

Abstract
Autosomal-dominant lateral temporal epilepsy (ADLTE) is a genetic epilepsy syndrome clinically characterized by focal seizures with prominent auditory symptoms. ADLTE is genetically heterogeneous, and mutations in LGI1 account for fewer than 50% of affected families. Here, we report the identification of causal mutations in reelin (RELN) in seven ADLTE-affected families without LGI1 mutations. We initially investigated 13 ADLTE-affected families by performing SNP-array linkage analysis and whole-exome sequencing and identified three heterozygous missense mutations co-segregating with the syndrome. Subsequent analysis of 15 small ADLTE-affected families revealed four additional missense mutations. 3D modeling predicted that all mutations have structural effects on protein-domain folding. Overall, RELN mutations occurred in 7/40 (17.5%) ADLTE-affected families. RELN encodes a secreted protein, Reelin, which has important functions in both the developing and adult brain and is also found in the blood serum. We show that ADLTE-related mutations significantly decrease serum levels of Reelin, suggesting an inhibitory effect of mutations on protein secretion. We also show that Reelin and LGI1 co-localize in a subset of rat brain neurons, supporting an involvement of both proteins in a common molecular pathway underlying ADLTE. Homozygous RELN mutations are known to cause lissencephaly with cerebellar hypoplasia. Our findings extend the spectrum of neurological disorders associated with RELN mutations and establish a link between RELN and LGI1, which play key regulatory roles in both the developing and adult brain.
Main Text
Temporal-lobe epilepsy is the most common type of focal epilepsy. It is sometimes associated with structural brain lesions, but genetic forms have also been described. Familial temporal-lobe epilepsy comprises two genetically distinct syndromes: familial mesial temporal-lobe epilepsy (FMTLE [MIM: 611630])1 and autosomal-dominant lateral temporal epilepsy (ADLTE [MIM: 600512]), also named autosomal-dominant partial epilepsy with auditory features (ADPEAF).2 ADLTE is a well-defined epileptic syndrome clinically characterized by focal seizures with prominent auditory and/or aphasic symptoms, normal brain MRI, and relatively benign evolution.2,3 Its inheritance pattern is autosomal dominant with reduced penetrance (around 70%). Mutations associated with ADLTE are found in leucine-rich, glioma inactivated 1 (LGI1 [MIM: 604619])4–6 in 30%–50% of ADLTE-affected families.3,7,8 Other genes harboring ADLTE-causing mutations are unknown. LGI1 is expressed mainly in neurons, particularly in the neocortex and limbic regions,4,9 and its protein product, LGI1, is secreted.9 LGI1 has been implicated in the transmission of K+ and AMPA synaptic currents10,11 and in the regulation of post-natal maturation of cortical excitatory synapses and dendrite pruning.12 However, it is not known which of these functions underlies ADLTE. Identification of additional genes whose mutations cause ADLTE will help to clarify the pathogenic mechanism leading to this disorder. Here we report the identification of ADLTE-causing heterozygous mutations in RELN (MIM: 600514), encoding the protein Reelin.
To identify genes harboring ADLTE-causing mutations, we performed linkage analysis of 16 ADLTE-affected families in whom Sanger sequencing had failed to detect LGI1 mutations; we did so by using a SNP array followed by whole-exome sequencing to identify candidate variants in the linkage peaks. Most of these families have been described previously.7 We reassessed the epilepsy phenotypes of affected members to confirm eligibility for linkage analysis on the basis of clinical features and intrafamilial clinical homogeneity. Each family contained two or more affected individuals with a history of focal epilepsy with auras characteristic of the syndrome, i.e., auditory (ringing, humming, sounds, voices, or music) and/or receptive-aphasic symptoms, and absence of identified structural or metabolic insults to the CNS. Written informed consent was obtained from all family members participating in the study. The study was approved by local, Italian League Against Epilepsy (LICE) or Columbia University ethics committees. Affected and unaffected members of these families (143 DNAs in total, 50 affected persons) were genotyped with the high-density HumanOmni1-Quad v.1.0 (Illumina) beadchip, and genome-wide linkage analysis was performed with the Merlin program,13 under the assumption of autosomal-dominant inheritance with 70% penetrance, a disease-allele frequency of 0.001, and a phenocopy rate of 0.0. To avoid inflated linkage values, we performed two independent analyses by using two different SNP subsets selected on the basis of different linkage-disequilibrium parameters, r2 < 0.4 (180,047 SNPs) and r2 < 0.2 (62,887 SNPs). In both analyses, the highest linkage peak (heterogeneity logarithm of odds [HLOD] = 2.349; alpha = 0.289) was at chromosome 7q22.1 (Figure S1), a region encompassing 5.70 Mb (chr7: 101,977,695–107,685,645) between SNPs rs803118 and rs1990158. Two relatively large families, shown in Figure 1, were the main contributors to this HLOD peak: F31 (LOD score = 2.03) and F14 (LOD score = 1.92).
Figure 1.

Pedigrees of Families in Whom RELN Mutations Segregate with Disease
Colors of symbols refer to the indicated diagnostic categories. Open symbols indicate healthy family members. Circles indicate females, and squares indicate males. Families F14 and F33, previously described by Michelucci et al.,7 were recently reassessed, and new affected individuals were ascertained. Individuals with a mutation in RELN are indicated by m/+; individuals tested for mutations and found to be negative are indicated by +/+.
