

Summary
Increased levels of the mitochondria-shaping protein Opa1 improve respiratory chain efficiency and protect from tissue damage, suggesting that it could be an attractive target to counteract mitochondrial dysfunction. Here we show that Opa1 overexpression ameliorates two mouse models of defective mitochondrial bioenergetics. The offspring from crosses of a constitutive knockout for the structural complex I component Ndufs4 (Ndufs4−/−), and of a muscle-specific conditional knockout for the complex IV assembly factor Cox15 (Cox15sm/sm), with Opa1 transgenic (Opa1tg) mice showed improved motor skills and respiratory chain activities compared to the naive, non-Opa1-overexpressing, models. While the amelioration was modest in Ndufs4−/−::Opa1tg mice, correction of cristae ultrastructure and mitochondrial respiration, improvement of motor performance and prolongation of lifespan were remarkable in Cox15sm/sm::Opa1tg mice. Mechanistically, respiratory chain supercomplexes were increased in Cox15sm/sm::Opa1tg mice, and residual monomeric complex IV was stabilized. In conclusion, cristae shape amelioration by controlled Opa1 overexpression improves two mouse models of mitochondrial disease.
Graphical Abstract
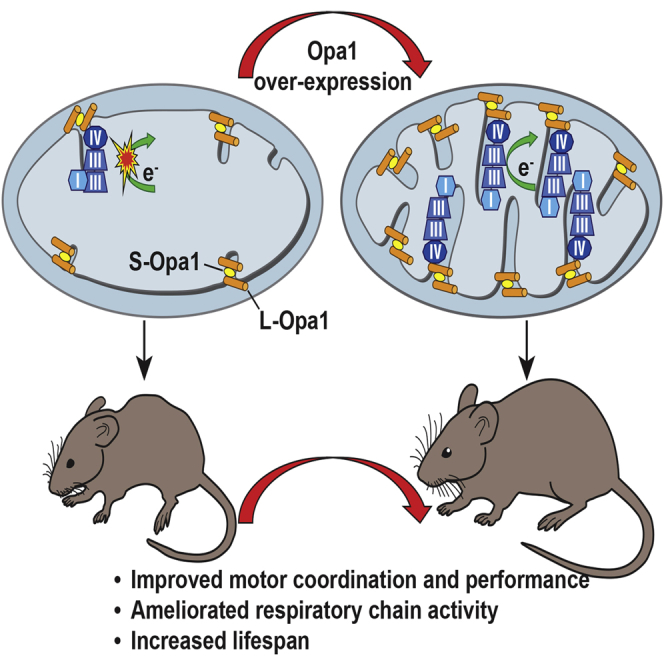
Highlights
-
•
Opa1 overexpression improves the phenotype of Ndufs4−/− and Cox15sm/sm mice
-
•
O2 consumption rate and respiratory chain activities are increased in double mutants
-
•
Mitochondrial ultrastructure is corrected by Opa1 overexpression in Cox15sm/sm
-
•
Respiratory complexes and supercomplexes are stabilized in Cox15sm/sm::Opa1tg
Although mitochondrial diseases have diverse causes, they are all characterized by defective oxidative phosphorylation. Civiletto et al. show that overexpression of the mitochondria-shaping protein OPA1, which improves respiratory chain efficiency, improves the phenotypes of two pre-clinical models of defective mitochondrial bioenergetics.
Introduction
Mutations in mitochondrial DNA (mtDNA), and in the vast repertoire of nuclear genes that converge on the formation and function of the mitochondrial respiratory chain (MRC), are responsible for primary “mitochondrial disorders,” a group of highly heterogeneous conditions, hallmarked by faulty oxidative phosphorylation (OXPHOS), that can affect any organ, at any age, and by any mode of transmission (Koopman et al., 2012). When taken as a whole, mitochondrial disorders are among the most frequent genetic diseases, affecting >1 in 5,000 individuals in the European population (Elliott et al., 2008). Despite substantial progress in mitochondrial medicine, the complexity of mitochondrial biology and genetics still constitutes a major challenge for understanding the mechanistic basis of mitochondrial disorders and explains, at least in part, their huge clinical and biochemical variability, which is also a major hurdle toward effective treatment. However, the development of “general” therapeutic strategies, extendable to diverse disease-associated OXPHOS defects, is now a realistic goal, based on rapidly expanding knowledge of the molecular mechanisms underpinning mitochondrial biogenesis, quality control, and signaling pathways. Several of these mechanisms are related to the control of mitochondrial shape and organization of the mitochondrial network.
Mitochondria are highly dynamic organelles that fuse and divide to adapt their structure and morphology to the energetic needs of the cell (Kasahara and Scorrano, 2014). Dynamin-related GTPases located on the inner and outer mitochondrial membranes (IMMs and OMMs) control the fission and fusion processes (Griparic and van der Bliek, 2001). Mitochondrial fission and fusion regulate a number of cellular processes, including organelle distribution during cell proliferation, bioenergetics proficiency, mitochondrial calcium flux, mitochondrial apoptosis, autophagy, and even complex morphogenetic processes such as the formation of dendritic spines (Kasahara and Scorrano, 2014). Fission is regulated by Dynamin-Related Protein 1 (DRP1) and by its OMM partners Fission 1 (FIS1), Mitochondrial Fission Factor (MFF), and Mitochondrial Division (MID) 49 and 51; fusion is controlled by the OMM proteins Mitofusins 1 and 2 (MFN1 and 2) and by the IMM protein Optic Atrophy 1 (OPA1). In humans, eight OPA1 isoforms are produced by alternative splicing of a single gene (Delettre et al., 2001), which are further processed by at least three proteases to form several long (L) and short (S) forms of OPA1 (Anand et al., 2014; Ehses et al., 2009; Griparic et al., 2007; Head et al., 2009; Ishihara et al., 2006; Song et al., 2007), which oligomerize to form functionally active quaternary structures (Frezza et al., 2006). In addition to its role in mitochondrial fusion, OPA1 controls the cristae remodelling arm of mitochondrial apoptosis (Frezza et al., 2006) and the physical and functional organization of the MRC complexes in MRC supercomplexes (Cogliati et al., 2013). These functional quaternary structures increase electron flow channelling through MRC complexes during respiration, thus minimizing electron leaks (Acin-Perez and Enriquez, 2014; Enriquez and Lenaz, 2014), and stabilize individual MRC complexes such as Complex III, CIII (Acín-Pérez et al., 2004). Consequently, OPA1 levels directly affect mitochondrial respiratory efficiency (Cogliati et al., 2013) and proteolytic inactivation of OPA1 has been shown to occur in cells from mitochondrial disease patients (Duvezin-Caubet et al., 2007). However, the role of OPA1 in vivo and the potential of its stabilization as a strategy to combat mitochondrial dysfunction and disease remains unclear. Genetic ablation of Opa1 is lethal during mouse embryonic development (Rahn et al., 2013) and causes massive dysfunction also in postmitotic tissues (L.S., unpublished data). On the other hand, high levels of OPA1 overexpression are toxic in cells, leading to paradoxical mitochondrial fragmentation (Cipolat et al., 2004). In order to avoid these effects, we have used X-chromosome-targeted Opa1 transgenesis (Opa1tg) to obtain moderate, ubiquitous overexpression in a mouse line (Cogliati et al., 2013). Remarkably, Opa1tg mice are fertile and viable, have normal lifespan, and are protected from a panoply of tissue insults, including denervation-induced skeletal muscle atrophy, brain and heart ischemia-reperfusion damage, and liver failure caused by Fas-ligand-induced apoptosis (see the accompanying paper by Varanita et al., 2015). We therefore reasoned that Opa1 overexpression could be exploited to correct the biochemical impairment and mitigate the pathology associated with genetically determined OXPHOS failure in available mouse models of mitochondrial disease. To this aim, we crossed the Opa1tg mouse with (i) a constitutive knockout mouse for Ndufs4 (Ndufs4−/−) (Karamanlidis et al., 2013; Kruse et al., 2008; Quintana et al., 2010; Quintana et al., 2012), encoding the 18 kDa subunit of complex I (cI), and (ii) a skeletal-muscle-specific knockout mouse for Cox15, in which the Cre-recombinase-driven ablation of Cox15 is dependent upon the muscle-specific actin gene promoter (Cox15sm/sm) (Viscomi et al., 2011). Cox15 encodes a key enzyme in the biosynthesis of heme a, an essential catalytic redox component of complex IV ([CIV]; cytochrome c oxidase [COX]) (Bareth et al., 2013; Barros et al., 2001; Barros and Tzagoloff, 2002; Glerum et al., 1997; Petruzzella et al., 1998; Wang et al., 2009). In humans, mutations in NDUFS4 are associated with early-onset fatal Leigh syndrome due to severe Complex I (CI) deficiency (Petruzzella et al., 2001; Scacco et al., 2003; van den Heuvel et al., 1998). Mutations in COX15 have been reported in children with severe isolated cardiomyopathy, encephalopathy, or cardioencephalomyopathy (Alfadhel et al., 2011; Antonicka et al., 2003; Bugiani et al., 2005). Here we report that the mild Opa1 overexpression achieved in the Opa1tg mice corrects these two models of CI and CIV deficiency, the latter to a greater extent.
