additional file 8). The nucleotide skew statistics for the entire majority strand of eight individuals indicated a weak A skew and a moderate C skew. In the protein coding genes, both the second and the third positions had negative AT-skew and GC-skew, with the first position having positive AT-skew and GC-skew; overall, PCGs had a negative AT-skew and a positive GC-skew (Additional file 8). The nucleotide skew statistics for the tRNA genes of the eight Dendrolimus individuals indicated a weak A skew and a moderate C skew. The rrnL gene of Dendrolimus is more than 60 bp longer than that of other Bombycoidea species, and the nucleotide skew statistics for rrnL of the eight individuals showed higher A- and C-skews than other Bombycoidea.
Codon usage and RSCU results of our eight moths and the other Bombycoidea species are also given (Additional file 9). Codon usage bias has been detected in currently sequenced mitochondrial genomes. Excluding stop codons, there were 3726 codons in both D. spectabilis and D. tabulaeformis,. One additional codon was detected in D. punctatus. The codon families show very similar behavior among all 20 species considered (Additional file 9). In the eight newly sequenced genomes, eight codon families (Phe, Leu2, Ile, Met, Ser2, Tyr, Asn, Gly) had at least 50 codons per thousand codons (CDs), and two codon families (Leu2, Ile) had at least 100 codons per thousand CDs; other Bombycoidea species had three codon families (Phe, Leu2, Ile) with at least 100 codons per thousand CDs.
RSCU results of Bombycoidea showed that codons exhibit a strong AT-bias in the third codon position, and GC-rich codons are less preferred among the usage of both four- and twofold degenerate codons [63] (Additional file 9). In our eight specimens, many codons were absent, with AGG absent in D. punctatus wenshanensis03, D. punctatus wenshanensis 06, D. punctatus04, D. tabulaeformis06 and D. tabulaeformis38; UGC and AGG were absent in D. spectabilis02 and D. spectabilis13 (Additional file 9).
Evolutionary relationships based on phylogenetic analyses
The phylogenetic analyses conducted in this study yielded the same topological relationships for the 13PCGs and 37gene data matrices in both ML and BI trees (Figure 2). The BI analyses met the three criteria simultaneously to ensure accurate results. Phylogenetic trees showed that in the genus Dendrolimus, the morphospecies D. spectabilis formed a stable clade distinct from other Dendrolimus, and that D. punctatus (including D. punctatus wenshanensis) and D. tabulaeformis formed a separate monophyletic clade. Thus from a phylogenetic point of view, we conclude that D. spectabilis has evolved as an independent lineage distinct from that of D. punctatus and D. tabulaeformis.
Figure 2.
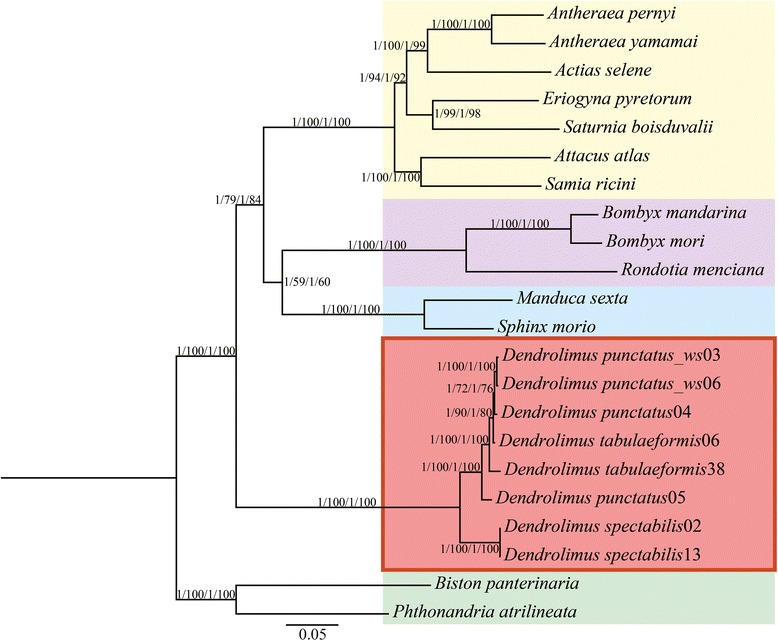
Phylogenetic tree (ML and BI) of Bombycoidea species with outgroups constructed with (1) 13 protein coding genes (13PCGs); (2) 37 genes (13 protein-coding genes + 22 transfer RNA genes + 2 ribosomal RNA genes, 37gene). Numbers above or below branches indicate bootstrap values, in the order of presentation 13PCGs-BI/13PCGs-ML/37gene-BI/37gene-ML. Clades with different colors indicate different families. Specimens within the red box are the newly sequenced genomes of this study.