We then performed whole-exome sequencing on two affected members each from 13 of the families analyzed by linkage. Whenever possible, we chose second- or third-degree relatives to reduce the fraction of the genome inherited by chance. Exome sequencing was performed at IGA Technology Services with the SureSelect 50-Mb v.2.0 Capture Kit (Agilent Technologies). Enriched libraries were sequenced on an Illumina HiSeq2000. Reads were aligned to the UCSC Genome Browser hg19 reference sequence with the Burrows-Wheeler Aligner,14 and variant calling and genotyping were performed with the Genome Analysis Toolkit.15 Variants were annotated by ANNOVAR16 and filtered with NCBI dbSNP v.135, the 1000 Genomes Project catalog, and AVSIFT.17 An average of 4.545 Gb of sequence was generated per affected individual; 99.8% of the total bases passed quality assessment and were aligned to the human reference sequence, and 80% mapped to the targeted exons with a mean coverage of 39×. At this depth of coverage, 95% of the targeted bases were sufficiently covered to pass our threshold for variant calling (≥10×). We considered only heterozygous nonsynonymous variants, splice-site changes, and short indels causing frameshifts that were shared by the two affected individuals analyzed in each family, and then filtered out those occurring in the 1000 Genomes Project catalog and the NHLBI Exome Sequencing Project Exome Variant Server (EVS) with frequency > 0.01, those in genome segmental duplications, and those classified as benign, tolerated, or not damaging by different prediction software tools, such as SIFT and PolyPhen-2 (see examples in Tables S1 and S2). Finally, we extracted the gene variants lying in the chromosome 7q22.1 linkage interval from the annotation file of potential candidate variants that passed the filtering steps. Three families (F31, F33, and F14) exhibited only one heterozygous mutation in the 7q22.1 candidate region (Table S1), and all three missense mutations, c.2392C>A (p.His798Asn), c.2531C>T (p.Pro844Leu), and c.8347G>T (p.Gly2783Cys), occurred in RELN (GenBank: NM_005045.3). These mutations were validated by Sanger sequencing (Figure S2), and each of them co-segregated with the disease in the corresponding family (Figure 1). Subsequently, we performed whole-exome sequencing on the probands of four additional ADLTE-affected families who had not been analyzed by genome-wide linkage and found an additional heterozygous RELN mutation, c.2288A>G (p.Asp763Gly), in one of them. Moreover, massive parallel sequencing of RELN exons in the probands of 11 small ADLTE-affected families, performed on the Illumina MiSeq System with the MiSeq Reagent Kit v.3, revealed three additional missense mutations—c.2015C>T (p.Pro672Leu), c.2168A>G (p.Tyr723Cys), and c.9526G>A (p.Glu3176Lys)—each in a single family. These mutations, all validated by Sanger sequencing, were also detected in the additional available affected members of the families (Figure 1). In total, we identified seven RELN mutations, each in a different ADLTE-affected family. The main clinical features of the families affected by RELN mutations are summarized in Table 1. None of the seven mutations was detected in 270 healthy control individuals of matched ancestry, in 174 simplex-case individuals with lateral temporal epilepsy, or in the 1000 Genomes Project catalog or EVS. The c.2015C>T (p.Pro672Leu) mutation, which was detected by targeted RELN sequencing, was present in dbSNP v.138 and the ExAC Browser (frequency = 0.0001), whereas c.2168A>G (p.Tyr723Cys) was present in the ExAC Browser only (frequency = 0.000024). All amino acid substitutions affect highly conserved residues (Figure S3) and are predicted to be deleterious by the PolyPhen-2, SIFT, and MutationTaster prediction tools (Table 1). Many potentially harmful RELN variants, most of which await Sanger confirmation, are reported in the EVS. However, all mutations identified in this study affect structurally important amino acids and/or result in amino acid changes that most likely perturb protein folding (see below). In the families we studied, 20/33 (60%) of individuals carrying one of these mutations have epilepsy, suggesting a relatively low penetrance of RELN mutations, and there are no affected persons who do not carry one of these mutations. All together, the above findings support a pathogenic effect of the reported mutations. In our cohort of Italian ADLTE-affected families, RELN mutations account for disease in 7/40 (17.5%) families, a frequency comparable with that of LGI1 mutations (12/40 families; 30%). We detected no RELN mutations in 17 FMTLE-affected families, suggesting that mutations in this gene might specifically cause ADLTE.
Table 1.