Results
Generation of Ndufs4−/−::Opa1tg and Cox15sm/sm::Opa1tg Mouse Lines
All procedures on animals were conducted under the UK Animals (Scientific Procedures) Act, 1986, and approved by the local ethical review committee. To obtain transgenic overexpression of Opa1 in OXPHOS defective mouse lines, we crossed the Opa1tg mouse with either Ndufs4−/− or Cox15sm/sm mice. The constitutive Ndufs4−/− mouse shows severe CI deficiency, and the accumulation of a catalytically inactive 830 kDa CI assembly intermediate, associated with the onset of a rapidly progressive syndrome, dominated by neurological impairment starting approximately 40 days after birth (Calvaruso et al., 2012; Kruse et al., 2008). The Cox15sm/sm mouse shows profound skeletal muscle COX deficiency, leading to muscle wasting, severe motor performance impairment, and markedly reduced survival (Viscomi et al., 2011).
Ndufs4−/−::Opa1tg Mice Live Longer and Show Better Motor Coordination than Ndufs4−/− Mice
Western blot analysis on two animals for each genotype showed tissue-specific increases in the levels of long and short forms of OPA1 in brain (Figure S1A), skeletal muscle (Figure S1B), and heart (Figure S1C) in Opa1tg and Ndufs4−/−::Opa1tg relative to their Ndufs4−/− and wild-type (WT) littermates. Motor coordination, measured as latency to fall by weekly performed rotarod test, showed steady downhill in both Ndufs4−/−::Opa1tg and Ndufs4−/− groups, each composed of six animals. However, the scores, which were identical in the two groups at the beginning of the observation (week 4 after birth), showed significant differences in subsequent measurements, with the Ndufs4−/−::Opa1tg group performing consistently better than the Ndufs4−/− group in weeks 5 (latency to fall: 556 ± 39 versus 388 ± 46 s) and 6 (latency to fall: 384 ± 56 versus 123 ± 50 s). At week 7, both groups were virtually unable to perform the test at any rate, due to collapse of neurological conditions or death (Figure 1A). Furthermore, Kaplan-Meier analysis showed that survival probability was moderately, but significantly, prolonged in Ndufs4−/−::Opa1tg versus Ndufs4−/− littermates (survival median: 62.0 versus 53.5 days; log-rank test p = 0.015) (Figure 1B). The amelioration in motor coordination and prolongation of the lifespan was associated with a ∼1.5-fold increase in glutamate-malate-dependent state-3 oxygen consumption rate of isolated brain mitochondria (reflecting CI-driven respiration), which nevertheless remained well below the values obtained in brain mitochondria of WT and Opa1tg littermates (Figure 1C). Accordingly, CI activity, measured by spectrophotometric assay (Figure 1D), was ∼2.5-fold increased (p < 0.05) in Ndufs4−/−::Opa1tg versus Ndufs4−/− isolated brain mitochondria. Several MRC complex subunits were analyzed by western blotting on SDS-PAGE of isolated brain mitochondria. Notably, the two tested CI subunits (NDUFA9 and NDUFB8), as well as the UQCRC2 subunit of CIII, were increased ∼1.3-fold in Ndufs4−/−::Opa1tg versus Ndufs4−/− brain mitochondria (Figure 1E). The p values of two-tail, unpaired Student’s t test of densitometric analysis obtained from four independent samples/genotype was p = 0.02 for NDUFA9, p = 0.04 for NDUFB8, and p = 0.01 for UQCRC2. Contrariwise, no significant change was observed for SDHA (CII), COX1 (CIV), and ATP5A (CV) subunits. Blue native gel electrophoresis (BNGE)-based in-gel activity of CI and CI-containing supercomplexes (sc) showed hardly any reactive band in both Ndufs4−/−::Opa1tg double mutants and Ndufs4−/− littermates (Figure S1D). Western blot analysis using antibodies against NDUFB8 (specific to CI), UQCRC1 (specific to CIII), and COX1 (specific to CIV) showed, in the same genotypes, a catalytically active 200 kDa assembly intermediate as well as the catalytically inactive 830 kDa CI and 830 kDa+CIII2 assembly intermediates previously reported in the Ndufs4−/− mouse model (Calvaruso et al., 2012), but neither CI holocomplex nor CI supercomplexes containing CI+CIII2 or CI+CIII2+CIV (Figure S1E). However, electron microscopy analysis of forebrain (Figure 1F) and cerebellum (not shown) suggested amelioration in cristae architecture, with no change in mitochondrial shape and dimensions.
Figure 1.

Improved Motor Function and Extended Lifespan in Ndufs4−/−::Opa1tg Double Mutants
(A) Rotarod analysis (n = 6/group). Dashed blue: WT; dashed red: Opa1tg; solid blue: Ndufs4−/−; solid red: Ndufs4−/−::Opa1tg. Error bars represent SEM. The asterisks represent the significance levels calculate by unpaired, two-tail Student’s t test, ∗p < 0.05, ∗∗∗p < 0.005, and refer to the comparison between Ndufs4−/−::Opa1tg and Ndufs4−/−.
(B) Kaplan-Meier survival probability. Significance was assessed by the log rank test. Solid blue: Ndufs4−/− (n = 16); solid red: Ndufs4−/−::Opa1tg (n = 9).