Evolutionary relationships based on genetic distance: individual genes and regions versus whole mitochondrial genome analysis
In most genes and regions (such as intergenic spacers, tRNA genes, rRNA genes, whole genome, atp6, cox1, nd2, nd4, nd4l, nd5 and nd6), the average K2P distances of D. punctatus to D. tabulaeformis were less than the K2P intraspecies distances of D. punctatus to D. punctatus or the distances of D. punctatus to its subspecies D. punctatus wenshanensis (Additional file 4). But for the atp8, cox2, cox3, cytb, nd1 and nd3 genes, the average genetic distances of D. punctatus to D. tabulaeformis were higher than the average intraspecific distances of D. punctatus and distances of D. punctatus to D. punctatus wenshanensis (Figure 1). However, for most regions and genes, especially for the complete genome, genetic distances between D. punctatus and D. tabulaeformis were less than the intraspecific differences of D. punctatus. These findings suggest that D. punctatus and D. tabulaeformis are extremely similar. The average K2P interspecific distances of D. punctatus and D. spectabilis, based on whole genomes or genes or regions, were higher than the K2P intraspecific distances of D. punctatus and distances of D. punctatus to D. punctatus wenshanensis (Figure 1). These results therefore suggest that D. spectabilis is a relatively distinct lineage from D. punctatus and D. tabulaeformis.
Another dataset, comprising many more specimens of D. punctatus, D. spectabilis and D. tabulaeformis, but only assessed for the barcoding gene region COI, was also examined (Figure 3). The average K2P genetic distance of D. spectabilis and D. punctatus (0.0491) was significantly higher than the intraspecific distance of D. punctatus (0.0126) and the genetic distances of D. punctatus to D. tabulaeformis (0.0132) and to D. punctatus wenshanensis (0.0133). The COI genetic distance between D. punctatus and D. tabulaeformis is similar to the distance between D. punctatus and its subspecies D. punctatus wenshanensis, demonstrating that D. tabulaeformis has, if anything, a closer relationship to D. punctatus than does D. punctatus wenshanensis.
Figure 3.
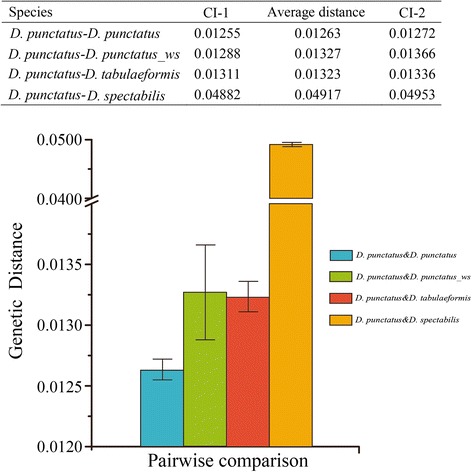
Average intraspecific K2P genetic distances of D. punctatus and genetic distances of D. tabulaeformis to D. punctatus, D. punctatus wenshanensis to D. punctatus and D. spectabilis to D. punctatus for the COI barcoding region of cox1. CI-1 and CI-2 indicate 95% confidence intervals. The length of line in the bar graph shows the confidence intervals. Note that large numbers of specimens were used for this assessment.
Note that these analyses were also repeated with the best fitting nucleotide substitution models (see Additional files 10 and 11), and these yielded the same conclusions as the K2P model [64].
However, there were some differences between the two datasets, the one using just eight samples but sequenced for the entire cox1 gene and the other examining many more samples but only for the COI region of cox1. In the mitochondrial genome dataset, the results from the cox1 gene show that the average genetic distance of D. punctatus to D. tabulaeformis was higher than the average genetic distance of D. punctatus to D. punctatus wenshanensis but less than the intraspecific distance of D. punctatus. In the larger COI dataset, the average interspecific distance of D. punctatus and D. tabulaeformis was higher than average intraspecific distance of D. punctatus but less than the genetic distance of D. punctatus to D. punctatus wenshanensis. There are two possible causes of this discrepancy: firstly, sample sizes are small for the cox1 dataset so that confidence intervals cannot be estimated and compared, and secondly, the cox1 gene in the mitochondrial genome dataset was 1,531 bp long, much longer than the length of the COI barcode sequence examined here (588 bp).
