

Abstract
Fanconi anemia (FA) is a rare genetic disorder characterized by genome instability, increased cancer susceptibility, progressive bone marrow failure (BMF), and various developmental abnormalities resulting from the defective FA pathway. FA is caused by mutations in genes that mediate repair processes of interstrand crosslinks and/or DNA adducts generated by endogenous aldehydes. The UBE2T E2 ubiquitin conjugating enzyme acts in FANCD2/FANCI monoubiquitination, a critical event in the pathway. Here we identified two unrelated FA-affected individuals, each harboring biallelic mutations in UBE2T. They both produced a defective UBE2T protein with the same missense alteration (p.Gln2Glu) that abolished FANCD2 monoubiquitination and interaction with FANCL. We suggest this FA complementation group be named FA-T.
Main Text
Fanconi anemia (FA) is a rare genetic disease characterized by genome instability, cancer predisposition, progressive bone marrow failure (BMF), and various developmental abnormalities that often include radial ray anomalies, short stature, and visceral malformations.1 FA cells are hypersensitive to DNA interstrand crosslink damage (ICL) and various types of damage due to endogenous aldehydes.2–5 FA is caused by mutations in any one of 16 genes that together comprise the FA pathway. These genes include FANCA (MIM: 617139), FANCB (MIM: 300515), FANCC (MIM: 613899), FANCD1 (BRCA2) (MIM: 600185), FANCD2 (MIM: 613984), FANCE (MIM: 613976), FANCF (MIM: 603467), FANCG (XRCC9) (MIM: 600901), FANCI (MIM: 611360), FANCJ (BRIP1) (MIM: 614082), FANCL (PHF9) (MIM: 614083), FANCN (PALB2) (MIM: 610832), FANCO (RAD51C) (MIM: 613390), FANCP (SLX4) (MIM: 613951), FANCQ (XPF) (MIM: 615272), and FANCS (BRCA1) (MIM: 113705). A recent study indicated that biallelic mutations in FA-related FANCM (MIM: 609644) do not cause an FA phenotype in humans,6 raising a concern whether this nomenclature is appropriate or not. In the upstream part of the pathway, the FA core E3 ligase complex consisting of eight gene products and other associated proteins monoubiquitinates FANCD2 and FANCI, resulting in chromatin accumulation/focus formation of FANCD2 that probably recognizes stalled replication forks upon ICL or aldehyde damage. This is the critical event that regulates recruitment of structure-specific nucleases and subsequent incision/unhooking of fork-blocking lesions, mobilizing the downstream repair pathway components.2,3 UBE2T (MIM: 610538) encodes an E2 ubiquitin conjugating enzyme (EC: 6.3.2.19) which has been implicated in this monoubiquitination reaction both in vivo7–9 and in vitro.10–13
We previously analyzed the ALDH2 genotypes in 64 Japanese FA-affected individuals with the approval of the Research Ethics Committee of the Tokai University Hospital and Kyoto University and obtained informed consent from the families of all subjects involved.14 Our report included two case subjects in which mutations in the genes previously associated with FA were excluded by whole exome sequencing (WES) (listed as numbers 60 and 61 in Table S1 in Hira et al.14) (Figure S1). Serendipitously, UBE2T mutations were found in both of them (Figures 1A–1C). The two persons are hereafter designated PNGS-252 (family 1-II-1 in Figure 1D) and PNGS-255 (family 2-II-1 in Figure 1D) (Table 1). They were from unrelated families (Figure 1D) living in different geographic locations in Japan. Both individuals displayed typical FA phenotypes, with malformations and hematological abnormalities that necessitated hematopoietic stem cell transplantation (Table 1; see Supplemental Data). Chromosome fragility in lymphocytes (described in Table S2 in Hira et al.14) was consistent with the diagnosis of FA.
Figure 1.
