

Abstract
Myoclonus-dystonia (M-D) is a rare movement disorder characterized by a combination of non-epileptic myoclonic jerks and dystonia. SGCE mutations represent a major cause for familial M-D being responsible for 30%–50% of cases. After excluding SGCE mutations, we identified through a combination of linkage analysis and whole-exome sequencing KCTD17 c.434 G>A p.(Arg145His) as the only segregating variant in a dominant British pedigree with seven subjects affected by M-D. A subsequent screening in a cohort of M-D cases without mutations in SGCE revealed the same KCTD17 variant in a German family. The clinical presentation of the KCTD17-mutated cases was distinct from the phenotype usually observed in M-D due to SGCE mutations. All cases initially presented with mild myoclonus affecting the upper limbs. Dystonia showed a progressive course, with increasing severity of symptoms and spreading from the cranio-cervical region to other sites. KCTD17 is abundantly expressed in all brain regions with the highest expression in the putamen. Weighted gene co-expression network analysis, based on mRNA expression profile of brain samples from neuropathologically healthy individuals, showed that KCTD17 is part of a putamen gene network, which is significantly enriched for dystonia genes. Functional annotation of the network showed an over-representation of genes involved in post-synaptic dopaminergic transmission. Functional studies in mutation bearing fibroblasts demonstrated abnormalities in endoplasmic reticulum-dependent calcium signaling. In conclusion, we demonstrate that the KCTD17 c.434 G>A p.(Arg145His) mutation causes autosomal dominant M-D. Further functional studies are warranted to further characterize the nature of KCTD17 contribution to the molecular pathogenesis of M-D.
Main Text
Dystonias are a clinically and genetically heterogeneous group of non-neurodegenerative movement disorders, mainly characterized by involuntary muscle contractions leading to abnormal postures or movements of body segments.1 Mutations in a growing number of genes (recently reviewed by our group)2 are responsible for Mendelian forms of dystonia. The identification of these genes allowed the recognition of different cellular pathways involved in the molecular pathogenesis of dystonia, including perturbed synaptic transmission and plasticity, abnormal transcription and cell-cycle regulation, and endoplasmic reticulum (ER) dysfunction.3
The association with additional movement disorders identifies a sub-group of dystonias, defined as combined dystonias.4 Myoclonus-dystonia (M-D [MIM 159900]), one of the combined dystonia syndromes, is a very rare condition with a suggested prevalence of about two per million in Europe.5 M-D is clinically characterized by a variable combination of non-epileptic myoclonic jerks, mainly affecting the upper body, and mild to moderate dystonia, usually in the form of cervical dystonia or writer’s cramp.6 There is often a dramatic improvement of myoclonus after alcohol consumption.7 Psychiatric co-morbidities (e.g., depression, anxiety, and obsessive-compulsive disorder) are frequently described.8
Mutations in SCGE [MIM 604149], coding for ε-sarcoglycan, represent a major cause of inherited autosomal dominant M-D.9 SGCE mutations are detected in 30%–50% of familial M-D cases, suggesting genetic heterogeneity and the existence of mutations in other genes responsible for this condition.10–14
We used a combination of genome-wide linkage analysis and whole-exome sequencing to investigate a previously unpublished dominant British pedigree (shown in Figure 1A) with multiple individuals affected with M-D, in which SCGE mutations (both point mutations and copy number variants) had been excluded.
Figure 1.
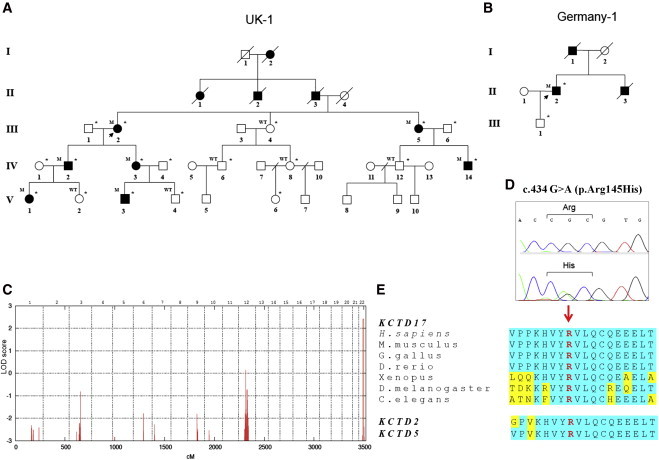
Family Trees, Linkage Analysis, and KCTD17 Mutation Analysis
(A and B) Pedigree of the British (A) and German (B) families with the KCTD17 c.434 G>A p.(Arg145His) mutation. Open symbols indicate unaffected family members, and solid black symbols indicate affected members. Individuals marked with an asterisk were clinically examined. The following abbreviations are used: WT, homozygous wild-type alleles; and M, heterozygous mutation carrier.
(C) LOD score plot for genome-wide linkage analysis in the British index pedigree showing a single linkage peak on chromosome 22q13 with a maximum LOD score of 2.4. An autosomal dominant model was specified with an estimated allele frequency of 0.00001 and 90% penetrance.
(D) Sanger sequencing confirmation of the KCTD17 c.434G>A p.(Arg145His) mutation.
(E) Multiple-sequence alignment showing complete conservation of protein sequence across species and human paralogs (KCTD2 and KCTD5) in the region of exon 4 of KCTD17, in which the disease-segregating mutation p.(Arg145His) was found.
Out of 19 living family members from the index family, 14 were clinically assessed. Assessment included a detailed medical interview and a full-videotaped neurological examination, with focus on movement disorders. All videos were reviewed by two experts in movement disorders, blinded to disease status.
The proband (III-2) developed involuntary jerky movements of her arms during childhood. In her late forties she developed constant head jerks and head deviation to the left. In her sixties her speech became involved. On examination at the age of 69 she had spasmodic dysphonia, facial myoclonus, blepharospasm, left torticollis, and frequent irregular dystonic head jerks. There was dystonic hand posturing and low amplitude brief myoclonus. When she walked she presented trunk and bilateral foot dystonia (Movie S1, section 1).
Six other family members displayed signs of dystonia and/or myoclonus, and were accordingly categorized as affected (Table 1). Age of onset of movement disorder symptoms ranged from 5 to 20 years. All affected family members initially presented with jerks or a jerky tremor, with mild dystonic features presenting later in life. All cases fulfilled the currently proposed clinical criteria for a definite diagnosis of M-D,15 except individual IV-3, who upon examination displayed isolated cervical dystonia, although she reported intermittent jerky arm tremor. Myoclonus involved predominantly the arms (Movie S1, section 2). Dystonia predominantly affected the cranio-cervical region and upper limbs. Older individuals (>60 years; III-2 and III-5) were more severely affected and also showed laryngeal involvement. None of the affected subjects reported improvement of symptoms with alcohol. Subject IV-3 had anxiety and social phobia and subject IV-14 had obsessive traits and suffered from depression. No other individuals presented with psychiatric symptoms.
