

Abstract
Background
Lung inflammation is a key factor in the pathogenesis of bronchopulmonary dysplasia (BPD). Stromal derived factor-1 (SDF-1) and its receptor chemokine receptor 4 (CXCR4) modulate the inflammatory response. Whether antagonism of CXCR4 will alleviate lung inflammation in neonatal hyperoxia-induced lung injury is unknown.
Objective
To determine whether CXCR4 antagonism would attenuate lung injury in rodents with experimental BPD by decreasing pulmonary inflammation.
Methods
Newborn rats exposed to normoxia (RA) or hyperoxia (FiO2=0.9) from postnatal day 2 (P2)-P16 were randomized to receive the CXCR4 antagonist, AMD3100 or placebo (PL) from P5 to P15. Lung alveolarization, angiogenesis, and inflammation were evaluated at P16.
Results
As compared to RA, hyperoxic-PL pups had a decrease in alveolarization, reduced lung vascular density and increased lung inflammation. In contrast, AMD3100-treated hyperoxic pups had improved alveolarization and increased angiogenesis. This improvement in lung structure was accompanied by a decrease in bronchoalveolar lavage fluid macrophage and neutrophil count and reduced lung myeloperoxidase activity.
Conclusion
CXCR4 antagonism decreases lung inflammation and improves alveolar as well as vascular structure in neonatal rats with experimental BPD. These findings suggest a novel therapeutic strategy to alleviate lung injury in preterm infants with BPD.
Keywords: CXCR4 blockade, AMD3100, bronchopulmonary dysplasia, angiogenesis, hyperoxia
BACKGROUND
Bronchopulmonary dysplasia (BPD) is characterized by an arrest of alveolar and vascular development [1]. Inflammation plays a major role in the pathogenesis of BPD [2]. This inflammatory response is believed to be triggered antenatally by intrauterine infection and augmented postnatally by factors such as hyperoxia and systemic infections [2]. Preterm infants at various stages in the development of BPD have increased numbers of inflammatory cells in their tracheal aspirate [3]. These inflammatory cells recruited to the lung in the earliest phase of lung injury initiate a cascade of injurious events which increase pulmonary microvascular edema and suppress lung growth.
Chemokines are peptides which orchestrate the migration of cells involved in inflammatory responses. Initially cloned from bone marrow stromal cells in 1993, the chemokine stromal derived factor-1 (SDF-1) is secreted by several tissues, with its major cellular sources being bone marrow stromal cells, macrophages, neutrophils, vascular endothelial cells, and fibroblasts [4]. Its cognate receptor, CXCR4 is a G-protein coupled receptor that is widely expressed on several tissues, including endothelial cells, fibroblasts, neutrophils, monocytes, hematopoietic and tissue committed stem cells [5]. Although the role of CXCR4/SDF-1 in BPD pathogenesis is unclear, Deng et al demonstrated increased CXCR4 positive bone marrow-derived fibroblasts in the lungs of rodents exposed to hyperoxia and these cells appeared to migrate to the lung under the direction of SDF-1[6].
CXCR4 blockade is a strategy to reduce lung inflammation and repair the injured lung. AMD3100 is a symmetric bicyclam potent non-peptide CXCR4 antagonist [7]. This compound was first utilized to block entry of the HIV virus into cells [7]. Although current clinical use of AMD3100 is restricted to adjunctive cancer therapy, accumulating pre-clinical evidence suggest that CXCR4 blockade with AMD3100 facilitates organ repair by decreasing tissue inflammation and increasing progenitor cell migration to areas of injury [8]. CXCR4 antagonism has been shown to decrease cockroach allergy-induced airway inflammation and bleomycin-induced pulmonary inflammation in rodents [9, 10]. In addition, a single dose of AMD3100 administered to mice with myocardial infarction, reduced fibrosis and inflammatory cell incorporation [8].
This study sought to ascertain whether CXCR4 blockade would attenuate lung injury in neonatal rats exposed to hyperoxia (HILI). We demonstrate that CXCR4 antagonism decreases lung inflammation in neonatal rats with HILI and this is accompanied by an improvement in lung vascular density and alveolarization. These findings suggest that CXCR4 blockade may be a potential strategy to reduce BPD in preterm neonates.
METHODS
Animals
Pregnant Sprague-Dawley rats were purchased from Charles River Laboratories (Wilmington, MA) and cared for according to NIH guidelines for use and care of animals during the experimental protocol. Rats were housed in a temperature- regulated room. Their chambers were cleaned twice weekly and food as well as water replaced as needed.