(C) Oxygen consumption measurements (nmol O2/min/mg of protein). Solid blue: WT (n = 5); blue outline: Opa1tg (n = 5); solid red: Ndufs4−/− (n = 5); red outline: Ndufs4−/−::Opa1tg (n = 6). The asterisks represent the significance levels calculate by unpaired, two-tail Student’s t test ∗p < 0.05.
(D) MRC activities (nmol/min/mg of protein) in isolated brain mitochondria. Solid blue: WT (n = 8); blue outline: Opa1tg (n = 5); solid red: Ndufs4−/− (n = 7); red outline: Ndufs4−/−::Opa1tg (n = 8). Error bars represent SEM. The asterisks represent the significance levels calculate by unpaired, two-tail Student’s t test ∗p < 0.05.
(E) Western blot analysis of subunits from CI (NDUFA9 and NDUFB8), CII (SDHA), CIII (UQCRC2), CIV (COX1), and CV (ATP5A) in brain isolated mitochondria. VDAC is taken as an internal control.
(F) Electron microscopy of brain specimens from the different genotypes. Note that mitochondrial morphology is well preserved in Ndufs4−/−::Opa1tg versus Ndufs4−/−. Scale bar 1 μm.
These findings suggest that moderate Opa1 overexpression partially rescues the phenotype of a CI-deficient mouse model by improving cristae morphology.
Survival and Functional Improvement in the Double Mutant Cox15sm/sm::Opa1tg Mice
Similar to Cox15sm/sm::Opa1tg, male and female Cox15sm/sm::Opa1tg mice were smaller than WT or Opattg littermates (p < 0.005; n = 4 for each genotype; data not shown). To test muscle performance, we monthly monitored motor endurance by a standard treadmill test starting at 2 months of age. Albeit still significantly lower than WT (n = 8) and Opa1tg (n = 9) littermates, the scores of the Cox15sm/sm::Opa1tg double mutant group (n = 8) were ∼2- to 3-fold higher than those of the Cox15sm/sm group (n = 10) (p < 0.005 at each time point; Figure 2A). On average, the distance covered by Cox15sm/sm::Opa1tg animals was three times that of the Cox15sm/sm littermates (169 ± 20 versus 58 ± 6 m, p = 2.59 × 10−6) (Figure S2A). This improvement was recorded in both homozygous females and hemizygous males, with no significant gender difference (Figure S2B). Interestingly, Opa1Tg performed better than WT littermates (890 ± 50 versus 803 ± 27 m, p < 0.001; Figure S2A). We followed the animals up to 6 months of age. Notably, Cox15sm/sm::Opa1tg double mutants showed a remarkable increase in survival probability compared to their Cox15sm/sm littermates (log-rank test p = 0.0003). Only 1/17 (7%) Cox15sm/sm::Opa1tg animal died during our observation period, versus 12/23 (50%) Cox15sm/sm littermates (Figure 2B).
Figure 2.

Improved Motor Function and Extended Lifespan in Cox15sm/sm−::Opa1Tg Animals
(A) Treadmill analysis of motor performance. Dashed blue: WT (n = 8); dashed red: Opa1tg (n = 9); solid blue: Cox15sm/sm (n = 10); solid red Cox15sm/sm::Opa1tg (n = 8). Error bars represent SEM. The asterisks represent the significance levels calculate by unpaired, two-tail Student’s t test, ∗∗∗p < 0.005, and refer to the comparison between Cox15sm/sm::Opa1tg and Cox15sm/sm.
(B) Kaplan–Meier survival probability. Significance was assessed by the log rank test. Solid blue: Cox15sm/sm (n = 23); solid red: Cox15sm/sm::Opa1tg (n = 17).
These findings indicate that controlled Opa1 overexpression ameliorates life expectancy and motor function of a mouse model of muscle CIV deficiency.
Amelioration of Mitochondrial Morphology and Biochemistry in Cox15sm/sm::Opa1tg Muscle
H&E staining of the mid-portion of gastrocnemius muscle (Figure 3A) showed numerous centralized nuclei in both Cox15sm/sm and Cox15sm/sm::Opa1tg, the proportion of which did not significantly differ between the two groups (Figure 3B). However, the average cross-sectional area (CSA) of muscle fibers was doubled in the Cox15sm/sm::Opa1tg relative to Cox15sm/sm muscles (p < 0.05), although still smaller (∼50%) than the areas measured in WT and Opa1tg muscles (p < 0.05) (Figure 3C). We also tested whether the muscle hypotrophic and dystrophic features of the Cox15sm/sm mice were associated with increased apoptosis or altered autophagy. TUNEL staining in muscle sections did not reveal hardly any positive fiber in either Cox15sm/sm or Cox15sm/sm::Opa1tg, similar to WT and Opa1Tg (not shown), suggesting that apoptosis is not a major player in the pathogenesis of Cox15sm/sm myopathy. Immunostaining of muscle sections with Lamp1 and P62 antibodies revealed increased reactions in Cox15sm/sm samples compared to WT and Opa1Tg, which, however, did not change in Cox15sm/sm::Opa1tg (not shown). These data indicate that apoptosis and autophagy are unlikely to contribute to the amelioration observed in Cox15sm/sm::Opa1tg mice.
Figure 3.

Improved Morphological Features of Cox15sm/sm::Opa1Tg Skeletal Muscle
(A) H&E staining of skeletal muscles from mice of the different genotypes. Scale bar 100 μm.
(B) Quantification of the number of centralized nuclei in the different genotypes. 600 fibers/sample were analyzed (n = 4/genotype). Solid blue: WT; blue outline: Opa1tg; solid red: Cox15sm/sm; red outline: Cox15sm/sm::Opa1tg. Statistical significance was calculated by unpaired, two-tail Student’s t test: ∗∗∗p < 0.005. Error bars represent SEM.
(C) Cross-sectional area of muscle fibers of gastrocnemius. 600 fibers/sample were analyzed (n = 4/genotype). Solid blue: WT; blue outline: Opa1tg; solid red: Cox15sm/sm; red outline: Cox15sm/sm::Opa1Tg. Statistical significance was calculated by unpaired, two-tail Student’s t test: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.005. Error bars represent SEM.
Next, we performed ultrastructural analysis of muscle fibers (Figure 4). Examination of tibialis anterior muscle revealed marked improvement of mitochondrial and myofibrillary morphology in Cox15sm/sm::Opa1tg compared to Cox15sm/sm. While Cox15sm/sm muscle mitochondria showed profound cristae disorganization and prominent vacuolar degeneration of the inner mitochondrial compartment, the Cox15sm/sm::Opa1tg cristae were organized in an ordered and parallel array and were tightly folded, similar to those observed in WT and Opa1tg littermate specimens (Figure 4A). No hyperfused organelles were observed in Opa1Tg muscle. Likewise, myofibrils, which in Cox15sm/sm appeared disorganized and separated from each other by the accumulation of membranous and granular material, showed a compact orderly array in Cox15sm/sm::Opa1tg, again resembling those of WT and Opa1tg specimens (Figure 4A). Quantification of area, perimeter, length, and width of muscle mitochondria showed significantly increased values (1.5- to 5-fold) in Cox15sm/sm, relative to WT and Opa1tg specimens, indicating mitochondrial ballooning. These parameters were partially corrected in Cox15sm/sm::Opa1tg samples (1.3- to 1.8-fold versus WT) (Figure 4B).
Figure 4.

