

Abstract
Cancer progression requires a significant reprogramming of cellular signaling to support the essential tumor-specific processes that include hyperproliferation, invasion (for solid tumors) and survival of metastatic colonies. NEDD9 (also known as CasL and HEF1) encodes a multi-domain scaffolding protein that assembles signaling complexes regulating multiple cellular processes relevant to cancer. These include responsiveness to signals emanating from the T and B cell receptors, integrins, chemokine receptors, and receptor tyrosine kinases, as well as cytoplasmic oncogenes such as BCR-ABL and FAK- and SRC-family kinases. Downstream, NEDD9 regulation of partners including CRKL, WAVE, PI3K/AKT, ERK, E-cadherin, Aurora-A (AURKA), HDAC6, and others allow NEDD9 to influence functions as pleiotropic as migration, invasion, survival, ciliary resorption, and mitosis. In this review, we summarize a growing body of preclinical and clinical data that indicate that while NEDD9 is itself non-oncogenic, changes in expression of NEDD9 (most commonly elevation of expression) are common features of tumors, and directly impact tumor aggressiveness, metastasis, and response to at least some targeted agents inhibiting NEDD9-interacting proteins. These data strongly support the relevance of further development of NEDD9 as a biomarker for therapeutic resistance. Finally, we briefly discuss emerging evidence supporting involvement of NEDD9 in additional pathological conditions, including stroke and polycystic kidney disease.
Keywords: NEDD9, CAS-L, HEF1, CAS family protein, human malignancy, protein function, signal transduction, prognosis
1. Introduction
NEDD9 (neural precursor cell expressed, developmentally down-regulated 9) is a non-catalytic scaffolding protein [1–3]. Based on the current understanding of NEDD9 function, it assembles complexes involving oncogenic kinases, including focal adhesion kinase (FAK), ABL, SRC, Aurora-A (AURKA), and others, and thereby regulates the magnitude and duration of cell signaling cascades that controls multiple processes that are crucial for tumorigenesis and metastases. These include apoptosis and cell cycle, migration, adhesion, invasion and chemotaxis [4–7]. Because of its pleiotropic function, in many human cancers, altered expression of NEDD9 has emerged as predictive of poor outcome, metastatic potential, and resistance to chemotherapy, while altered NEDD9 function has more recently been linked to additional non-malignant pathological conditions, such as stroke and polycystic kidney disease. Neither overexpression nor gene loss of NEDD9 induces tumorigenesis in the absence of other driver lesions. However, as described below, there is now strong evidence that NEDD9 is an important physiological modifier of multiple stages of cancer initiation and progression for many types of malignancies. In this article, we summarize the current preclinical and clinical data describing the action of NEDD9 in cancer, placing this work in the context of specific mechanisms by which altered expression of NEDD9 supports the disease process.
2. Regulation and action of NEDD9
A detailed discussion of NEDD9 gene and protein structure and function are beyond the scope of this article, but this topic has recently been reviewed in detail [4–7]. In very brief summary, the NEDD9 promoter contains a retinoic acid response element (RARE) [8, 9] that is specifically bound by a retinoid X receptor (RXR)/retinoic acid receptor (RAR) heterodimer [10], as well as xenobiotic responsive elements [11] and binding sites for transcription factors FoxC1 [12], Ngn2 [13], HIF-1α [14], TCF [15], SAFB1 [16], NF-Kβ, STAT5A, ER-α, GATA and others (web-links http://www.sabiosciences.com/chipqpcrsearch.php?app=TFBS and http://www.ncbi.nlm.nih.gov/gene/4739 provide useful resources) (Fig. 1A). Additionally, NEDD9 is regulated by miR-145, which binds the NEDD9 3′-untranslated region [17–19]. A non-coding RNA, named B2, extending from 10 kb upstream of Nedd9 exon 1 to exon 4 has been described, but the functional role for this ncRNA is not yet clear [20]. It is possible that a SNP located in an intronic region of NEDD9 contributes to predisposition to late onset Alzheimer’s disease [21–25], although this is not yet proven [24], as is discussed more fully in [26].