Evolutionary relationships based on species delimitation
The PTP model identified a total of 14 putative species from the 20 Bombycoidea genomes (Figure 4). Except for the eight newly sequenced genomes of Dendrolimus, they all formed independent entities. Two individuals of D. spectabilis clustered together forming a monophyletic group, while the other Dendrolimus species clustered in a separate clade as a single species (Figure 4). Coincident with the results of phylogenetic analyses, D. punctatus and D. tabulaeformis can be considered as one putative species and D. spectabilis as another.
Figure 4.
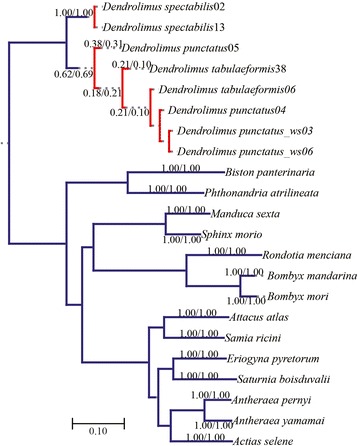
Species-delimitation using the PTP model conducted with (1) 13 protein coding genes (13PCGs) and (2) 37 genes (13 protein-coding genes + 22 transfer RNA genes + 2 ribosomal RNA genes, 37gene). Numbers above or below branches indicate support, in the order of presentation 13PCGs/37gene.
Evolutionary relationships with a mitochondrial genome feature: intergenic spacers
Mitochondrial genomes possess many organizational characteristics. The intergenic regions, for example, do not encode genes and as a result are under more relaxed selection and evolve faster than coding genes [19,20]. Consequently, intergenic regions are expected to be more variable than coding genes, and therefore could be a useful feature when studying the relationships of closely related species.
There were seventeen intergenic spacers in the eight sequenced mitochondrial genomes (Additional file 12). Six were longer than 10 bp: trnM-nd2, trnY-cox1, atp6-cox3, trnA-trnR, trnN-trnS (AGN) and nd4-nd4l. Comparing these six regions of the eight individuals, many differences can be observed (Additional file 13). The intergenic spacer trnM-nd2 was the largest at 58 bp, and is common to most lepidopteran mitochondrial genomes. In this spacer, the two individuals of D. spectabilis had identical nucleotide patterns. Except for one specimen, D. punctatus, D. punctatus wenshanensis and D. tabulaeformis were also identical, but differed by two nucleotides from D. spectabilis. The exception was D. punctatus04, with two different bases. In the other five intergenic spacers, the two D. spectabilis always showed an identical nucleotide distribution, which differed from those of D. punctatus, D. punctatus wenshanensis and D. tabulaeformis. The latter taxa present a mainly similar nucleotide pattern, but D. punctatus04 and D. punctatus05 often showed some differences. In the trnY-cox1 region, D. punctatus05 has 11 nucleotide differences, and for the atp6-cox3, trnA-trnR, trnN-trnS(AGN) and nd4-nd4l regions, D. punctatus04 showed one to three nucleotides difference. D. tabulaeformis always showed an identical nucleotide pattern to D. punctatus wenshanensis. These findings further support the conclusions that D. tabulaeformis has as close a relationship with D. punctatus as has D. punctatus wenshanensis, and that D. spectabilis is distinct.
Conclusions and Discussion
In this study, whole mitochondrial genomes were sequenced using conventional Sanger sequencing with the Primer-Walking method being applied to long fragment sequencing. Sequences were corrected manually to improve the accuracy of sequence determination, and assembled using overlap regions to obtain full-length mitochondrial genomes. Even though earlier laboratory hybridization studies suggested that both the current morphospecies D. spectabilis and D. tabulaeformis are subspecies of D. punctatus [26,27], there is as yet no consensus on the exact relationships of the three taxa [29-31]. After assessing the whole mitochondrial genome data in multiple ways, we find that D. spectabilis forms an independent lineage from D. punctatus, while D. tabulaeformis has a very close relationship with D. punctatus. Like D. punctatus wenshanensis, D. tabulaeformis is likely to be a subspecies of D. punctatus, and this conclusion is also supported by an earlier DNA barcoding based study [35].