Experimental Design
All animal experiments were performed according to guidelines set forth by the University of Miami Animal Care and Use Committee. At delivery, rat pups (n=44, 4 litters in total) were randomly separated into four groups. The rat pups were exposed to either normobaric hyperoxia (FiO2=0.9) or room air (RA; FiO2=0.21) from postnatal day (P) 2 to P16. The rat moms were rotated every 48 hours between the hyperoxia and normoxic chambers to prevent oxygen toxicity and standardized nutrition was provided to each litter. There were no deaths in the RA groups. There was however 1 death in each of the hyperoxia groups.
AMD3100 Administration
Rat pups exposed to hyperoxia or normoxia from P2-P16 were randomly assigned to receive daily subcutaneous injections of AMD3100 (240 μg/kg; Sigma-Aldrich, Saint Louis, MO) or vehicle (sterile water) as placebo (PL) from P5-P15. The dose was chosen based on previous studies that showed efficacy with this dose [11]. Animals were studied on P16 (Figure 1).
Figure 1. Experimental Design.
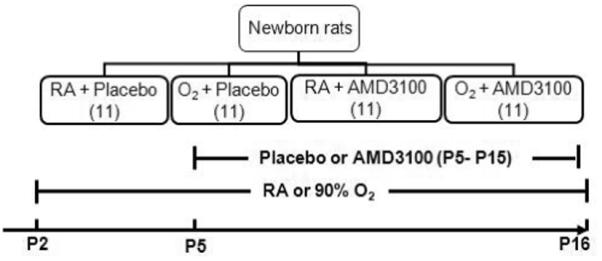
Newborn pups (P2) exposed to room air (RA) or hyperoxia (90% O2) were randomly assigned to received AMD3100 or placebo (PL) from postnatal day (P)2-P15. Pups were evaluated on P16.
Assessment of Pulmonary Hypertension
Right ventricular systolic pressure (RVSP) was measured as a surrogate of pulmonary artery pressure. The weight ratio of right ventricle to left ventricle and septum (RV to LV+S) was utilized as an index of right ventricular hypertrophy.
Assessment of Lung Alveolarization
Lung morphometric analysis was performed as previously described [12]. Serial paraffin-embedded lung sections five micrometers (μm) thick taken from the upper and lower lobes were stained by standard hematoxylin and eosin (H&E). Alveolarization was determined by measuring the mean linear intercept (MLI) and septal density. Images from five randomly selected, non-overlapping parenchymal fields were acquired from lung sections of each animal (n=10/group) at 20 X magnification.
Assessment of Vascular Density
Mid lung sections five μm thick of the upper and lower lobes were deparaffinized, rehydrated, and stained with polyclonal rabbit anti-human Von Willebrand Factor (vWF; Dako Corp, Carpinteri, CA). Six randomly selected, non-overlapping parenchymal fields were evaluated from lung sections of each animal (5-6/group). The number of vWF positive (vWFpos) blood vessels/hpf, (20-50 μm in diameter), were counted by a blinded observer.
Assessment of Pulmonary Vascular Remodeling
Paraffin embedded lung sections were stained with polyclonal rabbit anti-human vWF and monoclonal mouse anti-α-smooth muscle actin (α-SMA: 1:500, Sigma-Aldrich; St. Louis, MO). Medial wall thickness (MWT) of partially and fully muscular arteries (20-50 μm) was determined by using the formula: 2MT X 100/ED, where MT is the distance between the internal and external elastic laminas and ED is the external diameter. Approximately 20 randomly chosen arteries were evaluated per slide and all morphometric analyses were performed by a blinded observer.
Bronchoalveolar Lavage Fluid Analysis
Broncholaveolar lavage (BAL) fluid was obtained as previously described [13] and differential cell counts were performed on the cytospin preparations after Giemsa staining.
Western Blot
The protein expression of matrix metalloproteinase-9 (MMP-9), CXCR4 and vascular endothelial growth factor receptor 2 (VEGFR2) in lung homogenates was determined by Western Blot analysis. The polyclonal antibodies for CXCR4 (1: 500), MMP-9 (1:500) and VEGFR2 (1:200) were obtained from Abcam (Cambridge, MA) and Cell Signaling Technology (Danvers, MA) respectively. Lung homogenates were separated by 10% SDS-PAGE, transferred to nitrocellulose membranes, and blocked overnight at 4°C in 5% bovine serum albumin. Immunodetection was performed by incubating the membranes with the primary antibodies diluted in blocking buffer for 1 hour at room temperature. After washing, a semilumiscent horseradish peroxidase substrate was diluted in blocking buffer and applied for 60 minutes. Band intensity was quantified with Quantity One software (Bio-Rad, Hercules, CA).
