

Abstract
The use of genetically modified (GM) mice to assess carcinogenicity is playing an increasingly important role in the safety evaluation of chemicals. While progress has been made in developing and evaluating mouse models such as the Trp53+/−, Tg.AC and the rasH2, the suitability of these models as replacements for the conventional rodent cancer bioassay and for assessing human health risks remains uncertain. The objective of this research was to evaluate the use of accelerated cancer bioassays with GM mice for assessing the potential health risks associated with exposure to carcinogenic agents. We compared the published results from the GM bioassays to those obtained in the National Toxicology Program’s conventional chronic mouse bioassay for their potential use in risk assessment. Our analysis indicates that the GM models are less efficient in detecting carcinogenic agents but more consistent in identifying non-carcinogenic agents. We identified several issues of concern related to the design of the accelerated bioassays (e.g., sample size, study duration, genetic stability and reproducibility) as well as pathway-dependency of effects, and different carcinogenic mechanisms operable in GM and non-GM mice. The use of the GM models for dose-response assessment is particularly problematic as these models are, at times, much more or less sensitive than the conventional rodent cancer bioassays. Thus, the existing GM mouse models may be useful for hazard identification, but will be of limited use for dose-response assessment. Hence, caution should be exercised when using GM mouse models to assess the carcinogenic risks of chemicals.
Keywords: Carcinogen, concordance, dose-response, hazard identification, mechanism, pathway-specificity, study duration, transgenic
Introduction
For many years, the two-species rodent cancer bioassay has been the primary in vivo test relied upon to identify agents capable of causing cancer in humans (Huff, 1999). In addition to identifying carcinogenic effects, this bioassay allows the identification of pathological changes, including both pre-neoplastic and non-neoplastic lesions in multiple tissues as well as the monitoring of clinical, hematological, and urinary biomarkers to identify early changes associated with neoplasia and other chronic non-cancer adverse effects induced by the test agent. The animal strains and/or stocks, protocols, and evaluation approaches used to conduct chronic animal bioassays have been selected and developed over the past 40+ years.
Since the establishment of the rodent cancer bioassay, there have been significant advances in our understanding of structure and nature of the human and test species genomes with regard to conserved genes, such as the proto-oncogenes (e.g. Ha-Ras) commonly mutated in many human and rodent tumors and loss of tumor suppressor genes (e.g. Trp53) with gain or loss of function affecting pathways involved in carcinogenesis. Two major types of genetically modified (GM) animals have been developed and utilized to provide insights into normal and disease-related processes (Gulezian et al., 2000). One type involves creation of GM animals that have been engineered by inserting one or more genes from the same or another species randomly into the host genome. The second type utilizes targeted insertion by homologous recombination in embryonic stem cells at the gene locus of one or both copies of a mouse or human gene of interest inactivated by either partial deletion or mutation at specified amino acid positions for loss or gain of function, respectively (Castrop, 2010). For simplicity, in this article the descriptor GM will be used interchangeably for both transgenic animals and other GM animals.
Over the past two decades, there has been considerable interest in the development and application of short-term or accelerated cancer bioassays using GM animals that exhibit high sensitivity to chemically induced cancers (Boverhof et al., 2011; Gulezian et al., 2000; Mahler et al., 1998; Pritchard et al., 2003; Tennant et al., 1999a; Yamamoto et al., 1998a). The basis for this approach is that rodents with an alteration in a conserved gene critical for tumorigenesis could be developed as test organisms for accelerated cancer bioassays. The genetic alteration by itself would be insufficient to induce cancer in the animals within the assay period (6–12 months), but exposure of the GM animals to potentially carcinogenic agents would result in a rapid induction of tumors or a decrease in latency yielding tumors within the assay period before being confounded by the appearance of spontaneous tumors not associated with treatment. Within the context of the multi-step process of chemical carcinogenesis, these animals would be considered “genetically initiated,” and with additional chemically induced genetic or epigenetic changes, the animals would respond in a shorter period of time, with potentially higher sensitivity and specificity towards carcinogens (French et al., 2001a; Tennant et al., 1995). In a number of ways, these assays are analogous to earlier accelerated cancer assays that have been previously developed, such as those employing the Strain A mouse or the initiation/promotion assays on mouse skin (Enzmann et al., 1998; IARC, 1999; Ito et al., 1992).
Within the risk assessment framework of the National Research Council (NRC), the results from the conventional chronic rodent bioassays play a particularly important role in the hazard identification and the dose-response steps of the risk assessment paradigm (NRC, 1983). Although much has been written about various GM animals and accelerated bioassays, the potential role of these new bioassays in the human health risk assessment process has not been thoroughly and critically evaluated. The objective of this review is to evaluate the potential use of GM animals and the recently developed accelerated cancer bioassays including the study protocols used to date, for the assessment of health risks associated with potential carcinogenic agents.
Genetically modified animals used for accelerated cancer bioassays
GM animals have been developed and are being used to complement or, in some cases, replace the chronic rodent bioassays that have traditionally been performed for assessing cancer risks (reviewed in Boverhof et al., 2011; French et al., 2001b; Pritchard et al., 2003; Storer et al., 2010; Tennant et al., 1995; Yamamoto et al., 1998a). A large number of GM animals have been used and/or are currently being developed to investigate the role of specific genes and signaling pathways in chemical carcinogenesis (Boverhof et al., 2011; Goldsworthy et al., 1994; Macleod & Jacks, 1999). Many of these models have been designed to address a narrow range of research hypotheses. However, a few of these GM models affect genes, pathways and processes involved in many different types of human and rodent cancers and, in theory, should be suitable models for the development of a bioassay to efficiently evaluate the carcinogenic potential of chemicals.
To serve as a suitable model for accelerated cancer testing, GM mice should ideally be phenotypically normal prior to chemical exposure, show genetic stability and exhibit a low level of spontaneous tumors (Van Zeller & Combes, 1999). After exposure to a genotoxic or nongenotoxic carcinogen, the GM animal should have increased sensitivity, decreased tumor latency as compared to the non-GM or wild-type animal, and express the genetic modification in a wide range of tissues allowing cancer and noncancer effects to be seen in all potential target sites (Van Zeller & Combes, 1999).
In recent years, mouse models, such as the heterozygous Trp53 (B6.129-Trp53tm1Brd, Trp53+/−, or Trp53def), the FVB/ N-Tg.AC or TG.AC (carrying the mouse mutant v-Ha-Ras oncogene), the CB6F1-rasH2 or rasH2 (carrying a reconstructed human HRAS proto-oncogene), and the xeroderma pigmentosum group A knockout (Xpa−/−) mice have been investigated by researchers as suitable models for accelerated cancer bioassays (Boverhof et al., 2011; Gulezian et al., 2000; Pritchard et al., 2003; Tennant et al., 1995, 1999a). It is critical to note that each of these GM mouse models was created from different genetic backgrounds and comparisons have not accounted for differences in heritable strain susceptibilities and penetrance. The characteristics of each of these models are summarized in Table 1 and are compared to the B6C3F1 inbred strain of mice used in the conventional 2-year bioassay. The GM models differ from the conventional 2-year bioassays in the genetic background and strain of mice, the duration and, for some, the routes of exposure employed, and their sensitivity to genotoxic and/or non-genotoxic carcinogens. The ensuing discussion will focus primarily on the Trp53+/−, the Tg.AC, and the rasH2 models because more information is available on the performance and results of these first generation GM models. However, it should be noted that at the present time the Tg.AC and the Xpa−/− models are not commonly used or recommended for use (Boverhof et al., 2011).
Table 1.
Summary of testing protocols for conventional and genetically modified mouse assays.
| Assay characteristics | 2-year bioassay | Trp53+/− | Tg.AC | rasH2 | Xpa−/− |
|---|---|---|---|---|---|
| Mouse strain used | B6C3F1 | B6.129-Trp53tm1Brd* | FVB/N-Tg.AC | CB6F1-Tg-Hras2 | B6.129-Xpatm1Hvs |
| Primary genetic background | C3HeN and C57B/6N | C57BL/6NTac | FVB/N | BALB/cByJJic and C57BL/6J | C57BL/6J |
| Key reference | Huff, 1999 | Donehower et al., 1992 | Leder et al., 1990 | Saitoh et al., 1990 | van Kreijl et al., 2001; van Steeg et al., 2001 |
| Zygosity | Heterozygous | Heterozygous | Hemizygous for a mutant v-Hras1 transgene | Hemizygous for human HRAS transgene | Homozygous |
| Age (at start of the expt.) | 6 weeks | 6–8 weeks | 7–9 weeks | 7–9 weeks | 6–8 weeks |
| No. male or female mice/dose | 50–60 | 15–20 | 15–20 | 20–25 | 15 |
| No. mice/cage | 10 | 5 | 1 | 5 | 1(male)/>1 (female) |
| Treatment duration | 24 months | 26 or 36 weeks | 26 or 36 weeks | ≤26 weeks | 39 weeks |
| Holding time | – | 2 weeks | 6 weeks | – | – |
| Route of exposure | Feed, gavage, inhalation, skin, drinking water, i.p. | Topical, diet, gavage | Topical or gavage | Gavage, diet, i.p., or water | Topical or gavage |
| Basis for dose selection | Maximum Tolerated Dose (MTD) | MTD from previous cancer bioassay or range finding | MTD from previous cancer bioassay | 2-year bioassay | 4 week dose range finding study with genetically modified mice |
| Number of tissues for necropsy | 30–40 tissues | 40 (nongenotoxic) and 12 (genotoxic) | 12 tissues | 40 tissues | 40 tissues |
| Common spontaneous tumors | Hepatocellular adenomas/carcinomas, malignant lymphomas/leukemias | Lymphomas, osteosarcomas, hemangio-sarcomas | Odontogenic tumors, forestomach and skin papillomas, lung adenomas | Hemangiosarcomas, lung adenocarcinomas, skin papillomas, Harderian gland adenocarcinomas and lymphomas. | Lymphomas, lung adenomas, adrenal tumors |
| Predominant chemicals tested | Wide range of agents | Genotoxic | Genotoxic & nongenotoxic; tumor promoters | Genotoxic & nongenotoxic | Genotoxic carcinogens; carcinogens in general |
The B6.129 denotes insertion of a 129 ESC gene sequence into a B6 embryo creating a congenic mouse.
Use of genetically modified mice in hazard identification
The importance and potential use of GM models for hazard identification of potential carcinogenic agents has been increasingly recognized. This is due in part to a widespread recognition that it is not feasible to conduct the standard two-species/ two-sex lifetime (2-year) carcinogenicity bioassays to assess the possible carcinogenicity of the many thousands of chemicals in commerce and under development. In the original proposed use of the GM models, carcinogens would be identified more efficiently using these sensitive animals resulting in the use of fewer animals (originally 15 per treatment), a more rapid completion of the study (within 26 weeks) and thereby greatly reducing the costs to test a chemical. Ultimately, this should allow a larger number of chemicals to be tested, and expedite the drug development or chemical evaluation process. In addition, it has been proposed that by using human genes (e.g. HRAS) or inactivating genes (e.g. Trp53), which are highly conserved across species, the bioassay results may be more relevant to humans than those obtained in the conventional cancer bioassays (Contrera & DeGeorge, 1998; Jaworski et al., 2005; Luo et al., 2001; Tennant et al., 1999a).
Summaries of the results from the three models (Trp53+/−, Tg.AC and rasH2) that have had substantial evaluation to date have been presented in several reviews (Jacobs, 2005; Pritchard et al., 2003; Spalding et al., 2000; Storer et al., 2010; Tennant et al., 1999a; Yamamoto et al., 1998a). In these reports, a variety of carcinogenic and noncarcinogenic chemicals have been tested with frequently encouraging results. The Trp53+/− model is reported to detect primarily genotoxic carcinogens whereas the Tg.AC and the rasH2 models exhibit a broader range of sensitivity detecting both mutagenic and nonmutagenic carcinogens (Tennant et al., 1999a; Yamamoto et al., 1998a). However, the results are often presented in summary form in categorical (positive or negative) and not quantitative terms. This often obscures key details that are needed to critically evaluate the responses of the GM models. In addition, the models are often evaluated on an individual basis rather than compared as a group. We believe that examining the three GM models simultaneously along with the results of the chronic rodent bioassay can lead to a more encompassing perspective of their usefulness in the hazard characterization process.
Evaluation/validation of genetically modified mouse models
The Trp53+/− and the Tg.AC models have been tested and evaluated using a variety of chemicals by the National Institute of Environmental Health Sciences/National Toxicology Program (NIEHS/NTP) in their carcinogenicity studies (Humble et al., 2005; Tennant, 1998; Tennant et al., 1995, 1996, 2001). NTP has also tested other models such as the chimeric B6.19-P16INK4A/p19ARF using benzene (NTP, 2007a), glycidol (NTP, 2007b) and phenolphthalein (NTP, 2007c). The rasH2 model was evaluated primarily by a group of Japanese organizations comprising the Central Institute for Experimental Animals (CIEA), the National Institute for Health Sciences and several pharmaceutical companies (Yamamoto et al., 1998a), and has been reviewed for its utility for preclinical testing (Storer et al., 2010). The Xpa−/− and/or Xpa/Trp53+/− knockout mouse models were tested by the National Institute of Public Health and the Environment in the Netherlands (van Kreijl et al., 2001; van Kreijl & van Steeg, 1998). The Alternatives to Carcinogenicity Testing (ACT) committee of the International Life Sciences Institute (ILSI) organized study groups comprising governmental institutes, private research groups and more than 30 pharmaceutical companies worldwide to evaluate these GM models. Twenty-one chemicals, mostly pharmaceutical agents, were tested in one or more of the four GM models (Forster, 1998; Gulezian et al., 2000) and the results of the collaborative efforts were published in late 2001 (Cohen et al., 2001; Goodman, 2001; Robinson & MacDonald, 2001). Although the ILSI effort is presented by many as a validation of these GM models (Sills et al., 2001; Tennant et al., 1999a; Yamamoto et al., 1998a), it is more appropriately described as an evaluation because a number of the critical criteria involved in the validation process were not met (NIEHS, 1997). Scientists at the United States Food and Drug Administration (US FDA), in an earlier proposal, suggested the use of a combination of the 2-year rat bioassay with a transgenic mouse assay (Jacobson-Kram et al., 2004). More recently, they have proposed the use of a combination of rodent transgenic assays instead of conducting the 2-year bioassay (Jacobson-Kram, 2010). Following the fourth International Conference on Harmonization (ICH4), the US FDA and its counterparts within the European Union (EU) and Japan have allowed sponsors to submit data for new drug applications based upon a GM mouse model as a replacement for testing in a second species. The European Union, under its Registration, Evaluation, Authorization and Restriction of Chemicals (REACH) program has also permitted the use of GM animal bioassays as alternative test methods in supplementing the conventional 2-year rodent bioassays for hazard identification of carcinogens and noncarcinogens (Wells & Williams, 2009).
One of the initial and critical decisions that must be made in evaluating the ability of the GM models to identify carcinogens is the selection of the standard to which they are to be compared. The purpose of carcinogenicity testing is to identify agents that are potentially carcinogenic to humans, and ideally, the GM models would be compared with agents demonstrated to cause cancer in humans and not with other rodent-only carcinogens. While over 100 human carcinogens have been identified by the International Agency for Research on Cancer (IARC), only a portion of these agents have been evaluated in well-characterized and standardized 2-year animal bioassays. Further, many of these agents represent lifestyle or occupational exposure conditions that cannot feasibly be tested in experimental animal models. In addition, most of the human carcinogens identified to date represent potent agents belonging to a restricted range of chemical classes. Testing only these agents may not allow one to fully assess the ability of the GM models to detect less potent and more diverse types of carcinogenic agents, particularly agents that may be acting through different or novel mechanisms, to which humans are exposed. The common approach has been to compare the results of the GM mouse assays with those obtained from comprehensive 2-year rodent cancer bioassays, supplemented at times with IARC evaluations (Pritchard et al., 2003). This allows the performance of the GM models to be compared with agents that have been tested in rodents and allows direct comparisons to be made on dose levels, target site, species, etc.