Families Affected by ADLTE and Mutations in RELN
| Family | No. of Affected (Deceased) Individuals | Age of Onset (Years) | No. of Affected Individuals with Auditory and/or Aphasic Seizures | LOD Score at 7q22.1 | Nucleotide Change | Amino Acid Change | PolyPhen-2 Scorea | SIFT Scoreb | MutationTaster Probability Valuec |
|---|---|---|---|---|---|---|---|---|---|
| F14 | 7 (3) | 18–40 | 3 | 1.92 | c.8347G>T | p.Gly2783Cys | 1.000 | 0.00 | 0.99 |
| F31 | 5 | 10–25 | 4 | 2.03 | c.2392C>A | p.His798Asn | 0.936 | 0.01 | 0.99 |
| F33 | 6 (1) | 8–30 | 3 | 0.83 | c.2531C>T | p.Pro844Leu | 1.000 | 0.01 | 0.99 |
| FIA | 4 (1) | 19–24 | 3 | – | c.2288A>G | p.Asp763Gly | 0.997 | 0.01 | 0.99 |
| CRR | 2 | 8–12 | 2 | – | c.2015C>T | p.Pro672Leu | 1.000 | 0.01 | 0.99 |
| F15 | 2 | 20–24 | 2 | – | c.2168A>G | p.Tyr723Cys | 0.999 | 0.04 | 0.99 |
| PAC | 2 | 17 | 2 | – | c.9526G>A | p.Glu3176Lys | 0.951 | 0.01 | 0.99 |
Benign, 0.00–0.15; possibly damaging, 0.16–0.85; probably damaging, 0.86–1.00.
Not tolerated, 0.00–0.05; potentially not tolerated, 0.06–0.20; tolerated, 0.21–1.00.
Probability of causing disease; values close to 1 indicate a high confidence of the prediction.
RELN, first identified in the homozygous-null mutant reeler mouse strain,18 comprises 65 exons and is mostly expressed in the brain. It encodes Reelin (a gigantic secreted protein of 3,460 amino acids), which consists of an N-terminal signal peptide, an F-spondin-like domain, a unique region, and eight tandem repeats of 350–390 residues termed “Reelin repeats” (Figure 2A).18,19 Each Reelin repeat comprises a central epidermal growth factor (EGF) or EGF-like module flanked by two subrepeats (A and B), each containing a 14-amino-acid Asp-box (bacterial neuraminidase repeat) motif20 (Figures 2A and 2B). Both the EGF-like and Asp-box modules support the compact, “horseshoe-like” structural arrangement of each Reelin repeat21 (Figure 3). Some ADLTE-related RELN mutations affect amino acid residues that are important for proper folding of Reelin repeats. The p.Pro672Leu substitution affects the first residue of the EGF-like module in Reelin repeat 1, a proline that most likely facilitates the abrupt bend of the protein main chain at the subrepeat A-EGF boundary,21 and its replacement with a leucine might result in local unfolding of repeat 1. Two amino acid substitutions, p.His798Asn and p.Gly2783Cys, affect residues in Asp-boxes 2 and 13, respectively (Figures 2B and 2C), most likely resulting in structural abnormalities. Particularly, the p.Gly2783Cys substitution replaces an invariant Gly residue at position 8 in the Asp-box consensus sequence20 with a cysteine (Figure 2C), most likely altering the conformation of Asp-box 13. To evaluate the potential impact of the ADLTE-related amino acid substitutions on the structure of Reelin repeats, we generated a 3D model of the three Reelin repeats harboring these amino acid changes. To do this, we manually aligned the Reelin sequence (UniProt: P78509) according to its component repeats by using Jalview.23 Two crystal structures (PDB: 2DDU, 2E26)24 were used as starting points for modeling with the HOMER server. The models were analyzed with the ConSurf25 server, and figures were drawn with ESPript26 for sequence alignments and with PyMol (DeLano Scientific) for structures. This 3D model supports the deleterious effects of p.Pro672Leu, p.His798Asn, and p.Gly2783Cys substitutions on the organization of Reelin repeats (see Figure S4). In addition, it predicts structural consequences for the p.Tyr723Cys replacement, which is due to exposure of an unpaired cysteine to the solvent, and for the p.Glu3176Lys substitution, which disrupts a structurally important electrostatic interaction between the negatively charged Glu3176 and the positively charged Arg3409 (Figure S4). This latter effect, in particular, might not be dramatic in the intracellular environment, where pH is neutral, but might destabilize Reelin repeat 8 when Reelin is exported to the extracellular milieu. Finally, the p.Asp763Gly substitution is predicted to perturb the horseshoe-like arrangement of Reelin repeat 1.
Figure 2.
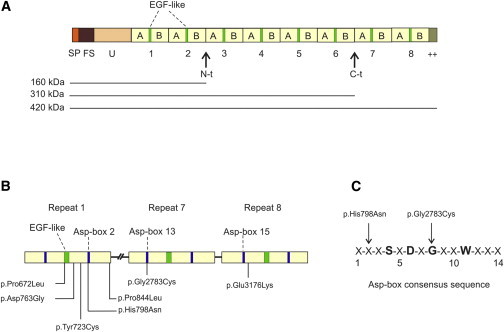
Schematic of Reelin and Localization of ADLTE-Causing Variants
(A) Multidomain structure of Reelin. Numbers 1–8 indicate Reelin repeats, each consisting of subrepeats A and B and an intervening EGF-like module. Horizontal lines indicate the three isoforms (160, 310, and 420 kDa) that were detected by immunoblotting with antibodies to N-terminal antigens. Abbreviations are as follows: SP, signal peptide; FS, F-spondin-like domain; U, unique region; ++, positively charged C-terminal region; N-t, N-terminal cleavage site; C-t, C-terminal cleavage site.