Geographically, D. spectabilis is distributed in northern China, and its range does not overlap with D. punctatus (central and southern China). Both genetic and distributional data strongly suggest that D. spectabilis has evolved as a separate lineage or species, perhaps recently. In contrast, D. tabulaeformis is mostly distributed in central China and geographically has not totally separated from D. punctatus. The eight specimens that formed the basis of this study were selected from different locations (Additional file 1): two individuals of D. punctatus from Baise, Guangxi, two D. punctatus wenshanensis from Wenshan Shilin, Yunnan, two D. tabulaeformis from Chengde, Hebei, D. spectabilis02 from Tongliaokulun, Neimenggu and D. spectabilis13 from Taian, Shandong. D. punctatus, D. punctatus wenshanensis and D. tabulaeformis came from different sampling sites, but still have very high sequence similarity, and genetically cannot be unambiguously distinguished. However, the two individuals of D. spectabilis, although from different sampling sites, showed a high degree of sequence identity to one another and significant differences from D. punctatus, D. punctatus wenshanensis and D. tabulaeformis.
The main goal of this study was to explore the phylogenetic relationships of closely related species in the genus Dendrolimus. The mitochondrial DNA genome was selected because of its relatively faster rate of evolution compared with nuclear markers. However, we note that our study does have several limitations. Firstly, the sample size of each species is relatively small compared with studies where a few molecular markers but many specimens are used. We only studied the whole mitochondrial genome from two specimens of each taxon due to high sequencing costs. An increased sample size would be desirable in future studies. However, we did analyse many more individuals using the COI barcoding region alone, coming to similar conclusions. Secondly, the mitochondrial genome only represents the evolution of maternal lineages, and misleading conclusions may be drawn when there are inconsistencies between mitochondrial and nuclear genealogy. Therefore, the introduction of many, not just one or two, nuclear markers in future studies is also highly desirable. Furthermore, with the development of next-generation sequencing technology, more and more mitochondrial genomes and transcriptomes are being examined to infer phylogenetic relationships [65-67]. We plan to include nuclear markers, including transcriptome analysis, in a future study of this taxon group.
Acknowledgments
This research was supported by China National Funds for Distinguished Young Scientists (to Zhang, Grant No. 31425023), by the Natural Science Foundation of China (to Zhang, Grant No. 31071963 and 31272340), by a Funding Project for Academic Human Resources Development in Institutions of Higher Learning Under the Jurisdiction of Beijing Municipality (to Zhang, Grant No. PHR201107120), by the National High-tech Research and Development Project (863) of China (2012AA021601), the Program for Changjiang Scholars and Innovative Research Team in University (IRT13081), and a Science and Technology Foundation Project (2012FY110803).
Additional files
Sample localities of Dendrolinus in China, geographical coordinates and altitudes of collecting sites.
Mitochondrial DNA regions and their primers used in the present study.
Partition scheme used in the present study.
Kimura 2-parameter distance measures for whole mitochondrial genomes and different components.
(a). Annotation of the mitochondrial genome of D. spectabilis 02; (b). Annotation of the mitochondrial genome of D. spectabilis 13; (c). Annotation of the mitochondrial genome of D. punctatus 04; (d). Annotation of the mitochondrial genome of D. punctatus 05; (e). Annotation of the mitochondrial genome of D. punctatus wenshanensis 03; (f). Annotation of the mitochondrial genome of D. punctatus wenshanensis 06; (g). Annotation of the mitochondrial genome of D.tabulaeformis 06; (h). Annotation of the mitochondrial genome of D.tabulaeformis 38.
Start codons and stop codons of the eight sequenced individuals.
Numbers of mismatched base pairs in the eight sequenced genomes.
Gene content of our eight sequenced specimens and other published Bombycoidea mitochondrial genomes.
(a). Codon usage of eight Dendrolimus mitochondrial genomes. Numbers above the column refer to the number of codons. The total number of codons of each individual is given. CDspT stands for codons per thousand codons. Codon Families are provided on the x axis. (b). The relative synonymous codon usage (RSCU) of eight Dendrolimus mitochondrial genomes. Codon Families are provided on the x axis. Codons that are absent in the mitochondrial genomes are marked at the top of columns. (c). Codon usage of Bombycoidea mitochondrial genomes that are published in GenBank. Numbers above the column refer to the number of codons. The total number of codons of each individual is given. CDspT stands for codons per thousand codons. Codon Families are provided on the x axis. (d). Codon usage of Bombycoidea mitochondrial genomes that are published in GenBank. Codon Families are provided on the x axis. Codons that are absent in the mitochondrial genomes are marked at the top of columns.