Table 1.
Clinical Features of Cases Harboring the KCTD17 c.434 G>A p.(Arg145His) Mutation
| Subject | III-2 | III-5 | IV-2 | IV-3 | IV-14 | V-1 | V-3 | II.2 (Germany) |
|---|---|---|---|---|---|---|---|---|
| Sex | F | F | M | F | M | F | M | M |
| Age at last examination (y) | 70 | 65 | 46 | 44 | 35 | 20 | 19 | 62 |
| Age at onset (y) | 10 | 6 | 10 | 20 | 10 | 5 | 5 | 10 |
| Presenting symptoms | Jerks; Arms | Jerks; Arms | Jerks; Arms | Jerky tremor; Arms | Jerky tremor; Arms | Jerks; Arms | Jerks; Arms | Jerks, Arms |
| Dystonic features | Yes | Yes | Yes | Yes | Yes | Yes | No | Yes |
| Involved areas | Cranial, Cervical, Brachial, Truncal, Crural | Cranial, Cervical, Brachial | Brachial | Cervical | Cervical, Brachial | Brachial | NA | Cranial, Cervical, Brachial, Truncal |
| Myoclonus | Yes | Yes | Yes | No | Yes | Yes | Yes | Yes |
| Involved areas | Cranial, Cervical, Brachial | Cranial, Brachial | Brachial | NA | Cranial, Brachial | Brachial | Cervical, Brachial | Brachial |
Case IV-12 had strabismus and benign congenital nystagmus, but no signs of M-D. The remaining individuals were asymptomatic and had an entirely normal neurological examination.
Samples were collected with the written consent of participants and formal ethical approval by the relevant research ethics committee (UCLH Project ID number 06/N076). DNA of 13 family members and 4 spouses was extracted from blood lymphocytes.
A genome-wide linkage analysis was subsequently performed in 7 affected individuals (III-2, III-5, IV-1, IV-3, IV-14, V-1, and V-3), 5 unaffected (III-4, IV-6, IV-8, IV-12, and V-4) and 4 spouses using the HumanCytoSNP-12 DNA Analysis BeadChip Kit (Illumina). The unaffected subject V-2 was not included as she was too young (17 when last examined) to exclude or confirm disease status.
Genome-wide multipoint parametric linkage analysis for an autosomal dominant model (estimated allele frequency 0.00001 and 90% penetrance) and haplotype reconstruction were performed with Simwalk2,16 using 24,000 informative single nucleotide polymorphisms (SNPs), equally spaced 0.1cM apart as described before.17
One single locus with a LOD score > 2 was identified on chromosome 22q13 (LOD score 2.4, the maximal expected value given the pedigree size; see Figure 1C).
Fine mapping identified a segregating haplotype delimitated by SNP markers rs926543 and rs3213584 and spanning 6.7 Mb (chr22:36989327–43716324; UCSC hg19 Genome Build), which contained 132 protein-coding genes. In addition, five other regions presented with uninformative multipoint LOD scores, ranging from −0.9 to +0.14, but haplotype analysis excluded segregation of these regions with the disease.
We subsequently performed whole-exome sequencing in the two most distantly related affected individuals (V-3 and IV-14). In short, paired-end sequence reads (TruSeq SBS chemistry sequenced on the Illumina HiSeq 2000) were aligned with Novoalign against the reference human genome (UCSC hg19). Duplicate read removal, format conversion, and indexing were performed with Picard. The Genome Analysis Toolkit (GATK) was used to recalibrate base quality scores, perform local realignments around possible indels, and to call and filter the variants. Annotated variant files were generated using ANNOVAR18 and included a comparison to publicly available databases of sequence variations (dbSNP version 129, 1000 Genomes project, NHLBI Exome Variant Server, and Complete Genomics 69). In silico prediction of pathogenicity was assessed using SIFT,19 PolyPhen2,20 MutationTaster,21 Provean,22 and CADD.23 Conservation of nucleotides involved by variants was scored using Genomic Evolutionary Rate Profiling (GERP).24 Interspecies alignment of protein sequences was generated using ClustalW2.25
In total, 83,572,847 (V-3) and 81,527,162 (IV-14) unique reads were generated. According to the Consensus Coding Sequences hg19 definition of the “TruSeq exome,” the average read depth of both exomes was >70, >95% of the target bases were covered at a read depth of 2×, and > 90% at a depth of 10×. A total of 22,857 (V-3) and 22,946 (IV-14) exonic/splicing variants were detected. We filtered out all synonymous changes and those not shared by the two affected individuals. Then, under the assumption that the mutation causing this rare autosomal dominant disease is extremely rare and not present in the general population, we also excluded variants that are present in the databases of sequence variations listed above. Furthermore, we excluded variants found in our own in-house exomes (n = 200) from individuals with unrelated diseases.
After applying filtering criteria, we were left with only 4 novel missense variants shared by the two affected individuals (see Table S1): c.10976 C>T p.(Ser3659Phe) in FLG (filaggrin; RefSeq NM_002016.1), c.1055 T>G p.(Phe352Cys) in OBSCN (obscurin; RefSeq NM_052843.3), c.1076 A>C p.(Lys359Thr) in LRRC6 (leucine rich repeat containing 6; RefSeq NM_012472.4) and c.434 G>A p.(Arg145His) in KCTD17 (potassium channel tetramerization domain containing 17; RefSeq NM_001282684.1). Of these variants, only the missense change in KCTD17 was located within the linked chromosomal locus on chromosome 22q. We did not detect any shared rare copy-number variants in exome sequencing data using the Exome depth algorithm.26
Sanger sequencing in all available family members confirmed perfect co-segregation of the KCTD17 variant with the disease-phenotype, being the nucleotide change present in all affected individuals and absent in all unaffected (including subject V-2, initially excluded from the linkage analysis). The variant is absent in over 3,700 individuals of European origin without movement disorders, who were exome sequenced by the UCL-exomes consortium, and in a further >61,000 individuals listed in the Exome Aggregation Consortium database (last accessed in March 2015).
Although the KCTD17 p.(Arg145His) substitution falls in a functionally uncharacterized portion of the protein, it lies in an extremely conserved amino acid motif, not only completely conserved down to invertebrate species, but also identical in the KCTD17 human paralogs KCTD2 and KCTD5 (Figure 1E). All in silico tools consistently predicted a deleterious effect of the substitution (Table S1).