Figure 1. NEDD9 gene and protein.

A. Schematic representation of NEDD9 gene (transcript variant NM_006403, 7 exons) and mRNA. Relative sizes of promoter, exons and introns are not to scale. Inside the red arrow are factors inducing transcription of NEDD9, with upstream regulatory factors indicated to right. The RNA B2 homology region begins 10 bp upstream of exon 1, and ends at the intron between exons 4 and 5 of NEDD9. The intronic region between exons 6 and 7 contains a C>G SNP (rs760678), discussed in text. A miR-145 binding region is located at 3′UTR of the NEDD9 mRNA. The encoded protein is 834 aa in humans, arising from 7 coding exons (pale blue boxes): numbers of amino acids encoded by each exon are indicated within boxes, with functionally defined domains encoded by exons indicated below. B. Domain structure and upstream regulators of NEDD9 protein. SH3 - N-terminal SH3 domain, SD - substrate domain, SRR - serine rich region, FAT - focal adhesion targeting domain. The T-cell and B-cell receptors (TCR and BCR), integrins and chemokine receptors [108] provide upstream activation signals. In T cells and B cells, this process requires NEDD9 interaction with NSP family scaffold proteins via the NEDD9 C-terminal domain [111, 112]. NEDD9 coordinates signaling between integrins and RTKs such as EGFR through interactions with NSP proteins [111, 112] and the adaptor proteins SHC and GRB2 (reviewed in [27, 113]). Subsequent activation of FAK, SRC and ABL-family kinase (ABL1, ABL2) causes extensive phosphorylation of NEDD9 substrate domain that creates multiple binding sites for downstream effectors; among these sites, Y189, Y317, and Y279 have been functionally validated [41]. FAK phosphorylation of the DYDY (amino acids 628–631) motif in the NEDD9 C-terminus creates a binding site for SRC kinase, and is important for NEDD9 function in migration and dother signaling functions [114]{Tachibana, 1997 #193}{Iwata, 2005 #103}{Kondo, 2012 #79}. Y189 phosphorylation is implicated in focal adhesion; this is a proposed phosphorylation site for FAK and SRC kinases [114]. Phosphorylation of S296 by Aurora-A kinase is implicated in NEDD9 proteasomal degradation [115], cell spreading [116] and cell cycle control [30]. NEDD9 potentiates TGFβ signaling by recruiting the inhibitory SMAD6 and SMAD7 proteins, and preventing their interaction with TGFβ [63]. TGFβ induces NEDD9 mRNA [97]; conversely, the TGFβ receptor effector SMAD3 mediates NEDD9 degradation via the APC complex [34, 35]. ABL – Abelson murine leukemia viral oncogene homolog; AURKA – Aurora kinase A; RAFTK – protein tyrosine kinase 2 beta (PYK2, PTK2B); FAK – focal adhesion kinase; SRC – (short for sarcoma), proto-oncogene non-receptor tyrosine kinase Src; SHC1 – SH2-domain containing transforming protein; GRB2 – growth factor receptor-bound protein 2; SMAD – homolog of C. Elegans protein SMA (from gene sma for small body size) and Drosophila protein MAD (mothers against decapentaplegic); APC/C – anaphase-promoting complex/cyclosome.
