

Abstract
Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene cause late-onset, autosomal dominant Parkinson's disease (PD). The clinical and neurochemical features of LRRK2-linked PD are similar to idiopathic disease although neuropathology is somewhat heterogeneous. Dominant mutations in LRRK2 precipitate neurodegeneration through a toxic gain-of-function mechanism which can be modeled in transgenic mice overexpressing human LRRK2 variants. A number of LRRK2 transgenic mouse models have been developed that display abnormalities in dopaminergic neurotransmission and alterations in tau metabolism yet without consistently inducing dopaminergic neurodegeneration. To directly explore the impact of mutant LRRK2 on the nigrostriatal dopaminergic pathway, we developed conditional transgenic mice that selectively express human R1441C LRRK2 in dopaminergic neurons from the endogenous murine ROSA26 promoter. The expression of R1441C LRRK2 does not induce the degeneration of substantia nigra dopaminergic neurons or striatal dopamine deficits in mice up to 2 years of age, and fails to precipitate abnormal protein inclusions containing alpha-synuclein, tau, ubiquitin or autophagy markers (LC3 and p62). Furthermore, mice expressing R1441C LRRK2 exhibit normal motor activity and olfactory function with increasing age. Intriguingly, the expression of R1441C LRRK2 induces age-dependent abnormalities of the nuclear envelope in nigral dopaminergic neurons including reduced nuclear circularity and increased invaginations of the nuclear envelope. In addition, R1441C LRRK2 mice display increased neurite complexity of cultured midbrain dopaminergic neurons. Collectively, these novel R1441C LRRK2 conditional transgenic mice reveal altered dopaminergic neuronal morphology with advancing age, and provide a useful tool for exploring the pathogenic mechanisms underlying the R1441C LRRK2 mutation in PD.
Introduction
Parkinson's disease (PD) is a progressive neurodegenerative movement disorder that is characterized by the cardinal symptoms of bradykinesia, muscular rigidity, resting tremor and often postural instability (1, 2). Underlying these motor symptoms is the progressive degeneration of substantia nigra dopaminergic neurons and their axonal projections leading to reduced levels of dopamine in the caudate putamen. Additional neuronal populations also degenerate in PD producing a range of non-motor symptoms including olfactory, cognitive, psychiatric, autonomic and gastrointestinal dysfunction (1, 2). The hallmark neuropathology of PD is characterized by the appearance of intracytoplasmic proteinaceous inclusions, termed Lewy bodies, a major component of which is fibrillar α-synuclein protein (3). Although typically an idiopathic disorder, 5-10% of PD cases are inherited with mutations identified in at least eight genes (4).
Mutations in the leucine-rich repeat kinase 2 (LRRK2, PARK8, OMIM 607060) gene cause late-onset, autosomal dominant PD and represent the most common cause of familial PD (5-8). LRRK2 mutations have also been identified in apparent idiopathic PD cases in certain populations due to the incomplete penance of mutations (5, 9-12), whereas common variation in the LRRK2 gene is associated with increased PD risk (13-15). The clinical, neurochemical and, for the most part, neuropathological spectrum of LRRK2-linked PD is largely indistinguishable from idiopathic PD (16). LRRK2 mutations can give rise to heterogeneous pathology including brainstem-specific or diffuse Lewy bodies, tau-positive neurofibrillary tangles, ubiquitin-positive inclusions, or the distinct absence of proteinaceous inclusions (7, 17-21). However, LRRK2 mutations are predominantly associated with classical Lewy body pathology similar to idiopathic PD (18, 20). The LRRK2 gene encodes a large multi-domain protein belonging to the ROCO protein family consisting of a Ras-of-Complex (ROC) GTPase domain and a C-terminal of ROC (COR) domain, followed by a serine/threonine-directed kinase domain with similarity to the mixed-lineage and receptor-interacting protein kinase families (22, 23). The central catalytic region is surrounded by putative protein-protein interaction domains including N-terminal ankyrin and leucine-rich repeat regions and a C-terminal WD40-like repeat domain. At least seven mutations have been identified that segregate with disease in LRRK2-linked families thus proving their pathogenicity, including N1437H, R1441C, R1441G and R1441H within the ROC domain, Y1699C in the COR domain, and G2019S and I2020T in the kinase domain (22, 23). LRRK2 mutations have been shown to variably alter enzymatic activity including enhancing kinase activity or impairing GTPase activity (22-24). Mutant forms of LRRK2 have been shown to commonly enhance neuronal toxicity in cultures relative to wild-type (WT) LRRK2 through a mechanism that is dependent on intact kinase and GTPase activity (25-29). LRRK2 has also been shown to regulate neuronal process morphology with the overexpression of certain LRRK2 mutants reducing neurite complexity and LRRK2 deletion or silencing enhancing neurite complexity through a pathway that may involve autophagy (30-34). Therefore, mutant LRRK2 impairs the integrity and viability of cultured neuronal cells consistent with a toxic gain-of-function mechanism for these dominant familial mutations.
To explore the pathogenic effects of familial LRRK2 mutations in vivo, a collection of transgenic mice have been generated to model the actions of dominant mutations. Transgenic mice have been developed that express WT, R1441C, R1441G, G2019S or I2020T variants of human or mouse LRRK2 from bacterial artificial chromosome (BAC, LRRK2 gene promoter), inducible (tetracycline-regulatable promoter) or mini-gene (CMV-enhanced PDGFβ or CMV promoters) constructs (35-44). LRRK2 transgenic mice collectively display subtle phenotypes, including motor abnormalities, altered dopaminergic neurotransmission and abnormal processing of tau protein (35, 36, 38). We and others have demonstrated that G2019S LRRK2 expression from a CMV-enhanced PDGFβ promoter in transgenic mice leads to human LRRK2 expression within dopaminergic neurons of the substantia nigra that is sufficient to induce the progressive degeneration of nigral dopaminergic neurons (39, 43), similar to LRRK2-linked and idiopathic PD. The absence of frank neurodegeneration in BAC and inducible LRRK2 transgenic mice could potentially result from insufficient transgene expression in midbrain dopaminergic neurons. In addition to the reported PDGFβ-G2019S LRRK2 mice developed by our laboratory, we also similarly developed PDGFβ-R1441C LRRK2 mice (39). These R1441C LRRK2 mice revealed high-level yet restricted transgene expression confined to the cerebral cortex and cerebellum suggesting that transgene expression from this mini-gene construct is subjected to strong chromosome-position effects. Nevertheless, the PDGFβ-R1441C LRRK2 mice displayed reduced catecholamine levels and the accumulation of autophagic vacuoles in the cerebral cortex, a progressive impairment of locomotor activity presumably resulting from cortical dysfunction, but lacked degeneration of the nigrostriatal dopaminergic pathway due to a lack of transgene expression in the midbrain (39). Since this is the only R1441C LRRK2 transgenic mouse model reported thus far and also exhibits promising neuropathological phenotypes, albeit in extra-nigral brain regions, we sought to develop transgenic mice expressing R1441C LRRK2 in the nigrostriatal dopaminergic pathway to be able to explore the pathogenic effects of this intriguing familial mutation in a neuronal population directly relevant to the pathogenesis of PD. Here, we report the development and phenotypic characterization with age of conditional transgenic mice selectively expressing human R1441C LRRK2 in midbrain dopaminergic neurons.
Results
Development of Conditional Human R1441C LRRK2 Transgenic Mice
To be able to directly explore the pathogenic effects of R1441C LRRK2 on the nigrostriatal dopaminergic pathway, we developed conditional transgenic mice that selectively express human R1441C LRRK2 in midbrain dopaminergic neurons. Conditional transgenic mice were created that express untagged human R1441C LRRK2 from the endogenous murine ROSA26 promoter in a Cre-recombinase-dependent manner; referred to as R26-LRRK2 mice (Fig. 1). A cassette containing a human LRRK2 transgene preceded by a loxP-flanked transcriptional termination sequence (neo-tpA) was targeted to the ROSA26 locus by homologous recombination in embryonic stem (ES) cells (Fig. 1A). In this model, expression of human LRRK2 is under the control of the endogenous murine ROSA26 promoter following Cre-mediated excision of the loxP-flanked neo-tpA cassette. Southern blot analysis was employed to identify correctly targeted ES cells (Fig. 1B). Following blastocyst injection of targeted ES cells (clones K6, K8 and K18), we identified chimeric mice by PCR analysis of genomic DNA that successfully produce germ line transmission of the targeted allele. The resulting heterozygous mice (R26-LRRK2+/-, where + denotes the transgene insertion) were intercrossed to produce homozygous mice (R26-LRRK2+/+) for further analysis. R26-LRRK2+/+ mice are viable and fertile, display normal survival and generally exhibit normal behavior.
Figure 1. Generation of conditional R1441C LRRK2 transgenic mice.