Genetic distance used HKY85 model for whole mitochondrial genomes and different components.
Genetic distance used selected best-fitting GTR model measures for the COI barcoding region of the additional dataset.
Intergenic spacer locations and nucleotides of eight sequenced pine moths.
Six intergenic spacers in the eight sequenced mitochondrial genomes. The locations of the intergenic spacers are indicated on the top of the figure. The left side of figure is the abbreviated species name. Spectabilis02 stands for Dendrolimus spectabilis02, Spectabilis13 stands for Dendrolimus spectabilis13, Punctatus04 stands for Dendrolimus punctatus04, Punctatus05 stands for Dendrolimus punctatus05, Punctatus_ws03 stands for Dendrolimus punctatus wenshanensis03, Punctatus_ws06 stands for Dendrolimus punctatus wenshanensis06, Tabulaeformis06 stands for Dendrolimus tabulaeformis06, Tabulaeformis38 stands for Dendrolimus tabulaeformis38. Different colors represent different deoxynucleotides. Green represents adenine, purple guanine, blue cytosine and orange thymine. Dashes (-) indicate no nucleotide.
Footnotes
Competing interests
The authors declare that they have no conflict of interests.
Authors' contributions
ABZ conceived the study, JQ performed most of the data analyses, conducted examination of initial annotation and re-annotation, JQ, ABZ, YZZ and RDW drafted and finalized the manuscript. JQ carried out most of the experiments (including PCR, sequence check and assembly) and initial annotation; ABZ initiated, led the research, and supervised all laboratory work; YZZ, XZ, XBK, SJW participated in data analyses. All authors read and approved the final manuscript.
Contributor Information
Jie Qin, Email: qinjie0912@163.com.
Yanzhou Zhang, Email: zhangyz@ioz.ac.cn.
Xin Zhou, Email: xinzhou@genomics.cn.
Xiangbo Kong, Email: kongxbo@hotmail.com.
Shujun Wei, Email: shujun268@163.com.
Robert D Ward, Email: Bob.Ward@csiro.au.
Ai-bing Zhang, Email: zhangab2008@mail.cnu.edu.cn.
Reference
- 1.Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27(8):1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taanman J-W. The mitochondrial genome: structure, transcription, translation and replication. Biochimica et Biophysica Acta (BBA)-BioenergeticS. 1999;1410(2):103–123. doi: 10.1016/S0005-2728(98)00161-3. [DOI] [PubMed] [Google Scholar]
- 3.Simon C, Buckley TR, Frati F, Stewart JB, Beckenbach AT: Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annual Rev Ecol Evol Systemat. 2006;37:545-579.
- 4.Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann Entomol Soc Am. 1994;87(6):651–701. doi: 10.1093/aesa/87.6.651. [DOI] [Google Scholar]
- 5.Salvato P, Simonato M, Battisti A, Negrisolo E. The complete mitochondrial genome of the bag-shelter moth Ochrogaster lunifer (Lepidoptera, Notodontidae) BMC Genomics. 2008;9(1):331. doi: 10.1186/1471-2164-9-331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reyes A, Gissi C, Pesole G, Saccone C. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol Biol Evol. 1998;15(8):957–966. doi: 10.1093/oxfordjournals.molbev.a026011. [DOI] [PubMed] [Google Scholar]
- 7.Ingman M, Kaessmann H, PaÈaÈbo S, Gyllensten U. Mitochondrial genome variation and the origin of modern humans. Nature. 2000;408(6813):708–713. doi: 10.1038/35047064. [DOI] [PubMed] [Google Scholar]
- 8.Lin C-P, Danforth BN. How do insect nuclear and mitochondrial gene substitution patterns differ? Insights from Bayesian analyses of combined datasets. Mol Phylogenet Evol. 2004;30(3):686–702. doi: 10.1016/S1055-7903(03)00241-0. [DOI] [PubMed] [Google Scholar]
- 9.Gissi C, Iannelli F, Pesole G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity. 2008;101(4):301–320. doi: 10.1038/hdy.2008.62. [DOI] [PubMed] [Google Scholar]