We subsequently sequenced the 9 coding exons of KCTD17 (NM_001282684.1; primers available in Table S2) in a further 87 unrelated probands with familial M-D of British, German, and Italian origin. None of the cases harbored mutations in SGCE. Mutational screening of KCTD17 exon 4 (containing the c.434 G>A mutation) was performed in a further 358 sporadic M-D cases without mutations in SCGE.
This analysis revealed the presence of the same KCTD17 mutation, c.434 G>A p.(Arg145His), in the index case of a German family with autosomal dominant M-D (Figure 1B). No further likely pathogenic mutations were identified.
The clinical presentation of this case closely resembled that of III-2 and III-5, the older affected subjects from the British family. He reported arm jerks and difficulty writing, starting in childhood. Right torticollis and a jerky head tremor appeared around age 40, becoming progressively debilitating. There was no response to alcohol or psychiatric comorbidities. He underwent surgery for bilateral pallidal deep brain stimulation at age 58, which resulted in marked improvement of cervical dystonia and myoclonus of the upper limbs. Clinical examination at age 62 showed generalized dystonia, with prominent cranio-cervical involvement, and myoclonic jerks involving the upper limbs (Movie S1, section 3). His father was also affected with a movement disorder, presenting with perioral dyskinesia in his forties. The proband’s brother had M-D, with similar clinical features, including generalized jerks, cervical dystonia, and dysarthria. Unfortunately, DNA samples of the deceased father and brother were not available for segregation analysis. The 25-year-old proband’s only son, who had no signs upon examination, refused genetic testing.
Haplotype comparison between the two pedigrees with KCTD17 c.434 G>A p.(Arg145His) was performed with SNP markers located 0.5 Mb up- and downstream of the mutation. This analysis showed that different alleles are located at markers rs5756477 and rs228924, delimitating a small region of ∼100 Kb of a possibly shared haplotype (Table S3). A further analysis with a highly polymorphic microsatellite, located only 1.4 Kb upstream of the 5′ end of KCTD17, revealed that the two pedigrees have different alleles, possibly suggesting the absence of a shared ancestral haplotype and that the variant might have arisen independently in the two pedigrees. Of relevance, the absence of a shared haplotype between the two families would make unlikely that the KCTD17 c.434 G>A p.(Arg145His) mutation is in linkage disequilibrium with the actual causative mutation but not itself pathogenic.
We explored the regional distribution of KCTD17 expression in the normal adult human brain. As previously described, we used microarray data (Affymetrix Exon 1.0 ST) from human post-mortem brain tissue collected by the UK Human Brain Expression Consortium (UKBEC).27 KCTD17 mRNA expression throughout the course of human brain development was assessed using the data available in the Human Brain Transcriptome (HBT) database.28,29 KCTD17 mRNA expression was high across all brain regions, but it was highest in the putamen followed by the thalamus (Figure 2A). These findings are consistent with the data available in the HBT database, showing increasing KCTD17 brain mRNA levels in the striatum and the thalamus from early midfetal development to adolescence (Figure 2B). In light of the current view of the neuroanatomical bases of dystonia, which is thought to be a network disorder of the basal ganglia connections,30,31 this pattern of expression is highly relevant and supports the pathogenic role of KCTD17 in the pathogenesis of M-D.
Figure 2.
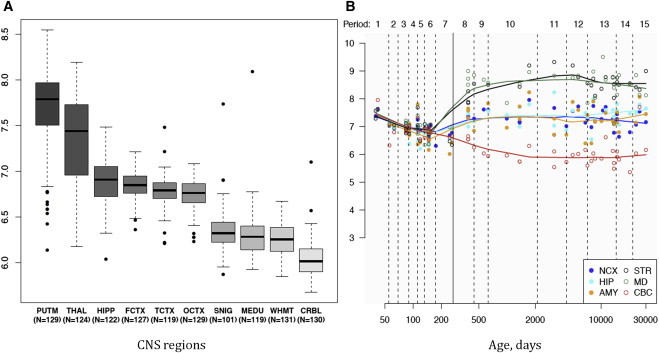
Summary of Brain Regional mRNA Expression Data
(A) Box plot of mRNA expression levels for KCTD17 in ten adult brain regions, based on exon array experiments and plotted on a log2 scale (y axis). This dataset was generated using Affymetrix Exon 1.0 ST Arrays and brain and CNS tissue originating from 134 control individuals, collected by the Medical Research Council (MRC) Sudden Death Brain and Tissue Bank, and the Sun Health Research Institute (SHRI), an affiliate of Sun Health Corporation, USA.27 The plot shows significant variation in KCTD17 transcript expression across the ten CNS regions analyzed: putamen (PUTM), frontal cortex (FCTX), temporal cortex (TCTX), hippocampus (HIPP), substantia nigra (SNIG), medulla (specifically inferior olivary nucleus, MEDU), intralobular white matter (WHMT), thalamus (THAL), and cerebellar cortex (CRBL). “N” indicates the number of brain samples analyzed to generate the results for each CNS region. KCT17 expression is higher in the putamen, followed by the thalamus. Whiskers extend from the box to 1.53 the interquartile range.
(B) Graph to show mRNA expression levels for KCTD17 in 6 brain regions during the course of human brain development, based on exon array experiments and plotted on a log2 scale (y axis).28,29 The brain regions analyzed are the striatum (STR), amygdala (AMY), neocortex (NCX), hippocampus (HIP), mediodorsal nucleus of the thalamus (MD), and cerebellar cortex (CBC). This shows increasing expression of KCTD17 mRNA during human brain development, particularly in the striatum and thalamus, from the early midfetal period to adolescence.