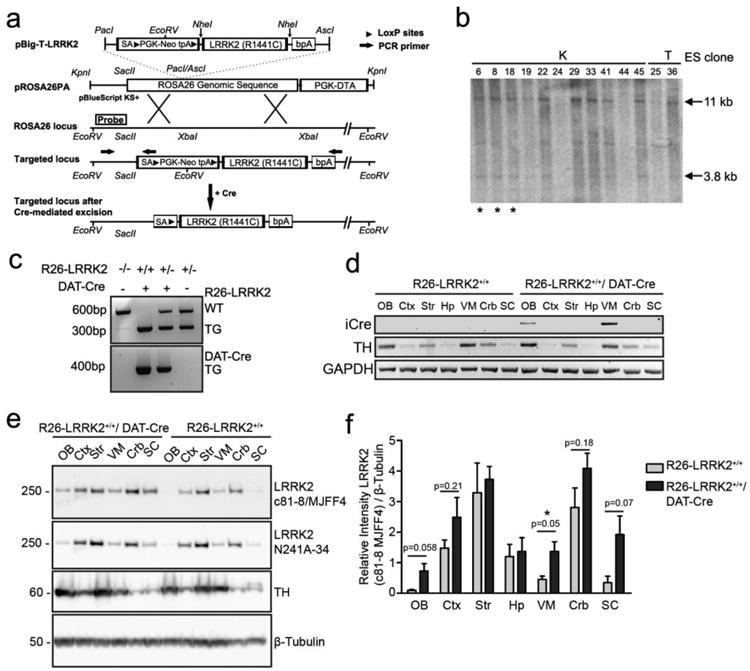
(A) Schematic diagram of the gene targeting strategy to generate R26-LRRK2 transgenic mice. Full-length human LRRK2 cDNA with the R1441C mutation is situated upstream of a bovine polyadenylation signal (bpA) and downstream of a SV40 splice acceptor (SA), neomycin resistance gene flanked by loxP sites (PGK-neo) and triple transcriptional termination sequence (tpA). The entire cassette was inserted into pROSA26PA via PacI and AscI restriction sites. pROSA26PA-LRRK2 construct was linearized and electroporated into ES cells where following homologous recombination the LRRK2-containing cassette was inserted into the ROSA26 locus downstream of the endogenous ROSA26 promoter. Cre-mediated excision of the neo-tpA stop cassette results in LRRK2 transgene expression driven by the endogenous ROSA26 promoter only in Cre-positive neurons. The location of PCR primers used for genotyping (arrows) and probe used for Southern blot analysis are indicated. (B) Southern blot analysis of EcoRV-digested genomic DNA isolated from ES cell clones. The 32P-labeled DNA probe hybridizes in the ROSA26 genomic locus as shown in (A) and detects the wild-type allele as 11 kb and the transgene targeted allele as 3.8 kb. Two different ES cell lines (K and T) and the clones selected for blastocyst microinjection (*) are shown. (C) PCR genotyping of R26-LRRK2/DAT-Cre mice to distinguish the targeted transgenic allele (∼300 bp) and wild-type allele (∼600 bp), as well as the DAT-Cre transgene (∼400 bp) from non-transgenic (no product), was performed in independent reactions with different primer primers. (D) Reverse transcription of total mRNA extracted from distinct brain regions: olfactory bulb (OB), frontal cortex (Ctx) striatum (Str), hippocampus (Hip), ventral midbrain (VM), cerebellum (Crb) and spinal cord (SC) followed by PCR using primers specific for iCre, mouse TH and GAPDH. (E) Western blot analysis of extracts from the same brain regions with anti-LRRK2 antibodies (c81-8/MJFF4 and N241A/34). Antibodies against tyrosine hydroxylase (TH) and β-tubulin were employed as controls for i) the dissection of brain regions containing dopaminergic neurons and ii) equal protein loading, respectively. Molecular mass markers are indicated in kilodaltons. (F) Densitometric analysis of LRRK2 levels (c81-8/MJFF4 antibody) in different brain regions normalized to β-tubulin levels was performed with NIH ImageJ software. Bars represent the mean ± SEM (n = 3 animals/genotype). Comparisons between genotypes for each brain region were assessed by unpaired, two-tailed Student's t-test P-values are indicated.
Cre-Dependent Expression of Human R1441C LRRK2
To induce the conditional expression of R1441C LRRK2 in the nigrostriatal dopaminergic pathway, R26-LRRK2+/+ mice were crossed with hemizygous BAC-DAT-iCre transgenic mice through a two-step breeding strategy to derive homozygous R26-LRRK2+/+ mice with or without Cre transgene expression. BAC-DAT-iCre mice express codon-improved Cre recombinase from the dopamine transporter (DAT) promoter from a transgenic BAC construct encompassing the entire mouse DAT (slc6) gene (45). Cre activity in these mice is largely restricted to central DAT-positive dopaminergic neuronal populations within the substantia nigra (A9), ventral tegmental area (A10), retrorubral field (A8) and the glomerular layer of the olfactory bulb (A16) (45). Cohorts of homozygous R26-LRRK2+/+/DAT-Cre mice and their R26-LRRK2+/+ littermate control mice were produced at expected frequencies, as verified by genomic PCR (Fig. 1C). RT-PCR analysis of total RNA extracted from various anatomic brain regions verified the expression of iCre mRNA restricted to the olfactory bulb and ventral midbrain of the R26-LRRK2+/+/DAT-Cre transgenic mice (Fig. 1D). To verify human LRRK2 expression in these mice we conducted quantitative RT-PCR analysis on total RNA derived from various anatomic brain regions of R26-LRRK2+/+/DAT-Cre and R26-LRRK2+/+ mice using a TaqMan primer/probe set specific for human LRRK2. Human LRRK2 mRNA expression is detected in the olfactory bulb, ventral midbrain and cerebellum of R26-LRRK2+/+/DAT-Cre mice relative to R26-LRRK2+/+ control mice (Fig. S1). To confirm human LRRK2 protein expression, protein extracts were prepared from brain tissue of R26-LRRK2+/+/DAT-Cre and R26-LRRK2+/+ mice followed by Western blot analysis with LRRK2-specific antibodies. Human LRRK2 protein is predominantly detected in the ventral midbrain, olfactory bulb and spinal cord, and at modest levels in cerebral cortex and cerebellum, of R26-LRRK2+/+/DAT-Cre mice compared to R26-LRRK2+/+ control mice using an antibody specific for LRRK2 (c81-8/MJFF4) (Fig. 1E and 1F). The increased levels of LRRK2 in R26-LRRK2+/+/DAT-Cre mice could also be confirmed using an alternative LRRK2 antibody (N241A/34) (Fig. 1E). The detection of human LRRK2 mRNA/protein in non-DAT-expressing regions (i.e. spinal cord, cerebral cortex and cerebellum) could reflect the localization of human LRRK2 to axonal projections or could indicate ectopic Cre recombinase activity. It was not possible to reliably detect human LRRK2 protein in the brains of R26-LRRK2+/+/DAT-Cre mice by immunohistochemistry using currently available antibodies potentially due to the low expression levels of human LRRK2 in this model (data not shown). Collectively, this data demonstrates that R26-LRRK2 mice express full-length human R1441C LRRK2 in a Cre recombinase-dependent manner.
Normal Motor and Olfactory Behavior in R1441C LRRK2 Transgenic Mice
To evaluate the potentially deleterious impact of R1441C LRRK2 expression on motor behavior, we conducted open field, rotarod and gait analysis on R26-LRRK2+/+/DAT-Cre mice compared to single R26-LRRK2+/+ littermate control mice at different time points as the mice aged (Fig. 2). In the open field quadrant, R26-LRRK2+/+/DAT-Cre mice exhibit similar locomotor activity to control mice at 10 and 20 months of age including measures of distance, speed and time spent in the center of the open field (Fig. 2D-E). Rotarod analysis using an accelerating paradigm does not reveal differences in motor coordination between R26-LRRK2+/+/DAT-Cre mice and control mice at 6 and 19 months of age (Fig. 2C). In the rotarod analysis, we observe an age-related decline in the performance of ROSA26-targeted mice relative to DAT-Cre mice suggesting that disruption of the ROSA26 genomic locus modestly compromises motor behavior at advanced age i.e. 19-20 months (data not shown). Gait analysis assessed using a digital Catwalk system does not reveal gait abnormalities in R26-LRRK2+/+/DAT-Cre mice at 11 and 20 months of age compared to control mice, including measures of cadence, base of support, footprint position, stepping pattern, speed and run duration (Fig. 2F-H). Since R1441C LRRK2 is also expressed in DAT-positive neurons of the olfactory bulb in R26-LRRK2+/+/DAT-Cre mice, we assessed olfactory function by measuring the latency to detect non-visible (i.e. buried) food (Fig. 2B). In this test, R26-LRRK2+/+/DAT-Cre mice exhibit a non-significant reduction in olfactory function when measured at 20 months of age compared to control mice. Finally, male and female R26-LRRK2+/+/DAT-Cre mice do not exhibit significantly altered body weight compared to R26-LRRK2+/+ control mice (Fig. 2A). Taken together, the expression of R1441C LRRK2 in nigrostriatal and olfactory dopaminergic neurons of mice does not influence motor or olfactory behaviors at advanced age.
Figure 2. Behavioral analysis of conditional R1441C LRRK2 transgenic mice.

(A) Body weight of male and female transgenic mice was measured at 20 months (n = 6 mice/genotype). (B) Olfactory function was assessed by an odor detection test on transgenic mice at 20 months whereby the latency to retrieve hidden food stimuli was recorded (n = 6 mice/genotype). (C) Motor coordination and balance of transgenic mice were evaluated by their performance on the rotarod at 6 (open circles) and 19 months (closed circles) of age. Testing was performed over 4 consecutive days using an accelerating rotation paradigm (5 to 50 rpm over 5 min). The latency of mice to fall from the rotating rod was recorded (n = 6 animals/genotype). (D-E) Transgenic mice at 10 (open rectangles) and 20 months (closed rectangles) were tested in the open field quadrant to assess their locomotor activity and anxiety-like behavior in a novel environment. Locomotor activity was measured by the total distance covered in the open field over 20 min as well as distance and speed every 5 minutes following introduction into the arena (n = 6 animals/genotype) (D). The time spent in the central zone of the open field arena was recorded as an index of anxiety (E). (F-H) Gait profiling of 11 and 20 month-old transgenic mice was performed with an automated gait analysis method. The temporal parameters calculated were the time for each animal to cross the walkway (F), the pace of walking (calculated as distance covered / sec), and variation in walking speed, as well as cadence which reflects the frequency of steps during the trial (G). (H) The relative spatial relationship between paws was assessed by calculating the base of support (the distance between the front paws or hind paws during walking) and print position (the distance between the hind paw and the previously placed ipsilateral front paw). The interlimb coordination is reflected on the step-sequence pattern that the mice employed while crossing the walkway (n = 6 animals/genotype). Data on all graphs represent the mean ± SEM. Comparisons between groups for each time-point were assessed by unpaired, two-tailed Student's t-test ns, non-significant.