- 10.Elson JL, Lightowlers RN. Mitochondrial DNA clonality in the dock: can surveillance swing the case? Trends Genet. 2006;22(11):603–607. doi: 10.1016/j.tig.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 11.Cameron SL. Insect mitochondrial genomics: implications for evolution and phylogeny. Annu Rev Entomol. 2014;59:95–117. doi: 10.1146/annurev-ento-011613-162007. [DOI] [PubMed] [Google Scholar]
- 12.Clayton DA. Replication of animal mitochondrial DNA. Cell. 1982;28(4):693–705. doi: 10.1016/0092-8674(82)90049-6. [DOI] [PubMed] [Google Scholar]
- 13.Cameron SL, Lambkin CL, Barker SC, Whiting MF. A mitochondrial genome phylogeny of Diptera: whole genome sequence data accurately resolve relationships over broad timescales with high precision. Syst Entomol. 2007;32(1):40–59. doi: 10.1111/j.1365-3113.2006.00355.x. [DOI] [Google Scholar]
- 14.Dowton M, Cameron SL, Austin AD, Whiting MF. Phylogenetic approaches for the analysis of mitochondrial genome sequence data in the Hymenoptera–a lineage with both rapidly and slowly evolving mitochondrial genomes. Mol Phylogenet Evol. 2009;52(2):512–519. doi: 10.1016/j.ympev.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 15.Fenn JD, Song H, Cameron SL, Whiting MF. A preliminary mitochondrial genome phylogeny of Orthoptera (Insecta) and approaches to maximizing phylogenetic signal found within mitochondrial genome data. Mol Phylogenet Evol. 2008;49(1):59–68. doi: 10.1016/j.ympev.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 16.Nardi F, Spinsanti G, Boore JL, Carapelli A, Dallai R, Frati F. Hexapod origins: monophyletic or paraphyletic? Science. 2003;299(5614):1887–1889. doi: 10.1126/science.1078607. [DOI] [PubMed] [Google Scholar]
- 17.Boore JL, Brown WM. Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr Opin Genet Dev. 1998;8(6):668–674. doi: 10.1016/S0959-437X(98)80035-X. [DOI] [PubMed] [Google Scholar]
- 18.Shao R, Barker S. Mitochondrial genomes of parasitic arthropods: implications for studies of population genetics and evolution. Parasitology. 2007;134(02):153–167. doi: 10.1017/S0031182006001429. [DOI] [PubMed] [Google Scholar]
- 19.Burger G, Gray MW, Franz Lang B. Mitochondrial genomes: anything goes. Trends Genet. 2003;19(12):709–716. doi: 10.1016/j.tig.2003.10.012. [DOI] [PubMed] [Google Scholar]
- 20.Ghikas DV, Kouvelis VN, Typas MA. Phylogenetic and biogeographic implications inferred by mitochondrial intergenic region analyses and ITS1-5.8 S-ITS2 of the entomopathogenic fungi Beauveria bassiana and B. brongniartii. BMC Microbiol. 2010;10(1):174. doi: 10.1186/1471-2180-10-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Torriani SF, Penselin D, Knogge W, Felder M, Taudien S, Platzer M, McDonald BA, Brunner PC. Comparative analysis of mitochondrial genomes from closely related Rhynchosporium species reveals extensive intron invasion. Fungal Genet Biol. 2014;62:34–42. doi: 10.1016/j.fgb.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 22.Arctander P. Comparison of a mitochondrial gene and a corresponding nuclear pseudogene. Proc R Soc London, Ser B. 1995;262(1363):13–19. doi: 10.1098/rspb.1995.0170. [DOI] [PubMed] [Google Scholar]
- 23.Krzywinski J, Grushko OG, Besansky NJ. Analysis of the complete mitochondrial DNA from Anopheles funestus: An improved dipteran mitochondrial genome annotation and a temporal dimension of mosquito evolution. Mol Phylogenet Evol. 2006;39(2):417–423. doi: 10.1016/j.ympev.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 24.Lopez JV, Cevario S, O'Brien SJ. Complete Nucleotide Sequences of the Domestic Cat (Felis catus) Mitochondrial Genome and a Transposed mtDNA Tandem Repeat (Numt) in the Nuclear Genome. Genomics. 1996;33(2):229–246. doi: 10.1006/geno.1996.0188. [DOI] [PubMed] [Google Scholar]
- 25.le Mellec A, Michalzik B. Impact of a pine lappet (Dendrolimus pini) mass outbreak on C and N fluxes to the forest floor and soil microbial properties in a Scots pine forest in Germany. Can J Forest Res. 2008;38(7):1829–1841. doi: 10.1139/X08-045. [DOI] [Google Scholar]