There are, however, a number of problems with the use of conventional animal bioassays to predict human cancer risk. These include questions of human relevance resulting from the chronic administration of high maximum tolerated doses, important physiological differences between rodents and humans, the use of rodent strains that spontaneously develop high incidences of tumors in some tissues (e.g. liver), while being ineffective at detecting carcinogen-induced tumors in other tissues (e.g. colon, pancreas and prostate), and changes in body weight and tumor incidence indicative of genetic drift that have occurred in common rodent strains over time. In spite of these concerns, the conventional rodent bioassay remains a critical tool used to evaluate the safety of chemicals. Many of the issues seen with the conventional rodent assays are present and may be compounded when GM animals are considered for use in assessing cancer risks.
The predictability of the chronic rodent bioassay has been estimated by comparing the results seen in mice with those observed in rats (Contrera et al., 1997; Gold et al., 1989; Gray et al., 1995). In these comparisons, approximately 70–75% of agents that are reported to be carcinogenic in mice are also positive for carcinogenicity in rats and vice versa. It has also been shown that interspecies correlations are improved by focusing on agents that induce tumors in multiple species, in both sexes, in multiple tissues, by more than one route of administration, and that are mutagenic or genotoxic (Gold et al., 1989; Gray et al., 1995).
Biologically, the most direct evaluation of these models would be to compare the results in the GM mouse models with the tumor data seen in non-GM mouse bioassays. To do this, we have compared the results obtained in the 2-year NTP mouse bioassay with those obtained in the accelerated bioassays using the Trp53+/−, the rasH2 and the Tg.AC mouse models (Table 2). The data from carcinogenic agents that were not tested by the NTP (e.g. cyclophosphamide, melphalan, diethylstilbestrol), or that were tested only in a rat model (e.g. mirex, oxymetholone), or were tested in mice infected with Helicobacter hepaticus (e.g. triethanolamine) were excluded from the comparison. Of the 52 chemicals compared in the NTP mouse bioassay, 37 were positive and 15 were inactive (including D-limonene which was positive only in the rat bioassay). Overall, as shown in Table 2, the carcinogenic agents ranged from multi-site and trans-species agents (e.g. benzene and 2,3,7,8-tetrachlorodibenzodioxin [TCDD]) to agents with a more limited range of species and targets (e.g. lauric acid diethanolamine, ethyl acrylate).
Table 2.
Summary results and evaluations for the 52 selected NTP-tested chemicals.
| Agent | CAS# | Mouse
|
Trp53+/− | Tg.AC | rasH2 | Rat
|
Human
|
||
|---|---|---|---|---|---|---|---|---|---|
| NTP | Sitea | NTP | Site | NTPb | |||||
| Positives | |||||||||
| 1,1,2-Trichloroethane | 79-00-5 | + | MS | 0 | 0 | − | − | NL | |
| 1,4-Dioxane | 123-91-1 | + | Li | 0 | 0 | e/+ | + | MS | RC |
| 2,4-Diaminotoluene | 95-80-7 | + | MS | e/− | e/+ | 0 | + | MS | RC |
| 4,4′-Thiodianiline | 139-65-1 | + | MS | 0 | 0 | + | + | MS | NL |
| 4-Vinyl-1-cyclohexene diepoxide | 106-87-6 | + | MS | e/+ | 0 | + | + | Sk | RC |
| 5-Nitro-o-toluidine | 99-55-8 | + | He, Li | 0 | 0 | + | − | NL | |
| 6-Nitrobenzimidazole | 94-52-0 | + | Li | 0 | 0 | − | − | NL | |
| 7,12-Dimethylbenzanthracene | 57-97-6 | + | Sk | + | + | 0 | 0 | NL | |
| Benzene | 71-43-2 | + | MS | + | + | + | + | MS | KC |
| Benzoyl peroxide | 94-36-0 | + | 0 | + | 0 | 0 | NL | ||
| Bromodichloromethane | 75-27-4 | + | Li, Ki | − | e/+ | 0 | + | Col, Ki | RC |
| Chloroform | 67-66-3 | + | Li | e/+ | − | − | + | Ki | RC |
| Chloroprene | 126-99-8 | + | MS | − | − | 0 | + | MS | RC |
| Coconut oil acid diethanolamine | 68603-42-9 | + | MS | − | − | 0 | − | NL | |
| Cupferron | 135-20-6 | + | MS | 0 | 0 | + | + | MS | RC |
| Di(2-ethylhexyl)phthalate | 117-81-7 | + | Li | − | − | + | + | Li | RC |
| Diethanolamine | 111-42-2 | + | MS | 0 | − | 0 | − | NL | |
| Dimethylvinyl chloride | 513-37-1 | + | MS | 0 | e/+ | 0 | + | MS | RC |
| Ethyl acrylate | 140-88-5 | + | FS | 0 | − | + | + | FS | DL |
| Ethylene thiourea | 96-5-7 | + | MS | 0 | 0 | + | + | Thy | RC |
| Furfural | 98-01-1 | + | Li | 0 | 0 | + | + | Bi | NL |
| Furfuryl alcohol | 98-00-0 | + | Ki | 0 | − | 0 | + | NOS | NL |
| Glycidol | 556-52-5 | + | MS | − | − | + | + | GS | RC |
| Lauric acid diethanolamine | 120-40-1 | e/+ | Li | − | + | 0 | − | NL | |
| Methylolacrylamide | 924-42-5 | + | MS | − | − | 0 | − | NL | |
| Methylphenidate | 298-59-9 | e/+ | Li | − | − | 0 | − | NL | |
| N-Methyl-N′-nitro-N-nitrosoguanidine | 70-25-7 | + | Sk | 0 | 0 | + | + | RC | |
| o-Benzyl-p-chlorophenol | 120-32-1 | + | Ki | 0 | + | 0 | − | Ki | NL |
| p-Cresidine | 120-71-8 | + | MS | + | + | e/+ | + | MS | RC |
| Pentachlorophenol | 87-86-5 | + | MS | − | + | 0 | + | MS | RC |
| Phenolphthalein | 77-09-8 | + | MS | + | 0 | − | + | MS | RC |
| Procarbazine | 366-70-1 | + | MS | 0 | 0 | e/+ | + | MS | RC |
| Pyridine | 110-86-1 | + | Li | − | − | 0 | + | Ki | NL |
| Reserpine | 50-55-5 | + | MS | − | 0 | 0 | + | Ad | RC |
| TCDD | 1746-01-6 | + | MS | − | + | 0 | + | MS | KC |
| Thio-TEPA | 52-24-4 | + | MS | 0 | 0 | + | + | MS | KC |
| Urethane | 51-79-6 | + | MS | + | + | + | 0 | RC | |
| Negatives | |||||||||
| 1-Chloro-2-propanol | 127-00-4 | − | − | − | 0 | − | NL | ||
| 2,6-Toluenediamine dihydrochloride | 820-40-5 | − | − | − | 0 | − | NL | ||
| 2-Chloroethanol | 107-07-3 | − | 0 | − | 0 | − | NL | ||
| 4-Nitro-o-phenylenediamine | 99-56-9 | − | 0 | 0 | e | − | NL | ||
| 8-Hydroxyquinoline | 148-24-3 | − | − | − | − | − | NL | ||
| Benzethonium chloride | 121-54-0 | − | 0 | e/− | 0 | − | NL | ||
| Diisopropylcarbodiimide | 693-13-0 | − | − | − | 0 | − | NL | ||
| D-Limonene | 5989-27-5 | − | − | 0 | 0 | + | Ki | ||
| D-Mannitol | 69-65-8 | − | − | 0 | − | − | NL | ||
| Oleic acid diethanolamine | 93-83-4 | − | − | e/− | 0 | − | NL | ||
| p-Anisidine | 90-04-0 | − | − | 0 | − | − | NL | ||
| Phenol | 108-95-2 | − | 0 | − | − | − | NL | ||
| Resorcinol | 108-46-3 | − | − | + | − | − | NL | ||
| Rotenone | 83-79-4 | − | − | + | − | − | NL | ||
| Xylenes (mixed) | 1330-20-7 | − | 0 | 0 | − | − | NL | ||
+ Positive; − negative; e/+, equivocal judged as a positive result; e/−, equivocal judged as a negative result; e, equivocal with no additional judgment made; 0, not tested.
i inadequate; Data were not considered reliable since the animals were infected with Helicobacter pylori.
MS, multiple sites; Li, liver; FS, forestomach; Ki, kidney; He, heart; GS, glandular stomach; NOS, nose; Th, thyroid; Sk, skin; Ad, adrenal gland; Bi, bile duct; Col, colon.
KC, known carcinogen; RC, reasonably anticipated to be a carcinogen; DL, de-listed; NL, not listed.
As shown in Table 2, only eight of the NTP-tested chemicals have been tested in all three GM models. Most were tested in only one or two of the GM models, and a moderate number of the GM assays produced equivocal results. The actual number of equivocal results may be higher as the actual test data were not available for many of the rasH2 studies. The equivocal studies were typically a case in which a positive trend was seen in the bioassay that did not attain statistical significance, but was judged to be biologically significant by the researchers. In other cases such as that of dimethylvinyl chloride, the number of tumor-bearing Tg.AC mice was not increased with treatment, but the compound was judged to be carcinogenic based upon an increase in the number of forestomach papillomas per mouse. Alternatively, in other equivocal cases, statistically significant increases were seen, which were deemed to be unrelated to the treatment, being primarily attributed to cage-effects and wounding in the Tg.AC animals. In each case, the investigators’ assessments have been used in comparing the results. However, it should be noted that in almost all cases, the decision made by the researchers, matches the expectation based upon the NTP bioassay results and suggests that prior knowledge may have influenced the interpretation of the data. As an example where prior knowledge clearly influenced the evaluation process, Maronpot and associates (2000) stated in the report of their collaborative study using the rasH2 mouse that “p-Cresidine was also considered positive, primarily because of the preneoplastic changes noted and its known carcinogenicity for urinary bladder (NCI, 1978) rather than the number of actual tumors diagnosed in the present study.”
Concordance between genetically modified and NTP bioassays
The degree of concordance of the three GM bioassays with different categories of chemicals is shown in Table 3. In the Trp53+/− model, the results were concordant for 57% (17 of 30) of the chemicals tested, of which it correctly identified 35% (7/20) of the mouse carcinogens and 100% (10/10) of the mouse noncarcinogens. If the comparison is restricted to those agents that were positive or negative in the NTP bioassays in both mice and rats (trans-species carcinogens/ noncarcinogens), a modest increase in the overall concordance is observed. In this case, the Trp53+/− assay detected 5 of 14 trans-species carcinogens and correctly identified 9 of 9 noncarcinogens for an overall concordance of 61% (14 of 23). For those agents listed by various authoritative bodies (IARC, NTP or the US EPA) as known or likely human carcinogens or reasonably anticipated to be carcinogenic, the Trp53+/− assay was positive for 6 of these 14 carcinogens (43%).
Table 3.
Concordance between genetically modified assays, the NTP bioassays and human evaluations.
| Model | Positive | Negative | Overall |
|---|---|---|---|
| Concordance with NTP mouse carcinogens | |||
| Trp53+/− | 7/20 (35%) | 10/10 (100%) | 17/30 (57%) |
| Tg.AC | 12/23 (52%) | 8/10 (80%) | 20/33 (61%) |
| rasH2 | 16/20 (80%) | 7/8 (88%) | 23/28 (82%) |
| Concordance with trans-species mouse and rat carcinogens | |||
| Trp53+/− | 5/14 (36%) | 9/9 (100%) | 14/23 (61%) |
| Tg.AC | 7/14 (50%) | 8/10 (80%) | 15/24 (63%) |
| rasH2 | 14/16 (88%) | 7/8 (88%) | 21/24 (88%) |
| Concordance with known or likely human carcinogens | |||
| Trp53+/− | 6/14 (43%) | ||
| Tg.AC | 8/12 (67%) | ||
| rasH2 | 12/14 (86%) | ||
For the Tg.AC model, overall concordance was 61% (20/33), which included a positive response for 52% of the mouse carcinogens and had a negative response for 80% of the noncarcinogens. Two chemicals, rotenone and resorcinol, which were negative in the NTP mouse bioassay exhibited strong positive responses in the Tg.AC model. These compounds were also negative in the NTP rat bioassay and the Salmonella reverse mutation assay (NTP, 1992) and are considered to be false positives. They are particularly interesting in that the responses were very strong with 67–100% of the Tg.AC mice exhibiting tumors. Again by restricting the comparison to trans-species carcinogens/ noncarcinogens, the concordance is improved slightly, with 63% of the results being in agreement, and 50% and 80% being classified as carcinogens and noncarcinogens, respectively. However, when the comparison is restricted to known or likely human carcinogens, ~67% of the carcinogens tested were detected by the Tg.AC assay.
Concordance was higher for the rasH2 model where similar results were seen for 23 of the 28 chemicals tested (82%), which included 80% of the carcinogens (16/20) and 88% of the noncarcinogens (7/8) being detected correctly. The results were modestly improved for the trans-species carcinogens/ noncarcinogens with a concordance of 88% (21/24). Twelve of the 14 known or likely human carcinogens (86%) showed positive responses in the rasH2 model. The two inactive compounds were phenolphthalein and chloroform, a multi-site and a single site carcinogen in both mice and rats, respectively.
The overall concordance between the GM assays and the NTP mouse bioassay ranged from 57% to 82%. By examining only trans-species carcinogens/noncarcinogens, the concordance improved with values ranging from 61% to 88%. There were 11 chemicals that induced tumors in the NTP mouse bioassays that were not detected as carcinogens by any of the GM models reviewed. It should be noted that none of the 11 were tested in all three GM models, and the majority were only tested in one of the models. Four of the 11 that were not detected in the GM models were trans-species carcinogens producing tumors in the NTP rat bioassay as well as in the mouse bioassay. Of the four agents, methylolacrylamide, diethanolamine, coconut oil acid diethanolamine and 1,1,2- trichloroethane, that appeared to be NTP mouse-specific carcinogens, all induced liver tumors in the B6C3F1 mouse, a tumor type that exhibits high spontaneous and variable frequencies in this strain of mouse. However, in all four cases, the increases in tumors were seen in both male and female mice, and were accompanied by a significant increase in tumors in at least one other organ. These are characteristics, which usually indicate agents with a higher likelihood of being carcinogenic in multiple species (Gray et al., 1995). Hence these do not easily fit into the category of being mouse liver-specific carcinogens, and are not easily dismissed from consideration. However, if some of these do represent strain-specific carcinogenic agents or if more thorough testing had been performed, then the concordance values for the GM models would likely be improved.
In its earlier initial evaluation of the GM models, the NIEHS/NTP estimated concordance between the three GM bioassays and the conventional bioassays to be 66% (25 of 38) (Bucher, 1998; Pritchard et al., 2003). In the NIEHS/NTP approach, concordance was defined as a positive result in any genetically altered model for a positive rodent or human carcinogen, or negative results in all genetically altered assays for a noncarcinogen. Using this definition, the data in Table 2 produces a similar albeit somewhat better concordance of 75% (39 of 52). This is expected because there is substantial overlap between the two data sets. From an examination of data in Tables 2 and 3, the GM models appear to be less efficient in detecting the carcinogenic agents and more consistent in classifying noncarcinogenic agents. It is also clear that no single model can be relied upon to detect all trans-species carcinogens. While the results for the rasH2 model look promising, it should be noted that the agents selected for testing in this model were investigator-selected and included more potent genotoxic agents than those tested in the other models. For a number of the rasH2 studies, the decisions appear to have been made based on categorical calls by professional judgment rather than on an actual statistical significance (see “Discussion” section), and many of the details, such as reproducibility, treatment and control incidences used to evaluate the data were not presented.
Of note, among the tested agents identified as human carcinogens or likely human carcinogens independent of the test agent’s genotoxicity or immunosuppression, 86% (12 of 14), 67% (8 of 14) and 43% (6 of 14) were detected as carcinogenic in the rasH2, the Tg.AC, and the Trp53+/− model, respectively.