Amelioration of Mitochondrial Ultrastructure in Cox15sm/sm::Opa1Tg Skeletal Muscle
(A) EM microphotographs of specimens from the different genotypes. Note that while there is accumulation of inter-myofibrillar amorphous material and disorganization of the mitochondrial cristae (inset) in the Cox15sm/sm, the myofibrillary array and cristae structure (inset) are both well preserved in the Cox15sm/sm::Opa1Tg. Mitochondria appear larger and with denser cristae in Opa1Tg than in WT. Scale bar 2 μm; scale bar of the inset: 200 nm.
(B) Morphometric quantification of the area, perimeter, length and width of mitochondria from the different genotypes. A total of 200 mitochondria from WT and Opa1Tg mice (n = 2) were analyzed, and 500 from Cox15sm/sm and Cox15sm/sm::Opa1Tg (n = 2).
We then tested whether the improvements in motor performance and muscle morphology observed in Cox15sm/sm::Opa1tg animals were associated with ameliorated biochemical parameters. Glutamate-malate dependent state-3 oxygen consumption rate in isolated muscle mitochondria (reflecting CI-driven respiration) was decreased in Cox15sm/sm animals (128 ± 37 nmol/min/mg protein), but increased significantly in Cox15sm/sm::Opa1tg (190 ± 48 nmol/min/mg; p < 0.05), up to lower normal values, not significantly different from those obtained in WT (250 ± 84 nmol/min/mg) and Opa1Tg (276 ± 101 nmol/min/mg) samples (Figure 5A). Similar results were obtained by measuring succinate- and TMPD/ascorbate-dependent state-3 oxygen consumption rate (reflecting CII- and CIV-driven respiration, respectively) (Figure 5A). We also analyzed the activity of the single respiratory complexes by spectrophotometric assays. The specific activity of CIV normalized to that of citrate synthase (CS) was significantly increased (p < 0.05) in Cox15sm/sm::Opa1tg (16 ± 3) versus Cox15sm/sm (10 ± 4) muscle homogenates, albeit still much lower than that of WT (47 ± 18) or Opa1Tg (44 ± 7) muscles (Figure 5B). These results were qualitatively confirmed by histochemical COX staining, which showed reduced COX-negative fibers in COX/SDH double reaction in Cox15sm/sm::Opa1tg versus Cox15sm/sm muscle fibers (Figure 5C).
Figure 5.

Increased Respiration and Mrc activities in Cox15sm/sm::Opa1Tg Skeletal Muscle
(A) Oxygen consumption rate (nmol O2/min/mg of protein). Solid blue: WT (n = 5); blue outline: Opa1tg (n = 5); solid red: Cox15sm/sm (n = 5); red outline: Cox15sm/sm::Opa1tg (n = 6). Error bars represent SEM. The asterisks represent the significance levels calculate by unpaired, two-tail Student’s t test, ∗p < 0.05.
(B) MRC complex activities (nmol/min/mg of protein) in skeletal muscle mitochondria shown as a percentage of CS activity. Solid blue: WT (n = 5); blue outline: Opa1tg (n = 5); solid red: Cox15sm/sm (n = 5); red outline: Cox15sm/sm::Opa1tg (n = 6). Error bars represent SD. The asterisks represent the significance levels calculate by unpaired, two-tail Student’s t test, ∗p < 0.05.
(C) COX/SDH histochemical double staining. Scale bar: 50 μm.
Overall, these data indicate that the ameliorated Cox15sm/sm::Opa1tg mitochondrial ultrastructure is accompanied by improved respiration and CIV activity.
Mitochondrial Protein and Transcriptional Analysis in Cox15sm/sm::Opa1tg Mice
In order to investigate the molecular bases of the observed correction of mitochondrial respiration in Cox15sm/sm::Opa1tg animals, we first analyzed the amount of subunits specific to each MRC complex by western blotting analysis of muscle homogenates separated by SDS-PAGE (Figure 6A). COX1, the mtDNA-encoded largest CIV subunit, which contains the two heme-a moieties and the CuB center, and COX4 (isoform 1), a nucleus-encoded early-assembled CIV subunit, were both reduced in Cox15sm/sm compared to WT and Opa1Tg samples, but did increase in those from Cox15sm/sm::Opa1tg animals (Figure 6B). In both Cox15sm/sm and Cox15sm/sm::Opa1tg samples, subunits of other MRC complexes were generally increased, particularly the UQRCR2 subunit of CIII, suggesting compensatory activation of MRC biogenesis induced by CIV deficiency. Interestingly, analysis of mitochondrial and nuclear transcripts of CIV subunits revealed a specific, marked reduction of mt-Cox1 mRNA in Cox15sm/sm samples, which was fully rescued in Cox15sm/sm::Opa1tg samples (Figure 6C). Conversely, other mtDNA- (mt-Cox2) and nucleus-encoded (Cox4I1 and Cox5a) transcripts were unchanged in Cox15sm/sm versus WT mice and were not affected by Opa1 overexpression (Figure 6C). We detected no quantitative change in any group of animals for transcripts Ppargc1a and Tfam, encoding PGC-1α and TFAM, respectively (Figure 6C). The mtDNA copy number, which was increased in Cox15sm/sm, returned to WT levels in Cox15sm/sm::Opa1tg (Figure S3A), suggesting normalization of the homeostatic control on mtDNA maintenance (Scarpulla et al., 2012). Importantly, the Cox15 transcript level measured in skeletal muscle of Cox15sm/sm was ∼9% of that present in WT and Opa1tg littermates and did not change significantly in Cox15sm/sm::Opa1tg (Figure S3B), ruling out a direct effect of Opa1 overexpression on Cox15 transcription.
Figure 6.

Molecular Analysis of Cox15sm/sm::Opa1tg Skeletal Muscle
(A) Western blot analysis of OPA1, SDHA (CII), ATP5A (CV), UQCRC2 (CIII), NDUFA9 (CI), COX1, and COX4 (CIV); GAPDH is taken as a loading reference.
(B) Densitometric analysis of MRC subunits. Solid blue: WT (n = 4); blue outline: Opa1tg (n = 4); solid red: Cox15sm/sm (n = 4); red outline: Cox15sm/sm::Opa1tg (n = 4). Error bars represent SEM. The asterisks represent the significance levels calculate by unpaired, two-tail Student’s t test. ∗∗p < 0.01; ∗∗∗p < 0.005.
(C) mRNA expression analysis of CIV- and mitochondrial-biogenesis-related genes. Gene transcripts, retrotranscribed into cDNA, were quantified by RT-PCR; normalized to the amount of the β-Actin gene transcript; taken as a standard; and expressed as time-fold variation relative to the WT. Error bars represent SEM. The asterisks represent the significance levels calculate by unpaired, two-tail Student’s t test. ∗∗p < 0.01; ∗∗∗p < 0.005.
In order to analyze the composition and amount of OPA1, we performed a western blot experiment using an anti-OPA1 antibody that specifically recognizes five OPA1 species in skeletal muscle, namely isoforms 1 and 7, and short forms 1–3, resulting from proteolytic cleavage (Figure 6A). Densitometric quantification showed a significant increase (∼1.4-fold) in the amount of all five detected OPA1 species in Opa1tg muscle samples relative to WT. The amount of OPA1 species was further increased (∼1.3- to 1.8-fold) in Cox15sm/sm versus WT and significantly more so in Cox15sm/sm::Opa1tg (∼1.5- to 2-fold), particularly for the short forms 1–3 (Figure S3C).