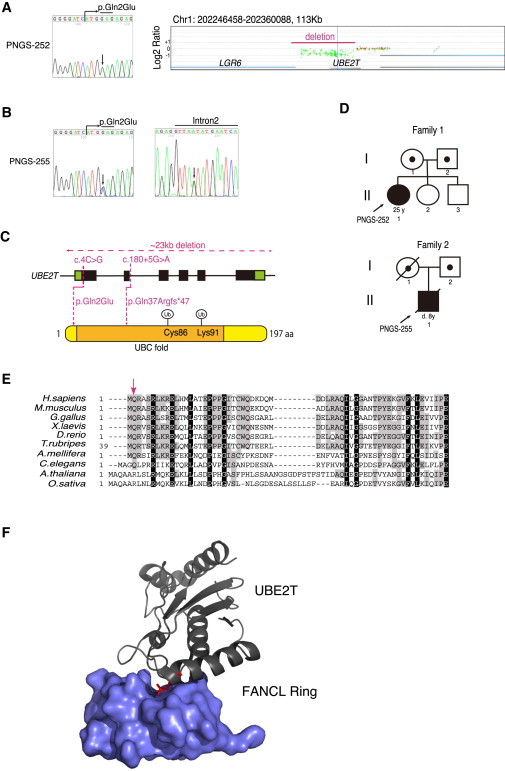
Identification of UBE2T Mutations
(A–C) Results of Sanger sequencing or array CGH of the individuals PNGS-252 (A) and PNGS-255 (B). In the array CGH, one of the deletion junctions was outside of our probes installed, and therefore it is impossible to see the whole deletion. A red line indicates the approximate region of the genome deletion detected by genome PCR (Figure S2). Schematic summary shown in (C).
Genomic DNA was isolated from PHA-stimulated lymphocytes via a Puregene (QIAGEN) kit. Genomic PCR was carried out using KOD-FX polymerase (TOYOBO) with primer pairs indicated in Table S1 and directly sequenced after ExoSAP-IT (Affymetrix) treatment or after purification from an agarose gel (Nucleospin, Takara). A custom CGH 4x180K array with a total of 179,673 50-mer probes in triplicate was designed using SureDesign (Agilent Technologies). The probes covered all of the known 16 genes associated with FA (including FANCM at the time of manufacturing) and related genes including UBE2T, NBS1, four RAD51 paralogs, FAAP20, FAAP24, and FAAP100. Appropriate control probes were also included based on the recommendations from Agilent Technologies. After CGH slides were manufactured by Agilent Technologies, fluorochrome labeling of genomic DNA from the FA-affected individuals and the reference subjects, hybridization with the CGH slides, scanning, and preliminary analysis were done by Takara Bio company. Further analysis was carried out using Genomic workbench software (Agilent). Cys86 in UBE2T is the ubiquitin acceptor site, and Lys91 is an auto-ubiquitination site.
(D) Pedigrees of the probands’ families.
(E) Alignments of the N-terminal UBE2T amino acid sequences across species. The following UBE2T amino acid sequences were aligned using Genetyx-Mac software with manual modifications (GenBank accession numbers shown): H. sapiens, NP_054895.3; M. musculus, NP_080300.1; G. gallus, XP_419230.2; X. laevis, NP_001080105.1; D. rerio, NP_001070763.1; T. rubripes, XP_003963365.2; A. mellifera, XP_003249077.1; C. elegans, NP_500272.2; A. thaliana, NP_566751.1; O. sativa, NP_001043518.1. The arrow indicates the Gln2 residue.
(F) Structure of the human FANCL Ring domain-UBE2T complex cited from Hodson et al.15 The Gln2 residue is highlighted by red lines.
Table 1.