R1441C LRRK2 Transgenic Mice Exhibit an Intact Nigrostriatal Dopaminergic Pathway
To determine whether the expression of R1441C LRRK2 could induce the dysfunction or degeneration of midbrain dopaminergic neurons, we assessed the integrity of the nigrostriatal dopaminergic pathway. The number of tyrosine hydroxylase (TH)- or vesicular monoamine transporter (VMAT)-positive dopaminergic and total Nissl-positive neurons in the substantia nigra pars compacta of R26-LRRK2+/+/DAT-Cre mice and littermate control mice at 12 and 22 months were assessed using unbiased stereological methodology. The selective expression of R1441C LRRK2 is not sufficient to induce nigral dopaminergic neuronal loss with advancing age (Fig. 3A-C). Dopaminergic neurons exhibit normal size and morphology in the substantia nigra of R26-LRRK2+/+/DAT-Cre mice (Fig. 3A). The density of TH-positive dopaminergic nerve terminals in the striatum is also not altered in R26-LRRK2+/+/DAT-Cre mice at 12 and 22 months of age (Fig. 3D-E). To evaluate the effect of R1441C LRRK2 expression on nigrostriatal dopaminergic function, we monitored the levels of biogenic amines in the striatum of homozygous R26-LRRK2+/+/DAT-Cre and control mice at 10-12 months of age by HPLC. We detect a modest yet non-significant increase of striatal dopamine and dopamine metabolite (DOPAC and HVA) levels, but normal dopamine turnover and serotonin (5-HT and 5-HIAA) levels, in R26-LRRK2+/+/DAT-Cre mice (Fig. 3F). Heterozygous R26-LRRK2+/-/DAT-Cre mice at 22-24 months of age similarly reveal a modest increase of striatal dopamine levels (Fig. 3G). Collectively, our data demonstrate that the expression of R1441C LRRK2 in DAT-positive dopaminergic neurons is not sufficient to induce the dysfunction or degeneration of the nigrostriatal dopaminergic pathway with advancing age.
Figure 3. Characterization of the nigrostriatal dopaminergic pathway of transgenic mice.

(A) Representative images of TH immunostaining in the substantia nigra of R26-LRRK2+/+ and R26-LRRK2+/+/DAT-Cre mice at 12 and 22 months. Scale bar: 100 μm. (B) Representative low power photomicrographs of TH staining from the ventral midbrain of transgenic mice at 22 months. Scale bar: 500 μm. (C) Stereological quantitation of dopaminergic (TH-positive or VMAT-positive) and Nissl-positive neurons in the substantia nigra at 12 and 22 months of R26-LRRK2+/+ and R26-LRRK2+/+/DAT-Cre mice. Bars represent mean ± SEM (n = 5 animals/genotype). Differences were assessed by unpaired, two-tailed Student's t-test, ns, nonsignificant. (D) Representative photomicrographs of TH immunostaining of nerve terminals in the striatum of R26-LRRK2+/+ and R26-LRRK2+/+/DAT-Cre mice at 12 and 22 months. Scale bar: 500 μm. (E) Quantitation of TH immunostaining in the striatum. Data represent the optical density of TH-positive signal (representing dopaminergic fibers) per area of tissue analyzed. Bars represent the mean ± SEM (n ≥ 4 animals/genotype). Differences between groups for each time-point were assessed by unpaired, two-tailed Student's t-test, ns, non-significant. (F) Striatal catecholamine levels were assessed by HPLC with electrochemical detection in 10 month-old homozygous R26-LRRK2+/+ and R26-LRRK2+/+/DAT-Cre mice. The concentration of each biogenic amine is expressed in picograms per μg of protein. The levels of dopamine and its metabolites: 3,4-dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA), 3 methoxytyramine (3-MT) and dopamine turnover rate ([DOPAC+HVA]/DA) are shown. The concentration of serotonin (5-HT) and its metabolite 5-hydroxyindoleacetic acid (5-HIAA) were also assessed. Bars represent the mean ± SEM (n = 5 animals/genotype). (G) Striatal catecholamine levels measured in 21-24 month-old heterozygous R26-LRRK2+/- and R26-LRRK2+/-/DAT-Cre mice (n = 5 animals/genotype). Comparisons between groups for each age were assessed by unpaired, two-tailed Student's t-test, ns, non-significant.
Increased Neurite Complexity of Cultured Midbrain Dopaminergic Neurons from R1441C LRRK2 Transgenic Mice
LRRK2 has been shown to regulate neuronal process morphology of cultured primary neurons (30, 39, 46). While the overexpression of G2019S LRRK2 consistently leads to reduced neuritic complexity compared to WT LRRK2 (39, 46), the effects of additional LRRK2 mutations are unclear. We have previously shown that G2019S LRRK2 expression reduces the complexity of dopaminergic neurites in primary midbrain cultures derived from PDGFβ-G2019S LRRK2 transgenic mice (39). To explore the effects of R1441C LRRK2 expression on this phenotype, primary midbrain cultures were prepared from the ventral mesencephalon of R26-LRRK2+/+/DAT-Cre and R26-LRRK2+/+ mice at post-natal day 1. These cultures typically contain ∼5% of TH-positive dopaminergic neurons (39). The assessment of neurite length reveals a modest yet non-significant increase in the length of dopaminergic axonal processes but no effect on dendritic processes from R26-LRRK2+/+/DAT-Cre mice (Fig. 4A-B). Sholl analysis was conducted to provide an overall measure of neuritic complexity. At days-in-vitro (DIV) 7 when dopaminergic neurite outgrowth is fully established, R26-LRRK2+/+/DAT-Cre mice exhibit a significantly increased complexity of dopaminergic neurites compared to R26-LRRK2+/+ control mice (Sholl coefficient), reflecting increased neurite branching (Fig. 4C-D and Table 1).
Figure 4. Dopaminergic neurons from R1441C LRRK2 transgenic mice exhibit increased neurite complexity.
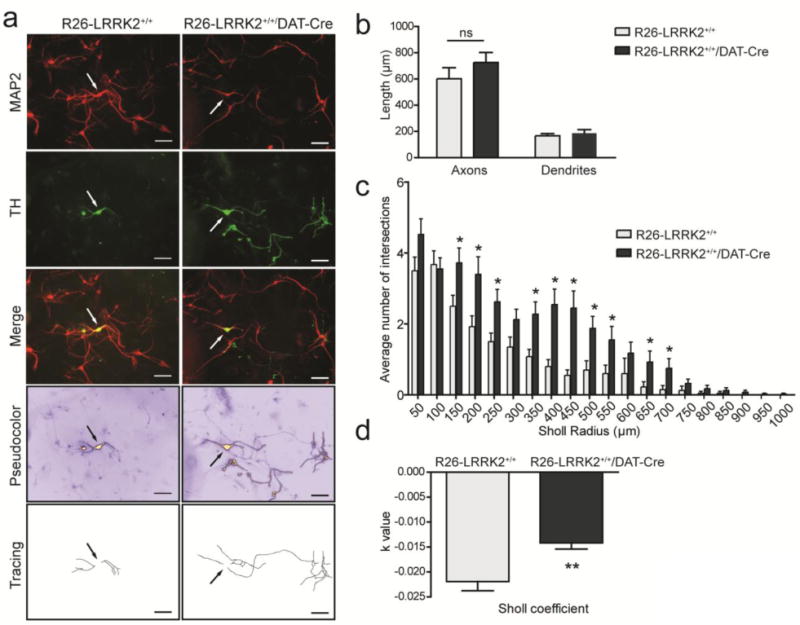
(A) Primary cultures prepared from the ventral midbrain of R26-LRRK2+/+/DAT-Cre and R26-LRRK2+/+ littermate controls at P1, fixed on DIV 7, and immunostained with antibodies against MAP2 and TH to label dopaminergic neurons. Representative fluorescent microscopic images are shown for each condition. ICA pseudocolor was employed to identify the dopaminergic neuronal soma (arrows) and neuritic processes for tracing. Scale bar: 500μm. (B) Quantitation of axonal and dendritic process length of TH-positive dopaminergic neurons in independent midbrain cultures derived from at least four mice per genotype. Bars represent neurite length (mean ± SEM, n = 80 neurons/genotype). (C) Sholl analysis of TH-positive dopaminergic neurites at DIV 7 to assess the mean number of dendritic intersections with concentric circles of increasing radii (μm) centered in the soma of each neuron. Bars represent mean ± SEM (n = 80 neurons/genotype). (D) Sholl regression coefficient (k value) is used to describe the changes in dendritic arborization with increasing distance from the neuronal soma. The value arises from the function: log10 (N/S) = -k × r + m, where N is the number of dendrite intersections for a circle of area S and radius r. The slope, k, of the regression line that results from this equation is decreasing with greater changes in the density of dendrites as the distance from the soma increases. Measurements were performed on independent cultures derived from at least four mice for each genotype. Data represent the mean ± SEM (n = 80 neurons/genotype). Comparisons between the two genotypes were done by unpaired, two-tailed Student's t-test (*P<0.01 or **P<0.0001). ns, non-significant.
Table 1. Sholl analysis of TH-positive neurons from R26-LRRK2+/+/DAT-Cre mice.
| Measurement | R26-LRRK2+/+ | R26-LRRK2+/+/DAT-Cre | |
|---|---|---|---|
| Sholl coefficient (-log slope of regression fit) | Measure of overall neurite complexity | -0.0219 ± 0.0018 | -0.0141 ± 0.0012* (P = 0.007) |
| Schoenen ramification index | Average number of branches formed per primary branch | 1.69 ± 0.168 | 2.15 ± 0.213 (P = 0.0910) |
| Critical radius (μm) | Sholl radius at which there is max number of intersections | 134.5 ± 21.7 | 231.0 ± 31.5* (P = 0.0137) |
indicates significant difference (unpaired, two-tailed Student's t-test)
Midbrain cultures derived from both mouse genotypes exhibit normal viability and comparable numbers of dopaminergic neurons (data not shown). Collectively, our data demonstrate that R1441C LRRK2 expression enhances the neuritic complexity of cultured dopaminergic neurons derived from transgenic mice.