The NEDD9 protein arises from 7 coding exons (Fig 1A). Defined protein motifs in NEDD9 include an N-terminal SH3 domain, an unstructured “substrate domain” (SD), a serine-rich region (SRR) that folds into a four-helix bundle, and a C-terminal domain which also folds into a four-helix bundle, and encompasses a focal adhesion targeting (FAT) function and a SRC-binding motif (Fig. 1B) [2, 4, 27, 28]. Interactions of cellular signaling proteins with these domains allow NEDD9 to execute biological functions at discrete cellular locations. Beyond the regulation at the level of transcription, NEDD9 expression is controlled at the post-transcriptional and post-translational level. Important sources of regulation include phosphorylation, which influences scaffolding activity and localization, and proteasomal degradation. For example, in interphase cells, the majority of NEDD9 localizes to focal adhesions [29]. However, some of the protein is also cytoplasmic, and small pools localize to the centrosome [30] and the basal body of cilia [31]. At mitotic entry NEDD9 moves along mitotic spindle, eventually localizing at the midbody at cytokinesis [30]. In most actively growing adherent cells, NEDD9 migrates as a doublet of 115 and 105 kDa, associated with distinct degrees of phosphorylation [32]. The conversion of p115 into p105 is activated by cell detachment through cytoskeletal regulation of phosphatase PP2A in interphase cells [33]. Serine/threonine hyper-phosphorylated p115 NEDD9 is also more common in G2/M phase cells [32], suggesting these modifications are associated with increased localization to centrosome and mitotic spindle. P115 is the primary target for proteasomal degradation of NEDD9 [33]. Proteasomal degradation of NEDD9 is triggered by a number of stimuli, including induction of transforming growth factor receptor beta (TGFβ) signaling [34]. An effector of the TGFβ receptor, SMAD3, may interact directly with APC subunit APC10 and thus recruit the APC complex. CDH1 subunit of the APC complex recognizes NEDD9 and regulates ubiquitination and subsequent degradation of NEDD9 [35]. NEDD9 is also degraded by the proteasome at the end of mitosis, following completion of activities with Aurora-A kinase and other factors that support mitotic progression [30, 36, 37].
As a scaffold, NEDD9 assembles protein complexes with diverse partners to activate multiple cellular functions, summarized in Fig. 2, 3 and 4. Hence, the overexpression of NEDD9 in cancer has the potential to simultaneously induce migration, in part by promoting focal adhesion turnover; induce invadopodia formation; stimulate proliferation-associated signaling; and contribute to genomic instability. These activities are reflected in a growing number of studies that recognize elevation of NEDD9 as a factor contributing to aggressive tumor behavior. Interestingly, in some cancer contexts, it is reduction or loss of NEDD9 rather than elevation that is associated with tumor growth. This may reflect underlying differences in the biology of distinct tumor types (for instance, solid versus liquid tumors), but alternatively or in addition may reflect dominant negative activity associated with loss of NEDD9, based on the disruption of signaling complexes for which it is an essential scaffold.
Figure 2. Signaling by NEDD9 relevant to the pathogenesis of BCR-ABL positive malignancies.

NEDD9 co-localizes and interacts with BCR-ABL and its adaptor protein CRKL [49, 51], likely serving as a hub [50] for an assembly of protein complexes between CRKL and its downstream effectors: SYK [53], C3G [42], BRAF, SAPK, JNK [47], and STAT5 [52]. SRC family kinase Syk induces the PI3K/AKT/MTOR pathway enhancing cell survival [117]; activation of the RAS/RAF/MEK/ERK pathway drives cell proliferation; transcription factors STAT5 and JNK activate transcription of a large number of target genes [47]. Signaling through B-cell receptor, chemokine receptor and/or integrins lead to NEDD9 activation by the FAK/SRC/PXN complex; active NEDD9, in turn, helps to support continuous activation of FAK and SRC [56]. Phosphorylated NEDD9 binds CRKL and promotes migration and chemotaxis through activation of WASP/ARP2/3 signaling (reviewed in [4, 54]). ABL-mediated tyrosine phosphorylation of NEDD9 and binding of C3G leads to activation of Rap1 and migration/chemotaxis of lymphoid cells [41]. ARP2/3 - protein complex regulating actin cytoskeleton; WASP - Wiskott-Aldrich syndrome family protein; CRKL - adaptor protein encoded by V-crk avian sarcoma virus CT10 oncogene homolog; PXN – paxillin; FAK – focal adhesion kinase; SRC – (short for sarcoma), proto-oncogene non-receptor tyrosine kinase Src; RAF – family of serine/threonine protein kinases controlling cell proliferation; activation of RAF kinases requires interaction with RAS-GTPases; SYK - spleen tyrosine kinase; JNK – c-Jun N-terminal kinase; SAPK – stress-activated protein kinase; STAT – signal transducers and activators of transcription.