Clinical Features of the Two FA-Affected Case Subjects
| Individual | PNGS-252 | PNGS-255 |
|---|---|---|
| Sex | female | male |
| Age at BMF (year)a | 7 | 3 |
| First allele | c.4C>G (p.Gln2Glu) | c.4C>G (p.Gln2Glu) |
| Second allele | ∼23 kb deletion (g.202288583_202309772del) | c.180+5G>A (p.Gln37Argfs∗47) |
| Physical abnormalities | left hypoplastic thumb, abnormalities of external genitalia, short stature | bilateral thumb polydactyly, abnormal shape of left ear, dysplasia of middle ear bone, deafness, facial nerve palsy |
| Hematological abnormalities | severe aplastic anemia | MDS (refractory anemia) evolving to AML |
| Age at HSCT (year)b | 13 | 8 |
| Outcome | alive and well 12 years after HSCT | died 5 months after HSCT |
| Solid tumors | none | none |
| Cells from JCRB Cell Bank | AP65P fibroblasts | not available |
| ALDH2 genotype | GA heterozygous | GA heterozygous |
The onset of BMF was defined as described.16
Haematopoietic stem cell transplantation.
WES and validation by Sanger sequencing in PNGS-252 revealed an apparent homozygous c.4C>G missense alteration (GenBank: NM_014176.3), resulting in the amino acid substitution p.Gln2Glu (Figure 1A). This mutation must be very rare, because this is not listed in the NHLBI Exome Sequencing Project or the Human Genetic Variation Browser databases. The glutamine residue (Gln2) is highly conserved in the homologs found from vertebrates to worms excluding plants (Figure 1E) and the mutation is rated as “damaging” by both SIFT and PolyPhen predictions. The Gln2 is located in the N-terminal helix of UBE2T, which constitutes part of the hydrophobic E3-E2 interaction surface, near the conserved E2 UBC fold15 (Figures 1C and 1F). Copy-number analysis using WES data suggested that there was a heterozygous deletion across the UBE2T locus in the PNGS-252 sample (data not shown). Indeed, our targeted array comparative genome hybridization (array CGH) revealed an area of reduced hybridization signal encompassing almost the entire UBE2T (Figure 1A). The deletion junction carried 3 bp of microhomology (Figures S2A–S2D), suggesting that the junction arose from microhomology-mediated repair.17 This person’s father carried the genomic deletion, and the mother had the heterozygous c.4C>G mutation (Figure S3). There was no family history of malformations, hematological abnormalities, or cancer predisposition.
In the individual PNGS-255, WES revealed the c.4C>G mutation as well as a splice donor site mutation (c.180+5G>A) (Figures 1B and 1C). Both alterations were heterozygous and on different chromosomes (Figure S4). Thus, this individual was compound heterozygous for the UBE2T mutations. In bone marrow fibroblasts, we found a small fraction of UBE2T transcripts with skipped exon 2, resulting in a frameshift and premature stop codon (p.Gln37Argfs∗47) (Figure S5). Family members of this person were not available for further evaluation. However, the results of SNP array analysis using the HumanOMni5 v.1.0 array (Illumina) suggested that a haplotype containing the c.4C>G mutation was shared by PNGS-252, her mother, and PNGS-255 (not shown). Thus, they might have a common ancestral origin.
We extended WES to AP65P FA fibroblasts provided by the JCRB Cell Bank and found the same UBE2T c.4C>G mutation. Moreover, 99.9% of the SNPs listed in dbSNP131 and identified in AP65P were identical to those in PNGS-252 (2,244 out of 2,247), demonstrating that AP65P was derived from PNGS-252 (Table 1). The AP65P individual has been reported as carrying no mutations in FANCA, FANCG, and FANCC.18 We transformed the cells with human TERT (hTERT) and termed them AP65P-hTERT. Unfortunately, we were unable to immortalize bone marrow fibroblasts from PNGS-255.