These data suggest that Opa1 overexpression leads to stabilization in CIV protein and transcript levels.
MRC Supercomplexes Are Increased in Cox15sm/sm::Opa1tg Mice
Individual stability of MRC complexes depends on their assembly in respiratory chain supercomplexes (sc) (Acín-Pérez et al., 2004). We therefore analyzed CI and CIV BNGE in-gel activities in digitonin-treated isolated muscle mitochondria. In Cox15sm/sm samples, the intensity of the band corresponding to individual CIV holocomplex was markedly reduced, and hardly any reaction was detected in the high-molecular-weight area harboring sc. However, individual CIV in-gel activity was increased, and CIV reactive high-molecular-weight bands were clearly detected in Cox15sm/sm::Opa1tg mitochondria (Figure 7A), indicating stabilization of CIV holocomplex and CIV-containing sc. Likewise, several high-molecular-weight CI-reactive bands, corresponding to CI- and CIV-containing sc, were virtually absent in the same Cox15sm/sm samples but were clearly, albeit weakly, visible in the Cox15sm/sm::Opa1tg samples (Figure 7B). Western blot analysis on BNGE samples using an anti-COX1-specific antibody confirmed the results of in-gel activity: a band corresponding to CIV holocomplex was increased in Cox15sm/sm::Opa1tg compared to Cox15sm/sm and high-molecular-weight Cox1-reactive bands, which were detected in WT and Opa1Tg samples, were absent, even upon long exposure, in Cox15sm/sm but clearly, albeit weakly, visible in Cox15sm/sm::Opa1tg samples (Figure 7C). Likewise, immunodetection of cI-containing sc with an anti-Ndufa9 antibody showed slight increased Ndufa9 amount in Cox15sm/sm::Opa1tg versus Cox15sm/sm (Figure 7D). These results indicate that increased OPA1 levels stabilize RCS and residual CIV in Cox15sm/sm mice.
Figure 7.

Stabilization of Respiratory Complexes and Supercomplexes in Cox15sm/sm::Opa1Tg Skeletal Muscle
(A) CIV-specific BNGE in-gel activity. Note the increased staining of the bands corresponding to CIV-holoenzyme and CIV-containing sc in Cox15sm/sm::Opa1tg versus Cox15sm/sm.
(B) CI-specific BNGE in-gel activity. Note the increased staining of the bands corresponding to the sc in Cox15sm/sm::Opa1tg versus Cox15sm/sm.
(C) Western blot analysis of MRC CIV from BNGE of digitonin-treated isolated mitochondria. The blot was immunodecorated with an anti-COX1 antibody.
(D) Western blot analysis of MRC CI- and CIV-containing sc’s from BNGE of digitonin-treated isolated mitochondria. The blot was immunodecorated with an anti-NDUFB8 antibody.
Discussion
The moderate overexpression of Opa1 achieved in the Opa1tg mouse model is well tolerated and compatible with normal development, fertility, and lifespan. No obvious detrimental effects were noted during the 6-month period of our experimental observation; although, as mentioned in the accompanying paper by Varanita et al. (2015), prolonged Opa1 overexpression may lead to increased strain-specific prevalence of cancer, a somehow expected result given the antiapoptotic role of OPA1. However, increased OPA1 amount in key tissues, including skeletal and cardiac muscle, brain, and liver, confer remarkable protection against a wide spectrum of experimental tissue damage models (accompanying paper by Varanita et al., 2015). Here, we demonstrate that this effect can be exploited to improve the motor performance, as well as biochemical and molecular phenotypes of two mouse models of genetically determined OXPHOS failure.
The Ndufs4−/− mouse is characterized by the complete absence of a structural component of CI. In the current map of mammalian CI, the NDUFS4 18 kDa subunit has been modeled into a density in a cleft between the 75 kDa subunit and the 49 kDa, 30 kDa, and NDUFB8 (TYKY) subunits (Vinothkumar et al., 2014). This localization may explain why disease mutations in the 18 kDa subunit lead to accumulation of late-stage interrupted-assembly intermediates lacking the NADH-dehydrogenase module (Calvaruso et al., 2012). The indispensable role of NDUFS4 in preserving the CI structure and redox activity can also explain the limited improvement in motor coordination, measured by rotarod, and biochemical phenotypes observed in Ndufs4−/−::Opa1tg double mutants. Rapamycin-induced inhibition of mTOR, which was recently shown to alleviate the phenotype, also failed to correct the CI defect of Ndufs4−/− mice (Johnson et al., 2013). However, the cristae-centric approach used here to treat CI deficiency improved survival to levels comparable to those achieved by every-other-day rapamycin administration (Johnson et al., 2013). It would be interesting to verify if the combination of Opa1 overexpression and mTOR inhibition is additive and can achieve even better amelioration. We also cannot exclude that OPA1-driven correction of OXPHOS failure may be less amenable in brain than in other tissues, e.g., skeletal muscle.
The Cox15sm/sm mouse model is based on muscle-specific ablation of Cox15, dependent on the activity of the Cre recombinase under the control of the skeletal-muscle-specific actin promoter. Although Cox15sm/sm animals display early-onset progressive mitochondrial myopathy, characterized by profound muscle weakness and wasting leading to early death, the genetic lesion underpinning this phenotype can be considered as a hypomorphic mutant allele, the effects of which on COX activity and MRC proficiency are severe but partial. Accordingly, Cox15 transcript was drastically decreased, but not absent, in Cox15sm/sm and Cox15sm/sm::Opa1tg muscles, explaining the presence of some residual COX activity in muscle of adult animals and the “mosaic”-like distribution of the histochemical reaction to COX, which is preserved in scattered muscle fibers (Figure 5C). The molecular Cox15sm/sm defect resembles that of the majority of mitochondrial disorders (i.e., partial rather than complete biochemical impairment) due to decreased but not abolished function of the mutant gene product. Incidentally, this category of mitochondrial diseases also includes the few COX15 mutant patients reported in the literature (Alfadhel et al., 2011; Antonicka et al., 2003; Bugiani et al., 2005). These considerations may explain the marked and persistent improvement of the motor performance observed in Cox15sm/sm::Opa1tg double mutants and suggest that OXPHOS impairment due to decreased, but not abolished, function of the mutant gene product is amenable to correction by potentiating the effects of OPA1 on mitochondrial cristae shape and MRC function.