KCTD17 encodes for a member of a recently identified family of 26 closely related and highly conserved proteins, the potassium channel tetramerization domain (KCTD)-containing proteins. KCTD proteins are characterized by the presence of a N-terminal bric-a-brack, tram-track, broad complex/poxvirus zinc finger (BTB/POZ) domain, homologous to the cytoplasmic domain T1 of voltage-gated potassium channels.32 The BTB/POZ domain is known to permit protein-protein interactions, either promoting self-oligomerization or facilitating interaction with other biological partners.33 KCTDs are small soluble proteins, which are not predicted by their structure to form transmembrane domains. Despite the homology reflected in their names, a direct interaction with potassium channels has not been shown for most members of the family and was explicitly excluded for KCTD5, a paralog 85% identical to KCTD17.34
KCTD proteins, despite the high level of sequence similarity, are involved in a surprisingly wide spectrum of cell functions, including regulation of cellular proliferation, gene transcription, cytosketelon organization, protein degradation targeting via the ubiquitin-proteasome system, and regulation of G protein-coupled receptors.35
Several members of the KCTD family have a primary role in the central nervous system36,37 and a growing number of neurological diseases have been linked to mutations in KCTD genes. KCTD7 [MIM 611725] mutations cause recessive progressive myoclonic epilepsy.38–40 Copy-number variants in KCTD13 [MIM 608947] have been associated with size of the head, autism disorder, and epilepsy.41 More recently, a homozygous missense mutation in KCTD3 [MIM 613272] was identified as the likely cause in a pedigree with severe psychomotor retardation, seizure, and cerebellar hypoplasia.42
The precise cellular localization and function of the KCTD17 protein are largely unknown. Recent work has shown that KCTD17 contributes to the ubiquitin-proteasome machinery, acting as an adaptor for the CUL3-RING E3 ligase and targeting substrates for degradation through polyubiquitinylation.43 Although most of the KCTD17-CUL3 substrates are currently unknown, CUL3 has been implicated in the elaboration of dendrite branching and neurite terminal morphogenesis in Drosophila models.44,45
Stably transfected SH-SY5 cells were generated by incorporating N- and C-terminally hemagglutinin (HA)-tagged wild-type and mutant KCTD17 cDNAs. KCTD17 staining with anti-HA primary monoclonal antibodies showed that the protein is diffusely distributed in the cytosol with fine reticular pattern and does not localize at the plasma membrane (Figure S1). Co-staining with ER, Golgi apparatus, mitochondria, and lysosomal markers failed to show any co-localization with KCTD17 (data not shown). We did not observe significant changes in subcellular localization of mutant versus wild-type KCTD17, indicating that the amino acid substitution does not lead to cellular mislocalization of the protein.
To gain further insight into the functional role of KCTD17 and identify molecular pathways possibly dysregulated by mutant KCTD17, weighted gene co-expression network analysis (WGCNA) was performed based on the UKBEC human brain mRNA expression data. In brief, this systems biology analytic approach uses brain regional whole-transcriptome gene expression data and establishes the degree of gene neighborhood sharing, as defined on the basis of co-expression relationships. This approach allows to identify in an unsupervised and unbiased manner modules of genes that are highly co-expressed and co-regulated and therefore likely to be functionally related.46 Microarray data on 19152 transcripts (corresponding to 17247 genes), generated from 101 brains, were used to create weighted modules of co-expressed genes for each analyzed brain regions. A detailed description of the methods used to generate the dataset is available in the manuscript of Forabosco and colleagues.47
KCTD17, in common with other genes associated with dystonia (e.g., ANO3 [MIM 610110] and GNAL [MIM 139312]), shows the highest expression in the putamen and this brain structure has an established role in the pathogenesis of dystonia. We therefore focused the analysis on the putamen module including KCTD17. This module contains 179 transcripts (equating to 172 genes; Table S4; see Figure 3 for a graphic representation of the module).
Figure 3.
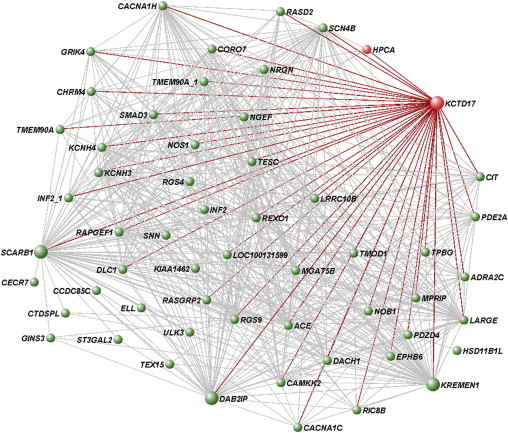
Network Representation of the Putamen KCTD17-Containing Gene Module
Array expression profiling of 788 brain samples obtained from 101 neuropathologically healthy individuals was performed and used for weighted gene co-expression network analysis (WGCNA). The WGCNA network was constructed for each brain region using a scale-free topology, as previously described.47 A dissimilarity matrix based on topological overlap measure was used to identify gene modules (i.e., densely interconnected and co-expressed genes) through a dynamic tree-cutting algorithm. Shown are all genes in the putamen KCTD17-containing module connected with a topological overlap measure exceeding 0.03. The dystonia genes in the module (KCDT17 and HPCA) and all the direct connections of KCTD17, based on topological overlap values, are highlighted in red. Larger circles represent the most interconnected genes in the module, including KCTD17.
We first assessed whether the module was enriched for genes linked to Mendelian forms of dystonia. We focused the analysis on the nine genes known to be associated with dystonia (TOR1A [MIM 605204], THAP1 [MIM 609520], SGCE, TUBB4A [MIM 602662], CIZ1 [MIM 611420], ANO3, GNAL, ATP1A3 [MIM 182350], PRKRA [MIM 603424])2 and HPCA [MIM 142622], a gene recently associated with autosomal recessive dystonia.48 We did not include in the analysis genes causing DOPA-responsive dystonia (GCH1 [MIM#600225], TH [MIM#191290], and SPR [MIM 182125]) as their established functional role in nigrostriatal dopamine synthesis, together with the specificity of the clinical presentation, clearly identifies them as a separate entity.
Importantly, the putamen KCTD17-module showed significant clustering of dystonia genes (KCTD17 and HPCA; Fisher’s exact test p = 5 × 10−3), suggesting the relevance of this gene network to the molecular pathogenesis of dystonia. The module was poorly preserved across other brain regions, indicating its specificity to the putamen (Figure S2). The brain regional specificity of this module might suggest why mutations in KCTD17 and HPCA manifest purely as a dysfunction of the basal ganglia (i.e., dystonia), in spite of the ubiquitous expression in the human brain.
To infer the biological and functional relevance of the putamen KCTD17 gene network, we then carried out functional annotation enrichment analysis using the online tool g:Profiler.49 This analysis allowed the identification of over-represented genes assigned to specific Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, namely “Circadian entrainment” (KEGG:04713; p = 5.13 × 10−3) and “Dopaminergic synapse” (KEGG:04728; p = 2.71 × 10−2). This suggests the involvement of the genes in the module with these molecular pathways.