Figure 3. NEDD9 signaling mechanisms analyzed in breast cancer models.

NEDD9 is activated by FAK and SRC kinases, and also helps support continuous activation of these kinases [56]. NEDD9 mediates TGFβ effects through a positive feedback loop [61]. Transcription factors SRF (serum response factor) and SMAD induce NEDD9 expression; NEDD9 scaffolding connects TGFβ/SMAD and Rho-actin-SRF signals to coordinate the expression of genes involved in tumorigenesis [61]. NEDD9 binding to the DOCK3 GEF activates RAC, which signals through WAVE/SCAR to initiate ARP2/3-dependent actin branching, lamellipodia formation, and MMP activation [69]. NEDD9 and SRC negatively regulate expression of E-cadherin through upregulation of the SLUG and SNAIL transcription factors [60], and also by regulating SRC-dependent removal of E-cadherin from adherens junctions [68]. NEDD9 promotes trafficking of ligand-bound integrins from early to late endosomes and degradation of ligand/integrin complexes by inactivating caveolin (CAV1), which mediates recycling of ligand-bound integrins from early endosomes back to the plasma membrane [65]. NEDD9 regulates turnover of MMP14 and its enzymatic recovery through the late endosomes by suppressing Arf6 dependent trafficking of MMP14/TIMP2 complexes from early/sorting endosomes back to the surface. MMP14 cleaves pro-MMP2 to produce active MMP2, degrading type IV collagen of basal membrane and promoting invasion [66]. FAK – focal adhesion kinase; SRC – (short for sarcoma), proto-oncogene non-receptor tyrosine kinase Src; SRF - Serum response factor; SMAD – homolog of C. Elegans protein SMA (from gene sma for small body size) and Drosophila protein MAD (mothers against decapentaplegic); DOCK3 – Dedicator of cytokinesis 3; SLUG, SNAIL – members of SNAIL family zinc finger transcription factors; ARP2/3 – protein complex regulating actin cytoskeleton; WAVE member of the Wiskott-Aldrich syndrome family protein, activates ARP2/3 complex; MMP - matrix metalloproteinase TIMP2 – metallopeptidase inhibitor 2; ARF6 – ADP-ribosylation factor 6; CAV1 – caveolin 1.
Figure 4. Interaction of NEDD9 with Aurora-A (AURKA) mediates resorption of cilia and mitosis.
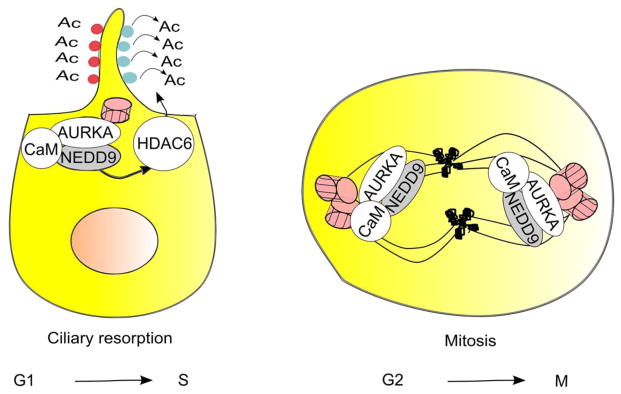
In G1 or G0 cells, transient NEDD9 induction in response to serum, Ca2+-bound calmodulin (CaM), or serum-derived growth factors allow NEDD9-dependent activation of AURKA kinase [31, 106, 107]. This induces resorption of the primary cilium, based in part on AURKA phosphorylation and activation of HDAC6, which deacetylates tubulin in the ciliary axoneme [31]. In mitosis, NEDD9 colocalizes with and binds Aurora-A and CaM at the centrosomes: these interactions support Aurora-A activation. Later in mitosis, NEDD9 and CaM move with Aurora-A to the spindle, and NEDD9 protects AURKA from degradation [37]. AURKA – Aurora kinase A, HDAC6 - histone deacetylase 6.