Challenges and concerns with genetically modified mouse models
A number of potential problems and concerns have been recognized with the use of GM animals as a primary method to identify carcinogenic agents (Boverhof et al., 2011; French et al., 2010; Jacobson-Kram, 2010; Lynch et al., 2007; Pritchard et al., 2003). While many studies have investigated the feasibility of using GM models to expedite the identification of carcinogens, there are a number of good reasons to exercise caution in their use and interpretation. Several major challenges and concerns are briefly described in the following sections.
Genetic background
One of the challenges in evaluating and comparing the GM models against each other and comparing them with non-GM cancer models is that, in addition to the genetic modification utilized, there are genetic background differences between the animals used in conventional and short-term cancer studies. For example, most NTP cancer bioassays have been conducted using the (C57BL/6N×C3HeN)F1 or B6C3F1 inbred mouse and the Fischer 344/N rat from independent production colonies. In contrast, the Trp53+/− mouse used in the accelerated bioassay was derived from a Trp53 null mutation introduced by homologous recombination in AB1 murine embryonic stem cells, which were derived from a black agouti 129Sv mouse. The embryonic stem cells were then introduced into a C57BL/6J embryo to produce a B6.129-Trp53tm1Brd congenic chimera that was back-crossed from 5 to 12 generations to create the B6.129-Trp53tm1Brd congenic mouse used in the short-term cancer bioassays (Donehower et al., 1992). Similarly, the rasH2 mice are the F1 offspring of BALB/cByJ female mice outcrossed with GM C57BL/ 6JJic-Tg(HRAS)2Jic hemizygous male mice to produce the CByB6F1-Tg(HRAS)2Jic hemizygous mouse (Yamamoto et al., 1998a). The FVB/NTac-Tg(Hba-x-v-Ha-ras)TG.ACLed mice were created and have been maintained on a FVB/N background (Spalding et al., 2000). Thus, each of the GM models is derived from a different genetic background and as a result may exhibit strain-related differences in response to chemical agents as compared to the NTP B6C3F1 strain, and result in potentially different type 1 and type 2 error rates for predicting human risk for carcinogenesis. As a result of these strain differences, it is difficult to determine if differences in the results reflect the genetic modification itself, the strain, or another factor that varies between the GM and conventional mouse models employed. Similarly, differences in genetic background can also complicate the interpretation of results obtained in the conventional rodent bioassays as well.
Route and type of exposure
A number of other differences can frequently be seen between the NTP bioassays and those conducted using GM animals. For example, the route of exposure may often be different affecting toxicokinetics; the primary route used for exposure in the Tg.AC assay is topical application of the chemical to the skin whereas, in contrast, only approximately 9% of the NTP bioassays have utilized the dermal route (Huff, 1999). While the Tg.AC mouse appears to be sensitive to a range of carcinogens and develops skin tumors in response, it may not detect carcinogens that require organ-specific metabolic activation or that affect specific molecular targets. Although there is some evidence that the dermal application of test agents to the skin of Tg.AC mice may result in tumors in other tissues (Tennant et al., 1996), this has not been rigorously studied. In addition, the doses administered dermally are often much greater than those administered by other routes, and it is not clear how one would relate skin responses following topical exposure with very high doses to those anticipated through other exposure routes. It is also not clear in this model, whether the test agent is activating the transgene or whether there is a selection of cells that constitutively express the mutant RAS protein based upon increased cell proliferation (promotion) alone. For many of the Tg.AC assays, only the skin, the site of application and primary target site, has been examined grossly and for histopathology. Pathological evaluations do not appear to have been performed for other tissues in either the treated or the untreated animals. While this does reflect the screening nature of this model and its intended usage, it may underestimate the sensitivity of the model and limit one’s ability to make accurate target tissue comparisons between the various models.
Of note, the induction and nature of tumors induced appears to vary at times substantially by the route of exposure in the various GM models. For example, the response and types of tumor induced by benzene, a systemic toxicant in humans, is dependent upon the route of exposure in the Tg.AC model. A strong increase in skin tumors was seen with topical treatment whereas no increase was seen by gavage. In contrast, in the Trp53+/− model the administration of benzene by gavage resulted primarily in sarcomas whereas inhalation exposure resulted in lymphomas. As another example of an exposure-related effect, administration of bromodichloromethane by gavage in the NTP 2-year bioassay resulted in kidney tumors in male mice and liver tumors in female mice (NTP, 1987). However, bromodichloromethane did not induce any tumors in Trp53+/− or TG.AC transgenic mice when administered by gavage (NTP, 2007d). Of note, bromodichloromethane was inactive in both the 2-year and GM bioassays when administered in drinking water (NTP, 2006, 2007d). The differences between the GM and conventional models make it difficult to determine whether the differences in assay results are due to the genetic modification or due to factors, such as the type and route of exposure or the genetic background.
Mechanistic basis of bioassay results and relevance to humans
Although experience has shown that these mouse models respond rapidly to many carcinogenic compounds, the mechanistic basis underlying the results and their relevance to humans in many cases is not clear. Carcinogenesis is a highly complex biological process occurring by numerous mechanisms and affecting many different tissues and organs (Johnson, 1999). While genetic modifications may enhance susceptibility and decrease the time required for detecting certain lesions in specific tissues in certain genotypes, it is not clear how such restricted gains in susceptibility translate to overall predictions that are in any way as complete or accurate as the results obtained from natural whole (non-GM) animal systems.
The integration and copy number of transgenes can alter gene expression not only for the transgene but also for other wild-type genes (Rudmann & Durham, 1999). This is not surprising as the two most common genes targeted in the GM models, RAS and TP53, are master regulatory genes that control and/or influence many other genes and pathways within the cell (Malumbres & Barbacid, 2003; Menendez et al., 2009; Pylayeva-Gupta et al., 2011). One of the concerns expressed in the application of GM models for hazard identification is the possibility of undesirable effects of the genetic modification on endogenous genes and their expression (Gollapudi et al., 1998). As a consequence, the observed increases in tumors or a lack of tumorigenic response may be due to alterations in the mouse occurring secondary to the transgene expression or the gene inactivation. Even though the Trp53+/− mouse is seen as having a relevant mechanistic basis, many of the tumors induced in this model do not always exhibit the expected loss of the wild-type allele, suggesting that loss of another tumor suppressor gene allele or another mechanism related to haploinsufficiency may be responsible for many of the induced tumors (French et al., 2001b; Venkatachalam et al., 1998, 2001).
Uncertainty has also been expressed about the role and significance of integration of 5 or 6 copies of the reconstructed human HRAS gene into the CB6F1 mouse with respect to the relevance of this model to human carcinogenesis (Blain et al., 1998). In addition, point mutations within the HRAS transgene or the native murine Hras1 or Kras genes, the presumed basis for the enhanced sensitivity of these animals, do not appear to play an important role in treatment-induced lung tumors in the genetically modified rasH2 mice whereas mutations in Kras do occur frequently in tumors induced by the same chemical in non-GM mice from the same litters (Mitsumori et al., 1997; Yamamoto et al., 1998a). This indicates that, in some cases, the mechanisms underlying the chemically-induced cancers in the GM animals differ fundamentally from those occurring in non-GM mice, while in others they are very similar. Similarly, the ability of the mutated Tg.AC (vHras1) mice to respond to carcinogens that exhibit a high degree of organ specificity in non-GM animals, suggests that the skin tumors produced in this model may be occurring through entirely different mechanisms (Ashby, 1997). At this time, there is relatively little known about the nature of the genetic changes in tumors induced by many of the chemicals active in the GM models. Thus, mechanistic differences in responses between GM and non- GM animals can create serious problems from a risk assessment perspective, and emphasize the need for further research on carcinogenic mechanisms in the GM mouse models.
Pathway-specificity of response
Another major concern identified by a number of investigators (Bucher, 1998; Clarke, 2000; Goldsworthy et al., 1994) stems from the nature of the model. The rapid onset of carcinogenesis in these modified animal strains is largely determined by the specific genetic alteration in the test animal. Although the Hras and Trp53 genes are clearly altered in tumors of various organs in both rodents and humans, many tumors arise without alterations in these genes. Chemicals that act through pathways independent of Hras or Trp53 may not cause an accelerated tumor onset in these GM models. This is illustrated using the Trp53+/− mouse as an example, but similar arguments can be made for models involving Kras or other cancer-related proto-oncogenes or tumor suppressor genes.
Although a key mutation in the Trp53 gene accelerates the cancer process in the Trp53 protein haploinsufficient mice, the mutation or the resulting haploinsufficiency can affect other cellular processes (e.g. metabolism, DNA repair, gene expression), which in turn could result in a different spontaneous or carcinogen-induced response in the p53+/− mice as compared to wild-type mice. For example, studies by Carmichael et al. (2001) have shown that the short-term administration of the synthetic estrogen diethylstilbestrol (DES) induced higher levels of DNA adducts as well as higher cytochrome P450 (CYP450) enzyme levels in Trp53+/− mice as compared to similarly treated wild-type mice (Carmichael et al., 2001). The CYP450 levels in untreated Trp53+/− mice were also increased when compared to those of wild-type mice. Similarly, the expression of genes involved in cell cycle regulation, signal transduction, apoptosis, and transcription also appear to be significantly different between DES-treated p53 heterozygous mice as compared to DES-treated wild-type mice (Salleh et al., 2004). Other genotoxic and other cellular responses (e.g. telomere length) have also been reported to differ to some extent between Trp53+/− and wild-type mice following treatment with agents, such as benzene, melphalan and azidothymidine (AZT) (Dobrovolsky et al., 2007; Healy et al., 2001; Sgura et al., 2008).
TP53 gene mutations are the most common alterations detected in human neoplasia affecting approximately 50% of human cancers. However, this implies that 50% of human cancers arise through TP53-independent pathways (as well as pathways in which the involvement of TP53 may be indirect). Similarly, tumors in the Trp53+/− mice can arise through both Trp53-dependent and independent pathways and approximately 50% of the tumors arising spontaneously in Trp53+/− mice appeared to retain an intact functional Trp53 allele (Venkatachalam et al., 2001). This was illustrated by loss of heterozygosity (LOH at the Trp53 locus) in almost all tumors following treatment with some carcinogens (i.e. benzene and phenolphthalein), but rarely with other agents (i.e. p-cresidine). Their preliminary data suggested that “LOH is dependent on both the mechanism of genotoxicity of the agent utilized and the tissue type targeted” (Venkatachalam et al., 2001). As a result, agents inducing cancer through these other pathways may not be detected in an accelerated bioassay using the Trp53+/− model. For example, in the 2-year bioassay in the B6C3F1 mouse, phenolphthalein was shown to increase the incidence of both lymphomas and ovarian tumors (NTP, 1996). Alterations in the Trp53 protein (detected immunochemically) were seen in the thymic lymphomas of the phenolphthalein-treated B6C3F1 mice indicating an involvement of the Trp53 gene in the development of this tumor (Dunnick et al., 1997). However, similar effects were not seen in the ovarian tumors of the treated mice. When tested in the Trp53+/− model, thymic lymphomas readily developed in the phenolphthalein-treated animals. However, no increase in ovarian tumors was observed in the 26-week accelerated cancer bioassay. These results support the hypothesis that the Trp53+/− mouse is efficient at detecting agents inducing tumors through a Trp53- dependent pathway but may not identify those developing along Trp53-independent pathways.
Similarly, studies by Medina and associates (2002) indicated that some carcinogenic agents that act through a Trp53-independent mechanism will not produce an accelerated response in the Trp53+/− model (Goldsworthy et al., 1994; Jerry et al., 1994). Female B6.129 chimeric Trp53+/− and sibling wild-type mice administered 7,12-dimethylbenzanthracene (DMBA) by gavage and given pituitary isografts to provide additional hormonal stimulation, developed mammary tumors and other tumors at approximately the same rate. Haploinsufficiency of Trp53 did not alter the incidence or the latency of the DMBA-induced tumors. Moreover, Southern blot analysis detected no gross alterations in the wild-type Trp53 allele in the mammary tumors from the Trp53+/− mice. Single-strand conformation polymorphism analysis of Trp53 exons 5–8 identified a mutation in only one of seven mammary tumors analyzed. These data indicate that Trp53 is not a common target for the mutagenic effects of DMBA in this mammary carcinogenesis model. Although in this case the carcinogenic effects of DMBA were detected in both the GM and the non-GM animals, it is possible that a less potent carcinogen acting through the same pathway would not be detected in an accelerated bioassay using the Trp53+/− model. Similar results have been seen with other agents in the Trp53+/− and the rasH2 models where the genetic modification in the animals did not seem to contribute to the tumorigenic effects (Donehower, 1996; Kemp, 1995; Yamamoto et al., 1998a,b). However, in most cases where comparisons have been made, the genetic alteration does seem to play a critical role in the tumorigenic effects (Yamamoto et al., 1998a,b).
In the initial evaluation of the GM models, Bucher (1998) expressed concern as to “why the models seem to be ‘blind’ to carcinogens producing tumors at certain sites.” He continued by stating “This seeming deficiency of the new models may actually be an advantage if the tumors missed are, for any reason, not predictive of human responses. Alternatively, the findings may suggest critical insensitivities of the new models to potentially important human health risks” (Bucher, 1998). For most agents, the number of organs showing increases in tumors is markedly lower in the genetically modified B6.129-Trp53tm1Brd mouse than in the NTP B6C3F1 mouse bioassay. Other investigators have raised concerns that the new GM models may be insensitive to chemicals inducing tumors in organs, such as the mammary gland, the lung, and the liver in the Trp53+/− model (Dass et al., 1999; Kemp, 1995) and the liver in the rasH2 model (Yamamoto et al., 1998a) due to differences in inbred strain susceptibilities. Similar information is not available for the Tg.AC model, as histopathology is typically not performed on tissues other than the skin. It is not clear at this point whether the insensitivity of these models to tumors arising in specific organs is a result of strain-related background differences in heritable susceptibility of target tissues to the test agents, the involvement of genes other than Trp53 or Hras, differences in the extent of histopathological examination, or other factors.
Uncertainty of the genetically modified bioassay results due to methodological issues
Another concern with the GM models involves the equivocal or ambiguous outcomes often seen in the accelerated cancer bioassays. For example, in the summary of the rasH2 bioassay results presented by Yamamoto et al. (1998a), 34% of the bioassay results were not definitive. For five of the agents, at least one type of tumor in the treated mice had an incidence of >13% but less than 25%. Another seven chemicals showed tumor incidences of ≥25%, but which were not significantly increased as compared to their respective controls. In other cases, additional factors, such as the presence of hyperplasia, a pre-neoplastic lesion in the presumed target tissue, played a role in the decision for agents, such as p-cresidine (Maronpot et al., 2000). As indicated previously, this needs to be considered when evaluating the various models, as different interpretations of the data can significantly alter the concordance seen between the assays. Equivocal findings are also a significant problem for the Tg.AC model, affecting approximately 20% of the assay results. The equivocal results in the GM assays seem to arise primarily from factors, such as inadequate sample sizes affecting sensitivity, decreased sensitivity due to shortened study duration, spontaneous tumor frequencies, genetic instability of the GM models, and lack of study-to-study reproducibility. Each of these will be briefly discussed below.