Recent work in Drosophila strongly reinforces the results of WGCNA and further suggests a relevant contribution of KCTD17 to regulation of dopaminergic transmission in the putamen. Insomniac (the KCTD17 fly ortholog) is an essential regulator of sleep homeostasis through the control of the dopaminergic arousal pathways.50,51 More specifically, insomniac seems to regulate dopaminergic signaling at the post-synaptic level, possibly controlling the turnover of dopamine receptors or their downstream effectors.51 Interestingly, abnormal post-synaptic dopaminergic signaling in the basal ganglia is one of the main themes in molecular dystonia pathogenesis, a concept recently strengthened by the identification of mutations in GNAL causing dystonia.52 Fitting well with this model, all the genes in the putamen KCTD17 module assigned to the KEGG “dopaminergic synapse” pathway (CACNA1C [MIM 114205], PPP1R1B [MIM 604399], PPP2R2C [MIM 605997], AKT1 [MIM 164730], GNAO1 [MIM 139311], and GNB2 [MIM 139390]) localize and act at the post-synaptic level.
Disruption of calcium (Ca2+) homeostasis has been recently implicated in the pathogenesis of several genetic forms of dystonia (e.g., TOR1A, ANO3, HPCA).48,53,54 As the putamen KCTD17-module included HPCA, a gene with an established role in intracellular Ca2+-dependent signaling,55 we hypothesized that the KCTD17 p.(Arg145His) substitution might have a significant impact on intracellular Ca2+ homeostasis. For this purpose, fibroblasts were isolated from a skin biopsy taken from a subject with the KCTD17 p.(Arg145His) variant (index family, III-2). Two unrelated age- and passage-matched controls were selected from in-house fibroblast lines. The expression of KCTD17 in fibroblasts was confirmed by RT-PCR (data not shown). Calcium homeostasis was assessed using the ratio-metric Ca2+ dye, Fura-2, AM (Molecular Probes), which indicates intracellular Ca2+ concentration and allows recordings of Ca2+ fluxes upon application of different pharmacological stimuli. We observed that stimulation with ATP (50 μM), which stimulates P2Y receptors and releases Ca2+ from the ER via IP3 receptors, resulted in significantly reduced and delayed cytosolic Ca2+ signal (p < 0.01; Figures 4A and 4B) in cells carrying the p.(Arg145His) mutation when compared to both control cells, indicating a smaller calcium pool within the ER. To further prove this finding, we subsequently carried out a second round of experiments using thapsigargin (1 μM) in Ca2+-free medium (plus 0.5 mM EGTA). Thapsigargin is an inhibitor of the ER calcium ATPase (SERCA) and induces the release of calcium from the ER to the cytosol, allowing an estimation of the ER Ca2+-pool. Ca2+ was then added at the end of the experiment to stimulate elevation of cytosolic Ca2+ through opening of store operated calcium channels. Thapsigargin stimulation resulted in a significantly smaller Ca2+signal in fibroblasts bearing the p.(Arg145His) mutation when compared to controls (p < 0.01; Figures 4C and 4D), confirming that the Ca2+ pool in the ER of mutation-carrying fibroblasts is reduced. Furthermore, stimulation of the store-operated Ca2+ channels induced a smaller Ca2+ influx in mutated fibroblasts (Figure 4C), possibly suggesting an insufficient Ca2+ influx across the plasma membrane in response to the fall in Ca2+ concentration within the ER lumen. Interestingly, we recently showed very similar defects of ER Ca2+ storage in fibroblasts bearing a pathogenic mutation in ANO3.53 This indicates that defective ER calcium signaling might represent a converging pathogenic mechanism in genetically unrelated forms of dystonia.
Figure 4.
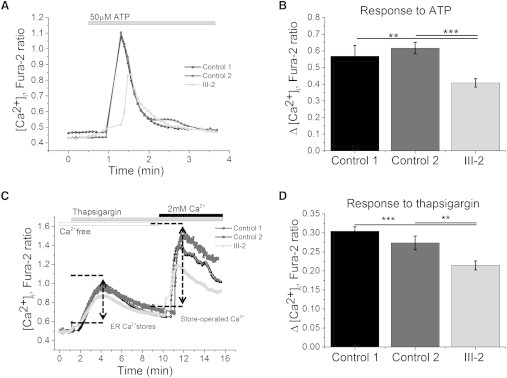
Functional Studies Showing Abnormalities of Endoplasmic Reticulum Calcium Signaling in KCTD17 p.(Arg145His) Substitution Bearing Fibroblasts
After obtaining informed consent, fibroblasts were isolated from a skin biopsy taken from a subject with the KCTD17 p.(Arg145His) substitution (index family, III-2). Two unrelated age- and passage-matched controls (ctrl 1 and 2) were selected from in-house cell lines. The fibroblasts were cultured in Dulbecco’s modified Eagle’s medium supplemented with GlutaMAX, 10% heat-inactivated fetal bovine serum, and 1% penicillin-streptomycin. Cytosolic calcium ([Ca2+]c) was measured with Fura-2, AM. Fibroblasts were loaded at room temperature for 30 min with 5M Fura-2 AM and 0.005% Pluronic in HBSS. Fluorescence measurements were obtained on an epifluorescence inverted microscope equipped with a 20× fluorite objective. [Ca2+]c was monitored in single cells using excitation light provided by a Xenon arc lamp, with the beam passing monochromator centered at 340 and 380 nm (Cairn Research). Emitted fluorescence light was reflected through a 515 nm long-pass filter to a cooled CCD camera (Retiga, QImaging). All experiments were carried out in triplicate. Data are represented as the mean ± SEM “n” indicates the total number of cells analyzed. The asterisks indicate p < 0.05 (∗), p < 0.01 (∗∗) and p < 0.001 (∗∗∗).
(A) Typical trace of [Ca2+]c in control and KCTD17 mutant fibroblasts in response to the application of 50 mM ATP.
(B) Histograms showing a significantly decreased [Ca2+]c response upon ATP stimulation in mutation-bearing fibroblasts (n = 56) versus controls (control 1 n = 41; control 2 n = 35), as measured by changes in Fura-2 fluorescence intensity.
(C) Typical trace of [Ca2+]c in control and KCTD17 mutant fibroblasts in response to the application of thapsigargin (1 μM), and subsequent Ca2+ challenge (2 mM).
(D) Histograms demonstrating a significant reduction in ER calcium pool in response to thapsigargin in mutation-bearing fibroblasts (n = 53) versus controls (control 1 n = 51; control 2 n = 65), as measured by changes in Fura-2 fluorescence intensity.
In conclusion, we demonstrate that a missense mutation in KCTD17, c.434 G>A p.(Arg145His), represents a rare genetic cause for inherited autosomal dominant M-D.