(B) Schematic structure of three Reelin repeats harboring ADLTE-causing variants. The approximate locations of EGF-like and Asp-box modules and positions of the substitutions are indicated.
(C) Consensus sequence of the Asp-box motif. The amino acids Ser (S), Asp (D), Gly (G), and Trp (W) at positions 4, 6, 8, and 11, respectively, are highly conserved in Asp-box motifs of different Asp-box-containing protein families and support the beta-hairpin folding of Asp-box motifs. X indicates any amino acid residue. Arrows indicate the positions of two ADLTE-causing missense variants in the Asp-box consensus sequence.
Figure 3.

Overall Ribbon Representation of the 3D Structure of a Generic Reelin Repeat
The repeat structure, formed by two subdomains, subrepeat A (left) and subrepeat B (right), is colored in light brown, residues forming the Asp-boxes are colored in blue, and the EGF-like domain is presented in green. Magenta spheres represent substitutions affecting repeat 1 (p.Pro672Leu, p.Asp763Gly, p.Tyr723Cys, p.His798Asn, and p.Pro844Leu), repeat 7 (p.Gly2783Cys), and repeat 8 (p.Glu3176Lys). Pathological substitutions are found at the interaction interface between the two subdomains, affect the EGF-like motif, or are exposed to the solvent. Zoom panels represent areas showing the wild-type intra-molecular bonding dynamics. The coloring of frames corresponds to different mutation-prone residues, which are presented in magenta and shown in stick models, whereas gray represents surrounding residues within 3.5Å. The figure was prepared with UCSF Chimera.22
Reelin is synthesized and secreted in several organs other than the brain and is found in blood serum, where it undergoes post-translational processing qualitatively similar to that observed in the brain.27 Blood-serum Reelin, most of which is secreted from the liver, has been examined for the investigation of the effects of RELN mutations on extracellular Reelin levels in humans28–30and reeler mice.27 In addition to the full-length protein (420 kDa), two smaller isoforms (160 and 310 kDa), resulting from enzymatic cleavage of the full-length protein, were detected by immunoblot (see schematic in Figure 2A). In the serum, the 310-kDa isoform accounts for most of the signal (>80%), whereas only trace amounts of the 160-kDa isoform are present.27 We analyzed blood sera samples from affected individuals carrying the Reelin substitutions p.Asp763Gly, p.His798Asn, p.Gly2783Cys, and p.Glu3176Lys by immunoblot. As shown in Figure 4A, the amount of the 310-kDa Reelin isoform was remarkably lower in the sera from these affected individuals than in the sera from control individuals, and there was no increase in the 420-kDa isoform. This trend was significant in four different experiments (p = 0.028; Figure 4B). Notably, in persons carrying the p.Asp763Gly, p.His798Asn, and p.Gly2783Cys substitutions, the amount of serum Reelin was approximately 50% of that in control individuals. Because missense mutations are unlikely to affect protein synthesis, lower levels of serum Reelin most likely result from impaired secretion of the altered proteins from hepatocytes, and this mechanism could also decrease the amount of extracellular Reelin in the brain. Serum Reelin levels might be influenced by a polymorphic GGC repeat located immediately 5′ of the RELN initiation codon. In vitro studies showed that compared to 8- to 10-repeat alleles, rare, “long” 12- to 14-repeat alleles might result in 50%–60% less Reelin expression,31 and subjects with at least one long allele were found to have an average of 25% less serum Reelin expression than were individuals with common 8- to 10-repeat alleles.29 Together, 8- and 10-repeat alleles account for >95% of all alleles in the Italian population overall.32 We detected only these two common alleles in our affected individuals and control individuals (data not shown), ruling out any influence of this polymorphism on serum Reelin levels in the study subjects.
Figure 4.

Reduced Reelin Levels in Blood Sera from Individuals with RELN Mutations
(A) The top panel shows representative immunoblot analysis of serum Reelin levels in individuals carrying the p.Asp763Gly, p.His798Asn, p.Gly2783Cys, or p.Glu3176Lys substitutions and in healthy control individuals, one male (Cm) and two females (Cf1 and Cf2). The three control individuals are representative of a group of ten unrelated healthy individuals exhibiting similar serum Reelin levels and carrying the most common alleles (8 or 10 repeats) at the GGC repeat polymorphism in the 5′ UTR of RELN. The same alleles were also detected in the affected individuals (see text). The two detectable isoforms of 420 and 310 kDa are indicated by arrows. The bottom panel shows quantification of the 310-kDa Reelin isoform values obtained from the representative immunoblot. Values were normalized to the serum levels of the amyloid precursor protein (APP), as reported previously.29 APP is an endogenous blood component that migrates in serum predominantly as a 180-kDa band. The 310-kDa isoform was substantially reduced in individuals carrying the substitutions p.Asp763Gly, p.His798Asn, and p.Gly2783Cys. Total protein amounts in human sera were determined via bicinchoninic acid assay. Samples, consisting of 100 μg of serum proteins, were fractionated on NuPAGE 3%–8% Tris-acetate gels and transferred to polyvinylidene flouride membrane. Blots were incubated with mouse anti-Reelin antibody 142 (dilution 1:1,000; Millipore) in 5% BSA for 75 min and then with secondary polyclonal antibody rabbit anti-mouse immunoglobulins conjugated with horseradish peroxidase (HRP) for 1 hr. Signals were detected with the ECL Prime Western Blotting Detection Reagent (GE Healthcare). Subsequently, the same membranes were immunoblotted, without stripping, with an anti-APP antibody (ab2072, dilution 1:500; Abcam) and polyclonal antibody goat anti-rabbit immunoglobulins conjugated with HRP. Densitometric analysis of 310-kDa Reelin bands was performed with the ImageJ software (NIH), and data were normalized against APP signals.