Interestingly, AP65P-hTERT cells displayed roughly similar protein levels of UBE2T as normal control cells (48BR), indicating that the p.Gln2Glu substitution does not significantly destabilize UBE2T protein (Figure 2A). We also detected the auto-monoubiquitinated form of UBE2T as previously described,7,8 suggesting that the mutant protein is able to receive activated ubiquitin from the E1 enzyme (Figure 2A). However, only faint amounts of long-form ID proteins were observed, even after MMC stimulation (Figure 2A). As expected, AP65P-hTERT cells transduced with lentivirus encoding normal UBE2T, but not with the mutant, clearly restored the MMC-induced long form of FANCD2 (Figure 2A) as well as FANCD2 foci formation (Figure 2B). Furthermore, both the increased levels of MMC-induced chromosome breakage (Figure 2C) and the MMC sensitivity (Figure 3A) in AP65P-hTERT cells were suppressed by exogenous wild-type UBE2T but not with UBE2T carrying p.Gln2Glu. Taken together, these results firmly established that the FA phenotype in these individuals is caused by the UBE2T mutations.
Figure 2.
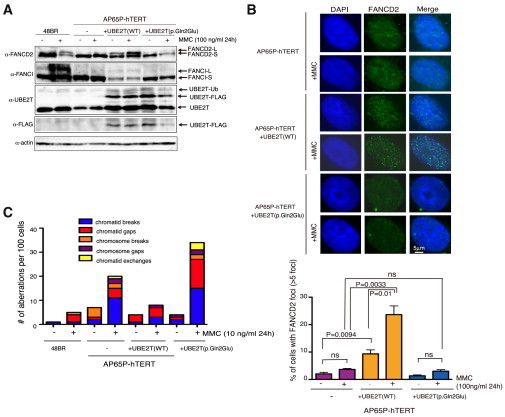
Functional Characterization of the Mutant UBE2T Protein
(A) Immunoblotting of cell lysates from normal fibroblasts (48BR) and from PNGS-252 (AP65P-hTERT) with or without indicated lentiviral transduction. AP65P primary fibroblasts (designated KURB1562 in JCRB) were obtained from the JCRB Cell Bank and transduced with an hTERT expressing retroviral vector pMSCV-hTERT-puro. For lentiviral production, HEK293T cells were transfected with CSII-CMV-MCS-IRES2-Bsd encoding human UBE2T-FLAG (either wild-type or the p.Gln2Glu mutant) together with packaging plasmids pCAG-HIVgp and pCMV-VSV-G-RSV-Rev, using Lipofectamine 2000. After 48 hr in culture, the 293T supernatants were filtered and added to the sparsely seeded AP65P-hTERT cells. Blastocidin S selection was started 2 days later (3 μg/ml). For immunoblotting, collected cells were lysed in RIPA buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1% NP-40, 5 mM EDTA, 20 mM β-glycerophosphate, 50 mM NaF, 1× protease inhibitor [Complete EDTA-free tablet Roche], 50 U/ml Benzonase, 1 mM PMSF). Samples were separated by SDS-polyacrylamide gel electrophoresis, transferred to a membrane, and detected with indicated antibodies and ECL reagents (GE Healthcare) using a LAS4000 Mini apparatus (GE Healthcare). The antibodies used in this study are summarized in Table S3.
(B) FANCD2 foci formation. Cells were grown on coverslips and subjected to 100 ng/ml MMC for 24 hr. Cells were then permeabilized with 0.5% Triton X-100/PBS for 10 min on ice and fixed with 3% paraformaldehyde/2% sucrose at room temperature for 30 min followed by blocking with 2% BSA/0.05% Triton X-100/PBS for 30 min at room temperature and staining with primary antibodies diluted in PBS containing 2% BSA and 0.05% Triton X-100 overnight at 4°C. Primary antibodies were detected by anti-rabbit Alexa 488 (Molecular Probes). Nuclei were stained with Prolong Gold mounting agent (Life Tech). Immunofluorescence images were captured with Keyence Biorevo BZ-9000. The mean and SD from three independent experiments are shown.
(C) Number of chromosomal aberrations induced by MMC treatment. MMC-exposed cells (10 ng/ml for 24 h) were arrested at M phase with Colcemid (100 ng/ml) for 3 hr, harvested, and further treated with 2 ml hypotonic 0.9% sodium citrate for 22 min. Carnoy’s solution (4 ml) was added, followed by centrifugation at 1,200 rpm for 10 min. Cell pellets were washed once with 10 ml Carnoy’s solution and resuspended in 1 ml Carnoy’s solution. In each condition, 100 cells were scored.