The clinical features of the KCTD17-mutated cases, although fully consistent with a clinical diagnosis of M-D, were distinct in many ways from the usual phenotype of subjects with SGCE mutations. Dystonia dominated the clinical picture and showed a progressive course, worsening over time and spreading to other sites (including speech involvement), a course unusual for SGCE-related M-D. Myoclonus, despite being the presenting symptom in most cases, was overall mild and not as disabling as in SGCE-mutated subjects.
These phenotypic differences might be explained not only by the different functions of the two genes but also by the clearly distinct patterns of brain regional expression. SGCE is highly expressed in the cerebellum, whereas its expression is low to moderate in putamen and globus pallidus.56 On the other hand, KCTD17 expression is high in the putamen and thalamus but relatively low in the cerebellum. Intriguingly, this could be the explanation for the scarce response to alcohol consumption in KCTD17-mutated cases, as alcohol probably exerts its beneficial effect in M-D secondary to SGCE mutations by modulating cerebellar activity.56
Preliminary data suggest an involvement of KCTD17 in dopaminergic signaling and an effect of the p.(Arg145His) substitution on ER-derived Ca2+ homeostasis. Further insight into the physiological role of KCTD17 and a better understanding of the pathogenic effect of the p.(Arg145His) substitution will shed light onto the mechanisms leading to abnormal neuronal activity underlying M-D. Furthermore, the identification of KCTD17 interactors will possibly highlight new potential pharmacological targets for the treatment of dystonia.
Mutational screening of additional cohorts of M-D cases will help to define the frequency and the spectrum of KCTD17 mutations. KCTD17 mutations should be considered in cases without mutations in SGCE presenting with myoclonus, dystonia, or a combination of both, particularly if there is predominant cranio-cervical and laryngeal involvement.
Acknowledgments
We would like to extend our thanks to the individuals whose participation made this research possible. This work was supported financially by a Medical Research Council/Wellcome Trust Strategic Award (WT089698/Z/09/Z) and a grant from the Bachman-Strauss Dystonia Parkinsonism Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The work was undertaken at University College London Hospitals (UCLH) and University College London (UCL), who receive support from the Department of Health’s NIHR Biomedical Research Center’s funding streams. E.L. and T.G. are supported by a grant from the Dystonia Medical Research Foundation (DMRF). C.K. is the recipient of a career development award from the Herman and Lilly Schilling Foundation. A.M.P. is funded by the Reta Lila Weston Trust. S.B.C. held a FPU fellowship from the Spanish Ministry of Education and Science jointly with a short-term stay grant by Cei-BioTic and University of Granada. Next Generation Sequencing was performed at the UCL Institute of Neurology Sequencing Facility. We thank the UCL-Exomes Consortium for providing the exome sequencing data of UK population controls. Expression data was provided by the UK Human Brain Expression Consortium (UKBEC), which comprises J.H., M.R., Michael Weale, D.T., Adaikalavan Ramasamy, Colin Smith, and Robert Walker. UKBEC members are affiliated with UCL Institute of Neurology (J.H., M.R., D.T.), King’s College London (M.R., Adaikalavan Ramasamy, and Michael Weale), and the University of Edinburgh (Colin Smith and Robert Walker). We thank Miss Elisavet Preza for providing help with the skin biopsy preparation.
Published: May 14, 2015
Contributor Information
Kailash P. Bhatia, Email: k.bhatia@ucl.ac.uk.
Nicholas W. Wood, Email: n.wood@ucl.ac.uk.
Supplemental Data
Index case (III.2): This segment shows the index case of the British family. Note the generalized dystonia with severe cranial and cervical involvement, including spasmodic dysphonia. Trunk and leg involvement are more evident while walking. Superimposed brief myoclonus are seen in the face and arms. Case V.3: This segment shows another member of the British family. He has frequent brief myoclonus involving the head and the arms. Myoclonus becomes more intense and frequent while talking. Index case German family: This segment shows the index case of the German family. He has generalized dystonia. There is marked cranial involvement, including dysarthria, and tongue dystonia. Trunk involvement is more evident while walking. He also displays arm myoclonus.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
ClustalW2 - Multiple Sequence Alignment, http://www.ebi.ac.uk/Tools/msa/clustalw2/
Complete Genomics cg69 database: http://www.completegenomics.com/public-data/69-Genomes
ExAC Browser, http://exac.broadinstitute.org/
G-profiler: http://biit.cs.ut.ee/gprofiler/index.cgi
Human Brain Transcritptome database: http://hbatlas.org/
Merlin: http://www.sph.umich.edu/csg/abecasis/merlin/index.html
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
PROVEAN, http://provean.jcvi.org/index.php
References
- 1.Albanese A., Bhatia K., Bressman S.B., Delong M.R., Fahn S., Fung V.S., Hallett M., Jankovic J., Jinnah H.A., Klein C. Phenomenology and classification of dystonia: a consensus update. Mov. Disord. 2013;28:863–873. doi: 10.1002/mds.25475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Charlesworth G., Bhatia K.P., Wood N.W. The genetics of dystonia: new twists in an old tale. Brain. 2013;136:2017–2037. doi: 10.1093/brain/awt138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ledoux M.S., Dauer W.T., Warner T.T. Emerging common molecular pathways for primary dystonia. Mov. Disord. 2013;28:968–981. doi: 10.1002/mds.25547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fung V.S., Jinnah H.A., Bhatia K., Vidailhet M. Assessment of patients with isolated or combined dystonia: an update on dystonia syndromes. Mov. Disord. 2013;28:889–898. doi: 10.1002/mds.25549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asmus F., Gasser T. Inherited myoclonus-dystonia. Adv. Neurol. 2004;94:113–119. [PubMed] [Google Scholar]
- 6.Asmus F., Zimprich A., Tezenas Du Montcel S., Kabus C., Deuschl G., Kupsch A., Ziemann U., Castro M., Kühn A.A., Strom T.M. Myoclonus-dystonia syndrome: epsilon-sarcoglycan mutations and phenotype. Ann. Neurol. 2002;52:489–492. doi: 10.1002/ana.10325. [DOI] [PubMed] [Google Scholar]