(B) Quantification of the 310-kDa Reelin isoform mean values obtained from four independent experiments. Values from each experiment were normalized to the serum levels of APP. Statistical analysis of difference in band densities between affected (n = 4) and control (n = 3) subgroups was performed with the nonparametric Wilcoxon-Mann-Whitney test. The overall quantitative difference between RELN-mutated persons and healthy control individuals is statistically significant (p = 0.028).
To get a preliminary insight into a possible relationship between Reelin and LGI1, we analyzed rat brain sections by double immunofluorescence and confocal microscopy. For LGI1, we used the antibody ab30868, whose specificity has previously been demonstrated.33 The temporal cortex and hippocampus of immature (3 weeks old) and mature (2 months old) rats were examined. As shown in Figure 5, a subset of neurons in both brain areas were immunoreactive for both Reelin and LGI1. Intense double labeling was observed in some cortical and hippocampal neurons of mature rats (Figures 5B–5D); other cortical neuronal cells displayed weak immunoreactivity for both proteins (Figure 5A), whereas double immunoreactivity was barely detected in hippocampal neurons of immature rats (not shown). Moreover, under non-permeabilized conditions, punctate co-immunostaining was detected outside the cell bodies and displayed a linear alignment in some cases, as if along neuronal projections (Figure S5). Overall, the neuronal co-localization of Reelin and LGI1 suggests that the two proteins might be involved in a common molecular pathway.
Figure 5.

Co-localization of Reelin and LGI1 in Rat Brains
Representative confocal images of double immunofluorescence analyses performed with the anti-Reelin antibody 142 (green) and the anti-LGI1 antibody ab30868 (red) in brain sections of immature (3-week-old) and mature (2-month-old) rats. The merged images show co-localization of Reelin with LGI1 in distinct neuronal cells in various brain areas: (A) temporal-lobe cortex (layer II) of immature rats; (B) temporal-lobe cortex (layers V and VI) of mature (adult) rats; (C) adult-rat hippocampus; and (D) adult-rat dentate gyrus. Abbreviations are as follows: CA1, region 1 of the Ammon’s horn; C.L. II, cortical layer II; C.L. V/VI, cortical layers V and VI; G.C., granule cells of dentate gyrus; S.O., stratum oriens; S.P., stratum pyramidale; S.R., stratum radiatum. The scale bar represents 20 μm. Rat procedures were approved by the ethics committee of the University of Padova, Italy. Coronal cortical-hippocampal slices were obtained from 3-week-old and 2-month-old Wistar rats perfused with cold 4% paraformaldehyde in PBS. Brains were post-fixed in the same fixative and cryoprotected in a sucrose solution (30% sucrose in PBS). 50-μm-thick coronal sections were obtained with a Leica vibratome VT1000S, air-dried, and then rinsed in PBS with or without 0.05% Triton. Sections were blocked and then incubated with both Reelin 142 (MAB5366, Chemicon) and LGI1 (ab30868, Abcam) primary antibodies for 72 hr at 4°C. After PBS washings, sections were incubated for 3 hr with goat anti-mouse Alexa 488 (Life Technologies) and goat anti-rabbit Cy3-conjugated (Jackson Immunoresearch Laboratories) and then covered with coverslips. Sections were examined with a 20× water objective, and images were captured with a Leica SP5 laser confocal microscope.
In this paper, we report the identification of heterozygous missense Reelin mutations in seven families affected by ADLTE and provide evidence supporting their pathogenicity. Particularly, we show that four amino acid substitutions—p.Asp763Gly, p.His798Asn, p.Gly2783Cys, and p.Glu3176Lys—significantly reduce serum levels of Reelin, suggesting a loss-of-function effect due to impaired secretion of altered proteins. Support for this suggestion comes from our 3D protein model, which predicts that these amino acid changes result in misfolding of Reelin repeats and most likely lead to degradation of the altered proteins by the endoplasmic reticulum quality-control machinery. Reduced levels of serum Reelin are observed in heterozygous reeler mice,27 which have apparently normal brains but display functional and molecular defects at the synapse.34 Similar synaptic defects might also be associated with the heterozygous RELN mutations detected in our subjects and might eventually give rise to ADLTE.
Previous work revealed that homozygous RELN mutations cause lissencephaly with cerebellar hypoplasia (LCH [MIM: 257320]), a recessive disorder characterized by severe and widespread neuronal-migration defects, delayed cognitive development, and seizures,28 a phenotype similar to that of homozygous reeler mice.35 Three small consanguineous LCH-affected families in whom RELN mutations segregate with disease have been reported so far; heterozygote individuals in these families exhibited reduced levels of Reelin in their sera and were reported as clinically normal.28,30 The apparent normal phenotype of these individuals is consistent with the relatively low penetrance of RELN mutations observed in this study and the possible occurrence of subtle epilepsy signs, not infrequent in ADLTE, which might remain undiagnosed in some individuals. Thus, as in other genetic epilepsy syndromes,36 RELN mutations might result in different clinical phenotypes, depending on mode of inheritance: heterozygous mutations might cause ADLTE, and homozygous mutations might result in the more severe LCH.