Absence of Neuropathology or Abnormal Autophagy in R1441C LRRK2 Transgenic Mice
PD brains harboring LRRK2 mutations are associated with heterogeneous neuropathology, including classical Lewy bodies, neurofibrillary tau pathology, ubiquitin inclusions and in some cases the absence of proteinaceous inclusions (7, 17, 18, 21). While the common G2019S mutation is predominantly associated with Lewy body pathology (18, 20), the R1441C mutation can cause diverse neuropathology even between related members of the same family (7, 19). In PDGFβ-LRRK2 R1441C transgenic mice we previously observed an accumulation of autophagic vacuoles in the cerebral cortex whereas BAC-LRRK2 R1441G transgenic mice exhibit abnormal tau hyperphosphorylation (36, 39). To investigate potential neuropathology in R26-LRRK2+/+/DAT-Cre mice, we conducted immunohistochemical analysis with a number of pathological markers in the ventral midbrain, striatum, olfactory bulb and cerebellum at 12 and 22 months of age (Fig. 5). Compared to their R26-LRRK2+/+ control littermates, R26-LRRK2+/+/DAT-Cre mice do not reveal abnormalities in the distribution or levels of α-synuclein (total: Syn1; phosphorylated: pSer129), ubiquitin, tau (total: Tau5; phosphorylated: PHF1 and CP13; conformation-specific: MC1), glial markers (GFAP and Iba1) or autophagy markers (LC3 and p62). Silver staining also confirms the absence of neuronal degeneration throughout the brains of R26-LRRK2+/+/DAT-Cre mice at 12 and 22 months of age (Fig. 5). Taken together, our data demonstrate the distinct absence of neuropathology in the brains of R26-LRRK2+/+/DAT-Cre mice with advancing age.
Figure 5. Analysis of neuropathology in conditional R1441C LRRK2 transgenic mice.
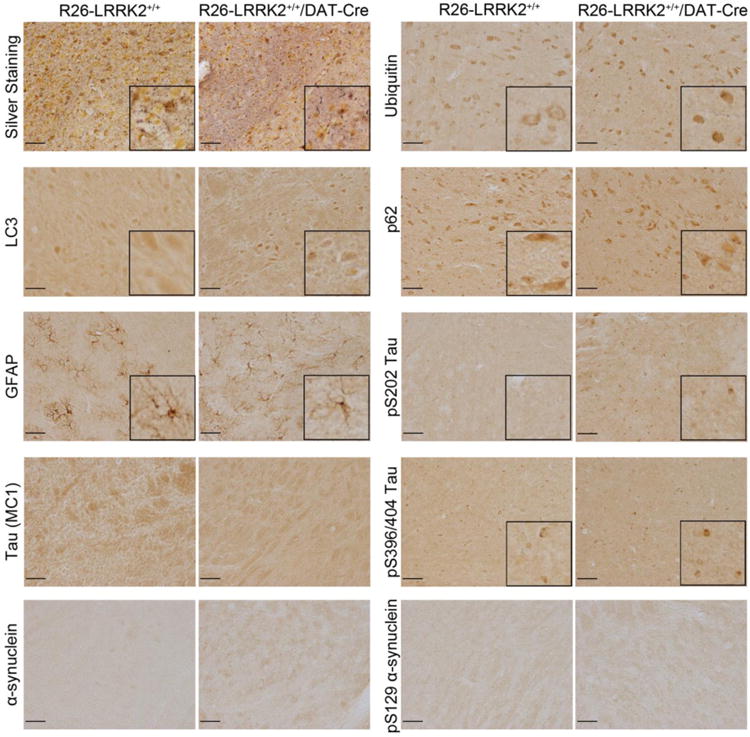
Representative photomicrographs from immunohistochemical analysis of the substantia nigra pars compacta of R26-LRRK2+/+/DAT-Cre and R26-LRRK2+/+ mice at 22 months of age. Sections were processed with Gallyas silver stain or antibodies detecting ubiquitin, the autophagosome marker LC3, the autophagy substrate p62/sequestosome, astrocytic marker GFAP, tau phosphorylated at Ser202 (clone CP13), tau pathological conformation (clone MC1), tau phosphorylated at Ser396/Ser404 (clone PHF-1), total α-synuclein, and α-synuclein phosphorylated at Ser129 (P-α-synuclein). Insets display higher magnification images. Scale bar: 50 μm.
Nuclear Abnormalities in R1441C LRRK2 Transgenic Mice
LRRK2 has recently been suggested to induce defects in nuclear architecture in neuronal cells derived from induced pluripotent stem cells of G2019S mutation carriers (47). In order to investigate the effects of R1441C LRRK2 expression on this phenotype, nuclear morphology was assessed in the substantia nigra of aged transgenic mice (20-21 months) by transmission electron microscopy (TEM). Nuclei of neurons sampled from the substantia nigra pars compacta region were assessed for their size and circularity. Neuronal nuclei from R26-LRRK2+/+/DAT-Cre mice display irregular shape and exhibit numerous invaginations of the nuclear envelope compared to R26-LRRK2+/+ littermate mice (Fig. 6A). Quantitative analysis of TEM images confirms abnormal nuclear morphology with significantly reduced nuclear circularity together with increased nuclear perimeter and invaginations in substantia nigra neurons from R26-LRRK2+/+/DAT-Cre mice, although total nuclear area is normal (Fig. 6B).
Figure 6. Abnormal nuclear morphology in aged R1441C LRRK2 transgenic mice.
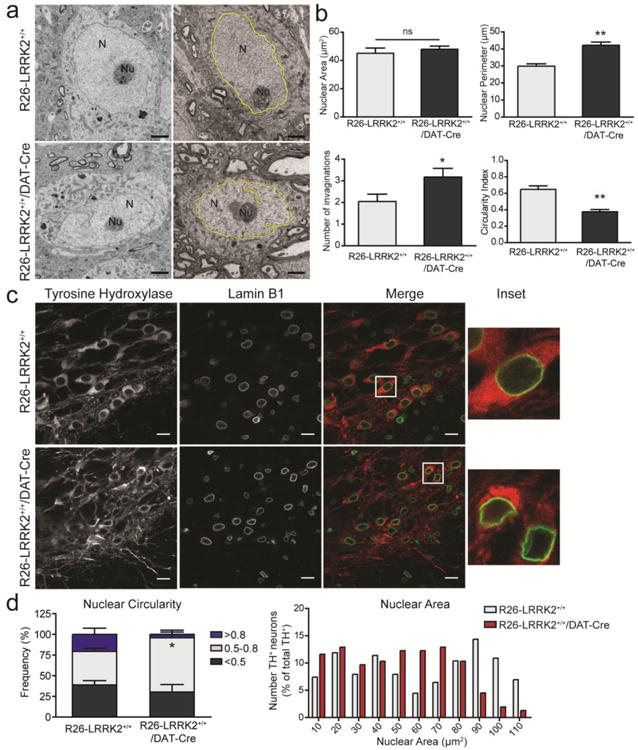
(A) Transmission electron microscopic analysis of ventral midbrain tissue from 20-22 month-old R26-LRRK2+/+/DAT-Cre and R26-LRRK2+/+ mice. The nucleus (N) and nucleolus (Nu) are indicated in representative images of neurons for each mouse genotype. Examples of nuclear traces used for quantification are highlighted in yellow. Scale bar: 2μm. (B) Quantitation of the nuclear area (μm2), nuclear perimeter (μm), average number of invaginations per nuclei and circularity index. Data represent the mean ± SEM (n ≥ 10 neurons/animal) sampled from at least 2 animals/genotype. (C) Immunofluorescence analysis of the substantia nigra from transgenic mice at 22 months co-labeled with antibodies to TH and Lamin B1 to determine nuclear morphology in dopaminergic neurons. Insets represent enlargement of the boxed areas in merge images. Scale bar: 15 μm. (D) The morphology of the nuclear envelop was assessed using a circularity index ranging from 0 (irregular shape) to 1 (perfect circle). The frequency distribution of nuclear circularity (left) and area (right) from dopaminergic neurons are shown in each graph. Data represent the mean ± SEM (n = 220, R26-LRRK2+/+, or n = 158, R26-LRRK2+/+/DAT-Cre, neurons) sampled from 4 mice/genotype. Statistical analysis was performed by unpaired, two-tailed Student's t-test (*P<0.05 or **P<0.001). ns, non-significant.
In order to confirm that the nuclear abnormalities identified by TEM occur in substantia nigra dopaminergic neurons we employed immunofluorescence analysis of the nuclear envelope marker, lamin B1, specifically in TH-positive neurons (Fig. 6C). Confocal analysis of neuronal lamin B1 immunofluorescence in 20-22 month-old mice and quantitative assessment of nuclear morphology reveals that dopaminergic neurons of R26-LRRK2+/+/DAT-Cre mice display abnormal nuclei with significantly reduced nuclear circularity compared to R26-LRRK2+/+ control mice (Fig. 6C-D). In younger R26-LRRK2+/+/DAT-Cre mice (at 5 months), nuclear morphology (lamin B1) within dopaminergic neurons is normal indicating that nuclear abnormalities occur in a progressive manner with advancing age (Fig. S2). Consistent with this observation, nuclear circularity in dopaminergic neurons is reduced at 22 months compared to 5 months for both mouse genotypes (Fig. 6D and S2). To further explore the impact of the R1441C LRRK2 mutation compared with other familial mutations (Y1699C and G2019S), nuclear morphology (lamin B1) was assessed in cultured rat primary cortical and midbrain dopaminergic neurons transiently transfected with each human LRRK2 variant. However, nuclear envelope circularity is not significantly altered by mutant LRRK2 expression in cortical neurons at DIV 14 (Fig. S3) or midbrain dopaminergic neurons at DIV 7 (Fig. S4), further suggesting that abnormal nuclear architecture may occur in dopaminergic neurons of R26-LRRK2+/+/DAT-Cre mice as a feature of advanced age. Taken together, these data demonstrate that selectively expressing human R1441C LRRK2 in dopaminergic neurons is not sufficient to produce neuronal degeneration within the lifespan of mice but instead induces nuclear abnormalities reminiscent of neuronal aging.