- 7.Nardocci N. Myoclonus-dystonia syndrome. In: Vinken P.J., Bruyn G.W., editors. Volume 100. Elsevier; 2011. pp. 563–575. (Handbook of Clinical Neurology). [DOI] [PubMed] [Google Scholar]
- 8.Peall K.J., Smith D.J., Kurian M.A., Wardle M., Waite A.J., Hedderly T., Lin J.P., Smith M., Whone A., Pall H. SGCE mutations cause psychiatric disorders: clinical and genetic characterization. Brain. 2013;136:294–303. doi: 10.1093/brain/aws308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zimprich A., Grabowski M., Asmus F., Naumann M., Berg D., Bertram M., Scheidtmann K., Kern P., Winkelmann J., Müller-Myhsok B. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat. Genet. 2001;29:66–69. doi: 10.1038/ng709. [DOI] [PubMed] [Google Scholar]
- 10.Schüle B., Kock N., Svetel M., Dragasevic N., Hedrich K., De Carvalho Aguiar P., Liu L., Kabakci K., Garrels J., Meyer E.M. Genetic heterogeneity in ten families with myoclonus-dystonia. J. Neurol. Neurosurg. Psychiatry. 2004;75:1181–1185. doi: 10.1136/jnnp.2003.027177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tezenas du Montcel S., Clot F., Vidailhet M., Roze E., Damier P., Jedynak C.P., Camuzat A., Lagueny A., Vercueil L., Doummar D., French Dystonia Network Epsilon sarcoglycan mutations and phenotype in French patients with myoclonic syndromes. J. Med. Genet. 2006;43:394–400. doi: 10.1136/jmg.2005.036780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carecchio M., Magliozzi M., Copetti M., Ferraris A., Bernardini L., Bonetti M., Defazio G., Edwards M.J., Torrente I., Pellegrini F. Defining the epsilon-sarcoglycan (SGCE) gene phenotypic signature in myoclonus-dystonia: a reappraisal of genetic testing criteria. Mov. Disord. 2013;28:787–794. doi: 10.1002/mds.25506. [DOI] [PubMed] [Google Scholar]
- 13.Peall K.J., Kurian M.A., Wardle M., Waite A.J., Hedderly T., Lin J.P., Smith M., Whone A., Pall H., White C. SGCE and myoclonus dystonia: motor characteristics, diagnostic criteria and clinical predictors of genotype. J. Neurol. 2014;261:2296–2304. doi: 10.1007/s00415-014-7488-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grünewald A., Djarmati A., Lohmann-Hedrich K., Farrell K., Zeller J.A., Allert N., Papengut F., Petersen B., Fung V., Sue C.M. Myoclonus-dystonia: significance of large SGCE deletions. Hum. Mutat. 2008;29:331–332. doi: 10.1002/humu.9521. [DOI] [PubMed] [Google Scholar]
- 15.Kinugawa K., Vidailhet M., Clot F., Apartis E., Grabli D., Roze E. Myoclonus-dystonia: an update. Mov. Disord. 2009;24:479–489. doi: 10.1002/mds.22425. [DOI] [PubMed] [Google Scholar]
- 16.Sobel E., Sengul H., Weeks D.E. Multipoint estimation of identity-by-descent probabilities at arbitrary positions among marker loci on general pedigrees. Hum. Hered. 2001;52:121–131. doi: 10.1159/000053366. [DOI] [PubMed] [Google Scholar]
- 17.Hersheson J., Mencacci N.E., Davis M., MacDonald N., Trabzuni D., Ryten M., Pittman A., Paudel R., Kara E., Fawcett K. Mutations in the autoregulatory domain of β-tubulin 4a cause hereditary dystonia. Ann. Neurol. 2013;73:546–553. doi: 10.1002/ana.23832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 20.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwarz J.M., Cooper D.N., Schuelke M., Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods. 2014;11:361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 22.Choi Y., Sims G.E., Murphy S., Miller J.R., Chan A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davydov E.V., Goode D.L., Sirota M., Cooper G.M., Sidow A., Batzoglou S. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput. Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 26.Plagnol V., Curtis J., Epstein M., Mok K.Y., Stebbings E., Grigoriadou S., Wood N.W., Hambleton S., Burns S.O., Thrasher A.J. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28:2747–2754. doi: 10.1093/bioinformatics/bts526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trabzuni D., Ryten M., Walker R., Smith C., Imran S., Ramasamy A., Weale M.E., Hardy J. Quality control parameters on a large dataset of regionally dissected human control brains for whole genome expression studies. J. Neurochem. 2011;119:275–282. doi: 10.1111/j.1471-4159.2011.07432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnson M.B., Kawasawa Y.I., Mason C.E., Krsnik Z., Coppola G., Bogdanović D., Geschwind D.H., Mane S.M., State M.W., Sestan N. Functional and evolutionary insights into human brain development through global transcriptome analysis. Neuron. 2009;62:494–509. doi: 10.1016/j.neuron.2009.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang H.J., Kawasawa Y.I., Cheng F., Zhu Y., Xu X., Li M., Sousa A.M., Pletikos M., Meyer K.A., Sedmak G. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kimmich O., Molloy A., Whelan R., Williams L., Bradley D., Balsters J., Molloy F., Lynch T., Healy D.G., Walsh C. Temporal discrimination, a cervical dystonia endophenotype: penetrance and functional correlates. Mov. Disord. 2014;29:804–811. doi: 10.1002/mds.25822. [DOI] [PubMed] [Google Scholar]
- 31.Neychev V.K., Gross R.E., Lehéricy S., Hess E.J., Jinnah H.A. The functional neuroanatomy of dystonia. Neurobiol. Dis. 2011;42:185–201. doi: 10.1016/j.nbd.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Z., Xiang Y., Sun G. The KCTD family of proteins: structure, function, disease relevance. Cell Biosci. 2013;3:45. doi: 10.1186/2045-3701-3-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stogios P.J., Downs G.S., Jauhal J.J., Nandra S.K., Privé G.G. Sequence and structural analysis of BTB domain proteins. Genome Biol. 2005;6:R82. doi: 10.1186/gb-2005-6-10-r82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dementieva I.S., Tereshko V., McCrossan Z.A., Solomaha E., Araki D., Xu C., Grigorieff N., Goldstein S.A. Pentameric assembly of potassium channel tetramerization domain-containing protein 5. J. Mol. Biol. 2009;387:175–191. doi: 10.1016/j.jmb.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skoblov M., Marakhonov A., Marakasova E., Guskova A., Chandhoke V., Birerdinc A., Baranova A. Protein partners of KCTD proteins provide insights about their functional roles in cell differentiation and vertebrate development. BioEssays. 2013;35:586–596. doi: 10.1002/bies.201300002. [DOI] [PubMed] [Google Scholar]
- 36.Matsui A., Tran M., Yoshida A.C., Kikuchi S.S., U M., Ogawa M., Shimogori T. BTBD3 controls dendrite orientation toward active axons in mammalian neocortex. Science. 2013;342:1114–1118. doi: 10.1126/science.1244505. [DOI] [PubMed] [Google Scholar]