Quantitative Real-time PCR
RNA from lung tissue was extracted (RNeasy Midi Kit, Qiagen, Inc. Valencia, CA) and reverse-transcribed. The specific cDNA for IL-6 was quantified by real time RTPCR using SuperArray (Frederick, MD) following the Real-Time Gene Expression Assay protocol. Primers for IL-6 and GAPDH (as an internal control) were pre-developed by SuperArray. The relative quantity IL-6 was normalized to GAPDH expression.
VEGF and SDF-1 ELISA
Lung vascular endothelial growth factor (VEGF-A) and SDF-1 tissue content were quantified using ELISA kits obtained from R&D Systems (Minneapolis, MN).
Myeloperoxidase Activity Assay
Lung myeloperoxidase (MPO) activity was determined using a specific MPO Colorimetric Activity Assay Kit as per manufacturer specifications (Biovision; Mountainview, CA).
Assessment of Lung Fibrosis
Lung sections were stained with Maason’s Trichrome stain. Lung collagen content was determined by performing a Sircol Collagen Assay as per manufacturer specifications (Biocolor; Carrickfergus, Northern Ireland).
Statistics
Results are reported as mean ± SD. Data were analyzed by two-way ANOVA followed by a post-hoc analysis (Holm-Sidak). Values of p<0.05 were considered statistically significant.
RESULTS
Lung CXCR4 Expression is increased in neonatal HILI
We first sought to ascertain whether hyperoxia exposure would affect the protein expression of CXCR4 in the lungs of neonatal pups. Whole lung lysates were obtained from newborn rat pups exposed to normoxia or hyperoxia (90% O2) for 14 days. The protein expression of CXCR4 was determined by Western blot. As compared to normoxic pups, there was an approximate 2-fold increase (p <0. 002; n=5/group) in the protein expression of CXCR4 in lung lysates obtained from hyperoxic pups (Figure 2). There was however no change in the lung tissue content of SDF-1 (0.396 ± 0.06 versus 0.459 ± 0.06 ng/ml; normoxia versus hyperoxia; p=0.07; n=5/group) following 14 days of hyperoxia.
Figure 2. Increased Lung CXCR4 Expression in Hyperoxia-Induced Lung Injury (HILI).
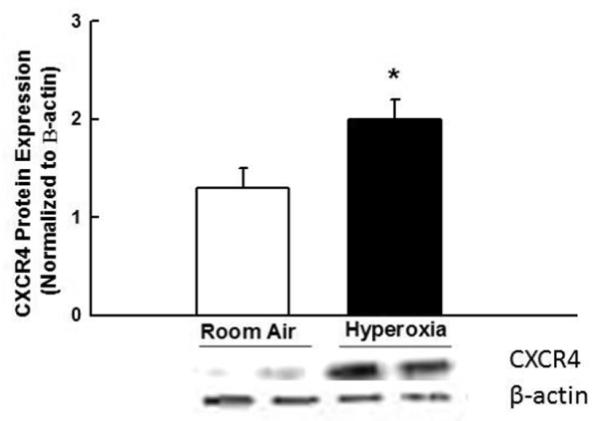
Increased lung CXCR4 protein expression in newborn rats exposed to 14 days of hyperoxia, (*P < 0.002, room air versus (vs.) hyperoxia; n = 5/group). CXCR4 expression is normalized to β-actin. A representative Western blot is shown in the lower panel.
CXCR 4 Blockade Improves Alveolarization in Neonatal HILI
There was no difference in the degree of alveolarization between the room air groups, (Figure 3A). Hyperoxia exposed animals showed marked simplification of the alveoli evidenced by larger alveoli with increased alveolar diameters and decreased septation, (Figure 3A). Furthermore, as compared to room air animals, there was an increase in MLI (43 ± 5 vs. 66 ± 5 μm; RA-PL vs. hyperoxia-PL; p<0.05; n=10/group), and a decrease in alveolar septation in the hyperoxia-exposed animals (42 ± 3 vs. 32 ± 2 septa/hpf; RA-PL vs. hyperoxia-PL; p<0.0001; n=10/group), Figures 3B and 3C. In contrast, administration of AMD3100 significantly improved alveolarization, as evidenced by increased secondary septation, (32 ± 2 vs. 45 ± 6 septa/hpf; hyperoxia-PL vs. hyperoxia-AMD3100; p<0.002; n=10/group) and decreased MLI (66 ± 5 vs. 55 ± 5 μm; hyperoxia-PL vs. hyperoxia-AMD3100; p<0.007; n=10/group), Figures 3B and 3C.
Figure 3. CXCR4 Blockade Improves Alveolarization in Hyperoxia-induced Lung injury (HILI).