Sample size effects on sensitivity
As initially proposed, the GM bioassays employed 15 animals per treatment, a 70% reduction from the 50 animals used per treatment and sex in the NTP and other conventional cancer bioassays (Eastin et al., 1998). As animals of only one sex are often used, this represented an additional reduction in the number of animals tested per treatment. A number of factors seem to have led to the use of the limited number of animals. Based on initial studies with potent carcinogens and the fact that the animals were already “genetically initiated,” it was believed that the GM animals would exhibit greatly increased sensitivity and that fewer animals would be required. In addition, cost considerations and the limited number of GM animals available as testing began, supported the move towards smaller sample sizes. However, these limited sample sizes often lead to less-than-definitive study results and will likely create problems during the hazard identification and dose-response stages of the risk assessment process. For example, consider the case with 2,4-diaminotoluene, a likely possible human carcinogen and mutagenic agent in Salmonella. When tested by the National Cancer Institute (NCI), this agent was a fairly potent carcinogen in the B6C3F1 female mouse inducing hepatocellular carcinomas in 0/19, 13/47 and 18/46 and lymphomas in 2/19, 29/47 and 11/46 of the treated animals at the 0, 79 and 171 ppm doses, respectively (NCI, 1979). In the GM bioassays, this agent was weakly active in the Trp53+/− mouse with 1 of 15 males and 2 of 15 female animals exhibiting tumors (lymphomas) in the two treatment groups versus 0 of 30 in the controls (Eastin et al., 1998). It was also judged as weakly positive in the Tg.AC mouse due to the occurrence of squamous cell carcinomas of the skin that were seen in 2 of 15 treated male animals – a lesion not seen in historical control males. The combined incidence of squamous cell papillomas and carcinomas in the male Tg.AC animals was 0/15 in the controls and 3/15 in the 30 mg/kg dose group. This increase is not statistically significant (p=0.11, 1-tail Fisher exact test). In the absence of the conventional bioassay data, it is not clear how these GM mouse results would be viewed in making a hazard identification decision because significant increases in tumors were not seen. In contrast, if the same frequency of tumors were seen in a bioassay employing a larger number of animals (e.g. 0/45 in the controls versus 9/45 in the treated), the results would have been clearly significant. In this case an increase in sample size would support the researchers’ conclusions. However, for others, such as 2-chloroethanol (Spalding et al., 1999), phenol (Spalding et al., 1993), 2-(chloromethyl)pyridine, 4-nitro-o-phenylenediamine, and tetraethylthiuram disulfide (Yamamoto et al., 1998a), an increase in sample size may have led to results differing from those seen in conventional rodent bioassays. As emphasized by Gulezian and colleagues, “there is a clear need to incorporate sound statistical principles in the design of carcinogenicity studies using GM animals to ensure that the study yields reliable and meaningful results” (Gulezian et al., 2000).
To address this problem, the NTP and other groups have begun using 25 GM animals per dose. For example, in the recent studies with AZT alone and in combination with other antiviral drugs (NTP, 2012a) and Senna (NTP, 2012b), the NTP used a sample size of 25 GM mice per treatment. In the AZT combination studies, the increased sample size did facilitate the detection of liver tumors induced by AZT in the Trp53+/− mice. However, with other treatments the observed increases in tumors were still considered to be equivocal due to sample size limitations combined with the modest size of the observed tumor increases when compared with the controls.
Decreased sensitivity due to shortened study duration
One of the appealing aspects of the accelerated bioassays is that they would require only a six-month exposure rather than the 24 months associated with the conventional rodent cancer bioassay. In addition to reducing the time and costs to conduct the assay, the reduced study duration would also reduce confounding due to spontaneous tumors affecting the mouse model. Spontaneous tumors inherent to a particular strain, such as liver tumors seen in the B6C3F1 mouse, generally develop late in the lifetime of the animal. Because carcinogenesis is enhanced in the GM models, tumors induced by a test agent would be identified with minimal interference from mouse-specific spontaneous tumors (Tennant et al., 1999a). An approximation of the time-response curves for spontaneous and chemically induced cancer in GM and non-GM mice is shown in Figure 1. With potent carcinogens, the induction of tumors often occurs earlier with the time-response curve for induced tumors being shifted to the left. The induction of tumors by less potent carcinogens or at lower doses may take longer and the time-response curve would occur closer to that of the spontaneous tumors. The initial tests of the GM assays were conducted with potent model carcinogens and the induced tumors often appeared within several months of treatment. Some of the earlier GM assays used very short exposure periods (Spalding et al., 1999), and the duration of the Trp53+/−, Tg.AC, and rasH2 bioassays was standardized to 26 weeks (Tennant et al., 1999a; Yamamoto et al., 1998a). However, with additional experience, it appears that the 26-week exposure period may not be sufficient to confidently identify a significant number of carcinogenic agents in the Trp53+/− and rasH2 models (Dass et al., 1999; Finch et al., 1998; Maronpot et al., 2000; Marsella et al., 1997). Increasing the study duration has been recommended by these and other investigators (Yamamoto et al., 1998b). As a result, in recent years the NTP has often conducted both 26 and 39-week (or longer) studies when using GM animals in its expedited cancer bioassays (NTP, 2012a). However, increasing study duration may prove to be problematic as the incidence of spontaneous tumors may increase with longer study duration and interfere with the sensitivity of the assay.
Figure 1.
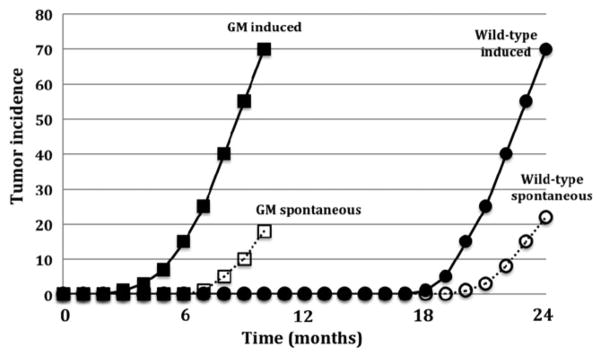
Generalized time-response curves for spontaneous and chemically-induced tumors in genetically modified and wild type mouse bioassays. (Adapted from Tennant et al., 1999a. Used with permission from the CIIT Centers for Health Research (CIIT) and Byron E. Butterworth).
The time course for induced versus spontaneous tumors is a critical parameter for the Trp53+/− model (Tennant et al., 1999a). Yet, few studies have been completed in the intermediate 6–12 month time frame. Fundamental time course studies are needed to maximize the response to weaker carcinogens and detect tumors occurring at lower doses while still avoiding interference by background tumors, and thus better define optimal exposure times for the assay. Changing the duration of the GM assays will require a significant change in the assay as it will necessitate deriving new data sets on the incidences of spontaneous tumors at these later time points. However, it should be emphasized that some less potent genotoxic carcinogens or non-genotoxic carcinogens may have latency periods longer than 26 weeks, and assay durations will need to be extended to confidently identify these carcinogens (Tennant et al., 1999a).
Spontaneous tumor frequencies
Another factor that can often lead to equivocal or ambiguous results relates to the high frequency of tumors occasionally seen in untreated control mice. Although the incidence of spontaneous tumors is generally low for the GM models at the end of a 26-week assay, elevated frequencies of tumors in some organs have been seen (Gulezian et al., 2000; Mahler et al., 1998; Mitsumori et al., 1998). In particular, elevated frequencies of skin tumors have been commonly seen in the Tg.AC model, and appear to stem from wounds caused by fighting among cage mates and the establishing of dominance in the cage (Spalding et al., 1999; Tennant et al., 1999a). While untreated, singly housed Tg.AC mice infrequently develop spontaneous skin tumors, it was common practice when the assay was first used to group house the female mice in the accelerated bioassays. In the studies of Tg.AC mice, skin tumors due to wounding among cage mates have been a frequent occurrence, at times affecting 50% or more of the untreated animals. For example, Spalding et al. (1999) reported control skin tumor incidence in two sets of acetone-treated control groups to be 60% (3/5) and 29% (4/ 14), where as in an ethanol-treated control mice it was 13% (2/15). Among cage mates that fight, the frequencies of skin tumors have been reported to be 57% (8/14) in the controls and approached 100% (15/15) among 12-O-tetradecanoylphorbol-13-acetate (TPA)-treated female Tg.AC mice (Tennant et al., 1999a). This exceeds the historical incidence of skin tumors, which has been reported to be 15%±20% in the controls and 95%±4% among the TPA-treated mice (Tennant et al., 1999a). These high and variable frequencies create significant difficulties in interpreting the bioassay results. However, these were considered to be due to wounding and the test agents were judged to be noncarcinogenic by the investigators (Spalding et al., 2000; Spalding et al., 1999). Another case, a study in which 50% of the dermally treated mice developed skin tumors, was concluded to be non-tumorigenic presumably for the same reason (Tennant et al., 1995). Similarly, control and treated animals dosed by gavage exhibit high frequencies of forestomach tumors which are believed to be due to chronic irritation caused by the gavage procedures (Tennant et al., 1999b). As a consequence, additional factors, such as the number of tumors per animal and the average latency period are used to determine whether a bioassay is considered positive (Dunson et al., 2000; Spalding et al., 2000). In some cases, agents have been judged to be carcinogenic in the Tg.AC assay based on these other factors even when high and similar frequencies of tumors have been seen in both the control and treated animals (Cannon et al., 2000).
Hematopoietic tumors, such as malignant lymphoma and soft tissue sarcomas are common in untreated Trp53+/− mice. Interestingly, recent reviews have noted that there is considerable difference in the spontaneous tumor spectrum between heterozygous Trp53+/− mice as compared to homozygous Trp53+/+ wild-type and Trp53−/− null mice (Donehower & Lozano, 2009). As highlighted in the review, Trp53+/− mice mainly showed B-cell lymphomas, osteosarcomas, soft tissue sarcomas, and a few carcinomas, while Trp53−/− mice exhibited primarily thymic T-cell lymphomas and soft tissue sarcomas. The wild-type Trp53+/+ mice showed predominantly B-lymphomas, but also exhibited to a lesser degree the three other types of tumors seen in the heterozygous Trp53+/− mice.
Although not as common, concerns were initially raised about the incidence of spontaneous tumors (which ranged between 10% and 20%) in the rasH2 model (Van Zeller & Combes, 1999). Hemangiosarcomas of the spleen (7.5% in females and 3.9% in males) and lung adenomas/adenocarcinomas (7.5% in females and 7.2% in males) were the most common spontaneous tumors to appear within the 26-week study period (Mitsumori et al., 1998). However, more recent 26-week studies have indicated that in most cases the spontaneous tumor incidences in the individual tissues are relatively low (typically under 5%) and have been remarkably stable over time (Nambiar et al., 2012).
It should be noted that the use of small numbers of animals combined with the relatively short assay duration increase the likelihood of obtaining negative results, particularly when testing weaker carcinogens, lower doses of potent carcinogens, or non-genotoxic carcinogens. In addition, elevated frequencies of spontaneous tumors decrease the power of the assay and consequently increase the likelihood that treatment-related increases will go undetected. While high and variable levels of spontaneous tumors could also increase the number of false positives, this could be offset by a dismissal due to concerns about relevance for treatment-related tumor increases in organs known to have high and variable spontaneous tumor levels.
Genetic instability of the genetically modified models
Another serious concern involves the genetic stability of a GM models created by random insertion of a concatenated multiple copy transgene. Changes in transgene copy number is a critical issue for experimental animals to be used in a standardized short-term bioassay and has been a particular problem with the Tg.AC mouse. In early studies, a subset of Tg.AC mice, hemizygous for the v-H-Ras transgene, were found to be non-responsive to TPA, a strong tumor promoter used as a positive control, which induces high frequencies of skin tumors in this mouse upon dermal application (Thompson et al., 1998; Weaver et al., 1998). Subsequent studies revealed two separate types of alterations in these and other non-responding mice (Honchel et al., 2001). The group of non-responding hemizygous mice had lost a diagnostic 2-kb BamH1 fragment of the transgene containing the zeta-globin promoter sequence. Another group of non-responding homozygous mice had a small asymmetric deletion in the same critical 2-kb BamH1 zeta-globin promoter fragment. It appears that the deleted regions form part of a palindromic structure that is critical for the responder phenotype and which may also underlie the genetic instability observed in this mouse as well, as other uncharacterized non-responding variants that have also been seen (Honchel et al., 2001). These results indicate that genetic instability is likely to be an inherent characteristic of this model and that vigilant monitoring of offspring will be needed for the breeding colonies of Tg.AC mice. Additionally, it was originally reported that the Tg.AC mice had 3 to 10 copies of the transgene per allele (Leder et al., 1990). However, subsequent studies have indicated that approximately 40 copies of the transgene per allele were present in the Tg.AC mice (Honchel et al., 2001; Thompson et al., 1998). It is believed that the palindromic orientation of the transgene is also probably responsible for the amplification of transgene copy number, leading Honchel et al. (2001) to state that freezing embryos will be essential to preserve the phenotypic characteristics of the present genotype. The genetic instability of this model is a serious concern that will have to be managed carefully if this model is to be used as a standardized assay to identify new carcinogenic agents.
There has also been concern expressed about genetic variation and study-to-study reproducibility (Figure 2) due to incomplete inbreeding of the B6.129-Trp53+/− N5 mouse (backcrossed for five generations). Within the ILSI study, one laboratory observed no response in both the male and female Trp53+/− mice and another saw an increase in the males but not the females when using a positive control carcinogen, even though they were using the same protocol as laboratories where effects were observed (Storer et al., 2001; Van Zeller & Combes, 1999). The difference in response may be attributed to either genetic drift within the small population of p53 breeding stocks although other explanations such as differences in dosing or dose formulation are also possible. More recently, the NTP has worked to minimize genetic variation by increasing the animal numbers and the use of F1 hybrids. NTP has recently used an outcross between female C3H/ HeNTac (C3) mice and male B6.129-Trp53tm1Brd (N12), designated as C3B6.129F1-Tp53tm1Brd+/−, to create an improved GM model for studying carcinogenicity and perinatal effects. The advantages of this new model are that the mother is more hospitable, the litter size is greater and a broader tumor spectrum can be measured. When compared to the parental strain C3H/HeN or the wildtype B6C3F1, the C3B6F1 Tp53+/− model detects hepatocarcinogens with comparable sensitivity and was also able to differentiate between promotional hepatocarcinogens, peroxisome proliferator- activated receptor agonists, potent genotoxicants, and epigenetic agents (NTP, 2012a). These initial results with the C3B6.129F1-Tp53tm1Brd+/− mice, suggest that this GM model has promise for testing both carcinogenic and noncarcinogenic chemicals.
Figure 2.

Urinary bladder incidence in B6.129-Trp53tm1Brd haploinsufficient mice (N5) treated with aromatic amine, p-cresidine, at 400 mg/kg, 7x/week, by gavage, as a positive control showing variation observed within and between laboratories. All vehicle controls were negative. Black – Males; Grey – Females. Derived from (Storer et al., 2001).
Reproducibility concerns
Another serious concern involves the lack of reproducibility that has at times been seen with the GM models. Although some laboratories have reported good reproducibility in results (Tennant et al., 1996), significant variability and, at times a lack of response, has been seen by others in a number of studies. This was particularly notable in the benzene bioassays where, in contrast to expectations and published reports (Table 4), some investigators observed reduced responses or no response using the Trp53+/− or the Tg.AC mice (Holden et al., 1998; Storer, 1998; van der Laan, 2000). In the Tg.AC mice, the difference was believed to be due, at least in part, to the use of hemizygous mice rather than mice homozygous for the transgene (Holden et al., 1998). However, other reports are not consistent with that explanation (Spalding et al., 1999). Significant variability in results has also been seen with the bladder carcinogen, p-cresidine, used in this Trp53+/− model as a positive control where significant intra-laboratory and inter-laboratory variability in tumor incidence has been seen (Figure 2) (Storer, 1998; Storer et al., 2001).
Table 4.