3. Altered NEDD9 expression in cancer
3.1 Leukemia and Lymphoma
In NEDD9-deficient mice, the marginal zone B-cell (MZB) population of spleen is almost absent, and the number of lymphocytes in bone marrow and thymus of NEDD9 knockout mice is also decreased [38]. Detailed study of the MZB phenotype indicated that it was due to failure of B-cells to adhere and/or migrate to MZ, which was in part attributable to a decreased response of Nedd9−/− B-cells to chemokines [38]. In non-transformed hematopoietic cells, NEDD9 is involved in T-cell and B-cell receptor signaling, integrin signaling, migration and chemotaxis (Fig. 2) [39–43]. Activation of chemokine receptors causes NEDD9 phosphorylation by ABL and ABL2 kinases [41]. Ligation of beta-integrin signaling causes NEDD9 tyrosine phosphorylation by FAK [44] and by the FAK related kinase RAFTK [40], and subsequently phosphorylation by SRC kinases [45]. Ligation of either β1 integrin or the B-cell receptor promotes interaction of phosphorylated NEDD9 with an SH2-domain containing adaptor protein CRKL (cellular regulator of kinase like), commonly deregulated in human malignancies and associated with tumor progression [40, 42, 46]. NEDD9 has a cluster of thirteen Tyr-x-x-Pro motifs which when phosphorylated serve as binding sites for CRKL and other SH2-domain containing proteins [47]. Stimulation of the T-cell receptor similarly induces phosphorylation of NEDD9 and its association with CRKL [43] and a guanine nucleotide exchange factor (GEF) C3G that activates the GTPase RAP1, mediating adhesion and migration of lymphocytes [41, 42].
Soon after the initial description of the protein, altered NEDD9 activity was associated with the pathogenesis of BCR/ABL-dependent tumors, such as the Philadelphia chromosome positive (Ph+) acute lymphoblastic leukemia (ALL) and chronic myelogenous leukemia (CML) [48, 49]. De Jong et al. showed that NEDD9 is hyperphosphorylated in P190BCR/ABL transgenic mice with pre-B cell ALL [49]. As discussed below, hyperphosphorylation is associated with increased protein-protein interaction capacity for NEDD9. In P190BCR/ABL transgenic mice, hyperphosphorylated NEDD9 was recovered from complexes with BCR/ABL and CRKL [49–51]. CRKL, a major in vivo binding partner and substrate of the deregulated BCR/ABL kinase, functions as a molecular link with other signaling proteins, such as BRAF [47], C3G [42], RAP1 [47], STAT5A [52], SYK, WASP [53], Pragmin [50], stress kinases SAPK and JNK [47], and others (Fig. 2). Although the exact role of NEDD9 in signal transduction of BCR/ABL positive tumors has not been fully explored, studies of BCR/ABL signaling network by an integrated proteomic approach (free quantitative protein complex and phosphorylation profiling by mass spectrometry) suggest that NEDD9 serves as a hub for an assembly of protein complexes between CRKL and its downstream effectors [50]. The clustering of multiple SH2-domain docking sites on NEDD9 allows the protein to function as intracellular assembly point for SH2-domain containing proteins, like CRKL, and their downstream signaling partners [47]. Additionally, direct interaction with NEDD9 has been shown for at least some proteins in the BCR/ABL downstream network. In B-cells, NEDD9 together with CRKL binds C3G, a guanine nucleotide exchange factor involved in RAS pathway [42]. ABL-mediated tyrosine phosphorylation of NEDD9 and binding of C3G leads to activation of Rap1 and migration/chemotaxis of lymphoid cells [41]. The NEDD9/CRKL complex promotes actin polymerization and migration and chemotaxis through activation of WASP/ARP2/3 signaling (reviewed in [4, 54]).