Reelin is localized almost exclusively in GABAergic interneurons in the post-natal brain,37 and two types of neurons have been identified by immunofluorescence analysis, i.e., neurons with intense Reelin immunoreactivity and neurons with weak immunoreactivity.37,38 The former neurons produce and secrete Reelin; the latter are targeted by Reelin molecules released from other cells.38 LGI1 is localized mainly in pyramidal neurons, but a detailed analysis of its distribution in the brain is missing. Notably, our results show that some cortical and hippocampal neurons intensely labeled by Reelin antibodies are also heavily immunoreactive for LGI1, suggesting that these neurons might synthesize and secrete relatively high levels of both proteins, which might act in a coordinate manner to exert particular functions possibly related to epilepsy. Future work aimed at characterizing these cell types will help elucidate the cellular bases underlying ADLTE.
Reelin is an extracellular glycoprotein that is secreted by Cajal-Retzius cells during embryogenesis and by GABAergic neurons in the post-natal brain.39 Reelin serves a dual purpose in the mammalian brain: it is crucial to the correct cytoarchitecture of laminated structures during embryonic development18,40 and modulates dendritic growth and synaptic plasticity at post-natal and adult stages.41,42 These activities are, at least in part, mediated by binding to apolipoprotein E receptor 2 (ApoER2) and very low-density lipoprotein receptor (VLDLR),43–45 resulting in phosphorylation of the intracellular adaptor protein protein disabled homolog 1 (DAB1).46–48 Phosphorylated DAB1 activates several different signaling pathways involved in formation and plasticity of neuronal networks (for review, see ref.49). Recent work on conditional knockout mice suggests that LGI1, like Reelin, could serve different functions during brain development and adulthood.50 Given that Reelin and LGI1 co-localize to distinct neurons in both the immature and mature rat brain, it is conceivable that a functional interplay between the two proteins is necessary for proper regulation of various neuronal processes during development and in adult life. Future work will hopefully elucidate the type of interaction between Reelin and LGI1 and how their mutations lead to ADLTE.
Acknowledgments
We thank Chiara Romualdi for statistical analysis of immunoblot data. This work was supported by Telethon-Italy (grant GGP12078 to C.N.), Fondazione Cassa di Risparmio di Padova e Rovigo (grant to C.N.), the Genetics Commission of the Italian League against Epilepsy (funding to R.M and C. N.), and the NIH (grants R01 NS036319 and R01 NS078419 to R.O.). G.M. is an Associazione Italiana per la Ricerca sul Cancro research fellow.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org
ExAC Browser, http://exac.broadinstitute.org/
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SIFT and AVSIFT, http://sift.jcvi.org/
UniProt, http://www.uniprot.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.Berkovic S.F., McIntosh A., Howell R.A., Mitchell A., Sheffield L.J., Hopper J.L. Familial temporal lobe epilepsy: a common disorder identified in twins. Ann. Neurol. 1996;40:227–235. doi: 10.1002/ana.410400214. [DOI] [PubMed] [Google Scholar]
- 2.Ottman R., Risch N., Hauser W.A., Pedley T.A., Lee J.H., Barker-Cummings C., Lustenberger A., Nagle K.J., Lee K.S., Scheuer M.L. Localization of a gene for partial epilepsy to chromosome 10q. Nat. Genet. 1995;10:56–60. doi: 10.1038/ng0595-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michelucci R., Poza J.J., Sofia V., de Feo M.R., Binelli S., Bisulli F., Scudellaro E., Simionati B., Zimbello R., D’Orsi G. Autosomal dominant lateral temporal epilepsy: clinical spectrum, new epitempin mutations, and genetic heterogeneity in seven European families. Epilepsia. 2003;44:1289–1297. doi: 10.1046/j.1528-1157.2003.20003.x. [DOI] [PubMed] [Google Scholar]
- 4.Kalachikov S., Evgrafov O., Ross B., Winawer M., Barker-Cummings C., Martinelli Boneschi F., Choi C., Morozov P., Das K., Teplitskaya E. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat. Genet. 2002;30:335–341. doi: 10.1038/ng832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morante-Redolat J.M., Gorostidi-Pagola A., Piquer-Sirerol S., Sáenz A., Poza J.J., Galán J., Gesk S., Sarafidou T., Mautner V.F., Binelli S. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum. Mol. Genet. 2002;11:1119–1128. doi: 10.1093/hmg/11.9.1119. [DOI] [PubMed] [Google Scholar]
- 6.Nobile C., Michelucci R., Andreazza S., Pasini E., Tosatto S.C.E., Striano P. LGI1 mutations in autosomal dominant and sporadic lateral temporal epilepsy. Hum. Mutat. 2009;30:530–536. doi: 10.1002/humu.20925. [DOI] [PubMed] [Google Scholar]