Discussion
Here, we report the development and phenotypic assessment of novel conditional transgenic mice selectively overexpressing human R1441C LRRK2 in DAT-positive dopaminergic neurons. Similar to other LRRK2 transgenic models, the expression of R1441C LRRK2 for up to 2 years is not sufficient to induce the degeneration of nigrostriatal pathway dopaminergic neurons or reduce the levels of striatal dopamine and its metabolites. Furthermore, R1441C LRRK2 expression in dopaminergic neurons does not manifest neurobehavioral deficits in mice such as abnormal motor activity or olfactory function, or produce abnormal protein inclusion pathology. However, R1441C LRRK2 expression does elicit subtle changes including the abnormal nuclear morphology of substantia nigra dopaminergic neurons with age, and the enhanced neurite complexity of cultured midbrain dopaminergic neurons. Collectively, our study identifies subtle yet intriguing phenotypes caused by the expression of R1441C LRRK2 in midbrain dopaminergic neurons that manifest prior to the onset of frank neuronal degeneration. These new conditional transgenic mice will provide a useful tool for dissecting the early pathogenic mechanisms underlying PD-associated mutations in LRRK2 in vivo.
To date, only transgenic mouse models with high-level ectopic G2019S LRRK2 expression in the substantia nigra driven by the PDGFβ promoter exhibit an age-dependent degeneration of dopaminergic neurons (39, 43). Mice with endogenous patterns of mutant LRRK2 expression driven from BAC constructs do not generally exhibit nigral dopaminergic neurodegeneration (35, 36, 38), which may reflect insufficient levels of transgene expression (i.e. copy number) or insufficient expression within dopaminergic neurons themselves. We reasoned that since endogenous LRRK2 is normally expressed at extremely low levels in nigral dopaminergic neurons of the mouse brain (48-50), that even a small increase in expression of human LRRK2 above endogenous levels would be sufficient to produce neuronal deficits and/or degeneration over the lifespan of mice. For this reason, we developed conditional transgenic mice by gene targeting where R1441C LRRK2 expression could be selectively expressed in specific neuronal populations from the endogenous murine ROSA26 promoter at or near to physiological levels. This was insured by using a low transgene copy number i.e. 2 copies/alleles at the ROSA26 locus, and by the moderate strength of the endogenous ROSA26 promoter (51-54). However, we fail to observe dopaminergic neurodegeneration in mice up to 2 years of age or indeed any signs of PD-related neuropathology such as abnormal protein inclusions or aggregates assessed by immunohistochemical and electron microscopic analysis. The lack of frank degeneration could potentially be explained by i) insufficient sustained levels of R1441C LRRK2 expression within dopaminergic neurons throughout the mouse lifespan, ii) compensatory mechanisms that occur during development due to the timing of DAT and/or ROSA26 promoter expression in developing dopaminergic neurons, iii) a reduced pathogenicity of the R1441C mutation relative to the well-studied G2019S mutation in the brain (i.e. since only G2019S LRRK2 mice so far display late-onset neurodegeneration (39, 43)), and/or iv) a non-cell autonomous mechanism for LRRK2-induced dopaminergic neuronal degeneration. The latter point is of particular interest given the recent observation of microglial inflammatory responses regulated by LRRK2 (55-57). A future application of our mouse model to further evaluate cell autonomous effects would be to compare the pathological impact of mutant LRRK2 expression driven by broad neuronal-specific or glial-specific Cre drivers. It would also be of interest to use this mouse model to explore the selective vulnerability of different neuronal populations to R1441C LRRK2 expression, especially since our prior PDGFβ-R1441C LRRK2 transgenic mice exhibit catecholamine deficits and cytopathology within the cerebral cortex accompanied by motor deficits (39).
Although nigral dopaminergic neurons do not degenerate in this mouse model, we observe signs that R1441C LRRK2 expression subtly alters the morphology of this neuronal population. R1441C LRRK2 mice display age-dependent abnormalities of nuclear morphology within nigral dopaminergic neurons. Abnormal nuclei display reduced circularity and an increased number of invaginations of the nuclear envelope although nuclear area was not altered. These observations are reminiscent of a recent study on neural stem cells derived from induced pluripotent stem cells (iPSCs) obtained from patients harboring a G2019S mutation in LRRK2 (47). Reduced nuclear circularity and increased nuclear area in these iPSC-derived neural stem cells became apparent in a passage-dependent manner suggesting that neuronal aging is potentially required. Prior studies have revealed that the integrity of the nuclear envelope is compromised with neuronal aging (58-62), an observation supported by the present study when comparing the nuclear envelope of dopaminergic neurons at early and late ages (Fig. 6 and S2). Therefore, dopaminergic neurons within conditional R1441C LRRK2 mice could potentially exhibit signs of enhanced neuronal aging that may represent one of earliest derangements that occurs prior to neuronal degeneration. Whether or not early alterations in nuclear architecture are sufficient or necessary for subsequent neuronal degeneration induced by R1441C LRRK2 remains to be determined, and whether this pathological feature is selectively limited to dopaminergic neurons is also not yet clear. Impairments of the nuclear envelope have previously been identified in hippocampal neurons of G2019S LRRK2 PD brains suggesting that nuclear abnormalities may represent a widespread occurrence during disease (47). Midbrain dopaminergic neurons cultured from mice expressing R1441C LRRK2 also exhibit increased neuritic complexity that manifests largely as an increase in neuritic branching. This phenotype would appear to be specific to cultured neurons perhaps related to an increased rate of neurite outgrowth, since the nigrostriatal dopaminergic pathway appears morphologically intact in the brains of mice expressing R1441C LRRK2. Our prior studies have demonstrated that midbrain dopaminergic neurons derived from PDGFβ-G2019S LRRK2 transgenic mice display reduced neurite complexity (39), and transfection experiments with G2019S LRRK2 in neuronal cultures reveal a similar effect (30, 32-34, 46). Similar studies of neurite complexity for the R1441C mutation are rather limited to date (30, 32), and no such studies in R1441C LRRK2 transgenic mice have been reported. Our studies with the PDGFβ-R1441C LRRK2 mice reveal that primary cortical neurons similarly display modestly enhanced neurite complexity (data not shown). Therefore, different disease-associated mutations in LRRK2 may have diverse effects on regulating neurite morphology potentially mediated by different mechanisms and/or specific to different neuronal populations. Nevertheless, our data reveal that midbrain dopaminergic neurons appear to be sensitive to the effects of R1441C LRRK2 expression.
Our novel conditional LRRK2 transgenic mice may prove useful for exploring pathological differences between different neuronal populations caused by R1441C LRRK2 expression. However, our data would suggest that any such differences would be subtle and age-dependent. Alternatively, this mouse model may prove useful for exploring the susceptibility of dopaminergic neurons expressing R1441C LRRK2 to secondary stressful stimuli since our data already reveal subtle pathological alterations occurring within this neuronal population. It would be of interest to explore how R1441C LRRK2 expression alters the susceptibility of nigral dopaminergic neurons to mitochondrial toxins such as MPTP as well as inflammatory stimuli such as lipopolysaccharide both of which may be relevant to the etiology of PD. The susceptibility of LRRK2 transgenic mouse models to such PD-relevant stressors has not been reported but represents an important avenue of investigation given that most LRRK2-related phenotypes in the rodent brain are subtle. Our conditional transgenic mice demonstrate that nigral dopaminergic neurons are sensitive to R1441C LRRK2 expression and suggest that abnormal nuclear architecture may represent an important and early feature of LRRK2-induced neuronal degeneration in PD.
Materials and Methods
Animals
Mice and rats were maintained in a pathogen-free barrier facility and exposed to a 12 h light/dark cycle with food and water provided ad libitum. Pregnant female Sprague-Dawley rats were obtained from Charles River Laboratories (L'Arbresle Cedex, France) and resulting P1 rats were used for preparation of primary neuronal cultures. All animal experiments were approved by the SCAV (Service de la consommation et des affaires veterinaires) in the Canton de Vaud, Switzerland (Animal authorization No. 2293), and conducted in strict accordance with the European Union directive (2010/63/EU) for the care and use of laboratory animals.
Generation of conditional LRRK2 transgenic mice
Full-length human LRRK2 cDNA with the R1441C mutation was targeted to the ROSA26genomic locus in 129/SvJ embryonic stem (ES) cells. An NheI-flanked human LRRK2 cDNAwas subjected to site-directed mutagenesis to remove an internal PacI site, and was subclonedinto the NheI site of acceptor plasmid pBigT downstream of a loxP-flanked neo-tpAtranscriptional termination cassette as described previously (52, 53). The entire neo-tpA-LRRK2conditional transgene was excised and inserted into targeting plasmid pROSA26PA via PacI andAscI sites. The final pROSA26PA-LRRK2 construct was linearized with KpnI, gel purified withGELase reagent (Epicenter, Madison, WI) and used for electroporation of mouse 129/SvJ EScells. Following G418 selection, correctly targeted ES cell clones were identified by PCR andSouthern blot analysis as described (52, 53), and three clones (K6, K8 and K18) weremicroinjected into C57Bl/6J mouse blastocysts to derive chimeric mice. Chimeras were bredwith C57Bl/6J mice with at least one chimeric mouse (derived from ES clone K18) producinggermline transmission of the targeted allele to F1 progeny. ES cell selection, blastocystmicroinjection and chimera breeding was conducted by the University of Cincinnati Gene-Targeted Mouse Service. F1 heterozygous mice were routinely identified by PCR of tail genomicDNA with primers: P1, 5′-AAAGTCGCTCTGAGTTGTTAT-3′; P2, 5′- GCGAAGAGTTTGTCCTCAACC-3′; and P3, 5′-GGAGCGGGAGAAATGGATATG-3′; producing a ∼500 bp product from the WT allele and a ∼250 bp product from the transgenic allele (refer to Fig. 1A). Following intercrossing of F1 heterozygous mice, mice were further analyzed by PCR to verify correct targeting of the transgene at the ROSA26 locus. The ROSA26-LRRK2R1441C mice are available from the Jackson Laboratory (Bar Harbor, ME; http://www.jax.org); JAX stock number 022793.