Effect of route of exposure on the tumor nature by benzene in tested in the National Toxicology Program chronic rodent bioassays and those observed in short-term genetically modified bioassays.
| Sex | Route | Assay | Target tissue | Dosea | Responses | Resultb | Study |
|---|---|---|---|---|---|---|---|
| M | Gavage | 2-year | Zymbal gland | 0, 25, 50, 100 mg/kg | 0/43, 1/34, 4/40, 21/39 | + | NTP, 1986 |
| Lymphoma | 4/49, 9/48, 9/50, 15/49 | ||||||
| Lung | 10/49, 16/48, 19/50, 21/49 | ||||||
| Harderian gland | 0/49, 9/46, 13/49, 11/48 | ||||||
| Preputial gland | 0/21, 3/28, 18/29, 28/35 | ||||||
| F | Gavage | 2-year | Zymbal gland | 0, 25, 50, 100 mg/kg | 0/43, 0/32, 1/37, 3/31 | + | |
| Lymphoma | 5/49, 24/45, 24/50, 20/49 | ||||||
| Lung | 4/49, 5/42, 10/50, 13/49 | ||||||
| Ovary (mixed) | 0/47, 1/44, 12/49, 7/48 | ||||||
| Ovary (granulosa) | 1/47, 1/44, 6/49, 7/48 | ||||||
| Mammary (carcinoma) | 0/49, 2/45, 5/50, 10/49 | ||||||
| Mammary (carcinosarcoma) | 0/49, 0/45, 1/50, 4/49 | ||||||
| F | Topical | Tg.AC | Skin | 0, ≤400 μl/week | 77% in treated | + | Tennant et al., 1995 |
| M | 0, 300, 450 μl/week | 0/10, 0/10, 3/10 | ec | Holden et al., 1998 | |||
| F | 0/10, 1/10, 1/10 | ec | |||||
| F | 0, 400, 800, 1600 μl/week | 3/5, 7/10, 8/10, 10/10 | + | Spalding et al., 1999 | |||
| F | 0, 75, 150, 300, 450, 600, 750 μl/week | 0/19, 3/13, 3/14, 12/15, 14/14, 15/15, 15/15 | + | Tennant et al., 1999a | |||
| F | Leukemia (granulocytic) | 0, 450, 800 μl/week | 0/19, 4/14, 11/15 | + | Furst et al., 2000 | ||
| F | Gavage | Tg.AC | Forestomach | 0, 50, 100, 200 mg/kg | 15/20, 10/13, 19/19, 14/18 | − | Tennant et al., 1999a |
| M | Gavage | Trp53 | Sarcoma | 0, 100, 200 mg/kg | 0/29, 7/20, 16/39 | + | French et al., 2001a |
| Lymphoma | 0, 100, 200 mg/kg | 0/29, 1/20, 3/39 | ed | ||||
| Sarcoma | 100 mg/kg | 19/39 | + | Hulla et al., 2001 | |||
| M | Gavage | rasH2 | Lung | 0, 50, 100 mg/kg | not reported | + | Yamamoto et al., 1998a |
| F | Forestomach | ||||||
| M | Inhalation | Trp53 (C57BL/6) | Hematopoietic neoplasia (primarily lymphomas) | 0, 33, 100, 300 ppm | 9/24, 11/27, 9/25, 23/26, 6/24, 20/24, 24/24e | + | Kawasaki et al., 2009 |
| Trp53 (C3H/He) | 0, 100, 300 ppm | + |
Administered five times per week unless otherwise indicated.
+ positive; − negative; e equivocal.
In the report, male and female animals were pooled for the analysis. Reported as a positive response.
Not significantly increased but trend is consistent with increases seen by other investigators where benzene exposure occurred by inhalation (Boley et al., 2000).
Kawasaki et al. (2009) indicate that 25/24 (104.2%) of the mice developed hematopoietic neoplasia.
Fewer studies from different laboratories have been published for the rasH2 model but some inconsistency in results has also been seen. In one of the few inter-laboratory comparisons, good concordance between the Japanese and the U.S. labs was reported (Maronpot et al., 2000). However, in the case of cyclosporin A, the two groups saw a significant difference in response. While this might be expected given the number of comparisons performed, the overall evaluation changed from a positive (>25% of animals affected but not statistically significant) in previous reports (Yamamoto et al., 1998a, b) to a negative response (Maronpot et al., 2000).
It is likely that factors noted above, such as small sample size, short assay duration, frequency of spontaneous tumors, and genetic variability are likely to have contributed to the variability in the results seen. In addition, other factors, such as route of administration or vehicle may play a significant role. In the Tg.AC model, modifications in the solvent vehicle have been shown to have pronounced effects on the response to TPA (Furst et al., 2000; Stoll et al., 2001). The use of acetone-olive oil or acetone:dimethyl sulfoxide or DMSO (4:1) as solvent vehicles dramatically reduces, and in the case of acetone-olive oil, eliminates the skin tumor response in TPA-treated Tg.AC mice. This suggests that these or other solvent vehicles used for test chemicals may also significantly influence the response in this model. This would be a particular concern when working with previously untested chemicals.
Hazard identification issues
In spite of the concerns discussed above, the GM assays as have been performed to date under the conditions described, appear to be moderately efficient in detecting carcinogens and very effective in correctly classifying non-carcinogens. It is clear in many ways that new bioassays using these GM models require further development and validation against benchmark carcinogens acting through a broader range of mechanisms or modes of action. Some limitations such as the limited pathways affected by the genetic modification and the restricted numbers of tissues affected, are intrinsic to the model and related to differences in genetic background and susceptibility of different target tissues. However, others, such as sample size and study duration are methodological in nature. The methodological issues can be addressed relatively easily with changes in study design and protocol, and the NTP has begun to address these issues. However, it will be more difficult to minimize or eliminate the intrinsic weaknesses of the models. One approach which has been recommended (Tennant et al., 1995) is to conduct tests using two or more GM models. In reviewing the three different GM models (i.e. Trp53+/−, TG.AC and rasH2), it was reported that when Trp53+/− and rasH2 models were used in combination they were able to accurately identify up to 90% of the chemicals tested in the conventional 2-year rodent bioassay (Pritchard et al., 2003). By using models involving commonly altered pathways, which affect many tissues, the problems inherent in a specific GM model should be minimized. However, there is a concern that the tumors in the GM animals may arise from different mechanisms or be unique to a specific genetic background that differs from those occurring in non-GM animals.
Limitations in the use of genetically modified mice in dose-response assessment
While one can easily envision how GM models can contribute to the hazard identification process, the use of GM animal data in dose-response assessment and for quantitative risk estimation is more problematic. There are five major issues to be addressed in the dose-response stage of a risk assessment. These involve identifying the shape of the dose-response curve, variable tumor incidence in different animal models, a consideration of high dose to low dose extrapolation, extrapolation from one route of exposure to another, and animal to human extrapolation. Each of these is addressed briefly below.
Dose-response analysis
The complex molecular and cellular events that underlie cancer and noncancer toxicity are likely to be both linear and dose-transitional. The dose response analysis commonly involves a determination of whether the shape of the dose-response is linear (no threshold) or is non-linear and exhibits another shape such as one with an apparent threshold. For example, it could be proposed that phenolphthalein exhibits a threshold based on the shape of the dose-response curve in the treated Trp53+/− mouse (Figure 3). However, given the small sample sizes typically used in the accelerated bioassays, it is unlikely that such a conclusion could confidently be reached. In general, the fewer animals used for each treatment, the less likely the assay will be to detect an increase in tumors, and the more likely it will be to have a zero response, particularly in the low dose region. This might be used to erroneously conclude that the agent worked through a threshold mechanism when the phenomenon could be more related to variability inherent with small sample sizes. A similar situation can be seen with urethane in which the dose response curves for both hemangiosarcoma/hemangioma and lung tumors in the GM assay appeared to be strongly nonlinear with an apparent threshold (Figure 4). However, no evidence for a threshold was seen in the 2-year NTP mouse bioassay when the incidence of these same tumors was examined (see the Section “Discussion”).
Figure 3.

Phenolphthalein-induced tumors in female mice in the Trp53+/− and NTP bioassays (Dunnick et al., 1997; NTP, 1996).
Figure 4.
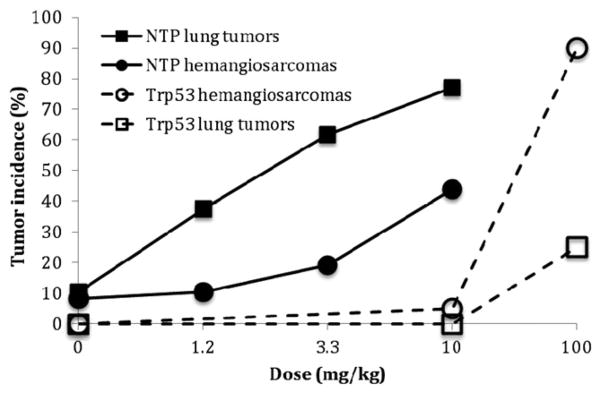
Hemangiosarcoma/hemangioma and lung tumors induced by urethane in male mice in the Trp+/− model when administered by gavage (Carmichael et al., 2000) and in the NTP 2-year mouse bioassay through the drinking water (NTP, 2004).
One of the appealing arguments for the use of the accelerated cancer bioassays is that with their increased sensitivity and reduced costs more doses can be evaluated, providing a more accurate estimation of the dose-response curve. In addition, because highly conserved genes have been modified in the GM animals, it has been proposed that the results may be more relevant for predicting effects in humans (Contrera & DeGeorge, 1998; Tennant et al., 1999a). However, these proposed benefits are based on the assumptions that the mechanisms by which the GM animals develop cancer is the same as that occurring in non-GM animals, and that the GM animals exhibit a similar response in 6 months as the non-GM animals do in 2 years. As indicated above, there are concerns about the validity of both of these assumptions. As described in the previous section, there is evidence that, at least for some tumors, the mechanisms underlying the carcinogenic response in the GM mice differ from those occurring in the non-GM animals (Ashby, 1997; Mitsumori et al., 1997; Yamamoto et al., 1998a). If this is the case, then the observations and conclusions reached in the GM models about the shape of the dose-response curves and whether an agent operates through a threshold-type of mechanism, may not be valid for assessing the effects in non-GM organisms.
Varying tumor incidence in different rodent models
There is considerable evidence that the induced tumor incidences observed in the GM models under standard study conditions differ significantly from those seen in two-year rodent bioassays. In some cases, the GM animals exhibit higher tumor incidences than those seen in the NTP bioassays. In others, the response is much weaker in the GM than in the conventional mice. As an example of the first case, the dose-response curves for the induction of thymic lymphomas by phenolphthalein in the Trp53+/− and NTP mouse models are shown in Figure 3. The histiocytic sarcoma data from the NTP mouse are also plotted for comparison. In this case, it is clear that the incidence of tumors is much greater in the Trp53+/− model than in the NTP mouse model.
In contrast, the skin tumor data for the Trp53+/− and NTP mice treated with 4-vinyl-1-cyclohexene diepoxide are shown in Figure 5. In this case, the response in the NTP bioassay is much stronger than in the Trp53+/− mouse. As illustrated in Figure 1, it is possible, although unknown, that if the duration of the accelerated bioassay had been extended, a larger and possibly more similar response might have been seen. Another informative example is urethane, which was administered in drinking water to male B6C3F1 mice in a 2-year NTP cancer bioassay at 0, 10, 30, and 90 ppm (estimated to be 0, 1.2, 3.3, and 10.1 mg/kg) (NTP, 2004). Urethane was also administered by gavage (0, 1, 10, and 100 mg/kg) to male Trp53+/− mice (Carmichael et al., 2000). In both models, urethane induced a dose-dependent increase in tumors in multiple tissues. However, the tumorigenic dose detected in the Trp53+/− model was 10×higher than the highest dose used in the NTP bioassay and 83×higher than the lowest tumorigenic dose seen in the NTP bioassay. Again, in this case the GM model was not nearly as sensitive as the conventional mouse model. It should be noted that the sensitivity and target organ specificity of urethane tumorigenesis in the GM models appear to be heavily influenced by the genetic background of the strain used (Ozaki et al., 2005), so strain differences as well as differences in the route/type of administration may have contributed to the different tumor frequencies seen.
Figure 5.

Skin tumors induced by 4-vinyl-1-cyclohexene diepoxide in male mice the Trp53+/− and NTP bioassays (NTP, 1989; Tennant et al., 1995).
In other cases, the responses in the GM and the NTP bioassays appear to be roughly equivalent (Figure 6). Since the sensitivity of the GM animal relative to what would be seen in a chronic bioassay would be unknown, the risk assessor would have to make an assumption as to the relative sensitivity of the GM model. In effect, this would add an additional extrapolation, a GM to wild-type animal extrapolation, to the evaluation process.
Figure 6.

Bladder tumors induced by p-cresidine in male mice in the Trp53+/− and NCI bioassays (NTP, 1979; Tennant et al., 1995).
Route and dose extrapolation
One of the considerations made in the dose-response assessment is to determine whether the dose and effects seen following exposure by one route would be comparable to those occurring through a different route of exposure. While one would expect that the responses in the GM and non-GM animals would be similar for most models and situations, the Tg.AC model poses special problems in this regard. It is not clear how one would relate skin responses following topical exposure, employing at times very high doses, to those anticipated through other exposure routes. For example, the induction of tumors by benzene, a systemic toxicant in humans, is dependent upon the route of exposure in the Tg.AC model (strong increase with topical treatment versus no increase by gavage). Similarly, the exposure route alters the types of tumors induced in the Trp53+/− model (primarily sarcomas by gavage versus lymphomas by inhalation). These route-of-exposure-dependent effects would complicate an assessment and increase the difficulty in making an accurate evaluation (Table 4). Differences in response due to route of administration can also be a problem in conventional rodent assays.
Animal to human extrapolation
Over the years, a number of conversion factors have been derived and generally accepted to allow doses in animals to be converted to human-equivalent doses and to adjust for less-than- lifetime studies. (Anderson, 1983; US EPA, 2011). In the absence of more specific toxicokinetic information, a default approach is often used. For example, to determine the human equivalent dose from an animal study, the animal dose in mg/kg-bw is often multiplied by the ratio of the animal body weight to the human body weight raised to the 1/4 power (Travis, 1993; US EPA, 2011). Similarly, if the duration of the cancer bioassay is less than the natural life-span of the test animal, commonly accepted as two years, then an adjustment [(lifetime of the animal/length of the experiment)3] is made to account for the exponential increase of tumors seen with age (Anderson, 1983). Since these conversions have been derived based on experiments in non-GM animals, it is not certain which of these would be applied to the GM assays. As the first conversion factor is based on the general observation that physiological processes tend to exhibit proportionality when scaled by body weight3/4, it is possible that it would still apply. However, it is also conceivable that the genetic modifications in some types of GM animals may fundamentally alter this relationship. In these cases, other scaling factors would have to be derived. The use of the current conversion factor for less-than-lifetime studies would not be appropriate for the GM assays, and an analogous factor might need to be derived. As illustrated in Figure 1, the end of the accelerated bioassay (6 months) often occurs before maximal tumor formation is seen in the GM animals. For bioassays where the maximal response was not seen at the end of the study, a conversion factor would most likely be needed to account for reduced tumor yield. However, without additional information, the risk assessor will not know how to distinguish those agents that have produced a maximal response within the 6-month study from those which have not.
Use of genetically modified animals in mode-ofaction studies
One area in the risk assessment process where current and new GM animals are likely to make significant contributions is in the evaluation of the mode-of-action (MOA) for individual chemicals. The MOA is an understanding of the key events and processes in the cell, tissue, and organ that lead to tumor development. However, it is understood that decisions on the MOA can be made without understanding the precise mechanistic details of how a chemical induces cancer. The development of targeted GM models provides a powerful approach for understanding the genes and processes involved in both spontaneous as well as chemically induced carcinogenesis. Significant progress has been made in recent years using these technologies (for reviews, see French et al., 2010; Gollapudi et al., 1998; Lynch et al., 2007; Macleod & Jacks, 1999; Pritchard et al., 2003; Rosenberg, 1997; Wells & Williams, 2009). GM animals are being actively used to investigate the role of specific xenobiotic metabolizing enzymes, DNA repair processes, cell-to-cell signaling, transcription factors, and gene expression, as well as cell cycle regulatory genes in tumor development (Boverhof et al., 2011; Gonzalez, 2001; Rosenberg, 1997). In most cases, the GM models which target specific genes and pathways differ substantially from the GM models described in this review which are primarily used to test or screen chemicals for carcinogenic effects. It has been shown that two GM mouse models, TG.AC and Trp53+/− did not differ significantly compared to their respective parent strains FVB/N and C57BL/6 mice in the metabolism of benzene, ethoxyquin and methacrylonitrile, suggesting that the toxicity testing and data interpretation would be comparable between GM and wild type models (Sanders et al., 2001). However, as indicated above, differences in metabolism, DNA adducts and other endpoints were seen for DES and other chemicals (Carmichael et al., 2001). Notable examples of the use of GM models to study different MOAs include the use of the peroxisome proliferator-activated receptor alpha (PPARα) knockout mouse to study mechanisms of peroxisome proliferation, the aryl hydrocarbon (Ah) receptor knockout mouse to investigate 2,3,7,8-tetrachlorodibenzodioxin (TCDD)- induced immunotoxicity, the cytochrome P450 class 2E1 (CYP2E1) knockout mouse to study the role of this enzyme in benzene metabolism and genotoxicity, and the CYP1B1 and microsomal epoxide hydrolase (mEH) knockout mice to study the metabolic pathways involved in DMBA carcinogenesis (Boverhof et al., 2011; Gonzalez, 2001, 2003; Nwosu et al., 2004; Rudmann & Durham, 1999; Valentine et al., 1996).