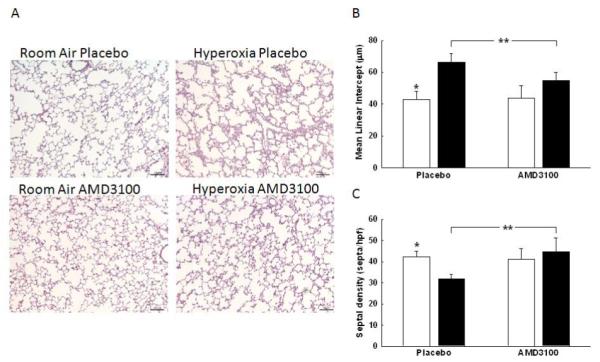
A. H&E stained lung sections demonstrating improved alveolar structure in hyperoxia exposed rats treated with AMD3100. Original magnification x 100, scale bars: 100 μm.
B. Decreased mean linear intercept (MLI) observed in hyperoxia-AMD3100 treated animals (*p<0.05; RA-PL vs. hyperoxia-PL; **p<0.007, hyperoxia-PL vs. hyperoxia-AMD3100; n=10/group). White bars represent RA animals and black bars represent hyperoxia animals.
C. Increased septal density in hyperoxic-AMD3100 treated rats (*p<0.0001; RA-PL vs. hyperoxia-PL; **p<0.002, hyperoxia-PL vs. hyperoxia-AMD3100; n=10/group).
CXCR4 Blockade Increases Vascular Density in Hyperoxia Induced Lung Injury
As compared to room air animals, hyperoxia exposed pups demonstrated decreased vascular density, (Figures 4A and 4B). In contrast, administration of AMD3100 to hyperoxic rats increased the lung vascular density by approximately 2-fold, (Figure 4B). These findings were associated with an increase in lung VEGF protein concentration (870 ± 18 vs. 1420 ± 61 pg/ml; hyperoxia-PL vs. hyperoxia-AMD3100; p<0.0001; n=5/group) and VEGFR2 protein expression (hyperoxia-PL vs. hyperoxia-AMD3100, p<0.02; n=5/group), Figures 4C-4D. There was no difference in the RVSP, RV/LV+S or the degree of pulmonary vascular remodeling (MWT) between the hyperoxic groups, (Figures 4E-4G).
Figure 4. CXCR4 Blockade Increases Lung Vascular Density in HILI.

A. Lung sections stained with vWF, red and 6-diamidino-2-phenylindole (DAPI; blue) demonstrating improved capillary density in hyperoxia exposed rats treated with AMD 3100. Original magnification x 100, scale bars: 100 μm.
B. Increased vascular density in hyperoxic-AMD3100 rats (*p<0.0001; RA-PL vs. hyperoxia-PL; **p<0.05; hyperoxia-PL vs. hyperoxia-AMD3100; n=10/group). White bars represent RA animals and black bars represent hyperoxia animals.
C. Increased lung VEGF concentration in hyperoxic-AMD3100 rats (*p<0.05; RA-PL vs. hyperoxia-PL; **p<0.0001; hyperoxia-PL vs. hyperoxia-AMD3100; n=5/group).
D. Increased lung VEGFR2 expression in hyperoxic-AMD3100 rats (*p<0.004; RA-PL vs. hyperoxia-PL; **p<0.02; hyperoxia-PL vs. hyperoxia-AMD3100; n=5/group). RA is room air and HYP is hyperoxia. VEGFR2 expression is normalized to β-actin.
E. Increased RVSP in hyperoxia groups (*p<0.05; RA-PL/AMD3100 vs. hyperoxia-PL/AMD3100; n=10/group). There was no difference in the RVSP between hyperoxia groups.
F. Increased RV/LV+S in hyperoxia groups (*p<0.05; RA-PL/AMD3100 vs. hyperoxia-PL/AMD3100; n=10/group). There was no difference in the RV/LV+S between hyperoxia groups.
G. Increased MWT in hyperoxia groups (*p<0.05; RA-PL/AMD3100 vs. hyperoxia-PL/AMD3100; n=10/group). There was no difference in the MWT between hyperoxia groups.