The Cox15sm/sm::Opa1tg animals displayed not only a dramatic increase in motor endurance but also a remarkable extension of survival up to, and in most cases well beyond, the 6-month period of observation of our experimental protocol. These effects were associated with robust increase of muscle mitochondrial respiration accompanied by a milder but significant increase of muscle COX activity. The discrepancy between the nearly complete reversion to normal in the respiratory rate and the modest increment of COX activity may be explained by increased stabilization of CIV holocomplex and CIV-associated supercomplexes, as indicated by results of BNGE in-gel activity and immunoblotting. The recovery in the motor performance and biochemical proficiency of the Cox15sm/sm::Opa1tg double mutants was associated with marked correction of the profound ultrastructural alteration in cristae morphology associated with Cox15 ablation. This effect is likely to be directly connected to the role of OPA1 in cristae shape and may be correlated to partial stabilization of the CIV holocomplex and supercomplex organization in the cristae membrane. Accordingly, both nucleus- and mitochondria-encoded MRC subunits were increased in Cox15sm/sm::Opa1tg versus Cox15sm/sm muscles, while the corresponding transcripts were unchanged, with the notable exception of mt-Cox1 mRNA. COX1 is the mtDNA-encoded catalytic CIV subunit that contains the heme-a moieties essential for COX1 stabilization (Bareth et al., 2013). The concordant decrease of both COX1 protein and mt-Cox1 mRNA in Cox15sm/sm was completely reverted in Cox15sm/sm::Opa1tg. Taken together, these results substantiate the possibility that in mammals, as in Saccharomyces cerevisiae, COX1 biosynthesis is regulated by a feedback mechanism linking translation and post-translation processing to transcription (Herrmann et al., 2013). In yeast, a key role in this homeostatic loop is carried out by the mt-Cox1-specific translation activators MSS51 and PET309 (Herrmann et al., 2013). The mammalian ortholog of PET309 has been identified as the leucine-rich pentatricopeptide repeat-containing (LRPPRC) protein, an RNA binding factor that is mutated in French-Canadian, COX defective Leigh-like syndrome in humans (Mootha et al., 2003). Interestingly, LRPPRC interacts with OPA1, at least in D. melanogaster (Banerjee and Chinthapalli, 2014). However, as there is no obvious mammalian ortholog of MSS51, and the 5′ UTR sequence of the yeast Cox1 mRNA, to which MSS51 binds, is virtually absent in the mammalian transcript, these feedback mechanisms are probably different in their molecular details between yeast and mammalian systems.
Finally, while apoptosis seems not to play a significant role in Cox15sm/sm myopathy and is not increased in Cox15sm/sm::Opa1tg samples, immunoanalysis suggested dysregulated autophagy in response to Cox15 ablation, but whether this is relevant to the pathogenesis of the disease remains to be further investigated.
Our study demonstrates that OPA1-dependent mitochondrial cristae and sc stabilization is effective in correcting mitochondrial disease conditions characterized by partial, however severe, MRC defects. OPA1 can even partially improve defects caused by complete lack of mitochondrial CI via an unknown mechanism. Our results indicate OPA1 as a new target for effective therapy of primary mitochondrial disorders. From a translational point of view, future work is warranted to elucidate the molecular details underpinning these remarkable effects, extend the experimental observation to additional OXPHOS-defective models, and eventually transfer this set of proof-of-principle observations into effective therapeutic approaches by, for example, selecting compounds or bioreactors able to control the expression or stability of OPA1 in suitable mammalian cell systems and animal models.
Experimental Procedures
Reagents and Materials
Antibodies (COX1, COX4, UQCRC2, NDUFA9, ATP5a, and SDHA) were from Mitoscience, GAPDH were from Millipore, P62 was from Sigma, LC3-I/II was from Cell Signaling, Lamp1 was from Sigma, and OPA1 was from BD Biosciences.
Animal Work
All procedures were conducted under the UK Animals (Scientific Procedures) Act, 1986, approved by Home Office license (PPL: 7538) and local ethical review. The mice were kept on a C57Bl6/129Sv mixed background, and WT littermates were used as controls. The animals were maintained in a temperature- and humidity-controlled animal-care facility with a 12 hr light/dark cycle and free access to water and food and were sacrificed by cervical dislocation.
Behavioral Analysis
A treadmill apparatus (Columbus Instruments) was used to measure motor exercise endurance according to the number of falls in the motivational grid during a gradually accelerating program with speed initially at 6.5 m/min and increasing by 0.5 m/min every 3 min. The test was terminated by exhaustion, defined as >10 falls/min into the motivational grid.
A rotarod apparatus (Ugo Basile) was used to assess coordination skills. After two acclimation sessions, the mice underwent three trial sessions at least 20 min apart, using a standard acceleration protocol pre-set by the constructor.
Oxygen Consumption Studies
Mouse brains were homogenized in 0.075 M sucrose, 0.225 M mannitol, 1 mM EGTA, and 0.01% fatty-acids-free BSA (pH 7.4); skeletal muscle were homogenized in 150 mM sucrose, 50 mM Tris-HCl, 50 mM KCl, 10 mM EDTA, 0.2% BSA (pH 7.4), and subtilisin 1 mg/g of muscle (Frezza et al., 2007). Mitochondria isolated by differential centrifugation and resuspended 25 mM sucrose, 75 mM sorbitol, 100 mM KCl, 0.05 mM EDTA, 5 mM MgCl2, 10 mM Tris-HCl (pH7.4), and 10 mM H3PO4 (pH 7.4) (Fernández-Vizarra et al., 2002).
For oxygraphic measurements, 250–500 μg of mitochondrial proteins were incubated in a buffer containing 225 mM sucrose, 75 mM mannitol, 10 mM Tris-HCl (pH 7.4), 10 mM KCl, 10 mM KH2PO4, 5 mM MgCl2, and 1 mg/ml fatty-acids-free BSA (pH 7.4). Oxygen consumption was evaluated by a Clark oxygen electrode (Hansatech, Instruments), using the following substrates and inhibitors concentrations: 5 mM glutamate and 2.5 mM malate for cI-dependent respiration, 5 mM succinate and 2 μM rotenone for cII-dependent respiration, 6 mM ascorbate, and 300 μM TMPD and antimycin A 0.25 μg/ml for cIV-dependent respiration. 100 μM ADP was added to stimulate ATP-coupled oxygen consumption. 100 μM NaCN was added to completely inhibit respiration (Frezza et al., 2007).
BNGE
For BNGE analysis, 250 μg of mitochondria isolated as described above were resuspended in native page buffer (Invitrogen), protease inhibitors, and 4% digitonin and incubated for 1 hr on ice before centrifuging at 20,000 × g at 4°C. 5% Coomassie G250 was added to the supernatant. 30 μg were separated by 3%–12% gradient BNGE and either stained with for in-gel activities or electroblotted on PVDF membranes for immunodetection (Nijtmans et al., 2002).
Morphological Analysis
For histochemical analysis, tissues were frozen in liquid-nitrogen-pre-cooled isopentane. 8-μm-thick sections were stained for COX and SDH, as described Sciacco and Bonilla (1996). Analysis of centralized nuclei and analysis was performed on H&E-stained sections using ImageJ on four samples/genotype (600 fibers/sample). For ultrastructural studies, samples were fixed with 2.5% glutaraldehyde in 10 mM phosphate buffer (pH 7.4). Quantification of mitochondrial morphometry (area, perimeter, long axis, and short axis) in electron micrographs was performed using ImageJ on 200 mitochondria from WT and Opa1tg muscles and 500 mitochondria from Cox15sm/sm and Cox15sm/sm::Opa1tg muscles.
Biochemical Analysis of MRC Complexes
Brain and skeletal muscle samples were snap-frozen in liquid nitrogen and homogenized in 10 mM phosphate buffer (pH 7.4). The spectrophotometric activity of CI, CII, CIII, and CIV, as well as CS, was measured as described in Bugiani et al. (2004).
Real-Time PCR
MtDNA content and transcripts analysis was carried out by SYBR Green real-time PCR, as described in Viscomi et al. (2011).