The functional role of NEDD9 in the development of hematologic malignancies has been explored in several mouse models. Seo et al. analyzed the consequence of deleting NEDD9 in a transgenic model for BCR-ABL leukemogenesis, and surprisingly found that the disease was more aggressive in mice lacking NEDD9. The authors proposed this was due to a role of NEDD9 in limiting extramedullary hyperplasia [55]. Additionally, in the same transgenic mouse model of CML, granulocytes of NEDD9 null mice (BCR-ABL;Nedd9−/−) showed decreased adhesion properties, compared to the granulocytes of BCR/-ABL;Nedd9+/+ controls [55].
Working with the same Nedd9 knockout mice, Izumchenko at al. showed that, at the age of 1 year, 9 out of 11 Nedd9−/− mice and 8 out of 11 Nedd9+/− mice developed reactive lymphoid hyperplasia affecting solid tissues, in contrast to only 2 out of 11 of Nedd9+/+ animals. Further, Nedd9 knockout resulted in a minor decrease in the concentration of B cells, but increased macrophages in the peripheral blood and spleen of Nedd9−/− in comparison to Nedd9+/+ mice. These findings suggest that a long term deficiency of NEDD9-dependent signaling is sufficient to trigger pro-inflammatory and/or spontaneous pre-neoplastic changes in lymphoid and myeloid cells [56].
Besides Ph+ leukemias, NEDD9 expression and phosphorylation is elevated in Ph- hematologic malignancies, such as HTLV-associated adult T-cell leukemia/lymphoma [57]. HTLV (human T-cell leukemia/lymphoma virus) can transform T-cells via production of an oncogenic TAX protein. TAX promotes the transcription of viral proteins in the nucleus, and regulates many human genes by changing the activity of signaling pathways including CREB/ATF, NF-κB, AP-1 and SRF [58]. Iwata et al. showed that NEDD9 co-precipitates and co-localizes with TAX, and that TAX may induce expression and tyrosine phosphorylation of NEDD9. Exogenous NEDD9 inhibited TAX-mediated transactivation of NF-kB pathway, while dominant-negative NEDD9 mutants inhibited TAX mediated increase in motility [57]. NEDD9 overexpression in adult T-cell leukemia/lymphoma is of special interest, because NEDD9 could contribute into infiltrative properties of leukemic cells in this disease, which is characterized by extensive infiltration of multiple organs and tissues by T-cells. The authors of this study hypothesized that NEDD9 could be “a double-edged sword”, because suppression of NF-kB-mediated transcription could act against leukemogenesis, whereas enhancement of cell motility could contribute to the invasive nature of leukemic cells. It is possible that NEDD9-mediated suppression of NF-kB transactivation by TAX must be overcome during leukemogenesis. The time needed for this process to occur might account for a characteristic long period of latency from initial viral infection until the development of leukemia [57].