Figure 3.

Cell Survival Curves after MMC or UV Treatment
(A) MMC sensitivity assay. AP65P-hTERT cells were exposed to varying concentrations of MMC for 24 hr, washed, and seeded into 6-well plates. After 5–7 days, surviving cells were counted using a LUNA digital automated cell counter (Logos Biosystems) or stained with 0.006% crystal violet/25% methanol solution. GM6914 (FA-A) and its complemented cells were included as control.
(B) Immunoblotting using the indicated antibodies (listed in Table S3). AP65P-hTERT cells were transfected with control (siLuc) or siRNAs targeting UBE2T (siUBE2T#1 and #2) (final 10 nM), seeded, and treated with MMC for 24 hr or UV irradiated. siRNAs (sequence listed in Table S2) were synthesized by Life Technologies and transfected using Lipofectamine RNAiMAX.
(C) MMC or UV sensitivity in AP65P-hTERT cells depleted of mutated UBE2T protein.
(D) UV sensitivity in AP65P-hTERT cells with or without UBE2T lentiviral transduction. The sensitivity assays were repeated at least three times, and a representative dataset with mean and SD of the triplicate cultures is shown.
How, exactly, does the UBE2T alteration affect the activity of the protein in promoting monoubiquitination of the ID complex? We hypothesized that the p.Gln2Glu substitution might disrupt the FANCL-UBE2T interaction. Indeed, the p.Gln2Glu alteration drastically reduced the signal intensity in a mammalian two-hybrid assay (Figure 4A). This was confirmed by a GST pull-down experiment using purified recombinant human or chicken GST-FANCL and wild-type or mutant UBE2T proteins (Figure 4B, 4C, and S6A). In an in vitro monoubiquitination assay,11 the mutated UBE2T protein displayed ∼3-fold less efficiency in promoting FANCD2 monoubiquitination in the presence (Figure 4D) or absence (Figure S6B) of stimulator DNA, whereas auto-ubiquitination was normal compared to control proteins (Figure S6C). The p.Gln2Glu substitution abrogated FANCL monoubiquitination in vitro (Figures 4D, S6B, and S6D); however, the FANCL-independent FANCI monoubiquitination was not affected (Figure S6D).12 These results are well explained by the specific disruption of the FANCL-UBE2T interaction by the p.Gln2Glu substitution.
Figure 4.
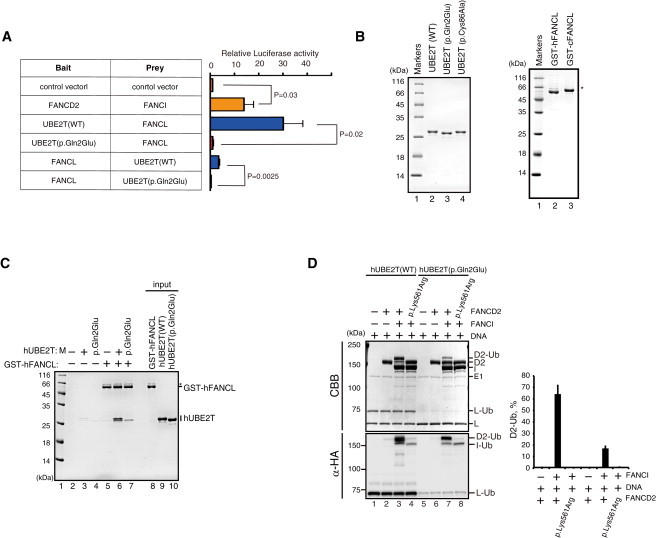
Functional Interaction between FANCL and UBE2T
(A) Mammalian two-hybrid assay. The assays were carried out as described.19 In brief, UBE2T and FANCL in the bait vector (pM) or prey vector (pVP16) were co-transfected into 293T cells with the reporter luciferase vector as well as an internal control (pRL Renilla Luciferase vector). Luminescence signals were quantified using a Dual-Glo Luciferase Reporter Assay System (Promega). The signal was first normalized to transfection efficiency using Renilla luciferase levels and further divided by the value obtained by the empty bait and prey vector. The mean and SD of more than three independent experiments are shown. Statistical analysis was done using Student’s t test, and p values are indicated.