CXCR4 Blockade Decreases Inflammation in HILI
Hyperoxia exposed rats showed increased numbers of BAL macrophages and neutrophils respectively compared to rats exposed to room air, (4 × 104 ± 1 × 103 vs. 32 × 104 ± 14 × 104 cells/ml; RA-PL vs. hyperoxia-PL; p<0.0001; n=5/group and 0.8 × 104 ± 0.2 × 103 vs. 3.5 × 104 ± 2 × 104 cells/ml; RA-PL vs. hyperoxia-PL; p<0.0001; n=5/group), Figures 5A and 5B. In contrast, hyperoxia exposed AMD3100 treated rats had markedly decreased BAL macrophage and neutrophil counts to near normoxic levels, (32 × 104 ± 14 × 104 vs. 5 × 104 ± 1 × 104 cells/ml; hyperoxia-PL vs. hyperoxia-AMD3100; p<0.0001; n=5/group and 3.5 × 104 ± 2 × 104 vs. 0.7 × 104 ± 0.6 × 104 cells/ml; hyperoxia-PL vs. hyperoxia-AMD3100; p<0.0001; n=5/group), Figures 5A and 5B. These findings were associated with a decrease in lung MPO activity (0.4 ± 0.17 vs. 0.01 ± 0 mU/ml; hyperoxia-PL vs. hyperoxia-AMD3100; p<0.02; n=5/group), MMP-9 expression (4-fold; hyperoxia-PL vs. hyperoxia-AMD3100; p<0.0001; n=5/group), Figures 5C and 5D and IL-6 gene expression (50-fold; hyperoxia-PL vs. hyperoxia-AMD3100; p<0.0001; n=5/group)
Figure 5. CXCR4 Blockade Decreases Inflammation in HILI.
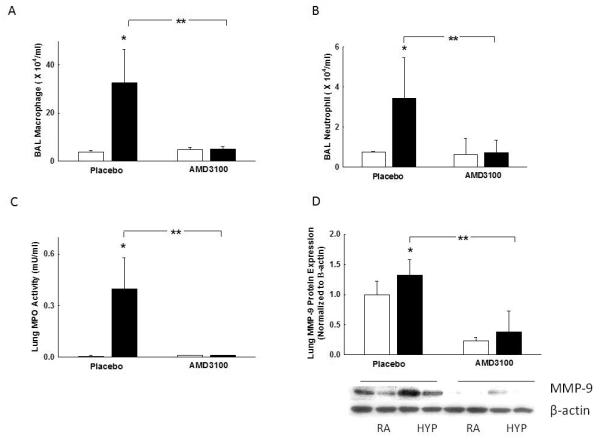
A. Reduced BAL macrophage counts in hyperoxia-AMD3100 rats (*p<0.0001; RA-PL vs. hyperoxia-PL; **p<0.0001; hyperoxia-PL vs. hyperoxia-AMD3100; n=5/group). White bars represent RA animals and black bars represent hyperoxia animals.
B. Decreased BAL neutrophil counts in hyperoxia-AMD3100 rats (*p<0.0001; RA-PL vs. hyperoxia-PL; **p<0.0001; hyperoxia-PL vs. hyperoxia-AMD3100, n=5/group).
C. Reduced lung myeloperoxidase (MPO) activity in hyperoxia-AMD3100 rats (*p<0.0001; RAPL vs. hyperoxia-PL; **p<0.02; hyperoxia-PL vs. hyperoxia-AMD3100; n=5/group).
D. Decreased lung MMP-9 protein expression in hyperoxia-AMD3100 rats (*p <0.05; RA-PL vs. hyperoxia-PL; **p<0.05; hyperoxia-PL vs. hyperoxia-AMD3100; n=5/group). RA is room air and HYP is hyperoxia. MMP-9 expression is normalized to β-actin.
CXCR4 Blockade Decreases Lung fibrosis in HILI
In order to determine the effects of AMD3100 on lung fibrosis, Masson’s Trichrome stained lung sections were evaluated. As compared to RA rats, hyperoxia-exposed rats had lung fibrosis and increased lung collagen, Figures 6A and 6B. In contrast, administration of AMD3100 to hyperoxic rats decreased lung fibrosis and collagen content, Figures 6A and 6B. There was no difference in lung collagen content between RA-PL and hyperoxia-AMD3100 groups.
Figure 6. CXCR4 Blockade Decreases Lung Fibrosis.
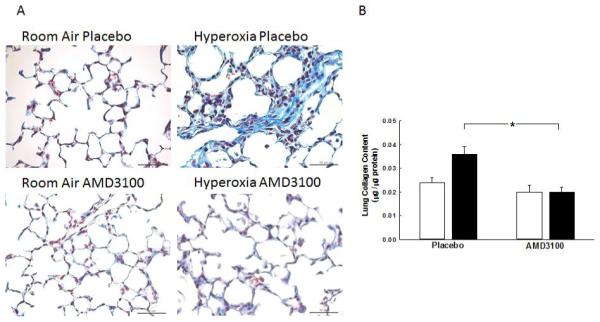
A. Lung sections stained with Maason’s Trichrome staining showing decreased lung fibrosis in hyperoxia- AMD3100 treated rats. Original magnification x 400, scale bars: 50 μm.
B. Decrease lung collagen in hyperoxia-AMD3100 treated rats (*p<0.0001, RA-PL vs. hyperoxia-PL, and hyperoxia-PL vs. hyperoxia-AMD3100, n=5/group). White bars represent RA animals and black bars represent hyperoxia animals.