Mechanistic follow-up studies using the Trp53+/− model are also beginning to formally contribute to the risk assessment process. For example, the observation that the thymic lymphomas induced by phenolphthalein in Trp53+/− mice exhibited losses of the normal wild-type Trp53 allele has been used by both the NTP and IARC to support the proposed involvement of a mutagenic mechanism in the tumorigenesis of this agent (IARC, 2000; NTP, 2011). Undoubtedly, data from GM animals will be proposed for use in future risk assessments to distinguish genotoxic from non-genotoxic agents, to identify effects unique to one species, and to support the use of either linear or non-linear models for extrapolation to the low-dose exposure region.
Risk characterization and general comments
During the risk characterization step of the risk assessment process, information from each of the hazard identification, dose response assessment, and exposure assessment steps is integrated to estimate the expected incidence of adverse effects in humans. As with information derived from other sources, information obtained from the GM models will undoubtedly contribute to the overall evaluation of a chemical’s safety and the evaluation of its risk to humans.
As indicated above, information from GM models has begun to contribute to the hazard identification and MOA evaluations of chemicals as evidenced by the use of results from GM models in recent NTP, IARC, FDA, and EUREACH evaluations (Cohen et al., 2001; Gollapudi et al., 1998; IARC, 1992; Jacobson-Kram, 2010; Jacobson-Kram et al., 2004; NTP, 2011; Pritchard et al., 2003; Wells & Williams, 2009). In most cases, these data are being used as supportive or additional information to bolster specific aspects of the evaluation. It is uncertain when these GM models will be sufficiently validated, and accepted to be relied upon as the primary evidence of carcinogenicity, or more importantly of non-carcinogenicity.
Different research groups have proposed a number of approaches for the use of GM animals in cancer risk assessment. Sills et al. (2001) outlined a combined use of GM models with mutations in tumor suppressor genes and reporter genes. Another group proposed a strategy that combines information on the genotoxicity and structure of the agent with a tiered use of the Trp53+/− and Tg.AC models to complement or replace the need for a conventional two-year bioassay (Tennant et al., 1995). The International Conference on Harmonization (ICH) recommended the use of one or more of the GM models as a second species to replace the mouse bioassay in standard testing requirements (ICH, 1997). This approach has largely been adopted by the US FDA with uncertain success (Jacobs, 2005), and advocated by Japanese investigators (Urano et al., 2012). Another approach would use these models as a “third species” to test specific hypotheses or as a follow-up assay to resolve contradictory results when observed in 2-year rodent bioassays. A similar approach, discussed by investigators at the NTP, is to use the GM models in pre-chronic testing (Bucher, 1998). If a chemical were positive in an accelerated bioassay, then there would be no need for the chronic bioassay. If the short-term test were negative, the test would still provide information on the appropriate doses that should be used in the chronic study. Later, it was also suggested that employing a combination of the 2-year rat bioassay and GM models might be useful in human health risk assessment after validation and standardization of the methods (Pritchard et al., 2003). A similar approach was proposed by the EU-REACH program. More recently, Jacobson-Kram (2010) has highlighted the potential use of such a mixed approach, which would help to definitively identify human carcinogens. He also suggested the use of a “carcinogenicity battery” that includes genetic toxicology studies, a GM mouse assay, and a 1-year rat study combined with an understanding of structure/activity relationships and a future use of “omics” platforms for the evaluation of carcinogens (Jacobson-Kram, 2010).
It is likely that because of varying institutional goals, different groups evaluating the carcinogenic risks of chemicals may use the GM models in different ways. For example, using an accelerated bioassay to rapidly determine whether a drug exhibits carcinogenic effects may make sense to a pharmaceutical company especially concerned with false positives and the time required to get an important product to market. In contrast, for the NTP, EPA, or other groups charged with protecting public health that are equally concerned with false negatives, a more cautious approach might be taken in the use of GM animal data.
It is evident from the information available from different accelerated bioassays that none of the GM models can be used as a stand-alone model for risk assessment due to their limitations. In particular, there is evidence that the route of exposure and the genetic background can significantly influence the outcome of the assay as in the case of benzene (Table 4). In order to minimize such problems, the GM animals could be used as a first tier in the hazard identification of chemicals of environmental concern as suggested previously (Pritchard et al., 2003; Tennant et al., 1995). Initially genetic toxicology information generated from a battery of short-term assays (such as those using human cells or cell lines) would be combined with mechanistic, pathway-related and omics data to identify potential effects of concern. Following the initial screening, a decision would be made to conduct follow-up testing using the most appropriate GM model, such as the Trp53+/−, TG.AC, or rasH2, for conducting an accelerated cancer bioassay (Pritchard et al., 2003). For example, if the chemical has been shown to be genotoxic in the in vitro battery of assays, a rasH2 model may be used for further screening. In theory the Trp53+/− model could also be used for follow-up testing. However, until its sensitivity to detect carcinogens is enhanced, it remains a less-than-ideal choice. If the compound is non-genotoxic by in vitro testing, it may be tested in either the TG.AC or rasH2 model. Based on the information available from the first tier screening and GM assay results, a comprehensive 2-year rodent bioassay could be used to confirm the earlier results and to establish dose-response relationships for both cancer and noncancer effects.
Future research needs
The GM mouse models initially employed were based on an understanding of the role of proto-oncogenes and tumor suppressor genes that were observed to be dysregulated due to mutations that altered protein functions and resulted in the loss of control over cell proliferation and the cell cycle in a variety of human cancers. This concept was best explained in Vogelstein’s colorectal cancer model (Fearon & Vogelstein, 1990). In addition, the role of individual susceptibility to heritable or carcinogen-induced cancers was not considered, although the heritability of cancer was clearly recognized (Berger et al., 2011). The prevailing belief was that carcinogen-induced genetic lesions would have a larger effect than the heritable genetic susceptibility. However, we now recognize that both endogenous and exogenous factors result in processes, such as DNA damage, epigenetic suppression, immunosuppression and loss of immune surveillance, and ligand–receptor interference with receptor-based mechanisms, that are associated with numerous diseases, including cancer. Consequently, in the future, a battery of GM models may be required to cover the breadth of chemicals and the variety of signaling pathways and cell functions that are dysregulated in cancer. Based upon our current knowledge, methodological issues with the GM assays can be addressed by increasing the assay sample sizes and the duration of the bioassays, as long as spontaneous neoplasia do not confound identification and analysis of tumors caused by recognizable endogenous or exogenous factors and as a result of genome ×environment interactions. Thus, the creation and maintenance of stable GM lines developed to answer specific questions arising from our expanding knowledge is critically important. As indicated, the NTP and other groups are actively working to address some of these issues. However, there are additional critical needs to be addressed such as understanding the mechanisms of tumorigenesis operating in the GM models, their relevance to cancer in humans, and the reasons for unexpected positive and negative tumor responses that occur based upon the selection of inbred strain(s), diet and other environmental factors, and how these may confound the testing of toxicants at low levels and at human relevant exposures.
There are several large-scale initiatives that will aid the development of GM models for research and testing of presumptive human toxicants and carcinogens in genetically diverse models for human disease. Some examples include: (1) the Knock-Out Mouse Project (KOMP), a trans-NIH initiative, which with other members of the International Knock-out Mouse Consortium, aims to generate a resource for the gene trapping and targeted mutation of all mouse protein-coding genes in the C57BL/6N mouse; and (2) The Collaborative Cross and Diversity Outbred mice project – an investigator- and NIH-sponsored creation of genetically diverse mouse populations of Advanced Intercross Recombinant Inbred Lines (AIRILs) (Aylor et al., 2011; Chesler et al., 2008; Churchill et al., 2004; Threadgill et al., 2011). Selected generations of breeders from the Collaborative Cross AIRILS have been used to create diversity outbred (DO) mice using a randomized breeding protocol (Svenson et al., 2012). Each DO mouse is genetically different from every other DO mouse from generation to generation and represents a degree of genetic diversity in the mouse genome that is equal to or greater than that of human populations. Increasing genetic diversity and the odds that a true positive can be identified in GM models are also critical needs. One way that might be used to address the diversity problem is to use the approach suggested by Hunter (2012) and incorporate more genetic diversity by crossing females of the desired GM model with a male of one or more of the Collaborative Cross advanced intercross recombinant inbred lines. In addition, the recent establishment of GM rat models is also a promising development that will provide additional strategies for testing carcinogens (Boverhof et al., 2011; Tsuda et al., 2005; van Boxtel & Cuppen, 2011; Yan et al., 2012). Concurrent development of GM rat models using the same targets would greatly enhance the two species approach, where positive outcomes in both species would greatly strengthen the evidence and increase the likelihood that the test substance would be a human carcinogen. Although considerable progress has been made in developing the various GM models for short-term cancer bioassays, it is apparent that additional research is needed to ensure that the models are sufficiently sensitive and reliable for the hazard identification and dose-response characterization necessary for regulatory purposes and to protect public health.
Summary
It is clear that GM animals are playing an increasingly important role in the safety evaluation and risk evaluation of chemical carcinogens. While significant progress has been made in evaluating GM mouse models such as the Trp53+/−, Tg.AC and the rasH2, much work remains to be to done to fully assess the suitability of these models for hazard identification and as replacement for the conventional rodent cancer bioassay. To date, the GM models have shown a moderate amount of success in the identification of carcinogens and non-carcinogens. The current models seem to be relatively inefficient in detecting carcinogenic agents, but are more consistent in classifying non-carcinogenic agents. From the studies to date, it is apparent that no single GM model can be relied upon to detect all trans-species and human carcinogens. By using more than one GM model, the efficiency of these models to detect carcinogens can be enhanced. However, there will almost certainly be trans-species and human carcinogens that operate through mechanisms, which will not be detected using the current GM approaches.
In addition, a number of significant concerns have been raised about the use of GM models. Conducting more studies and optimizing study designs and protocols can address some of these concerns, such as those related to sample size, study duration, genetic instability and reproducibility. Yet other concerns, such as those involving pathway-dependent effects and the different mechanisms of chemical tumorigenesis occurring in GM and non-GM animals, will be more challenging to overcome. Moreover, the use of these GM models in the dose-response analysis is problematic and it is not clear at this point how estimates of dose-response will be obtained from the GM results. Until these models are more thoroughly validated and the dose-response issues adequately addressed, caution should be exercised in the use of the current GM models to assess the carcinogenic risks of chemicals.
Footnotes
In memory of Tatsuji Nomura, M.D., Ph.D., visionary, developer, and promoter of the use of genetically modified animals for research and testing, who passed away on January 11, 2013.
Declaration of interest
The employment affiliations of the authors are shown on the cover page. This article is an updated and substantially modified version of an earlier report prepared by Dr. Eastmond for the US EPA. The updated paper was prepared by the authors during the normal course of their employment, and the authors declare that they have no conflict of interest. The analyses, interpretations and conclusions reported in the paper are exclusively those of the authors and do not necessarily reflect the views or policies of the US EPA or the NIEHS.