(B) Purified recombinant proteins detected by Coomassie brilliant blue (CBB) gel staining. Human UBE2T proteins with or without the p.Gln2Glu substitution were produced in E. coli and purified to homogeneity as previously described.11 WT, wild-type.
(C) In vitro pull-down assay between GST-human (h)FANCL and hUBE2T protein with or without the p.Gln2Glu substitution. Purified human UBE2T (6 μg) and GST-chicken FANCL or human FANCL (9 μg) were incubated at 37°C for 1 hr in 200 μl of reaction buffer containing 20 mM Tris-HCl (pH 7.5), 10% glycerol, 100 mM NaCl, 1 mM ZnOAc, 0.01% NP-40, and 5 mM 2-mercaptoethanol. Glutathione sepharose 4B beads (3 μl; GE Healthcare) were added to the reaction mixtures, and reaction mixtures were gently mixed at 25°C for 1 hr. The beads were then washed twice with 1 ml of reaction buffer. The proteins bound to the beads were separated by 15% SDS-PAGE and were visualized by Coomassie brilliant blue staining. Asterisk indicates read-through products.
(D) In vitro FANCD2 monoubiquitination assay in the presence of DNA. The assay was repeated three times, and the mean and SD of the percent of monoubiquitinated FANCD2 (D2-Ub) are shown in the graph. Execution of the in vitro FANCD2 ubiquitination reaction was as described.11 As a control, FANCD2 protein carrying a p.Lys561Arg substitution blocking monoubiquitination was included. CBB, Coomassie brilliant blue. Immunoblotting with anti-HA (α-HA) was used to detect HA-tagged ubiquitin.
In conclusion, we propose that UBE2T (FANCT) mutations define a FA subtype. This is also a rare example of a mutated E2 enzyme causing an inherited human disorder, like UBE2A.20 The p.Gln2Glu substitution is probably hypomorphic, as indicated by the fact that a siUBE2T knockdown made AP65P-hTERT cells more sensitive to MMC and completely eliminated the trace FANCD2 monoubiquitination that could still be observed in the siLuc control knockdown cells (Figures 3B and 3C). Finally, it is interesting to note a recent report suggesting that UBE2T functions with an unknown E3 in nucleotide excision repair.21 It is common for an E2 to function with a set of E3 ligases, since far fewer E2s (∼38) are encoded in the genome than E3 ligases (600–1,000).22 UBE2T might have a partner other than FANCL, such as BRCA123 or other E3s, as has been suggested by yeast two-hybrid assays,24,25 raising the possibility that UBE2T might have a function outside the FA pathway. Although a siUBE2T knockdown in AP65P-hTERT modestly sensitized cells to UV (Figure 3C), we detected only a marginal impact of UBE2T lentiviral transduction on UV survival (Figure 3D). These results suggest that the p.Gln4Glu substitution is a separation of function alteration that specifically reduces UBE2T function in the FA pathway but not in UV resistance. In line with this, neither of our FA-T-affected individuals experienced any photosensitivity. It thus remains unclear whether or how complete loss of UBE2T function would impact human phenotypes.