DISCUSSION
This study sought to ascertain whether CXCR4 blockade would attenuate neonatal hyperoxia-induced lung injury, an experimental model of BPD. We show that administration of the CXCR4 antagonist, AMD3100 to neonatal rodents with experimental BPD decreases lung inflammation, improves alveolarization and angiogenesis. Our findings suggest that strategies based on modulating the activity of the SDF-1/CXCR4 axis may be potentially efficacious in repairing the injured preterm lung.
We first demonstrate an increase in lung CXCR4 expression during hyperoxia. This finding is in keeping with those other several investigators who have found increased lung CXCR4 expression in hyperoxia and LPS-induced lung injury [6, 14]. Surprisingly, in our study, hyperoxia did not increase lung SDF-1 tissue content. While our findings are similar to those of Balasubramaniam et al [15], other investigators have found an increase in lung SDF-1 concentration during hyperoxia [6]. It is possible that the disparity between our findings and those of other investigators maybe secondary to differences in our animal model. Nonetheless, in agreement with other studies, the absence of an SDF-1 gradient did not affect the anti-inflammatory effects of AMD3100 [16].
In our present study, administration of AMD3100 to hyperoxic pups reduced lung inflammation as evidenced by decreased BAL inflammatory cells and lung MPO activity. Previous studies have shown that inflammation is a key component in the pathogenesis of BPD [2]. Moreover, preterm infants in whom BPD develop have elevated protein levels of inflammatory cytokines and increased numbers of inflammatory cells in their tracheal aspirates [17]. Hyperoxia is one of the most potent inducers of inflammation in these preterm patients. Our current finding that CXCR4 blockade reduces lung inflammation in a hyperoxic model of BPD is consistent with those of other investigators who showed that antagonism of the SDF-1/CXCR4 axis reduced lung neutrophil infiltration during lipopolysaccharide induced lung injury [14]. It is also possible that AMD3100 may have decreased lung inflammation by increasing the egress of neutrophils from the lung [18], decreased inflammatory cell trans-endothelial migration or by having negative functional effects on other chemokine receptors[19].
The improvement in lung inflammation in our study was associated with decreased lung MMP-9 expression. MMP-9 is expressed by several cells, including neutrophils and it works synergistically with SDF-1 to regulate the trans-endothelial migration of inflammatory cells [20]. We speculate that the decreased MMP-9 expression in the AMD3100 treated pups is not only due to the decrease in inflammatory cells in the hyperoxic group but this may also be secondary to reduced activation of SDF-1/CXCR4 down-stream signaling pathways which modulate MMP-9 expression [21].
AMD3100 also decreased lung collagen content in the hyperoxic pups. Increased total lung collagen content has been previously shown in the lungs of infants with BPD [22]. Moreover, Deng et al demonstrated increased CXCR4 positive fibroblasts in the lungs of rodents with hyperoxia-induced lung injury. Our present finding that CXCR4 blockade improves lung collagen content following hyperoxia-induced lung injury is consistent with other studies which have shown decreased lung collagen content in rodent models of bleomycin-induced lung fibrosis following administration of a CXCR4 antagonist [9].
Interestingly, in our present study, although there was an improvement in lung vascular density following AMD3100, there were no significant effects on RVSP, RVH or vascular remodeling. The negative findings in our present study may be due to the fact that although there was an improvement in lung vascular density in hyperoxic-AMD3100 rats, the number of intra-acinar vessels/hpf was still significantly lower than in the RA-PL rats. Interestingly, prior studies have shown decreased vascular remodeling in hypoxic rodent models of pulmonary hypertension following AMD3100 administration [23]. It is plausible that this disparity in the studies is secondary to the differences in CXCR4 signaling during hypoxia as compared to the hyperoxic conditions in our study, the timing of our intervention and the dynamic processes involved in repair.
Finally, there were also several limitations to our study. The hyperoxic rodent model utilized corresponds to the saccular -alveolar stages of human lung development model and most preterm infants who develop BPD are in the late cannalicular to early saccular stages of lung development. In addition, although hyperoxia is significant contributor to the pathogenesis of BPD, we utilized a relatively high oxygen concentration which mimics severe BPD. Indeed, most preterm infants are not exposed to this degree of postnatal hyperoxia and thus potentially the efficacy of our therapy may be altered. Finally, given in vitro data demonstrating that CXCR4 knock-down impeded alveolar epithelial cell wound healing, future studies evaluating the long term effect of CXCR4 blockade on alveolar epithelial cell homeostasis will need to be performed [24].