References
- Anderson EL. Quantitative approaches in use to assess cancer risk. Risk Anal. 1983;3:277–95. [Google Scholar]
- Ashby J. Identifying potential human carcinogens – the role of genetically altered rodents. Toxicol Pathol. 1997;25:241–3. doi: 10.1177/019262339702500220. [DOI] [PubMed] [Google Scholar]
- Aylor DL, Valdar W, Foulds-Mathes W, et al. Genetic analysis of complex traits in the emerging Collaborative Cross. Genome Res. 2011;21:1213–22. doi: 10.1101/gr.111310.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature. 2011;476:163–9. doi: 10.1038/nature10275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blain PG, Battershill JM, Venitt S, et al. Consideration of short-term carcinogenicity tests using transgenic mouse models. Mutat Res. 1998;403:259–63. doi: 10.1016/s0027-5107(98)00026-8. [DOI] [PubMed] [Google Scholar]
- Boley SE, Anderson EE, French JE, et al. Loss of p53 in benzene-induced thymic lymphomas in p53+/− mice: evidence of chromosomal recombination. Cancer Res. 2000;60:2831–5. [PubMed] [Google Scholar]
- Boverhof DR, Chamberlain MP, Elcombe CR, et al. Transgenic animal models in toxicology: historical perspectives and future outlook. Toxicol Sci. 2011;121:207–33. doi: 10.1093/toxsci/kfr075. [DOI] [PubMed] [Google Scholar]
- Bucher JR. Update on national toxicology program (NTP) assays with genetically altered or “transgenic” mice. Environ Health Perspect. 1998;106:619–21. doi: 10.1289/ehp.98106619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon RE, Graves S, Spalding JW, et al. Oral administration of dimethylvinyl chloride increases frequency of forestomach papillomas in Tg.AC mice. Mol Carcinog. 2000;29:229–35. doi: 10.1002/1098-2744(200012)29:4<229::aid-mc1005>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Carmichael NG, Debruyne EL, Bigot-Lasserre D. The p53 heterozygous knockout mouse as a model for chemical carcinogenesis in vascular tissue. Environ Health Perspect. 2000;108:61–5. doi: 10.1289/ehp.0010861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael PL, Mills JJ, Campbell M, et al. Mechanisms of hormonal carcinogenesis in the p53+/− hemizygous knockout mouse: studies with diethylstilbestrol. Toxicol Pathol. 2001;29:155–60. doi: 10.1080/019262301753178564. [DOI] [PubMed] [Google Scholar]
- Castrop H. Genetically modified mice – successes and failures of a widely used technology. Pflugers Arch. 2010;459:557–67. doi: 10.1007/s00424-009-0770-z. [DOI] [PubMed] [Google Scholar]
- Chesler EJ, Miller DR, Branstetter LR, et al. The Collaborative Cross at Oak Ridge National Laboratory: developing a powerful resource for systems genetics. Mamm Genome. 2008;19:382–9. doi: 10.1007/s00335-008-9135-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA, Airey DC, Allayee H, et al. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nat Genet. 2004;36:1133–7. doi: 10.1038/ng1104-1133. [DOI] [PubMed] [Google Scholar]
- Clarke AR. Manipulating the germline: its impact on the study of carcinogenesis. Carcinogenesis. 2000;21:435–41. doi: 10.1093/carcin/21.3.435. [DOI] [PubMed] [Google Scholar]
- Cohen SM, Robinson D, MacDonald J. Alternative models for carcinogenicity testing. Toxicol Sci. 2001;64:14–19. doi: 10.1093/toxsci/64.1.14. [DOI] [PubMed] [Google Scholar]
- Contrera JF, DeGeorge JJ. In vivo transgenic bioassays and assessment of the carcinogenic potential of pharmaceuticals. Environ Health Perspect. 1998;106:71–80. doi: 10.1289/ehp.98106s171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contrera JF, Jacobs AC, DeGeorge JJ. Carcinogenicity testing and the evaluation of regulatory requirements for pharmaceuticals. Regul Toxicol Pharmacol. 1997;25:130–45. doi: 10.1006/rtph.1997.1085. [DOI] [PubMed] [Google Scholar]
- Dass SB, Bucci TJ, Heflich RH, Casciano DA. Evaluation of the transgenic p53+/− mouse for detecting genotoxic liver carcinogens in a short-term bioassay. Cancer Lett. 1999;143:81–5. doi: 10.1016/s0304-3835(99)00196-2. [DOI] [PubMed] [Google Scholar]
- Dobrovolsky VN, Shaddock JG, Mittelstaedt RA, et al. Frequency of Hprt mutant lymphocytes and micronucleated erythrocytes in p53-haplodeficient mice treated perinatally with AZT and AZT in combination with 3TC. Environ Mol Mutagen. 2007;48:270–82. doi: 10.1002/em.20280. [DOI] [PubMed] [Google Scholar]
- Donehower LA. The p53-deficient mouse: a model for basic and applied cancer studies. Semin Cancer Biol. 1996;7:269–78. doi: 10.1006/scbi.1996.0035. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Donehower LA, Lozano G. 20 years studying p53 functions in genetically engineered mice. Nat Rev Cancer. 2009;9:831–41. doi: 10.1038/nrc2731. [DOI] [PubMed] [Google Scholar]
- Dunnick JK, Hardisty JF, Herbert RA, et al. Phenolphthalein induces thymic lymphomas accompanied by loss of the p53 wild type allele in heterozygous p53-deficient (+/−) mice. Toxicol Pathol. 1997;25:533–40. doi: 10.1177/019262339702500601. [DOI] [PubMed] [Google Scholar]
- Dunson DB, Haseman JK, van Birgelen AP, et al. Statistical analysis of skin tumor data from Tg.AC mouse bioassays. Toxicol Sci. 2000;55:293–302. doi: 10.1093/toxsci/55.2.293. [DOI] [PubMed] [Google Scholar]
- Eastin WC, Haseman JK, Mahler JF, Bucher JR. The National Toxicology Program evaluation of genetically altered mice as predictive models for identifying carcinogens. Toxicol Pathol. 1998;26:461–73. doi: 10.1177/019262339802600401. [DOI] [PubMed] [Google Scholar]
- Enzmann H, Iatropoulos M, Brunnemann KD, et al. Short- and intermediate-term carcinogenicity testing – a review. Part 2: available experimental models. Food Chem Toxicol. 1998;36:997–1013. doi: 10.1016/s0278-6915(98)00064-7. [DOI] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- Finch GL, March TH, Hahn FF, et al. Carcinogenic responses of transgenic heterozygous p53 knockout mice to inhaled 239PuO2 or metallic beryllium. Toxicol Pathol. 1998;26:484–91. doi: 10.1177/019262339802600404. [DOI] [PubMed] [Google Scholar]
- Forster R. A global collaboration on carcinogenicity screening in transgenic mouse models. Transgenic Res. 1998;7:485–7. doi: 10.1023/a:1008889427323. [DOI] [PubMed] [Google Scholar]
- French J, Storer RD, Donehower LA. The nature of the heterozygous Trp53 knockout model for identification of mutagenic carcinogens. Toxicol Pathol. 2001a;29:24–9. doi: 10.1080/019262301753178456. [DOI] [PubMed] [Google Scholar]
- French JE, Lacks GD, Trempus C, et al. Loss of heterozygosity frequency at the Trp53 locus in p53-deficient (+/−) mouse tumors is carcinogen-and tissue-dependent. Carcinogenesis. 2001b;22:99–106. doi: 10.1093/carcin/22.1.99. [DOI] [PubMed] [Google Scholar]
- French JE, Leblanc B, Long GG, et al. Panel discussion: alternative mouse models for carcinogenicity assessment. Toxicol Pathol. 2010;38:72–5. doi: 10.1177/0192623309352091. [DOI] [PubMed] [Google Scholar]
- Furst SM, Stoll RE, Mennear J. Dermal carcinogenicity in transgenic mice: effect of vehicle on responsiveness of hemizygous Tg.AC mice to phorbol ester (TPA). The Tg. AC Workgroup Newsletter. 2000;3:1–2. doi: 10.1080/019262301317226339. [DOI] [PubMed] [Google Scholar]
- Gold LS, Bernstein L, Magaw R, Slone TH. Interspecies extrapolation in carcinogenesis: prediction between rats and mice. Environ Health Perspect. 1989;81:211–19. doi: 10.1289/ehp.8981211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsworthy TL, Recio L, Brown K, et al. Transgenic animals in toxicology. Fundam Appl Toxicol. 1994;22:8–19. doi: 10.1006/faat.1994.1002. [DOI] [PubMed] [Google Scholar]
- Gollapudi BB, Stott WT, Yano BL, Bus JS. Mode of action considerations in the use of transgenic animals for mutagenicity and carcinogenicity evaluations. Toxicol Lett. 1998;102–103:479–84. doi: 10.1016/s0378-4274(98)00342-7. [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ. The use of gene knockout mice to unravel the mechanisms of toxicity and chemical carcinogenesis. Toxicol Lett. 2001;120:199–208. doi: 10.1016/s0378-4274(01)00296-x. [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ. Role of gene knockout and transgenic mice in the study of xenobiotic metabolism. Drug Metab Rev. 2003;35:319–35. doi: 10.1081/dmr-120026496. [DOI] [PubMed] [Google Scholar]
- Goodman JI. A perspective on current and future uses of alternative models for carcinogenicity testing. Toxicol Pathol. 2001;29:173–6. doi: 10.1080/019262301753178582. [DOI] [PubMed] [Google Scholar]
- Gray GM, Li P, Shlyakhter I, Wilson R. An empirical examination of factors influencing prediction of carcinogenic hazard across species. Regul Toxicol Pharmacol. 1995;22:283–91. doi: 10.1006/rtph.1995.0011. [DOI] [PubMed] [Google Scholar]
- Gulezian D, Jacobson-Kram D, McCullough CB, et al. Use of transgenic animals for carcinogenicity testing: considerations and implications for risk assessment. Toxicol Pathol. 2000;28:482–99. doi: 10.1177/019262330002800320. [DOI] [PubMed] [Google Scholar]
- Healy LN, Pluta LJ, James RA, et al. Induction and timedependent accumulation of micronuclei in peripheral blood of transgenic p53+/− mice, Tg.AC (v-Ha-ras) and parental wild-type (C57BL/6 and FVB/N) mice exposed to benzene by inhalation. Mutagenesis. 2001;16:163–8. doi: 10.1093/mutage/16.2.163. [DOI] [PubMed] [Google Scholar]
- Holden HE, Stoll RE, Spalding JW, Tennant RW. Hemizygous Tg.AC transgenic mouse as a potential alternative to the two-year mouse carcinogenicity bioassay: evaluation of husbandry and housing factors. J Appl Toxicol. 1998;18:19–24. doi: 10.1002/(sici)1099-1263(199801/02)18:1<19::aid-jat464>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Honchel R, Rosenzweig BA, Thompson KL, et al. Loss of palindromic symmetry in Tg.AC mice with a nonresponder phenotype. Mol Carcinog. 2001;30:99–110. [PubMed] [Google Scholar]
- Huff J. Value, validity, and historical development of carcinogenesis studies for predicting and confirming carcinogenic risks to humans. In: Kitchin KT, editor. Carcinogenicity: testing, predicting, and interpreting chemical effects. New York: Marcel Dekker; 1999. pp. 21–123. [Google Scholar]
- Hulla JE, French JE, Dunnick JK. Chromosome 11 allelotypes reflect a mechanism of chemical carcinogenesis in heterozygous p53- deficient mice. Carcinogenesis. 2001;22:89–98. doi: 10.1093/carcin/22.1.89. [DOI] [PubMed] [Google Scholar]
- Humble MC, Trempus CS, Spalding JW, et al. Biological, cellular, and molecular characteristics of an inducible transgenic skin tumor model: a review. Oncogene. 2005;24:8217–28. doi: 10.1038/sj.onc.1209000. [DOI] [PubMed] [Google Scholar]
- Hunter KW. Mouse models of cancer: does the strain matter? Nat Rev Cancer. 2012;12:144–9. doi: 10.1038/nrc3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC. Mechanisms of carcinogenesis in risk identification. IARC Working Group Meeting; 1991 June 11–18; Lyon: IARC Sci Publ; 1992. pp. 1–608. [PubMed] [Google Scholar]
- IARC. Consensus report. In: McGregor DB, Rice JM, Venitt S, editors. The use of short- and medium-term tests for carcinogens and data on genetic effects in carcinogenic hazard evaluation. Vol. 146. IARC Sci Publ; 1999. pp. 1–18. [PubMed] [Google Scholar]
- IARC. Phenolphthalein. IARC Monographs on the Evaluation of Carcinogenic Risks to Man. 2000;76:387–415. [Google Scholar]
- ICH. Guidance for Industry S1B Testing for Carcinogencity of Pharmaceuticals. International Conference on Harmonization, Expert Working Group on Safety; Geneva, Switzerland. 1997. [last accessed 23 Jul 2013]. [Online]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm074916.pdf. [Google Scholar]
- Ito N, Shirai T, Hasegawa R. Medium-term bioassays for carcinogens. IARC Sci Publ. 1992;116:353–88. [PubMed] [Google Scholar]
- Jacobs A. Prediction of 2-year carcinogenicity study results for pharmaceutical products: how are we doing? Toxicol Sci. 2005;88:18–23. doi: 10.1093/toxsci/kfi248. [DOI] [PubMed] [Google Scholar]
- Jacobson-Kram D. Cancer risk assessment approaches at the FDA/CDER: is the era of the 2-year bioassay drawing to a close? Toxicol Pathol. 2010;38:169–70. doi: 10.1177/0192623309351892. [DOI] [PubMed] [Google Scholar]
- Jacobson-Kram D, Sistare FD, Jacobs AC. Use of transgenic mice in carcinogenicity hazard assessment. Toxicol Pathol. 2004;32:49–52. doi: 10.1080/01926230490424761. [DOI] [PubMed] [Google Scholar]
- Jaworski M, Hailfinger S, Buchmann A, et al. Human p53 knockin (hupki) mice do not differ in liver tumor response from their counterparts with murine p53. Carcinogenesis. 2005;26:1829–34. doi: 10.1093/carcin/bgi142. [DOI] [PubMed] [Google Scholar]
- Jerry DJ, Butel JS, Donehower LA, et al. Infrequent p53 mutations in 7,12-dimethylbenz[a]anthracene-induced mammary tumors in BALB/c and p53 hemizygous mice. Mol Carcinog. 1994;9:175–83. doi: 10.1002/mc.2940090309. [DOI] [PubMed] [Google Scholar]
- Johnson FM. Carcinogenic chemical-response “fingerprint” for male F344 rats exposed to a series of 195 chemicals: implications for predicting carcinogens with transgenic models. Environ Mol Mutagen. 1999;34:234–45. [PubMed] [Google Scholar]
- Kawasaki Y, Hirabayashi Y, Kaneko T, et al. Benzene-induced hematopoietic neoplasms including myeloid leukemia in Trp53- deficient C57BL/6 and C3H/He mice. Toxicol Sci. 2009;110:293–306. doi: 10.1093/toxsci/kfp107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp CJ. Hepatocarcinogenesis in p53-deficient mice. Mol Carcinog. 1995;12:132–6. doi: 10.1002/mc.2940120304. [DOI] [PubMed] [Google Scholar]
- Leder A, Kuo A, Cardiff RD, et al. v-Ha-ras transgene abrogates the initiation step in mouse skin tumorigenesis: effects of phorbol esters and retinoic acid. Proc Natl Acad Sci USA. 1990;87:9178–82. doi: 10.1073/pnas.87.23.9178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo JL, Yang Q, Tong WM, et al. Knock-in mice with a chimeric human/murine p53 gene develop normally and show wild-type p53 responses to DNA damaging agents: a new biomedical research tool. Oncogene. 2001;20:320–8. doi: 10.1038/sj.onc.1204080. [DOI] [PubMed] [Google Scholar]
- Lynch D, Svoboda J, Putta S. Mouse skin models for carcinogenic hazard identification: utilities and challenges. Toxicol Pathol. 2007;35:853–64. doi: 10.1080/01926230701748131. [DOI] [PubMed] [Google Scholar]
- Macleod KF, Jacks T. Insights into cancer from transgenic mouse models. J Pathol. 1999;187:43–60. doi: 10.1002/(SICI)1096-9896(199901)187:1<43::AID-PATH246>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Mahler JF, Flagler ND, Malarkey DE, et al. Spontaneous and chemically induced proliferative lesions in Tg.AC transgenic and p53-heterozygous mice. Toxicol Pathol. 1998;26:501–1. doi: 10.1177/019262339802600406. [DOI] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–65. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- Maronpot RR, Mitsumori K, Mann P, et al. Interlaboratory comparison of the CB6F1-Tg rasH2 rapid carcinogenicity testing model. Toxicology. 2000;146:149–59. doi: 10.1016/s0300-483x(00)00168-2. [DOI] [PubMed] [Google Scholar]
- Marsella JM, Liu BL, Vaslet CA, Kane AB. Susceptibility of p53-deficient mice to induction of mesothelioma by crocidolite asbestos fibers. Environ Health Perspect. 1997;105:1069–72. doi: 10.1289/ehp.97105s51069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina D, Ullrich R, Meyn R, et al. Environmental carcinogens and p53 tumor-suppressor gene interactions in a transgenic mouse model for mammary carcinogenesis. Environ Mol Mutagen. 2002;39:178–83. doi: 10.1002/em.10064. [DOI] [PubMed] [Google Scholar]
- Menendez D, Inga A, Resnick MA. The expanding universe of p53 targets. Nat Rev Cancer. 2009;9:724–37. doi: 10.1038/nrc2730. [DOI] [PubMed] [Google Scholar]
- Mitsumori K, Koizumi H, Nomura T, Yamamoto S. Pathological features of spontaneous and induced tumors in transgenic mice carrying a human prototype c-Ha-ras gene used for six-month carcinogenicity studies. Toxicol Pathol. 1998;26:520–31. doi: 10.1177/019262339802600408. [DOI] [PubMed] [Google Scholar]