ROSA26-LRRK2 mice were maintained on a mixed genetic background (129/SvJ × C57Bl/6J) through backcrossing to C57Bl/6J mice for 3-4 generations and then intercrossing of heterozygous progeny. BAC-DAT-iCre mice were kindly provided by Francois Tronche via the European Mutant Mouse Archive (strain EM: 01738) and maintained on a C57Bl/6J background (45). To generate cohorts of mice for aging, homozygous ROSA26-LRRK2+/+ mice were first crossed with hemizygous BAC-DAT-iCre mice, and the F1 ROSA26-LRRK2+/-/CreTg progeny were crossed with homozygous ROSA26-LRRK2+/+ mice to produce F2 mice for further analysis. The F2 mice produced from these crosses were used throughout this study. Age-matched ROSA26-LRRK2+/- or ROSA26-LRRK2+/+ littermates without Cre were used as controls. BAC-DAT-iCre mice were genotyped by Cre-specific PCR with the primers: 5′-AAATGTTGCTGGATAGTTTTTACTGC-3′ and 5′-GGAAGGTGTCCAATTTACTGACCGTA-3′ to produce a 300 bp transgenic fragment (45).
Behavioral assessments
Behavioral testing was performed on male and female ROSA-LRRK2+/+, ROSA-LRRK2+/+/Cre and hemizygous BAC-DAT-iCre mice at ages indicated for each test.
Open field test
To assess novelty-induced locomotor activity, mice were individually placed in 40 cm2 arenas, which were separated virtually by the software into two separate regions: the central region (20 cm2 around the midpoint of the open field) and the periphery. Their activity (total distance traveled, speed of walking and time spent in the central region) over a period of 20 minutes was recorded and analyzed using an Ethovision XT video tracking system (Noldus Information Technology, Wageningen, Netherlands).
Rotarod
Mice were tested for their ability to maintain their balance on a rotating rod (Ugo Basile trendmill rotarod, Stoelting Co). The protocol consisted of three phases: habituation on the rotarod (day 1), training (day 2) and testing (days 3-4). During training and testing trials the mice had to run on the rod while it rotated at increasing speed from 5 to 50 rpm over a period of 5 min. Each mouse received three trials of 5 min with 30 min intervals between trials. The latency to fall from the rod and the maximum speed reached before falling was recorded for each mouse per trial.
Gait analysis
The CatWalk automated gait analysis system (Noldus Information Technology) was employed for assessment of gait and motor coordination. The apparatus consists of a 130 × 10 cm glass-floor walkway with fluorescence light beaming into the glass from the side which illuminates the animal's paws when they touch the glass producing a bright print image of the paws. Catwalk software (Noldus Information Technology) was used to record and process the position of footprints allowing quantitative analysis of gait. The experimental animals were habituated on the catwalk (day 1), trained (day 2) and data were acquired from three consecutive trials on day 3. Temporal (walk speed, maximum variation in running speed, cadence), relative spatial relationship between paws (base of support, print position) and interlimb coordination (step pattern) were amongst the parameters analyzed with the CatWalk system. The parameter cadence reflects the frequency of steps per second; base of support is the average distance between the two front or hind paws which is measured at max contact during each step cycle; print position reflects the distance between the position of the hind paw and the position of the previously placed front paw on the same side of the body at the same cycle. Regarding step pattern there are three categories of normal step-sequence patterns which are observed in rodents a) Cruciate (C) which can be either Ca: RF-LF-RH-LH or Cb: LF-RF-LH-RH, b) Alternate (A) which is either Aa: RF-RH-LF-LH or Ab: LF-RH-RF-LH and c) Rotate (R) which is either Ra: RF-LF-LH-RH or Rb: LF-RF-RH-LH.
Hidden food olfactory test
The hidden food test was performed as described previously (63). Briefly, mice were familiarized to sweetened cereal that was used as a food stimulus for three consecutive days before the test and it was confirmed that the cereal was consumed by the animals. For the test each individual mouse was placed into clean cage with fresh bedding and was left to acclimate for 5 min then the animal was temporarily removed from the cage while the food stimulus was buried under bedding. The animal was re-introduced to the cage and the latency to retrieve the buried food pellet was recorded.
RT-PCR and qPCR analysis
Brain regions used for this analysis were dissected from 4-5 month old R26-LRRK2+/+ and R26-LRRK2+/+/DAT-Cre mice. Total RNA was extracted with RNeasy mini kit (Qiagen) and digested with DNase I (Qiagen) to remove contaminating genomic DNA. RNAs were reverse transcribed using PrimeScript reverse transcriptase kit (Takara Bio Inc.) with oligo(dT). Equivalent mRNA-derived cDNAs (50 ng) were amplified by PCR with iCre-specific primers or mouse-specific primers for GAPDH and TH. For quantitative PCR (qPCR), TaqMan Universal PCR Master Mix (Applied Biosystems) was used to amplify cDNAs in triplicate for each transcript. Pre-designed TaqMan probe and primer sets specific for human LRRK2 (assay ID: Hs01115051_m1) and the housekeeping gene mouse Polr2a (assay ID: Mm00839502_m1) were obtained from Applied Biosystems. Quantitative PCR was performed using a 7900HT Fast Real-Time PCR system (Applied Biosystems). Relative mRNA expression levels were calculated using the comparative CT method (∆∆CT). For each sample the signal for human LRRK2 was normalized to Polr2a, and data were expressed as the ratio of R26-LRRK2+/+/DAT-Cre to R26-LRRK2+/+ mice to calibrate the samples with ‘1’ indicating no difference.
Tissue extraction and Western blot analysis
Following homogenization of brain samples in lysis buffer (10 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.5% NP-40, 1X Complete protease inhibitor cocktail [Roche Applied Sciences], 1X phosphatase inhibitor cocktails 2 and 3 [Sigma]), samples were subjected to ultracentrifugation (100,000g for 30 min) and protein concentration of soluble supernatant fractions (NP-40-soluble) was determined by BCA protein assay (Pierce Biotechnology). Proteins (50 μg) we resolved on 7% SDS-PAGE gels and transferred to nitrocellulose membranes. Blots were probed with monoclonal antibodies against LRRK2 (MJFF4/c81-8, Epitomics; N241A/34, NeuromAbs, UC Davis), rabbit polyclonal anti-TH (Novus Biologicals), and mouse monoclonal anti-β-tubulin (Sigma Aldrich). Secondary antibodies used were mouse monoclonal anti-rabbit IgG, light chain-specific (Jackson Immunoresearch) and goat anti-mouse IgG, light chain-specific (Jackson Immunoresearch). All secondary antibodies were HRP-conjugated and were detected using ECL reagent (Amersham). Western blot images were captured on a chemiluminescent image analyzer (LAS-4000, Fujifilm) and protein bands were quantified by densitometric analysis using NIH ImageJ software.
Measurement of biogenic amines by HPLC with electrochemical detection
Striatal tissue was homogenized by sonication in 25 volumes of 0.1 M perchloric acid. Catechols were resolved on a reverse-phase separation column (Eicompak SC-3ODS) connected to a HPLC-ECD system (HTEC-500, EICOM) and the chromatograms were analyzed with the EPC-500 powerchrome software. The resulting protein pellets were solubilized in 2% SDS/50 mM Tris pH 7.4 and protein concentration was determined using the BCA microassay kit (Pierce, Rockford, IL). Monoamine levels were normalized to protein concentration and expressed as picograms of analyte /μg of protein.
Immunohistochemistry
Animals were deeply anesthetized and transcardially-perfused with saline solution followed by 4% PFA in 0.1 M phosphate buffer (pH 7.3). Brains were cryoprotected in 30% sucrose solution before sectioning using a cryostat (Leica, Jung Frigocut 2800N). Heat-induced antigen unmasking and immunohistochemistry methods were conducted as described previously (39, 64). The tissues were stained with the following primary antibodies: rabbit anti-tyrosine hydroxylase (TH, NB300-109, Novus Biosciences), mouse anti-TH (T1299, Sigma), rabbit anti-VMAT2 (ab81855, Abcam), rabbit anti-glial fibrillary acidic protein (GFAP, DakoCytomation, Denmark), mouse anti-α-synuclein (clone 42, BD Biosciences), anti-phospho-Ser129 α-synuclein (clone pSyn#64; Wako Chemical Company), guinea pig anti-p62 (GP62, Progen Biotech), anti-LC3B (#2775; Cell Signaling), mouse anti-ubiquitin (clone P4D1, Cell signaling), rabbit anti-lamin B1 (ab16048, Abcam), mouse anti-Tau (total tau: clone Tau5, Invitrogen; phospho-tau: clones CP13/pSer202 and PHF-1/pSer396/404; conformation-specific tau: MC1, kindly provided by Prof. Peter Davies, Albert Einstein College of New York).
For bright-field microscopy, biotinylated secondary antibodies used were goat anti-mouse, anti-rabbit or anti-guinea pig (Vector Laboratories) followed by ABC reagent and DAB Peroxidase Substrate (Vector Laboratories). For fluorescence confocal imaging, secondary antibodies used were: anti-rabbit-IgG-AlexaFluor-488 and anti-mouse-IgG-AlexaFluor-546 (Life Technologies). Gallyas silver staining was performed using the FD NeuroSilver kit II according to the manufacturer's protocol (FD Neurotechnologies, Ellicott City, MD). Images were captured with the light microscopes AX70 (Olympus), Slide Scanner VS120-L100 (Olympus) and Zeiss LSM 700 Upright Scanning Laser confocal microscope (Carl Zeiss AG).