Nonetheless, our present study shows that CXCR4 antagonism reduces alveolar growth arrest and impaired angiogenesis in neonatal rodents with hyperoxia-induced BPD-like phenotype. Although further long-term studies will need to be performed to evaluate the effects of SDF-1/CXCR4 axis modulation on other developing organs, these findings suggest that modulation of the SDF-1/CXCR4 axis may be a potential strategy to treat BPD.
ACKNOWLEDGEMENTS
We wish to thank Dr. Claudia O. Rodrigues for critiquing this manuscript. This work is supported by a National Institute of Health KO8 Award, Florida Biomedical Research Award and Batchelor Research Foundation Award to KY.
Footnotes
COMPETING INTERESTS None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
AUTHORS’ CONTRIBUTIONS SD, SR, CS and KY were involved in the conception and design of experiments and wrote the manuscript. SD, SR, ET, JH, DH, CS and KY performed the experiments, analyzed the data, read and approved the final manuscript.
REFERENCES
- 1.Philip AG. Bronchopulmonary dysplasia: then and now. Neonatol. 2012;102:1–8. doi: 10.1159/000336030. [DOI] [PubMed] [Google Scholar]
- 2.Jobe AJ. The new BPD: an arrest of lung development. Pediatr Res. 1999;46:641–643. doi: 10.1203/00006450-199912000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Munshi UK, Niu JO, Siddiq MM, Parton LA. Elevation of interleukin-8 and interleukin-6 precedes the influx of neutrophils in tracheal aspirates from preterm infants who develop bronchopulmonary dysplasia. Ped Pulmonol. 1997;24:331–336. doi: 10.1002/(sici)1099-0496(199711)24:5<331::aid-ppul5>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 4.McGrath KE, Koniski AD, Maltby KM, McGann JK, Palis J. Embryonic Expression and Function of the Chemokine SDF-1 and Its Receptor, CXCR4. Develop Biol. 1999;213:442–456. doi: 10.1006/dbio.1999.9405. [DOI] [PubMed] [Google Scholar]
- 5.Ratajczak MZ, Majka M, Kucia M, Drukala J, Pietrzkowski Z, Peiper S, Janowska-Wieczorek A. Expression of functional CXCR4 by muscle satellite cells and secretion of SDF-1 by muscle-derived fibroblasts is associated with the presence of both muscle progenitors in bone marrow and hematopoietic stem/progenitor cells in muscles. Stem Cells. 2003;21:363–371. doi: 10.1634/stemcells.21-3-363. [DOI] [PubMed] [Google Scholar]
- 6.Deng C, Wang J, Zou Y, Zhao Q, Feng J, Fu Z, Guo C. Characterization of fibroblasts recruited from bone marrow-derived precursor in neonatal bronchopulmonary dysplasia mice. J Appl Physiol. 2011;111:285–294. doi: 10.1152/japplphysiol.00201.2010. [DOI] [PubMed] [Google Scholar]
- 7.Donzella GA, Schols D, Lin SW, Este JA, Nagashima KA, Maddon PJ, Allaway GP, Sakmar TP, Henson G, De Clercq E, Moore JP. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat Med. 1998;4:72–77. doi: 10.1038/nm0198-072. [DOI] [PubMed] [Google Scholar]
- 8.Jujo K, Hamada H, Iwakura A, Thorne T, Sekiguchi H, Clarke T, Ito A, Misener S, Tanaka T, Klyachko E, et al. CXCR4 blockade augments bone marrow progenitor cell recruitment to the neovasculature and reduces mortality after myocardial infarction. Proc Natl Acad Sci U S A. 2010;107:11008–11013. doi: 10.1073/pnas.0914248107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu J, Mora A, Shim H, Stecenko A, Brigham KL, Rojas M. Role of the SDF-1/CXCR4 axis in the pathogenesis of lung injury and fibrosis. Am J Respir Cell Mol Biol. 2007;37:291–299. doi: 10.1165/rcmb.2006-0187OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lukacs NW, Berlin A, Schols D, Skerlj RT, Bridger GJ. AMD3100, a CxCR4 Antagonist, Attenuates Allergic Lung Inflammation and Airway Hyperreactivity. Am J Pathol. 2002;160:1353–1360. doi: 10.1016/S0002-9440(10)62562-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Devine SM, Flomenberg N, Vesole DH, Liesveld J, Weisdorf D, Badel K, Calandra G, DiPersio JF. Rapid Mobilization of CD34+ Cells Following Administration of the CXCR4 Antagonist AMD3100 to Patients With Multiple Myeloma and Non-Hodgkin’s Lymphoma. J Clin Oncol. 2004;22:1095–1102. doi: 10.1200/JCO.2004.07.131. [DOI] [PubMed] [Google Scholar]