- 7.Michelucci R., Pasini E., Malacrida S., Striano P., Bonaventura C.D., Pulitano P., Bisulli F., Egeo G., Santulli L., Sofia V. Low penetrance of autosomal dominant lateral temporal epilepsy in Italian families without LGI1 mutations. Epilepsia. 2013;54:1288–1297. doi: 10.1111/epi.12194. [DOI] [PubMed] [Google Scholar]
- 8.Ottman R., Winawer M.R., Kalachikov S., Barker-Cummings C., Gilliam T.C., Pedley T.A., Hauser W.A. LGI1 mutations in autosomal dominant partial epilepsy with auditory features. Neurology. 2004;62:1120–1126. doi: 10.1212/01.wnl.0000120098.39231.6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Senechal K.R., Thaller C., Noebels J.L. ADPEAF mutations reduce levels of secreted LGI1, a putative tumor suppressor protein linked to epilepsy. Hum. Mol. Genet. 2005;14:1613–1620. doi: 10.1093/hmg/ddi169. [DOI] [PubMed] [Google Scholar]
- 10.Schulte U., Thumfart J.O., Klöcker N., Sailer C.A., Bildl W., Biniossek M., Dehn D., Deller T., Eble S., Abbass K. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron. 2006;49:697–706. doi: 10.1016/j.neuron.2006.01.033. [DOI] [PubMed] [Google Scholar]
- 11.Fukata Y., Adesnik H., Iwanaga T., Bredt D.S., Nicoll R.A., Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science. 2006;313:1792–1795. doi: 10.1126/science.1129947. [DOI] [PubMed] [Google Scholar]
- 12.Zhou Y.D., Lee S., Jin Z., Wright M., Smith S.E., Anderson M.P. Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat. Med. 2009;15:1208–1214. doi: 10.1038/nm.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 14.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu X., Jian X., Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011;32:894–899. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D’Arcangelo G., Miao G.G., Chen S.C., Soares H.D., Morgan J.I., Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 19.Ichihara H., Jingami H., Toh H. Three novel repetitive units of reelin. Brain Res. Mol. Brain Res. 2001;97:190–193. doi: 10.1016/s0169-328x(01)00307-2. [DOI] [PubMed] [Google Scholar]
- 20.Copley R.R., Russell R.B., Ponting C.P. Sialidase-like Asp-boxes: sequence-similar structures within different protein folds. Protein Sci. 2001;10:285–292. doi: 10.1110/ps.31901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nogi T., Yasui N., Hattori M., Iwasaki K., Takagi J. Structure of a signaling-competent reelin fragment revealed by X-ray crystallography and electron tomography. EMBO J. 2006;25:3675–3683. doi: 10.1038/sj.emboj.7601240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 23.Waterhouse A.M., Procter J.B., Martin D.M.A., Clamp M., Barton G.J. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berman H., Henrick K., Nakamura H., Markley J.L. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007;35:D301–D303. doi: 10.1093/nar/gkl971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ashkenazy H., Erez E., Martz E., Pupko T., Ben-Tal N. ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010;38 doi: 10.1093/nar/gkq399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gouet P., Robert X., Courcelle E. ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003;31:3320–3323. doi: 10.1093/nar/gkg556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smalheiser N.R., Costa E., Guidotti A., Impagnatiello F., Auta J., Lacor P., Kriho V., Pappas G.D. Expression of reelin in adult mammalian blood, liver, pituitary pars intermedia, and adrenal chromaffin cells. Proc. Natl. Acad. Sci. USA. 2000;97:1281–1286. doi: 10.1073/pnas.97.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong S.E., Shugart Y.Y., Huang D.T., Shahwan S.A., Grant P.E., Hourihane J.O., Martin N.D., Walsh C.A. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat. Genet. 2000;26:93–96. doi: 10.1038/79246. [DOI] [PubMed] [Google Scholar]
- 29.Lugli G., Krueger J.M., Davis J.M., Persico A.M., Keller F., Smalheiser N.R. Methodological factors influencing measurement and processing of plasma reelin in humans. BMC Biochem. 2003;4:9. doi: 10.1186/1471-2091-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaki M., Shehab M., El-Aleem A.A., Abdel-Salam G., Koeller H.B., Ilkin Y., Ross M.E., Dobyns W.B., Gleeson J.G. Identification of a novel recessive RELN mutation using a homozygous balanced reciprocal translocation. Am. J. Med. Genet. A. 2007;143A:939–944. doi: 10.1002/ajmg.a.31667. [DOI] [PubMed] [Google Scholar]
- 31.Persico A.M., Levitt P., Pimenta A.F. Polymorphic GGC repeat differentially regulates human reelin gene expression levels. J. Neural Transm. 2006;113:1373–1382. doi: 10.1007/s00702-006-0441-6. [DOI] [PubMed] [Google Scholar]