- 37.Schwenk J., Metz M., Zolles G., Turecek R., Fritzius T., Bildl W., Tarusawa E., Kulik A., Unger A., Ivankova K. Native GABA(B) receptors are heteromultimers with a family of auxiliary subunits. Nature. 2010;465:231–235. doi: 10.1038/nature08964. [DOI] [PubMed] [Google Scholar]
- 38.Kousi M., Anttila V., Schulz A., Calafato S., Jakkula E., Riesch E., Myllykangas L., Kalimo H., Topçu M., Gökben S. Novel mutations consolidate KCTD7 as a progressive myoclonus epilepsy gene. J. Med. Genet. 2012;49:391–399. doi: 10.1136/jmedgenet-2012-100859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krabichler B., Rostasy K., Baumann M., Karall D., Scholl-Bürgi S., Schwarzer C., Gautsch K., Spreiz A., Kotzot D., Zschocke J. Novel mutation in potassium channel related gene KCTD7 and progressive myoclonic epilepsy. Ann. Hum. Genet. 2012;76:326–331. doi: 10.1111/j.1469-1809.2012.00710.x. [DOI] [PubMed] [Google Scholar]
- 40.Van Bogaert P., Azizieh R., Désir J., Aeby A., De Meirleir L., Laes J.F., Christiaens F., Abramowicz M.J. Mutation of a potassium channel-related gene in progressive myoclonic epilepsy. Ann. Neurol. 2007;61:579–586. doi: 10.1002/ana.21121. [DOI] [PubMed] [Google Scholar]
- 41.Golzio C., Willer J., Talkowski M.E., Oh E.C., Taniguchi Y., Jacquemont S., Reymond A., Sun M., Sawa A., Gusella J.F. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485:363–367. doi: 10.1038/nature11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alazami A.M., Patel N., Shamseldin H.E., Anazi S., Al-Dosari M.S., Alzahrani F., Hijazi H., Alshammari M., Aldahmesh M.A., Salih M.A. Accelerating novel candidate gene discovery in neurogenetic disorders via whole-exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015;10:148–161. doi: 10.1016/j.celrep.2014.12.015. [DOI] [PubMed] [Google Scholar]
- 43.Kasahara K., Kawakami Y., Kiyono T., Yonemura S., Kawamura Y., Era S., Matsuzaki F., Goshima N., Inagaki M. Ubiquitin-proteasome system controls ciliogenesis at the initial step of axoneme extension. Nat. Commun. 2014;5:5081. doi: 10.1038/ncomms6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu S., Perez R., Pan M., Lee T. Requirement of Cul3 for axonal arborization and dendritic elaboration in Drosophila mushroom body neurons. J. Neurosci. 2005;25:4189–4197. doi: 10.1523/JNEUROSCI.0149-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Djagaeva I., Doronkin S. COP9 limits dendritic branching via Cullin3-dependent degradation of the actin-crosslinking BTB-domain protein Kelch. PLoS ONE. 2009;4:e7598. doi: 10.1371/journal.pone.0007598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oldham M.C., Horvath S., Geschwind D.H. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. Proc. Natl. Acad. Sci. USA. 2006;103:17973–17978. doi: 10.1073/pnas.0605938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Forabosco P., Ramasamy A., Trabzuni D., Walker R., Smith C., Bras J., Levine A.P., Hardy J., Pocock J.M., Guerreiro R. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol. Aging. 2013;34:2699–2714. doi: 10.1016/j.neurobiolaging.2013.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Charlesworth G., Angelova P.R., Bartolomé-Robledo F., Ryten M., Trabzuni D., Stamelou M., Abramov A.Y., Bhatia K.P., Wood N.W. Mutations in HPCA Cause Autosomal-Recessive Primary Isolated Dystonia. Am. J. Hum. Genet. 2015;96:657–665. doi: 10.1016/j.ajhg.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reimand J., Arak T., Vilo J. g:Profiler—a web server for functional interpretation of gene lists (2011 update) Nucleic Acids Res. 2011;39:W307–W315. doi: 10.1093/nar/gkr378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stavropoulos N., Young M.W. insomniac and Cullin-3 regulate sleep and wakefulness in Drosophila. Neuron. 2011;72:964–976. doi: 10.1016/j.neuron.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pfeiffenberger C., Allada R. Cul3 and the BTB adaptor insomniac are key regulators of sleep homeostasis and a dopamine arousal pathway in Drosophila. PLoS Genet. 2012;8:e1003003. doi: 10.1371/journal.pgen.1003003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuchs T., Saunders-Pullman R., Masuho I., Luciano M.S., Raymond D., Factor S., Lang A.E., Liang T.W., Trosch R.M., White S. Mutations in GNAL cause primary torsion dystonia. Nat. Genet. 2013;45:88–92. doi: 10.1038/ng.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Charlesworth G., Plagnol V., Holmström K.M., Bras J., Sheerin U.M., Preza E., Rubio-Agusti I., Ryten M., Schneider S.A., Stamelou M. Mutations in ANO3 cause dominant craniocervical dystonia: ion channel implicated in pathogenesis. Am. J. Hum. Genet. 2012;91:1041–1050. doi: 10.1016/j.ajhg.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iwabuchi S., Kakazu Y., Koh J.Y., Harata N.C. Abnormal cytoplasmic calcium dynamics in central neurons of a dystonia mouse model. Neurosci. Lett. 2013;548:61–66. doi: 10.1016/j.neulet.2013.05.047. [DOI] [PubMed] [Google Scholar]
- 55.Palmer C.L., Lim W., Hastie P.G., Toward M., Korolchuk V.I., Burbidge S.A., Banting G., Collingridge G.L., Isaac J.T., Henley J.M. Hippocalcin functions as a calcium sensor in hippocampal LTD. Neuron. 2005;47:487–494. doi: 10.1016/j.neuron.2005.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ritz K., van Schaik B.D., Jakobs M.E., van Kampen A.H., Aronica E., Tijssen M.A., Baas F. SGCE isoform characterization and expression in human brain: implications for myoclonus-dystonia pathogenesis? Eur. J. Hum. Genet. 2011;19:438–444. doi: 10.1038/ejhg.2010.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Index case (III.2): This segment shows the index case of the British family. Note the generalized dystonia with severe cranial and cervical involvement, including spasmodic dysphonia. Trunk and leg involvement are more evident while walking. Superimposed brief myoclonus are seen in the face and arms. Case V.3: This segment shows another member of the British family. He has frequent brief myoclonus involving the head and the arms. Myoclonus becomes more intense and frequent while talking. Index case German family: This segment shows the index case of the German family. He has generalized dystonia. There is marked cranial involvement, including dysarthria, and tongue dystonia. Trunk involvement is more evident while walking. He also displays arm myoclonus.