Acknowledgments
The use of FANCT as an alias for UBE2T was approved by the HUGO Gene Nomenclature Committee. We would like to thank the individuals PNGS-252 and -255 and their family members for making this work possible. We also thank Dr. Masao S. Sasaki (Professor Emeritus, Kyoto University) for his long-standing effort to collect Japanese FA samples, including AP65P fibroblasts; Dr. James Hejna (Graduate School of Biostudies, Kyoto University) for critical reading of the manuscript and English editing; Dr. Takayuki Yamashita (Gunma University) for GM6914 cells; Dr. Hiroyuki Miyoshi (RIKEN, currently at Keio University) and RIKEN Bio-resource Center (Tsukuba, Ibaragi, Japan) for a lentivirus construct (CSII-CMV-MCS-IRES-Bsd) and the packaging system; Dr. Settara C. Chandrasekharappa (NIH) for advice on the CGH array; Dr. Yoko Katsuki for advice on immunofluorescence; Ms. Tomoko Hirayama (JCRB) for a protocol for karyotyping in fibroblasts; Ms. Fumiko Tsuchida, Chinatsu Ohki, Akiko Watanabe, and Mao Hisano for expert technical help; and Drs. Toshiyasu Taniguchi and Agata Smogorzewska for advice on anti-FANCD2/FANCI immunoblotting. The AP65P cell line (KURB1562) was kindly provided by JCRB Cell Bank, National Institute of Biomedical Innovation (Saito, Ibaraki, Osaka). This work was supported in part by grants from the Ministry of Health, Labor and Welfare of Japan.
Accession Numbers
The WES sequencing data have been deposited in the European Genome-Phenome Archive (EGA) under the accession number EGA: EGAS00001001103.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
European Genome-phenome Archive (EGA), https://www.ebi.ac.uk/ega
Human Genetic Variation Database (HGVD), http://www.genome.med.kyoto-u.ac.jp/SnpDB/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
References
- 1.Alter B.P. Diagnosis, genetics, and management of inherited bone marrow failure syndromes. Hematology (Am Soc Hematol Educ Program) 2007;2007:29–39. doi: 10.1182/asheducation-2007.1.29. [DOI] [PubMed] [Google Scholar]
- 2.Kottemann M.C., Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature. 2013;493:356–363. doi: 10.1038/nature11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walden H., Deans A.J. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu. Rev. Biophys. 2014;43:257–278. doi: 10.1146/annurev-biophys-051013-022737. [DOI] [PubMed] [Google Scholar]
- 4.Langevin F., Crossan G.P., Rosado I.V., Arends M.J., Patel K.J. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature. 2011;475:53–58. doi: 10.1038/nature10192. [DOI] [PubMed] [Google Scholar]
- 5.Garaycoechea J.I., Crossan G.P., Langevin F., Daly M., Arends M.J., Patel K.J. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature. 2012;489:571–575. doi: 10.1038/nature11368. [DOI] [PubMed] [Google Scholar]
- 6.Lim E.T., Würtz P., Havulinna A.S., Palta P., Tukiainen T., Rehnström K., Esko T., Mägi R., Inouye M., Lappalainen T., Sequencing Initiative Suomi (SISu) Project Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet. 2014;10:e1004494. doi: 10.1371/journal.pgen.1004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Machida Y.J., Machida Y., Chen Y., Gurtan A.M., Kupfer G.M., D’Andrea A.D., Dutta A. UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol. Cell. 2006;23:589–596. doi: 10.1016/j.molcel.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 8.Alpi A.F., Pace P.E., Babu M.M., Patel K.J. Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol. Cell. 2008;32:767–777. doi: 10.1016/j.molcel.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Alpi A., Langevin F., Mosedale G., Machida Y.J., Dutta A., Patel K.J. UBE2T, the Fanconi anemia core complex, and FANCD2 are recruited independently to chromatin: a basis for the regulation of FANCD2 monoubiquitination. Mol. Cell. Biol. 2007;27:8421–8430. doi: 10.1128/MCB.00504-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajendra E., Oestergaard V.H., Langevin F., Wang M., Dornan G.L., Patel K.J., Passmore L.A. The genetic and biochemical basis of FANCD2 monoubiquitination. Mol. Cell. 2014;54:858–869. doi: 10.1016/j.molcel.