- 12.Thurlbeck WM. Measurement of pulmonary emphysema. Am Rev Respir Dis. 1967;95:752–764. doi: 10.1164/arrd.1967.95.5.752. [DOI] [PubMed] [Google Scholar]
- 13.Hummler SC, Rong M, Chen S, Hehre D, Alapati D, Wu S. Targeting Glycogen Synthase Kinase-3β to Prevent Hyperoxia-Induced Lung Injury in Neonatal Rats. Am J Resp Cell Mol Biol. 2013;48:578–588. doi: 10.1165/rcmb.2012-0383OC. [DOI] [PubMed] [Google Scholar]
- 14.Petty JM, Sueblinvong V, Lenox CC, Jones CC, Cosgrove GP, Cool CD, Rai PR, Brown KK, Weiss DJ, Poynter ME, Suratt BT. Pulmonary Stromal-Derived Factor-1 Expression and Effect on Neutrophil Recruitment during Acute Lung Injury. J Immunol. 2007;178:8148–8157. doi: 10.4049/jimmunol.178.12.8148. [DOI] [PubMed] [Google Scholar]
- 15.Balasubramaniam V, Mervis CF, Maxey AM, Markham NE, Abman SH. Hyperoxia reduces bone marrow, circulating, and lung endothelial progenitor cells in the developing lung: implications for the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1073–1084. doi: 10.1152/ajplung.00347.2006. [DOI] [PubMed] [Google Scholar]
- 16.Zuk A, Gershenovich M, Ivanova Y, MacFarland RT, Fricker SP, Ledbetter S. CXCR(4)antagonism as a therapeutic approach to prevent acute kidney injury. Am J Physiol Renal Physiol. 2014;307:F783–797. doi: 10.1152/ajprenal.00685.2013. [DOI] [PubMed] [Google Scholar]
- 17.Ogden BE, Murphy SA, Saunders GC, Pathak D, Johnson JD. Neonatal lung neutrophils and elastase/proteinase inhibitor imbalance. Am Rev Respir Dis. 1984;130:817–821. doi: 10.1164/arrd.1984.130.5.817. [DOI] [PubMed] [Google Scholar]
- 18.Devi S, Wang Y, Chew WK, Lima R, A-González N, Mattar CNZ, Chong SZ, Schlitzer A, Bakocevic N, Chew S, et al. Neutrophil mobilization via plerixafor-mediated CXCR4 inhibition arises from lung demargination and blockade of neutrophil homing to the bone marrow. J Exp Med. 2013;210:2321–2336. doi: 10.1084/jem.20130056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sohy D, Yano H, de Nadai P, Urizar E, Guillabert A, Javitch JA, Parmentier M, Springael JY. Hetero-oligomerization of CCR2, CCR5, and CXCR4 and the protean effects of “selective” antagonists. J Biol Chem. 2009;284:31270–31279. doi: 10.1074/jbc.M109.054809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang H, Trivedi A, Lee J-U, Lohela M, Lee SM, Fandel TM, Werb Z, Noble-Haeusslein LJ. Matrix Metalloproteinase-9 and Stromal Cell-Derived Factor-1 Act Synergistically to Support Migration of Blood-Borne Monocytes into the Injured Spinal Cord. J Neuro. 2011;31:15894–15903. doi: 10.1523/JNEUROSCI.3943-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu X, Huang Y, Collin-Osdoby P, Osdoby P. Stromal Cell-Derived Factor-1 (SDF-1) Recruits Osteoclast Precursors by Inducing Chemotaxis, Matrix Metalloproteinase-9 (MMP-9) Activity, and Collagen Transmigration. J Bone Mineral Res. 2003;18:1404–1418. doi: 10.1359/jbmr.2003.18.8.1404. [DOI] [PubMed] [Google Scholar]
- 22.Cherukupalli K, Larson JE, Rotschild A, Thurlbeck WM. Biochemical, clinical, and morphologic studies on lungs of infants with bronchopulmonary dysplasia. Ped Pulmonol. 1996;22:215–229. doi: 10.1002/(SICI)1099-0496(199610)22:4<215::AID-PPUL1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 23.Young KC, Torres E, Hatzistergos KE, Hehre D, Suguihara C, Hare JM. Inhibition of the SDF-1/CXCR4 axis attenuates neonatal hypoxia-induced pulmonary hypertension. Circ Res. 2009;104:1293–1301. doi: 10.1161/CIRCRESAHA.109.197533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh MC, Makena PS, Gorantla V, Sinclair SE, Waters CM. CXCR4 regulates migration of lung alveolar epithelial cells through activation of Rac1 and matrix metalloproteinase-2. A J Physiol - Lung Cell and Mol Physiol. 2012;302:L846–L856. doi: 10.1152/ajplung.00321.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]