- 26.Zhao QS, Wu WB, Lv GP, Yuan X, Li SK. Hybridization among pine moths species. Acta Entomol Sin. 1992;35(1):28–32. [Google Scholar]
- 27.Zhao QS, Wu WB, Lv GP, Chen TF, Lin QY. Hybridization among pine moths species and its genetic rule. Scientia Silvae Sinicae. 1999;35(4):45–50. [Google Scholar]
- 28.Zhang AB, Wang ZJ, Tan SJ, Li DM. Monitoring the masson pine moth, Dendrolimus punctatus Walker Lepidoptera: Lasiocampidae with synthetic sex pheromone-baited traps in Qianshan County. China JPN J Appl Entomol Z. 2003;38(2):177–186. doi: 10.1303/aez.2003.177. [DOI] [Google Scholar]
- 29.Zhang AB, Kong XB, Li DM. DNA finger printing evidence for the phylogenetic relationship of eight species and subspecies of Dendrolimus (Lepidoptera: Lasiocampidae) in China. Acta Entomol Sin. 2004;47(2):236–242. [Google Scholar]
- 30.Zhang AB, Zhang Z, Wang HB, Kong XB. Geographical distribution of Lasiocampidae in China and its relationship with environmental factors. J Beijing Forestry Uni. 2004;26(4):54–60. [Google Scholar]
- 31.Zhang AB, Li DM, Chen J. Geohistories of Dendrolimus punctatus Walker and its host plant pine genus in China. Chinese Bulletin Entomol. 2004;2:146–150. [Google Scholar]
- 32.Walker F, Gray JE. List of the specimens of lepidopterous insects in the collection of the British Museum: order of the Trustees. 1855. [Google Scholar]
- 33.Tsai P, Liu Y. A study of the genus Dendrolimus (Lasiocampidae) of China with descriptions of two new species and one new subspecies. Acta Entomol Sin. 1962;11(3):237–252. [Google Scholar]
- 34.Butler A. Descriptions of new species of Heterocera from Japan, Part 3, Geometries. Ann Mag Nat Hist. 1878;5:392–419. doi: 10.1080/00222937808682350. [DOI] [Google Scholar]
- 35.Dai Q-Y, Gao Q, Wu C-S, Chesters D, Zhu C-D, Zhang A-B. Phylogenetic reconstruction and DNA barcoding for closely related pine moth species (Dendrolimus) in China with multiple gene markers. PLoS One. 2012;7(4) doi: 10.1371/journal.pone.0032544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic acids symposium series. 1999;1999:95–98. [Google Scholar]
- 37.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):0955–0964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang X, Xue D, Han H. The complete mitochondrial genome of Biston panterinaria (Lepidoptera: Geometridae), with phylogenetic utility of mitochondrial genome in the Lepidoptera. Gene. 2013;515(2):349–358. doi: 10.1016/j.gene.2012.11.031. [DOI] [PubMed] [Google Scholar]
- 39.Yang L, Wei Z-J, Hong G-Y, Jiang S-T, Wen L-P. The complete nucleotide sequence of the mitochondrial genome of Phthonandria atrilineata (Lepidoptera: Geometridae) Mol Biol Rep. 2009;36(6):1441–1449. doi: 10.1007/s11033-008-9334-0. [DOI] [PubMed] [Google Scholar]
- 40.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC bioinformatics. 2004;5(1):113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vaidya G, Lohman DJ, Meier R. SequenceMatrix: concatenation software for the fast assembly of multi‐gene datasets with character set and codon information. Cladistics. 2011;27(2):171–180. doi: 10.1111/j.1096-0031.2010.00329.x. [DOI] [PubMed] [Google Scholar]
- 42.Lanfear R, Calcott B, Ho SY, Guindon S. PartitionFinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol Biol Evol. 2012;29(6):1695–1701. doi: 10.1093/molbev/mss020. [DOI] [PubMed] [Google Scholar]
- 43.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 44.Ronquist F, Huelsenbeck JP. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19(12):1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- 45.Gelman A, Rubin DB: Inference from iterative simulation using multiple sequences. Stat Sci 1992;7:457-472
- 46.Rambaut A, Drummond A: Tracer v1. 4. In.; 2007 [http://beast.bio.ed.ac.uk/Tracer].