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sato K., Toda K., Ishiai M., Takata M., Kurumizaka H. DNA robustly stimulates FANCD2 monoubiquitylation in the complex with FANCI. Nucleic Acids Res. 2012;40:4553–4561. doi: 10.1093/nar/gks053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Longerich S., Kwon Y., Tsai M.-S., Hlaing A.S., Kupfer G.M., Sung P. Regulation of FANCD2 and FANCI monoubiquitination by their interaction and by DNA. Nucleic Acids Res. 2014;42:5657–5670. doi: 10.1093/nar/gku198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Longerich S., San Filippo J., Liu D., Sung P. FANCI binds branched DNA and is monoubiquitinated by UBE2T-FANCL. J. Biol. Chem. 2009;284:23182–23186. doi: 10.1074/jbc.C109.038075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hira A., Yabe H., Yoshida K., Okuno Y., Shiraishi Y., Chiba K., Tanaka H., Miyano S., Nakamura J., Kojima S. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood. 2013;122:3206–3209. doi: 10.1182/blood-2013-06-507962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hodson C., Purkiss A., Miles J.A., Walden H. Structure of the human FANCL RING-Ube2T complex reveals determinants of cognate E3-E2 selection. Structure. 2014;22:337–344. doi: 10.1016/j.str.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Butturini A., Gale R.P., Verlander P.C., Adler-Brecher B., Gillio A.P., Auerbach A.D. Hematologic abnormalities in Fanconi anemia: an International Fanconi Anemia Registry study. Blood. 1994;84:1650–1655. [PubMed] [Google Scholar]
- 17.McVey M., Lee S.E. MMEJ repair of double-strand breaks (director’s cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–538. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamada T., Tachibana A., Shimizu T., Mugishima H., Okubo M., Sasaki M.S. Novel mutations of the FANCG gene causing alternative splicing in Japanese Fanconi anemia. J. Hum. Genet. 2000;45:159–166. doi: 10.1007/s100380050203. [DOI] [PubMed] [Google Scholar]
- 19.Unno J., Itaya A., Taoka M., Sato K., Tomida J., Sakai W., Sugasawa K., Ishiai M., Ikura T., Isobe T. FANCD2 binds CtIP and regulates DNA-end resection during DNA interstrand crosslink repair. Cell Rep. 2014;7:1039–1047. doi: 10.1016/j.celrep.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 20.Nascimento R.M.P., Otto P.A., de Brouwer A.P.M., Vianna-Morgante A.M. UBE2A, which encodes a ubiquitin-conjugating enzyme, is mutated in a novel X-linked mental retardation syndrome. Am. J. Hum. Genet. 2006;79:549–555. doi: 10.1086/507047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelsall I.R., Langenick J., MacKay C., Patel K.J., Alpi A.F. The Fanconi anaemia components UBE2T and FANCM are functionally linked to nucleotide excision repair. PLoS ONE. 2012;7:e36970. doi: 10.1371/journal.pone.0036970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ye Y., Rape M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009;10:755–764. doi: 10.1038/nrm2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ueki T., Park J.-H., Nishidate T., Kijima K., Hirata K., Nakamura Y., Katagiri T. Ubiquitination and downregulation of BRCA1 by ubiquitin-conjugating enzyme E2T overexpression in human breast cancer cells. Cancer Res. 2009;69:8752–8760. doi: 10.1158/0008-5472.CAN-09-1809. [DOI] [PubMed] [Google Scholar]
- 24.Sheng Y., Hong J.H., Doherty R., Srikumar T., Shloush J., Avvakumov G.V., Walker J.R., Xue S., Neculai D., Wan J.W. A human ubiquitin conjugating enzyme (E2)-HECT E3 ligase structure-function screen. Mol. Cell. Proteomics. 2012;11:329–341. doi: 10.1074/mcp.O111.013706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Wijk S.J.L., de Vries S.J., Kemmeren P., Huang A., Boelens R., Bonvin A.M.J.J., Timmers H.T.M. A comprehensive framework of E2-RING E3 interactions of the human ubiquitin-proteasome system. Mol. Syst. Biol. 2009;5:295. doi: 10.1038/msb.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.