Western Blot Analysis
Mouse tissues were homogenized in ten volumes of 10 mM potassium phosphate buffer (pH 7.4). Mitochondrial-enriched fractions were collected after centrifugation at 800 × g for 10 min in the presence of protease inhibitors, and frozen and thawed three times in liquid nitrogen. Protein concentration was determined by the Lowry method. Aliquots, 70 μg each, were run through a 4%–12% SDS-PAGE and electroblotted onto a nitrocellulose membrane, which was then immunodecorated with different antibodies.
Statistical Analysis
All numerical data are expressed as mean ± SEM. Student’s unpaired two-tail t test and Kaplan-Meier distribution were used for statistical analysis. Differences were considered statistically significant for p < 0.05.
Author Contributions
G.C. carried out the in vivo phenotype analysis and performed biochemical measurements and molecular analysis and wrote the manuscript; T.V. generated mouse colonies, carried out BNGE in-gel activity, western blot, and ultrastructural analysis and wrote the manuscript; T.G. and S.B. helped to carry out in vivo phenotype analysis; R.C., S.M., and C.L. carried out histological, histochemical, and morphometric analysis; C.V. supervised and participated in the experimental design, supervised the experimental work, and contributed to write the manuscript; L.S. and M.Z. designed the study and wrote the manuscript.
Acknowledgments
This work was supported by: Telethon-Italy GPP10005 (to M.Z. and L.S.), GGP11011 (to M.Z.), GGP12162, GGP14187A (to L.S.), AIRC IG 11716-2011 (to L.S.), Cariplo 2011-0526 (to M.Z.), ERC FP7-322424 (to M.Z.) and FP7-282280 (to L.S.), FP7 CIG PCIG13-GA-2013-618697 (to L.S.), Italian Ministry of Research FIRB RBAP11Z3YA_005 (to L.S.), and Italian Ministry of Health GR-2010-2306-756 (to C.V.). We are grateful to the personnel at Phenomics and ARES animal care facilities for skillful technical assistance.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Contributor Information
Luca Scorrano, Email: luca.scorrano@unipd.it.
Massimo Zeviani, Email: mdz21@mrc-mbu.cam.ac.uk.
Supplemental Information
References
- Acin-Perez R., Enriquez J.A. The function of the respiratory supercomplexes: the plasticity model. Biochim. Biophys. Acta. 2014;1837:444–450. doi: 10.1016/j.bbabio.2013.12.009. [DOI] [PubMed] [Google Scholar]
- Acín-Pérez R., Bayona-Bafaluy M.P., Fernández-Silva P., Moreno-Loshuertos R., Pérez-Martos A., Bruno C., Moraes C.T., Enríquez J.A. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol. Cell. 2004;13:805–815. doi: 10.1016/s1097-2765(04)00124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfadhel M., Lillquist Y.P., Waters P.J., Sinclair G., Struys E., McFadden D., Hendson G., Hyams L., Shoffner J., Vallance H.D. Infantile cardioencephalopathy due to a COX15 gene defect: report and review. Am. J. Med. Genet. A. 2011;155A:840–844. doi: 10.1002/ajmg.a.33881. [DOI] [PubMed] [Google Scholar]
- Anand R., Wai T., Baker M.J., Kladt N., Schauss A.C., Rugarli E., Langer T. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J. Cell Biol. 2014;204:919–929. doi: 10.1083/jcb.201308006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonicka H., Mattman A., Carlson C.G., Glerum D.M., Hoffbuhr K.C., Leary S.C., Kennaway N.G., Shoubridge E.A. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am. J. Hum. Genet. 2003;72:101–114. doi: 10.1086/345489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S., Chinthapalli B. A proteomic screen with Drosophila Opa1-like identifies Hsc70-5/Mortalin as a regulator of mitochondrial morphology and cellular homeostasis. Int. J. Biochem. Cell Biol. 2014;54:36–48. doi: 10.1016/j.biocel.2014.05.041. [DOI] [PubMed] [Google Scholar]
- Bareth B., Dennerlein S., Mick D.U., Nikolov M., Urlaub H., Rehling P. The heme a synthase Cox15 associates with cytochrome c oxidase assembly intermediates during Cox1 maturation. Mol. Cell. Biol. 2013;33:4128–4137. doi: 10.1128/MCB.00747-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barros M.H., Tzagoloff A. Regulation of the heme A biosynthetic pathway in Saccharomyces cerevisiae. FEBS Lett. 2002;516:119–123. doi: 10.1016/s0014-5793(02)02514-0. [DOI] [PubMed] [Google Scholar]
- Barros M.H., Carlson C.G., Glerum D.M., Tzagoloff A. Involvement of mitochondrial ferredoxin and Cox15p in hydroxylation of heme O. FEBS Lett. 2001;492:133–138. doi: 10.1016/s0014-5793(01)02249-9. [DOI] [PubMed] [Google Scholar]
- Bugiani M., Invernizzi F., Alberio S., Briem E., Lamantea E., Carrara F., Moroni I., Farina L., Spada M., Donati M.A. Clinical and molecular findings in children with complex I deficiency. Biochim. Biophys. Acta. 2004;1659:136–147. doi: 10.1016/j.bbabio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Bugiani M., Tiranti V., Farina L., Uziel G., Zeviani M. Novel mutations in COX15 in a long surviving Leigh syndrome patient with cytochrome c oxidase deficiency. J. Med. Genet. 2005;42:e28. doi: 10.1136/jmg.2004.029926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvaruso M.A., Willems P., van den Brand M., Valsecchi F., Kruse S., Palmiter R., Smeitink J., Nijtmans L. Mitochondrial complex III stabilizes complex I in the absence of NDUFS4 to provide partial activity. Hum. Mol. Genet. 2012;21:115–120. doi: 10.1093/hmg/ddr446. [DOI] [PubMed] [Google Scholar]
- Cipolat S., Martins de Brito O., Dal Zilio B., Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA. 2004;101:15927–15932. doi: 10.1073/pnas.0407043101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogliati S., Frezza C., Soriano M.E., Varanita T., Quintana-Cabrera R., Corrado M., Cipolat S., Costa V., Casarin A., Gomes L.C. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delettre C., Griffoin J.M., Kaplan J., Dollfus H., Lorenz B., Faivre L., Lenaers G., Belenguer P., Hamel C.P. Mutation spectrum and splicing variants in the OPA1 gene. Hum. Genet. 2001;109:584–591. doi: 10.1007/s00439-001-0633-y. [DOI] [PubMed] [Google Scholar]
- Duvezin-Caubet S., Koppen M., Wagener J., Zick M., Israel L., Bernacchia A., Jagasia R., Rugarli E.I., Imhof A., Neupert W. OPA1 processing reconstituted in yeast depends on the subunit composition of the m-AAA protease in mitochondria. Mol. Biol. Cell. 2007;18:3582–3590. doi: 10.1091/mbc.E07-02-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehses S., Raschke I., Mancuso G., Bernacchia A., Geimer S., Tondera D., Martinou J.C., Westermann B., Rugarli E.I., Langer T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009;187:1023–1036. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott H.R., Samuels D.C., Eden J.A., Relton C.L., Chinnery P.F. Pathogenic mitochondrial DNA mutations are common in the general population. Am. J. Hum. Genet. 2008;83:254–260. doi: 10.1016/j.ajhg.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enriquez J.A., Lenaz G. Coenzyme q and the respiratory chain: coenzyme q pool and mitochondrial supercomplexes. Mol. Syndromol. 2014;5:119–140. doi: 10.1159/000363364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Vizarra E., López-Pérez M.J., Enriquez J.A. Isolation of biogenetically competent mitochondria from mammalian tissues and cultured cells. Methods. 2002;26:292–297. doi: 10.1016/S1046-2023(02)00034-8. [DOI] [PubMed] [Google Scholar]