- 47.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24(8):1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 48.Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 1995;41(3):353–358. doi: 10.1007/BF01215182. [DOI] [PubMed] [Google Scholar]
- 49.Hebert PD, Cywinska A, Ball SL. Biological identifications through DNA barcodes. Proc R Soc London, Ser B. 2003;270(1512):313–321. doi: 10.1098/rspb.2002.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hebert PD, Ratnasingham S, de Waard JR. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc R Soc London, Ser B. 2003;270(Suppl 1):S96–S99. doi: 10.1098/rsbl.2003.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang J, Kapli P, Pavlidis P, Stamatakis A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics. 2013;29(22):2869–2876. doi: 10.1093/bioinformatics/btt499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pons J, Barraclough TG, Gomez-Zurita J, Cardoso A, Duran DP, Hazell S, Kamoun S, Sumlin WD, Vogler AP. Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst Biol. 2006;55(4):595–609. doi: 10.1080/10635150600852011. [DOI] [PubMed] [Google Scholar]
- 54.Huerta-Cepas J, Dopazo J, Gabaldón T. ETE: a python Environment for Tree Exploration. BMC bioinformatics. 2010;11(1):24. doi: 10.1186/1471-2105-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Negrisolo E, Babbucci M, Patarnello T. The mitochondrial genome of the ascalaphid owlfly Libelloides macaronius and comparative evolutionary mitochondriomics of neuropterid insects. BMC Genomics. 2011;12(1):221. doi: 10.1186/1471-2164-12-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cameron SL, Whiting MF. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene. 2008;408(1):112–123. doi: 10.1016/j.gene.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 57.Cao Y-Q, Ma C, Chen J-Y, Yang D-R. The complete mitochondrial genomes of two ghost moths, Thitarodes renzhiensis and Thitarodes yunnanensis: the ancestral gene arrangement in Lepidoptera. BMC Genomics. 2012;13(1):276. doi: 10.1186/1471-2164-13-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. 1981. [DOI] [PubMed] [Google Scholar]
- 59.Ojala D, Merkel C, Gelfand R, Attardi G. The tRNA genes punctuate the reading of genetic information in human mitochondrial DNA. Cell. 1980;22(2):393–403. doi: 10.1016/0092-8674(80)90350-5. [DOI] [PubMed] [Google Scholar]
- 60.Lee E-S, Shin KS, Kim M-S, Park H, Cho S, Kim C-B. The mitochondrial genome of the smaller tea tortrix Adoxophyes honmai (Lepidoptera: Tortricidae) Gene. 2006;373:52–57. doi: 10.1016/j.gene.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 61.Lavrov DV, Brown WM, Boore JL. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus. Proc Natl Acad Sci. 2000;97(25):13738–13742. doi: 10.1073/pnas.250402997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Masta SE, Boore JL. The complete mitochondrial genome sequence of the spider Habronattus oregonensis reveals rearranged and extremely truncated tRNAs. Mol Biol Evol. 2004;21(5):893–902. doi: 10.1093/molbev/msh096. [DOI] [PubMed] [Google Scholar]
- 63.Chai H-N, Du Y-Z, Zhai B-P. Characterization of the complete mitochondrial genomes of Cnaphalocrocis medinalis and Chilo suppressalis (Lepidoptera: Pyralidae) Int J Biol Sci. 2012;8(4):561. doi: 10.7150/ijbs.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16(2):111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 65.Kawahara AY, Breinholt JW. Phylogenomics provides strong evidence for relationships of butterflies and moths. Proc R Soc Lond B Biol Sci. 2014;281(1788):20140970. doi: 10.1098/rspb.2014.0970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Timmermans MJ, Lees DC, Simonsen TJ: Towards a mitogenomic phylogeny of Lepidoptera. Mol Phylogen Evol. 2014;79:169-178. [DOI] [PubMed]
- 67.Bazinet AL, Cummings MP, Mitter KT, Mitter CW. Can RNA-Seq Resolve the Rapid Radiation of Advanced Moths and Butterflies (Hexapoda: Lepidoptera: Apoditrysia)? An Exploratory Study. PLoS One. 2013;8(12) doi: 10.1371/journal.pone.0082615. [DOI] [PMC free article] [PubMed] [Google Scholar]