- Mitsumori K, Wakana S, Yamamoto S, et al. Susceptibility of transgenic mice carrying human prototype c-Ha-ras gene in a shortterm carcinogenicity study of vinyl carbamate and ras gene analyses of the induced tumors. Mol Carcinog. 1997;20:298–307. doi: 10.1002/(sici)1098-2744(199711)20:3<298::aid-mc6>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Nambiar PR, Turnquist SE, Morton D. Spontaneous tumor incidence in rasH2 mice: review of internal data and published literature. Toxicol Pathol. 2012;40:614–23. doi: 10.1177/0192623311436181. [DOI] [PubMed] [Google Scholar]
- NCI. Bioassay of p-cresidine for possible carcinogenicity. Bethesda, MD: National Cancer Institute, Department of Health, Education, and Welfare, Public Health Service, National Institutes of Health; 1978. [Google Scholar]
- NCI. Bioassay of 2,4-diaminotoluene for possible carcinogenicity. Natl Cancer Inst Carcinog Tech Rep Ser. 1979;162:1–139. [PubMed] [Google Scholar]
- NIEHS. A Report of the ad hoc Interagency Coordinating Committee on the Validation of Alternative Methods. Research Triangle Park, NC: 1997. [last accessed 23 Jul 2013]. Validation and Regulatory Acceptance. Available from: http://iccvam.niehs.nih.gov/docs/about_docs/validate.pdf. [Google Scholar]
- NRC. Risk assessment in the federal government: managing the process. Washington, D.C: National Research Council, National Academy Press; 1983. [PubMed] [Google Scholar]
- NTP. Bioassay of p-cresidine for possible carcinogenicity. Natl Cancer Inst Carcinog Tech Rep Ser. 1979;142:1–123. [PubMed] [Google Scholar]
- NTP. NTP toxicology and carcinogenesis studies of benzene (CAS No. 71-43-2) in F344/N rats and B6C3F1 mice (gavage studies) Natl Toxicol Program Tech Rep Ser. 1986;289:1–277. [PubMed] [Google Scholar]
- NTP. NTP toxicology and carcinogenesis studies of bromodichloromethane (CAS No. 75-27-4) in F344/N rats and B6C3F1 mice (gavage studies) Natl Toxicol Program Tech Rep Ser. 1987;321:1–182. [PubMed] [Google Scholar]
- NTP. Toxicology and carcinogenesis studies of 4-vinyl-1- cyclohexene diepoxide (CAS No. 106-87-6) in F344/N rats and B6C3F1 mice (dermal studies) Natl Toxicol Program Tech Rep Ser. 1989;362:1–249. [PubMed] [Google Scholar]
- NTP. Toxicology and carcinogenesis studies of resorcinol (CAS No. 108-46-3) in F344 rats and B6C3F1 mice (gavage studies) Natl Toxicol Program Tech Rep Ser. 1992;403:1–234. [PubMed] [Google Scholar]
- NTP. NTP toxicology and carcinogenesis studies of phenolphthalein (CAS No. 77-09-8) in F344/N rats and B6C3F1 mice (feed studies) Natl Toxicol Program Tech Rep Ser. 1996;465:1–354. [PubMed] [Google Scholar]
- NTP. Twelfth Report on Carcinogens. Research Triangle Park: 2011. [last accessed 23 Jul 2013]. Available from: http://ntp.niehs.nih.gov/?objectid=03C9AF75-E1BFFF40-DBA9EC0928DF8B15. [Google Scholar]
- NTP. Toxicology and carcinogensis. Studies of urethane, ethanol, and urethane/ethanol (urethane, CAS No. 51-79-6; ethanol, CAS No. 64-17-5) in B6C3F1 mice (drinking water studies) Natl Toxicol Program Tech Rep Ser. 2004;510:1–346. [PubMed] [Google Scholar]
- NTP. NTP Toxicology and carcinogenesis studies of bromodichloromethane (CAS No. 75-27-4) in male F344/N rats and female B6C3F1 mice (drinking water studies) Natl Toxicol Program Tech Rep Ser. 2006;532:1–248. [PubMed] [Google Scholar]
- NTP. NTP report on the toxicology and carcinogenesis study of benzene (CAS No. 71-43-2) in genetically modified haploinsufficient p16 Ink4a/p19 Arf mice (gavage study) Natl Toxicol Program Genet Modif Model Rep, GMM. 2007a;8:1–81. [PMC free article] [PubMed] [Google Scholar]
- NTP. Toxicology and carcinogenesis study of glycidol (CAS No. 556-52-5) in genetically modified haploinsufficient p16(Ink4a)/ p19(Arf) mice (gavage study) Natl Toxicol Program Genet Modif Model Rep, GMM. 2007b;13:1–81. [PMC free article] [PubMed] [Google Scholar]
- NTP. The toxicology and carcinogenesis study of phenolphthalein (CAS No. 77-09-8) in genetically modified haploinsufficient p16(Ink4a)/p19(Arf) mice (feed study) Natl Toxicol Program Genet Modif Model Rep, GMM. 2007c;12:1–85. [PMC free article] [PubMed] [Google Scholar]
- NTP. Toxicology studies of bromodichloromethane (CAS No. 75-27-4) in genetically modified (FVB Tg.AC hemizygous) mice (dermal, drinking water, and gavage studies) and carcinogenicity studies of bromodichloromethane in genetically modified [B6.129- Trp53tm1Brd (N5) haploinsufficient] mice (drinking water and gavage studies) Natl Toxicol Program Genet Modif Model Rep, GMM. 2007d;5:1–227. [PMC free article] [PubMed] [Google Scholar]
- NTP. NTP report on the toxicology and carcinogenicity study of mixtures of 3’-azido-3′-deoxythymidine (AZT), lamivudine (3TC), and nevirapine (NVP) (CAS NOS. 30516-87-1, 134678-17-4, 129618-40-2) in genetically modified C3B6.129F1-Trp53tm1Brd N12 haploinsufficient mice (In utero and postnatal gavage study) draft. Natl Toxicol Program Genet Modif Model Rep GMM. 2012a;16:1–260. [PMC free article] [PubMed] [Google Scholar]
- NTP. Toxicology study of Senna (CAS No. 8013-11-4) in C57BL/6NTAC mice and toxicology and carcinogenesis study of senna in genetically modified C3B6.129F1/Tac-Trp53tm1Brd N12 haploinsufficient mice (Feed Studies) Natl Toxicol Program Genet Modif Model Rep, GMM. 2012b;15:1–114. [PMC free article] [PubMed] [Google Scholar]
- Nwosu VC, Kissling GE, Trempus CS, et al. Exposure of Tg.AC transgenic mice to benzene suppresses hematopoietic progenitor cells and alters gene expression in critical signaling pathways. Toxicol Appl Pharmacol. 2004;196:37–46. doi: 10.1016/j.taap.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Ozaki M, Ozaki K, Watanabe T, et al. Susceptibilities of p53 knockout and rasH2 transgenic mice to urethane-induced lung carcinogenesis are inherited from their original strains. Toxicol Pathol. 2005;33:267–71. doi: 10.1080/01926230590908231. [DOI] [PubMed] [Google Scholar]
- Pritchard JB, French JE, Davis BJ, Haseman JK. The role of transgenic mouse models in carcinogen identification. Environ Health Perspect. 2003;111:444–54. doi: 10.1289/ehp.5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–74. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson DE, MacDonald JS. Background and framework for ILSI’s collaborative evaluation program on alternative models for carcinogenicity assessment. International Life Sciences Institute. Toxicol Pathol. 2001;29:13–19. doi: 10.1080/019262301753178438. [DOI] [PubMed] [Google Scholar]
- Rosenberg MP. Gene knockout and transgenic technologies in risk assessment: the next generation. Mol Carcinog. 1997;20:262–74. doi: 10.1002/(sici)1098-2744(199711)20:3<262::aid-mc2>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Rudmann DG, Durham SK. Utilization of genetically altered animals in the pharmaceutical industry. Toxicol Pathol. 1999;27:111–14. doi: 10.1177/019262339902700121. [DOI] [PubMed] [Google Scholar]
- Saitoh A, Kimura M, Takahashi R, et al. Most tumors in transgenic mice with human c-Ha-ras gene contained somatically activated transgenes. Oncogene. 1990;5:1195–200. [PubMed] [Google Scholar]
- Salleh MN, Ismail P, Abdullah AS, Taufiq-Yap YH. Gene expression profiling of p53(+/−) knockout and wild-type mice following diethylstilbestrol administration. IUBMB Life. 2004;56:409–16. doi: 10.1080/15216540400003425. [DOI] [PubMed] [Google Scholar]
- Sanders JM, Burka LT, Chanas B, Matthews HB. Comparative xenobiotic metabolism between Tg.AC and p53+/− genetically altered mice and their respective wild types. Toxicol Sci. 2001;61:54–61. doi: 10.1093/toxsci/61.1.54. [DOI] [PubMed] [Google Scholar]
- Sgura A, De Amicis A, Stronati L, et al. Chromosome aberrations and telomere length modulation in bone marrow and spleen cells of melphalan-treated p53+/− mice. Environ Mol Mutagen. 2008;49:467–75. doi: 10.1002/em.20405. [DOI] [PubMed] [Google Scholar]
- Sills RC, French JE, Cunningham ML. New models for assessing carcinogenesis: an ongoing process. Toxicol Lett. 2001;120:187–98. doi: 10.1016/s0378-4274(01)00293-4. [DOI] [PubMed] [Google Scholar]
- Spalding JW, French JE, Stasiewicz S, et al. Responses of transgenic mouse lines p53(+/−) and Tg.AC to agents tested in conventional carcinogenicity bioassays. Toxicol Sci. 2000;53:213–23. doi: 10.1093/toxsci/53.2.213. [DOI] [PubMed] [Google Scholar]
- Spalding JW, French JE, Tice RR, et al. Development of a transgenic mouse model for carcinogenesis bioassays: evaluation of chemically induced skin tumors in Tg.AC mice. Toxicol Sci. 1999;49:241–54. doi: 10.1093/toxsci/49.2.241. [DOI] [PubMed] [Google Scholar]
- Spalding JW, Momma J, Elwell MR, Tennant RW. Chemically induced skin carcinogenesis in a transgenic mouse line (TG.AC) carrying a v-Ha-ras gene. Carcinogenesis. 1993;14:1335–41. doi: 10.1093/carcin/14.7.1335. [DOI] [PubMed] [Google Scholar]
- Stoll RE, Furst SM, Stoltz JH, et al. Dermal carcinogenicity in transgenic mice: effect of vehicle on responsiveness of hemizygous Tg.AC mice to phorbol 12-myristate 13-acetate (TPA) Toxicol Pathol. 2001;29:535–40. doi: 10.1080/019262301317226339. [DOI] [PubMed] [Google Scholar]
- Storer R. Experience to date with the positive control compounds p-cresidine and benzene. The p53 Workgroup Newsletter. 1998;1:E7–8. [Google Scholar]
- Storer RD, French JE, Haseman J, et al. P53+/− hemizygous knockout mouse: overview of available data. Toxicol Pathol. 2001;29:30–50. doi: 10.1080/019262301753178465. [DOI] [PubMed] [Google Scholar]
- Storer RD, Sistare FD, Reddy MV, DeGeorge JJ. An industry perspective on the utility of short-term carcinogenicity testing in transgenic mice in pharmaceutical development. Toxicol Pathol. 2010;38:51–61. doi: 10.1177/0192623309351718. [DOI] [PubMed] [Google Scholar]
- Svenson KL, Gatti DM, Valdar W, et al. High-resolution genetic mapping using the Mouse Diversity outbred population. Genetics. 2012;190:437–47. doi: 10.1534/genetics.111.132597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant RW. Evaluation and validation issues in the development of transgenic mouse carcinogenicity bioassays. Environ Health Perspect. 1998;106:473–6. doi: 10.1289/ehp.98106473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant RW, Butterworth BE, Preston RJ. Genetically engineered cancer models: three viewpoints on issues from testing to risk assessment. CIIT Activities. 1999a;19:1–7. [Google Scholar]
- Tennant RW, French JE, Spalding JW. Identifying chemical carcinogens and assessing potential risk in short-term bioassays using transgenic mouse models. Environ Health Perspect. 1995;103:942–50. doi: 10.1289/ehp.95103942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tennant RW, Spalding J, French JE. Evaluation of transgenic mouse bioassays for identifying carcinogens and noncarcinogens. Mutat Res. 1996;365:119–27. doi: 10.1016/s0165-1110(96)90016-0. [DOI] [PubMed] [Google Scholar]
- Tennant RW, Stasiewicz S, Eastin WC, et al. The Tg.AC (v-Haras) transgenic mouse: nature of the model. Toxicol Pathol. 2001;29:51–9. doi: 10.1080/019262301753178474. [DOI] [PubMed] [Google Scholar]
- Tennant RW, Stasiewicz S, Mennear J, et al. Genetically altered mouse models for identifying carcinogens. IARC Sci Publ. 1999b;146:123–50. [PubMed] [Google Scholar]
- Thompson KL, Rosenzweig BA, Sistare FD. An evaluation of the hemizygous transgenic Tg.AC mouse for carcinogenicity testing of pharmaceuticals. II. A genotypic marker that predicts tumorigenic responsiveness. Toxicol Pathol. 1998;26:548–55. doi: 10.1177/019262339802600411. [DOI] [PubMed] [Google Scholar]
- Threadgill DW, Miller DR, Churchill GA, de Villena FP. The collaborative cross: a recombinant inbred mouse population for the systems genetic era. ILAR J. 2011;52:24–31. doi: 10.1093/ilar.52.1.24. [DOI] [PubMed] [Google Scholar]
- Travis CC. Interspecies extrapolation of toxicological data. In: Wang RGM, Knaak JB, Maibach HI, editors. Health risk assessment: dermal and inhalation exposure and absorption of toxicants. Boca Raton: CRC Press; 1993. pp. 387–410. [Google Scholar]
- Tsuda H, Fukamachi K, Ohshima Y, et al. High susceptibility of human c-Ha-ras proto-oncogene transgenic rats to carcinogenesis: a cancer-prone animal model. Cancer Sci. 2005;96:309–16. doi: 10.1111/j.1349-7006.2005.00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. EPA. Recommended Use of Body Weight¾ at the Default Method in Derivation of the Oral Reference Dose. Washington, DC: Office of Science Advisor; 2011. [last accessed 23 Jul 2013]. EPA/100/R11/0001, Available from: http://www.epa.gov/raf/publications/pdfs/recommended-use-of-bw34.pdf. [Google Scholar]
- Urano K, Tamaoki N, Nomura T. Establishing a laboratory animal model from a transgenic animal: RasH2 mice as a model for carcinogenicity studies in regulatory science. Vet Pathol. 2012;49:16–23. doi: 10.1177/0300985811430318. [DOI] [PubMed] [Google Scholar]
- Valentine JL, Lee SS, Seaton MJ, et al. Reduction of benzene metabolism and toxicity in mice that lack CYP2E1 expression. Toxicol Appl Pharmacol. 1996;141:205–13. doi: 10.1006/taap.1996.0277. [DOI] [PubMed] [Google Scholar]
- van Boxtel R, Cuppen E. Generation of genetically modified rodents using random ENU mutagenesis. Methods Mol Biol. 2011;693:295–308. doi: 10.1007/978-1-60761-974-1_18. [DOI] [PubMed] [Google Scholar]
- van der Laan JW. Current status and use of short/medium-term models for assessment of carcinogenicity of human pharmaceuticals: regulatory perspectives. Toxicol Lett. 2000;112–113:567–72. doi: 10.1016/s0378-4274(99)00228-3. [DOI] [PubMed] [Google Scholar]
- van Kreijl CF, McAnulty PA, Beems RB, et al. Xpa and Xpa/ p53+/− knockout mice: overview of available data. Toxicol Pathol. 2001;29:117–27. doi: 10.1080/0192623013014189281. [DOI] [PubMed] [Google Scholar]
- van Kreijl CF, van Steeg H. Transgenic mouse models for the identification of human carcinogens: a European perspective. Toxicol Pathol. 1998;26:757–8. doi: 10.1177/019262339802600607. [DOI] [PubMed] [Google Scholar]
- van Steeg H, de Vries A, van Oostrom C, et al. DNA repairdeficient Xpa and Xpa/p53+/− knock-out mice: nature of the models. Toxicol Pathol. 2001;29:109–16. doi: 10.1080/019262301753178519. [DOI] [PubMed] [Google Scholar]
- Van Zeller A-M, Combes RD. Transgenic mouse bioassays for carcinogenicity testing: a step in the right direction? ATLA. 1999;27:839–46. [Google Scholar]
- Venkatachalam S, Shi YP, Jones SN, et al. Retention of wild-type p53 in tumors from p53 heterozygous mice: reduction of p53 dosage can promote cancer formation. EMBO J. 1998;17:4657–67. doi: 10.1093/emboj/17.16.4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatachalam S, Tyner SD, Pickering CR, et al. Is p53 haploinsufficient for tumor suppression? Implications for the p53+/− mouse model in carcinogenicity testing. Toxicol Pathol. 2001;29:147–54. doi: 10.1080/019262301753178555. [DOI] [PubMed] [Google Scholar]
- Weaver JL, Contrera JF, Rosenzweig BA, et al. An evaluation of the hemizygous transgenic Tg.AC mouse for carcinogenicity testing of pharmaceuticals. I. Evidence for a confounding nonresponder phenotype. Toxicol Pathol. 1998;26:532–40. doi: 10.1177/019262339802600409. [DOI] [PubMed] [Google Scholar]
- Wells MY, Williams ES. The transgenic mouse assay as an alternative test method for regulatory carcinogenicity studies – implications for REACH. Regul Toxicol Pharmacol. 2009;53:150–5. doi: 10.1016/j.yrtph.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Urano K, Koizumi H, et al. Validation of transgenic mice carrying the human prototype c-Ha-ras gene as a bioassay model for rapid carcinogenicity testing. Environ Health Perspect. 1998a;106 (Suppl 1):57–69. doi: 10.1289/ehp.98106s157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Urano K, Nomura T. Validation of transgenic mice harboring the human prototype c-Ha-ras gene as a bioassay model for rapid carcinogenicity testing. Toxicol Lett. 1998b;102–103:473–8. doi: 10.1016/s0378-4274(98)00341-5. [DOI] [PubMed] [Google Scholar]
- Yan HX, Wu HP, Ashton C, et al. Rats deficient for p53 are susceptible to spontaneous and carcinogen-induced tumorigenesis. Carcinogenesis. 2012;33:2001–5. doi: 10.1093/carcin/bgs238. [DOI] [PMC free article] [PubMed] [Google Scholar]