- Frezza C., Cipolat S., Martins de Brito O., Micaroni M., Beznoussenko G.V., Rudka T., Bartoli D., Polishuck R.S., Danial N.N., De Strooper B., Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Frezza C., Cipolat S., Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat. Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- Glerum D.M., Muroff I., Jin C., Tzagoloff A. COX15 codes for a mitochondrial protein essential for the assembly of yeast cytochrome oxidase. J. Biol. Chem. 1997;272:19088–19094. doi: 10.1074/jbc.272.30.19088. [DOI] [PubMed] [Google Scholar]
- Griparic L., van der Bliek A.M. The many shapes of mitochondrial membranes. Traffic. 2001;2:235–244. doi: 10.1034/j.1600-0854.2001.1r008.x. [DOI] [PubMed] [Google Scholar]
- Griparic L., Kanazawa T., van der Bliek A.M. Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J. Cell Biol. 2007;178:757–764. doi: 10.1083/jcb.200704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head B., Griparic L., Amiri M., Gandre-Babbe S., van der Bliek A.M. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J. Cell Biol. 2009;187:959–966. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann J.M., Woellhaf M.W., Bonnefoy N. Control of protein synthesis in yeast mitochondria: the concept of translational activators. Biochim. Biophys. Acta. 2013;1833:286–294. doi: 10.1016/j.bbamcr.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Ishihara N., Fujita Y., Oka T., Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–2977. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S.C., Yanos M.E., Kayser E.B., Quintana A., Sangesland M., Castanza A., Uhde L., Hui J., Wall V.Z., Gagnidze A. mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science. 2013;342:1524–1528. doi: 10.1126/science.1244360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamanlidis G., Lee C.F., Garcia-Menendez L., Kolwicz S.C., Jr., Suthammarak W., Gong G., Sedensky M.M., Morgan P.G., Wang W., Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013;18:239–250. doi: 10.1016/j.cmet.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara A., Scorrano L. Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol. 2014;24:761–770. doi: 10.1016/j.tcb.2014.08.005. [DOI] [PubMed] [Google Scholar]
- Koopman W.J., Willems P.H., Smeitink J.A. Monogenic mitochondrial disorders. N. Engl. J. Med. 2012;366:1132–1141. doi: 10.1056/NEJMra1012478. [DOI] [PubMed] [Google Scholar]
- Kruse S.E., Watt W.C., Marcinek D.J., Kapur R.P., Schenkman K.A., Palmiter R.D. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 2008;7:312–320. doi: 10.1016/j.cmet.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha V.K., Lepage P., Miller K., Bunkenborg J., Reich M., Hjerrild M., Delmonte T., Villeneuve A., Sladek R., Xu F. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl. Acad. Sci. USA. 2003;100:605–610. doi: 10.1073/pnas.242716699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijtmans L.G., Henderson N.S., Holt I.J. Blue Native electrophoresis to study mitochondrial and other protein complexes. Methods. 2002;26:327–334. doi: 10.1016/S1046-2023(02)00038-5. [DOI] [PubMed] [Google Scholar]
- Petruzzella V., Tiranti V., Fernandez P., Ianna P., Carrozzo R., Zeviani M. Identification and characterization of human cDNAs specific to BCS1, PET112, SCO1, COX15, and COX11, five genes involved in the formation and function of the mitochondrial respiratory chain. Genomics. 1998;54:494–504. doi: 10.1006/geno.1998.5580. [DOI] [PubMed] [Google Scholar]
- Petruzzella V., Vergari R., Puzziferri I., Boffoli D., Lamantea E., Zeviani M., Papa S. A nonsense mutation in the NDUFS4 gene encoding the 18 kDa (AQDQ) subunit of complex I abolishes assembly and activity of the complex in a patient with Leigh-like syndrome. Hum. Mol. Genet. 2001;10:529–535. doi: 10.1093/hmg/10.5.529. [DOI] [PubMed] [Google Scholar]
- Quintana A., Kruse S.E., Kapur R.P., Sanz E., Palmiter R.D. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc. Natl. Acad. Sci. USA. 2010;107:10996–11001. doi: 10.1073/pnas.1006214107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana A., Morgan P.G., Kruse S.E., Palmiter R.D., Sedensky M.M. Altered anesthetic sensitivity of mice lacking Ndufs4, a subunit of mitochondrial complex I. PLoS ONE. 2012;7:e42904. doi: 10.1371/journal.pone.0042904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahn J.J., Stackley K.D., Chan S.S. Opa1 is required for proper mitochondrial metabolism in early development. PLoS ONE. 2013;8:e59218. doi: 10.1371/journal.pone.0059218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scacco S., Petruzzella V., Budde S., Vergari R., Tamborra R., Panelli D., van den Heuvel L.P., Smeitink J.A., Papa S. Pathological mutations of the human NDUFS4 gene of the 18-kDa (AQDQ) subunit of complex I affect the expression of the protein and the assembly and function of the complex. J. Biol. Chem. 2003;278:44161–44167. doi: 10.1074/jbc.M307615200. [DOI] [PubMed] [Google Scholar]
- Scarpulla R.C., Vega R.B., Kelly D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 2012;23:459–466. doi: 10.1016/j.tem.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciacco M., Bonilla E. Cytochemistry and immunocytochemistry of mitochondria in tissue sections. Methods Enzymol. 1996;264:509–521. doi: 10.1016/s0076-6879(96)64045-2. [DOI] [PubMed] [Google Scholar]
- Song Z., Chen H., Fiket M., Alexander C., Chan D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007;178:749–755. doi: 10.1083/jcb.200704110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varanita T., Soriano M.E., Romanello V., Zaglia T., Quintana-Cabrera R. The Opa1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic and ischemic tissue damage. Cell Met. 2015;21:834–844. doi: 10.1016/j.cmet.2015.05.007. this issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Heuvel L., Ruitenbeek W., Smeets R., Gelman-Kohan Z., Elpeleg O., Loeffen J., Trijbels F., Mariman E., de Bruijn D., Smeitink J. Demonstration of a new pathogenic mutation in human complex I deficiency: a 5-bp duplication in the nuclear gene encoding the 18-kD (AQDQ) subunit. Am. J. Hum. Genet. 1998;62:262–268. doi: 10.1086/301716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinothkumar K.R., Zhu J., Hirst J. Architecture of mammalian respiratory complex I. Nature. 2014;515:80–84. doi: 10.1038/nature13686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viscomi C., Bottani E., Civiletto G., Cerutti R., Moggio M., Fagiolari G., Schon E.A., Lamperti C., Zeviani M. In vivo correction of COX deficiency by activation of the AMPK/PGC-1α axis. Cell Metab. 2011;14:80–90. doi: 10.1016/j.cmet.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Wang Y., Hegg E.L. Regulation of the heme A biosynthetic pathway: differential regulation of heme A synthase and heme O synthase in Saccharomyces cerevisiae. J. Biol. Chem. 2009;284:839–847. doi: 10.1074/jbc.M804167200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.