Stereological assessment of dopaminergic neurons
Unbiased stereological quantitation of dopaminergic neurons was performed using the optical fractionator probe of the StereoInvestigator software (MicroBrightField Biosciences), as previously described (39, 53, 65). For this analysis every 4th serial coronal section of 35 μm thickness, covering the entire substantia nigra region was immunostained with anti-TH or anti-VMAT2 antibodies and counterstained with cresyl violet. Sampling was performed in a systematic random manner using a grid of 225 × 200 μm squares covering the substantia nigra overlayed on each section and applying an optical dissector consisting of a 75 × 75 × 10 μm square cuboid. All the analyses were performed by investigators blinded to each condition.
The optical density of TH-immunoreactive nerve terminals was quantified throughout the striatum as a measure of dopaminergic innervation. The rostral and caudal limits of the striatum used for analysis were 2.00 mm to -0.50 mm relative to bregma, and every 10th serial section within this region was examined. Following immunolabeling with anti-TH antibody (Novus Biologicals) the sections were scanned and quantified using NIH ImageJ software.
Primary midbrain cultures and analysis of neuritic complexity and nuclear morphology
Primary cortical and midbrain cultures were prepared from P1 rats as described previously (39). For analysis of neurite complexity midbrain cultures were fixed at DIV 7 with 4% PFA and immunostained with rabbit polyclonal anti-TH (Novus Biologicals) and mouse monoclonal anti-MAP2 (Sigma Aldrich) antibodies, and anti-rabbit IgG-AlexaFluor-488 anti-mouse IgG-AlexaFluor-633 secondary antibodies (Invitrogen). Fluorescent images were captured with an inverted fluorescence microscope (Evos) at 10× magnification. Neuronal process length measurements were performed on TH-positive neurons using a NeuronJ plugin of ImageJ software. Neuritic complexity was quantified with the advanced Sholl analysis plugin v2.1 of ImageJ software using the semi-log method.
To assess nuclear morphology, primary cortical (DIV 9) and midbrain (DIV 4) cultures were transiently transfected with plasmid DNAs using Lipofectamine 2000 (Life Technologies) according to manufacturer's recommendations. Cultures were transfected with full-length FLAG-tagged human LRRK2 (WT, R1441C, Y1699C and G2019S) or an empty control plasmid (pcDNA3.1) and neurons were fixed at DIV 14 (cortical) or DIV 7 (midbrain) with 4% PFA, and immunostained with mouse anti-FLAG M2 (F1804, Sigma), rabbit anti-Lamin B1 (ab16048, Abcam) and sheep anti-TH (NB300-110, Novus Biologicals) followed by anti IgG-AlexFluor-488, -546 or -633 (Life Technologies). Images were captured with a Zeiss LSM 700 upright scanning laser confocal microscope (Carl Zeiss AG) with a Plan-Aprochromat 40×/1.30 oil objective. Acquired images from a single z-plane (0.1 μm-thick) were analyzed with NIH ImageJ software to assess nuclear circularity, as described below.
Transmission electron microscopy
Mice were deeply anesthetized with an intraperitoneal injection of sodium pentobarbitone (30 mg/kg body weight), and then immediately perfused with 2.5% glutaraldehyde and 2% paraformaldehyde in phosphate buffer (0.1 M, pH 7.4). The brains were removed two hours after the perfusion and vibratome slices cut at 80 μm in the coronal plane through the substantia nigra. The slices were then postfixed with 1.5% potassium ferrocyanide and 1% osmium tetroxide, then with 1% osmium tetroxide alone, and finally in 1% aqueous uranyl acetate. They were dehydrated with increasing concentrations of alcohol, embedded in Durcupan resin, and hardened at 65° for 24 h. The region for thin sectioning was cut from the embedded sections, and mounted onto a blank resin block. Silver-grey sections were cut at 50 nm thickness with a diamond knife and mounted onto single slot copper grids with a pioloform support film. These were stained with lead citrate and uranyl acetate, and imaged at 80 kV in a TEM (Tecnai Spirit, FEI Company). Neuronal somata were recognized according to their morphology and each one was imaged if a significant (< 3 micron diameter) amount of the nucleus appeared in the section.
Nuclear morphology analysis
Nuclei imaged by TEM as well as randomly sampled dopaminergic nuclei immunostained with Lamin B1 were traced and measured with NIH ImageJ software. Nuclear circularity index is calculated with the formula: circularity = 4π × (area / perimeter2).
Supplementary Material
Highlights.
Development of conditional R1441C LRRK2 transgenic with Cre-dependent expression
R1441C LRRK2 does not induce dopaminergic neurodegeneration or motor deficits
R1441C LRRK2 increases neurite complexity of cultured midbrain dopaminergic neurons
R1441C LRRK2 induces nuclear abnormalities in aged nigral dopaminergic neurons
R1441C LRRK2 transgenic mice manifest early abnormalities in dopaminergic neurons
Acknowledgments
The authors are grateful to members of the EPFL Histology Core Facility, Center for Phenogenomics and BioImaging and Optics platform for technical assistance and advice.
Funding: This work was supported by funding from Parkinson Schweiz (D.J.M.), American Parkinson Disease Association (D.J.M.), the Ecole Polytechnique Fédérale de Lausanne (D.J.M.), Van Andel Research Institute (D.J.M.), National Parkinson Foundation (V.L.D. and D.J.M.), Swedish Research Council (D.G.), The Swedish Brain Power (D.G.) and NIH NS38377 (V.L.D. and T.M.D.). T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. The authors acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with The Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation's Parkinson's Disease Programs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lang AE, Lozano AM. Parkinson's disease. First of two parts. The New England journal of medicine. 1998;339:1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- 2.Lang AE, Lozano AM. Parkinson's disease. Second of two parts. The New England journal of medicine. 1998;339:1130–1143. doi: 10.1056/NEJM199810153391607. [DOI] [PubMed] [Google Scholar]
- 3.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 4.Gasser T. Mendelian forms of Parkinson's disease. Biochimica et biophysica acta. 2009;7:587–596. doi: 10.1016/j.bbadis.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 5.Healy DG, Falchi M, O'Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, et al. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson's disease: a case-control study. Lancet neurology. 2008;7:583–590. doi: 10.1016/S1474-4422(08)70117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 7.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Nichols WC, Pankratz N, Hernandez D, Paisan-Ruiz C, Jain S, Halter CA, Michaels VE, Reed T, Rudolph A, Shults CW, et al. Genetic screening for a single common LRRK2 mutation in familial Parkinson's disease. Lancet. 2005;365:410–412. doi: 10.1016/S0140-6736(05)17828-3. [DOI] [PubMed] [Google Scholar]
- 9.Ozelius LJ, Senthil G, Saunders-Pullman R, Ohmann E, Deligtisch A, Tagliati M, Hunt AL, Klein C, Henick B, Hailpern SM, et al. LRRK2 G2019S as a cause of Parkinson's disease in Ashkenazi Jews. The New England journal of medicine. 2006;354:424–425. doi: 10.1056/NEJMc055509. [DOI] [PubMed] [Google Scholar]
- 10.Gilks WP, Abou-Sleiman PM, Gandhi S, Jain S, Singleton A, Lees AJ, Shaw K, Bhatia KP, Bonifati V, Quinn NP, et al. A common LRRK2 mutation in idiopathic Parkinson's disease. Lancet. 2005;365:415–416. doi: 10.1016/S0140-6736(05)17830-1. [DOI] [PubMed] [Google Scholar]
- 11.Lesage S, Durr A, Tazir M, Lohmann E, Leutenegger AL, Janin S, Pollak P, Brice A French Parkinson's Disease Genetics Study, G. LRRK2 G2019S as a cause of Parkinson's disease in North African Arabs. The New England journal of medicine. 2006;354:422–423. doi: 10.1056/NEJMc055540. [DOI] [PubMed] [Google Scholar]
- 12.Hulihan MM, Ishihara-Paul L, Kachergus J, Warren L, Amouri R, Elango R, Prinjha RK, Upmanyu R, Kefi M, Zouari M, et al. LRRK2 Gly2019Ser penetrance in Arab-Berber patients from Tunisia: a case-control genetic study. Lancet neurology. 2008;7:591–594. doi: 10.1016/S1474-4422(08)70116-9. [DOI] [PubMed] [Google Scholar]
- 13.Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nature genetics. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 14.Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nature genetics. 2009;41:1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.International Parkinson Disease Genomics, C. Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, Saad M, Simon-Sanchez J, Schulte C, Lesage S, et al. Imputation of sequence variants for identification of genetic risks for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377:641–649. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Biskup S, West AB. Zeroing in on LRRK2-linked pathogenic mechanisms in Parkinson's disease. Biochimica et biophysica acta. 2009;7:625–633. doi: 10.1016/j.bbadis.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rajput A, Dickson DW, Robinson CA, Ross OA, Dachsel JC, Lincoln SJ, Cobb SA, Rajput ML, Farrer MJ. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology. 2006;67:1506–1508. doi: 10.1212/01.wnl.0000240220.33950.0c. [DOI] [PubMed] [Google Scholar]
- 18.Ross OA, Toft M, Whittle AJ, Johnson JL, Papapetropoulos S, Mash DC, Litvan I, Gordon MF, Wszolek ZK, Farrer MJ, et al. Lrrk2 and Lewy body disease. Annals of neurology. 2006;59:388–393. doi: 10.1002/ana.20731. [DOI] [PubMed] [Google Scholar]
- 19.Zimprich A, Muller-Myhsok B, Farrer M, Leitner P, Sharma M, Hulihan M, Lockhart P, Strongosky A, Kachergus J, Calne DB, et al. The PARK8 locus in autosomal dominant parkinsonism: confirmation of linkage and further delineation of the disease-containing interval. American journal of human genetics. 2004;74:11–19. doi: 10.1086/380647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giasson BI, Covy JP, Bonini NM, Hurtig HI, Farrer MJ, Trojanowski JQ, Van Deerlin VM. Biochemical and pathological characterization of Lrrk2. Annals of neurology. 2006;59:315–322. doi: 10.1002/ana.20791. [DOI] [PubMed] [Google Scholar]
- 21.Funayama M, Hasegawa K, Ohta E, Kawashima N, Komiyama M, Kowa H, Tsuji S, Obata F. An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Annals of neurology. 2005;57:918–921. doi: 10.1002/ana.20484. [DOI] [PubMed] [Google Scholar]