- 32.Persico A.M., D’Agruma L., Maiorano N., Totaro A., Militerni R., Bravaccio C., Wassink T.H., Schneider C., Melmed R., Trillo S., Collaborative Linkage Study of Autism Reelin gene alleles and haplotypes as a factor predisposing to autistic disorder. Mol. Psychiatry. 2001;6:150–159. doi: 10.1038/sj.mp.4000850. [DOI] [PubMed] [Google Scholar]
- 33.Fukata Y., Lovero K.L., Iwanaga T., Watanabe A., Yokoi N., Tabuchi K., Shigemoto R., Nicoll R.A., Fukata M. Disruption of LGI1-linked synaptic complex causes abnormal synaptic transmission and epilepsy. Proc. Natl. Acad. Sci. USA. 2010;107:3799–3804. doi: 10.1073/pnas.0914537107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ventruti A., Kazdoba T.M., Niu S., D’Arcangelo G. Reelin deficiency causes specific defects in the molecular composition of the synapses in the adult brain. Neuroscience. 2011;189:32–42. doi: 10.1016/j.neuroscience.2011.05.050. [DOI] [PubMed] [Google Scholar]
- 35.Goffinet A.M. The reeler gene: a clue to brain development and evolution. Int. J. Dev. Biol. 1992;36:101–107. [PubMed] [Google Scholar]
- 36.de Nijs L., Wolkoff N., Coumans B., Delgado-Escueta A.V., Grisar T., Lakaye B. Mutations of EFHC1, linked to juvenile myoclonic epilepsy, disrupt radial and tangential migrations during brain development. Hum. Mol. Genet. 2012;21:5106–5117. doi: 10.1093/hmg/dds356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramos-Moreno T., Galazo M.J., Porrero C., Martínez-Cerdeño V., Clascá F. Extracellular matrix molecules and synaptic plasticity: immunomapping of intracellular and secreted Reelin in the adult rat brain. Eur. J. Neurosci. 2006;23:401–422. doi: 10.1111/j.1460-9568.2005.04567.x. [DOI] [PubMed] [Google Scholar]
- 38.Campo C.G., Sinagra M., Verrier D., Manzoni O.J., Chavis P. Reelin secreted by GABAergic neurons regulates glutamate receptor homeostasis. PLoS ONE. 2009;4:e5505. doi: 10.1371/journal.pone.0005505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.D’Arcangelo G. Reelin in the Years: Controlling Neuronal Migration and Maturation in the Mammalian Brain. Adv. Neurosci. (Hindawi) 2014;2014:1–19. [Google Scholar]
- 40.Lambert de Rouvroit C., Goffinet A.M. The reeler mouse as a model of brain development. Adv. Anat. Embryol. Cell Biol. 1998;150:1–106. [PubMed] [Google Scholar]
- 41.Beffert U., Weeber E.J., Durudas A., Qiu S., Masiulis I., Sweatt J.D., Li W.-P., Adelmann G., Frotscher M., Hammer R.E., Herz J. Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron. 2005;47:567–579. doi: 10.1016/j.neuron.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 42.Niu S., Renfro A., Quattrocchi C.C., Sheldon M., D’Arcangelo G. Reelin promotes hippocampal dendrite development through the VLDLR/ApoER2-Dab1 pathway. Neuron. 2004;41:71–84. doi: 10.1016/s0896-6273(03)00819-5. [DOI] [PubMed] [Google Scholar]
- 43.D’Arcangelo G., Homayouni R., Keshvara L., Rice D.S., Sheldon M., Curran T. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24:471–479. doi: 10.1016/s0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- 44.Hiesberger T., Trommsdorff M., Howell B.W., Goffinet A., Mumby M.C., Cooper J.A., Herz J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24:481–489. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- 45.Strasser V., Fasching D., Hauser C., Mayer H., Bock H.H., Hiesberger T., Herz J., Weeber E.J., Sweatt J.D., Pramatarova A. Receptor clustering is involved in Reelin signaling. Mol. Cell. Biol. 2004;24:1378–1386. doi: 10.1128/MCB.24.3.1378-1386.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Howell B.W., Herrick T.M., Cooper J.A. Reelin-induced tyrosine [corrected] phosphorylation of disabled 1 during neuronal positioning. Genes Dev. 1999;13:643–648. doi: 10.1101/gad.13.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keshvara L., Benhayon D., Magdaleno S., Curran T. Identification of reelin-induced sites of tyrosyl phosphorylation on disabled 1. J. Biol. Chem. 2001;276:16008–16014. doi: 10.1074/jbc.M101422200. [DOI] [PubMed] [Google Scholar]
- 48.Ballif B.A., Arnaud L., Cooper J.A. Tyrosine phosphorylation of Disabled-1 is essential for Reelin-stimulated activation of Akt and Src family kinases. Brain Res. Mol. Brain Res. 2003;117:152–159. doi: 10.1016/s0169-328x(03)00295-x. [DOI] [PubMed] [Google Scholar]
- 49.Stranahan A.M., Erion J.R., Wosiski-Kuhn M. Reelin signaling in development, maintenance, and plasticity of neural networks. Ageing Res. Rev. 2013;12:815–822. doi: 10.1016/j.arr.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boillot M., Huneau C., Marsan E., Lehongre K., Navarro V., Ishida S., Dufresnois B., Ozkaynak E., Garrigue J., Miles R. Glutamatergic neuron-targeted loss of LGI1 epilepsy gene results in seizures. Brain. 2014;137:2984–2996. doi: 10.1093/brain/awu259. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.