- 22.Tsika E, Moore DJ. Mechanisms of LRRK2-mediated neurodegeneration. Current neurology and neuroscience reports. 2012;12:251–260. doi: 10.1007/s11910-012-0265-8. [DOI] [PubMed] [Google Scholar]
- 23.Tsika E, Moore DJ. Contribution of GTPase activity to LRRK2-associated Parkinson disease. Small GTPases. 2013;4 doi: 10.4161/sgtp.25130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greggio E, Cookson MR. Leucine-rich repeat kinase 2 mutations and Parkinson's disease: three questions. ASN Neuro. 2009;1 doi: 10.1042/AN20090007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nature neuroscience. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- 26.Smith WW, Pei Z, Jiang H, Moore DJ, Liang Y, West AB, Dawson VL, Dawson TM, Ross CA. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:18676–18681. doi: 10.1073/pnas.0508052102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, et al. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Human molecular genetics. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- 28.Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiology of disease. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 29.Xiong Y, Coombes CE, Kilaru A, Li X, Gitler AD, Bowers WJ, Dawson VL, Dawson TM, Moore DJ. GTPase activity plays a key role in the pathobiology of LRRK2. PLoS genetics. 2010;6:1000902. doi: 10.1371/journal.pgen.1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 31.Plowey ED, Cherra SJ, 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. Journal of neurochemistry. 2008;105:1048–1056. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramsden N, Perrin J, Ren Z, Lee BD, Zinn N, Dawson VL, Tam D, Bova M, Lang M, Drewes G, et al. Chemoproteomics-based design of potent LRRK2-selective lead compounds that attenuate Parkinson's disease-related toxicity in human neurons. ACS chemical biology. 2011;6:1021–1028. doi: 10.1021/cb2002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Biosa A, Trancikova A, Civiero L, Glauser L, Bubacco L, Greggio E, Moore DJ. GTPase activity regulates kinase activity and cellular phenotypes of Parkinson's disease-associated LRRK2. Human molecular genetics. 2013;22:1140–1156. doi: 10.1093/hmg/dds522. [DOI] [PubMed] [Google Scholar]
- 34.Stafa K, Trancikova A, Webber PJ, Glauser L, West AB, Moore DJ. GTPase activity and neuronal toxicity of Parkinson's disease-associated LRRK2 is regulated by ArfGAP1. PLoS genetics. 2012;8:9. doi: 10.1371/journal.pgen.1002526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Patel JC, Wang J, Avshalumov MV, Nicholson C, Buxbaum JD, Elder GA, Rice ME, Yue Z. Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson's disease mutation G2019S. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:1788–1797. doi: 10.1523/JNEUROSCI.5604-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Y, Liu W, Oo TF, Wang L, Tang Y, Jackson-Lewis V, Zhou C, Geghman K, Bogdanov M, Przedborski S, et al. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson's disease. Nature neuroscience. 2009;12:826–828. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, et al. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson's-disease-related mutant alpha-synuclein. Neuron. 2009;64:807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Melrose HL, Dachsel JC, Behrouz B, Lincoln SJ, Yue M, Hinkle KM, Kent CB, Korvatska E, Taylor JP, Witten L, et al. Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiology of disease. 2010;40:503–517. doi: 10.1016/j.nbd.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramonet D, Daher JP, Lin BM, Stafa K, Kim J, Banerjee R, Westerlund M, Pletnikova O, Glauser L, Yang L, et al. Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PloS one. 2011;6:0018568. doi: 10.1371/journal.pone.0018568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maekawa T, Mori S, Sasaki Y, Miyajima T, Azuma S, Ohta E, Obata F. The I2020T Leucine-rich repeat kinase 2 transgenic mouse exhibits impaired locomotive ability accompanied by dopaminergic neuron abnormalities. Molecular neurodegeneration. 2012;7:1750–1326. doi: 10.1186/1750-1326-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou H, Huang C, Tong J, Hong WC, Liu YJ, Xia XG. Temporal expression of mutant LRRK2 in adult rats impairs dopamine reuptake. Int J Biol Sci. 2011;7:753–761. doi: 10.7150/ijbs.7.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herzig MC, Bidinosti M, Schweizer T, Hafner T, Stemmelen C, Weiss A, Danner S, Vidotto N, Stauffer D, Barske C, et al. High LRRK2 levels fail to induce or exacerbate neuronal alpha-synucleinopathy in mouse brain. PloS one. 2012;7:15. doi: 10.1371/journal.pone.0036581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen CY, Weng YH, Chien KY, Lin KJ, Yeh TH, Cheng YP, Lu CS, Wang HL. (G2019S) LRRK2 activates MKK4-JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ. 2012;19:1623–1633. doi: 10.1038/cdd.2012.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tong Y, Pisani A, Martella G, Karouani M, Yamaguchi H, Pothos EN, Shen J. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14622–14627. doi: 10.1073/pnas.0906334106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Turiault M, Parnaudeau S, Milet A, Parlato R, Rouzeau JD, Lazar M, Tronche F. Analysis of dopamine transporter gene expression pattern -- generation of DAT-iCre transgenic mice. The FEBS journal. 2007;274:3568–3577. doi: 10.1111/j.1742-4658.2007.05886.x. [DOI] [PubMed] [Google Scholar]
- 46.Parisiadou L, Xie C, Cho HJ, Lin X, Gu XL, Long CX, Lobbestael E, Baekelandt V, Taymans JM, Sun L, et al. Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:13971–13980. doi: 10.1523/JNEUROSCI.3799-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu GH, Qu J, Suzuki K, Nivet E, Li M, Montserrat N, Yi F, Xu X, Ruiz S, Zhang W, et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature. 2012;491:603–607. doi: 10.1038/nature11557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, et al. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Annals of neurology. 2006;60:557–569. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- 49.West AB, Cowell RM, Daher JP, Moehle MS, Hinkle KM, Melrose HL, Standaert DG, Volpicelli-Daley LA. Differential LRRK2 expression in the cortex, striatum, and substantia nigra in transgenic and nontransgenic rodents. The Journal of comparative neurology. 2014 doi: 10.1002/cne.23583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Higashi S, Moore DJ, Colebrooke RE, Biskup S, Dawson VL, Arai H, Dawson TM, Emson PC. Expression and localization of Parkinson's disease-associated leucine-rich repeat kinase 2 in the mouse brain. Journal of neurochemistry. 2007;100:368–381. doi: 10.1111/j.1471-4159.2006.04246.x. [DOI] [PubMed] [Google Scholar]
- 51.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nature genetics. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 52.Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC developmental biology. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Daher JP, Ying M, Banerjee R, McDonald RS, Hahn MD, Yang L, Flint Beal M, Thomas B, Dawson VL, Dawson TM, et al. Conditional transgenic mice expressing C-terminally truncated human alpha-synuclein (alphaSyn119) exhibit reduced striatal dopamine without loss of nigrostriatal pathway dopaminergic neurons. Molecular neurodegeneration. 2009;4:34. doi: 10.1186/1750-1326-4-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gu X, Andre VM, Cepeda C, Li SH, Li XJ, Levine MS, Yang XW. Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington's disease. Molecular neurodegeneration. 2007;2:8. doi: 10.1186/1750-1326-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, DeSilva TM, Cowell RM, West AB. LRRK2 inhibition attenuates microglial inflammatory responses. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:1602–1611. doi: 10.1523/JNEUROSCI.5601-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gillardon F, Schmid R, Draheim H. Parkinson's disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience. 2012;208:41–48. doi: 10.1016/j.neuroscience.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 57.Kim B, Yang MS, Choi D, Kim JH, Kim HS, Seol W, Choi S, Jou I, Kim EY, Joe EH. Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PloS one. 2012;7:e34693. doi: 10.1371/journal.pone.0034693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Worman HJ, Ostlund C, Wang Y. Diseases of the nuclear envelope. Cold Spring Harbor perspectives in biology. 2010;2:a000760. doi: 10.1101/cshperspect.a000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Woulfe JM. Abnormalities of the nucleus and nuclear inclusions in neurodegenerative disease: a work in progress. Neuropathology and applied neurobiology. 2007;33:2–42. doi: 10.1111/j.1365-2990.2006.00819.x. [DOI] [PubMed] [Google Scholar]
- 60.Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–1063. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rodriguez S, Eriksson M. Evidence for the involvement of lamins in aging. Current aging science. 2010;3:81–89. doi: 10.2174/1874609811003020081. [DOI] [PubMed] [Google Scholar]
- 62.D'Angelo MA, Raices M, Panowski SH, Hetzer MW. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell. 2009;136:284–295. doi: 10.1016/j.cell.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang M, Crawley JN. Simple behavioral assessment of mouse olfaction. In: Crawley Jacqueline N, et al., editors. Current protocols in neuroscience. Chapter 8. 2009. Unit 8 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ramonet D, Podhajska A, Stafa K, Sonnay S, Trancikova A, Tsika E, Pletnikova O, Troncoso JC, Glauser L, Moore DJ. PARK9-associated ATP13A2 localizes to intracellular acidic vesicles and regulates cation homeostasis and neuronal integrity. Human molecular genetics. 2012;21:1725–1743. doi: 10.1093/hmg/ddr606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Daher JP, Pletnikova O, Biskup S, Musso A, Gellhaar S, Galter D, Troncoso JC, Lee MK, Dawson TM, Dawson VL, et al. Neurodegenerative phenotypes in an A53T alpha-synuclein transgenic mouse model are independent of LRRK2. Human molecular genetics. 2012;21:2420–2431. doi: 10.1093/hmg/dds057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.