3.2 Breast cancer
A number of studies suggest elevation of NEDD9 expression contributes to the development, progression, and metastasis of breast cancer. NEDD9 is abundantly expressed in many breast cancer cell lines [32]. Overexpression of NEDD9 promotes migration and invasion of breast tumor cells [56, 59, 60] through mechanisms that include activating proteins of the focal adhesion complex (FAK and SRC) [56], mediating effects of TGFβ [61–63], increasing the expression and activity of matrix metalloproteases (MMPs) [59, 64], regulating trafficking and enzymatic recovery of MMPs and integrins [65, 66] increasing synthesis of tumor-associated glycocalyx [67], and down-regulating E-cadherin [60, 68] (Fig. 3). Negative regulation of E-cadherin is a fundamental event in epithelial-mesenchymal transition (EMT) prior to formation of metastases. NEDD9 activates EMT in part by increasing the expression of EMT-inducing transcription factors SNAIL and SLUG, which bind and repress the E-cadherin promoter [60]. Additionally, NEDD9 signals through SRC to promote E-cadherin removal from the cell membrane and lysosomal degradation [68]. In parallel, NEDD9 in complex with the DOCK3 guanine exchange factor (GEF) activates the RAC GTPase. This activates a WAVE/SCAR complex to trigger ARP2/3-dependent actin branching, lamellipodia formation, and activation of matrix metalloproteases (MMPs) [69].
Multiple in vivo models confirmed a functional role for NEDD9 in breast cancer invasion and metastasis. Izumchenko et al. investigated the effect of a Nedd9−/− genotype on mammary cancer initiation in the MMTV-polyoma virus middle T (PyVmT) antigen mouse model [56]. MMTV-expressed PyVmT antigen induces mammary cancers due to its binding and activation of the proteins SHC, SRC, and PI3K, which are central effectors of HER2 signaling [70]. Lack of NEDD9 increased the latency until tumor appearance, and slowed the growth rate of mammary tumors. This activity was likely tumor intrinsic, because the Nedd9 genotype did not influence immune cell infiltration or cause gross changes in the stroma or angiogenesis. Slow-growing MMTV-PyVmT;Nedd9−/− tumors that arose had decreased expression or activity of a number of proteins directly bound or indirectly regulated by NEDD9, including AKT, SRC, FAK, and ERK1/2 [56]. However, cell lines selected from these MMTV-PyVmT;Nedd9−/− tumor cells were more aggressive than MMTV-PyVmT;Nedd9+/+ cells, based on orthotopic xenograft and tail vein injections, and had elevated activity of AKT and ERK1/2, most likely due to the development of compensatory mutations or epigenetic changes [71]. Additionally, based on studies indicating NEDD9 is necessary for EMT [69], it is possible that the increased metastatic potential of MMTV-PyVmT;Nedd9−/− tumors in some mouse models may reflect the fact that tumor cells injected directly into the tail vein do not need to migrate through a rigid extracellular matrix surrounding breast primary tumor, where NEDD9 overexpression would be beneficial. In these settings, a switch from elongated cells (a mesenchymal-type mode of invasion) to amoeboid cells (a cell deforming mode of invasion) may actually promote lung colonization [69, 72].
In a subsequent study, using a different mouse model, Little et al. found that Nedd9−/− mice were remarkably resistant to MMTV-neu (HER2) induction of mammary tumors [73]. MMTV-HER2/neu;Nedd9−/− mice had a dramatic reduction in tumor incidence (18 versus 80%), and a significantly increased latency until tumor appearance. In this mouse model, there was a significant reduction of lung colonization following tail vein injection of MMTV-HER2/neu;Nedd9−/− tumor cells comparing to MMTV-HER2/neu;Nedd9+/+ cells [73]. The MMTV-neu;Nedd9−/− genotype reduced the number and colony-forming potential of mammary luminal epithelial progenitor cells, while not affecting basal epithelial progenitors. MMTV-neu;Nedd9−/− mammospheres had striking defects in morphology and cell polarity that were associated with depressed expression of FAK, and with increased sensitivity to small molecule inhibitors of FAK and SRC [73].
The reduced metastatic potential of breast tumors lacking NEDD9 was confirmed and extended in a study by McLoughlin et al. [64]. Reduction of NEDD9 expression by inducible short-hairpin RNAs (shRNA) in breast cancer xenograft models led to a drastic decrease in a number of circulating tumor cells, resulting in a decrease in the overall number and size of pulmonary metastases. This was mediated in part by inactivation of MMP14, and confirmed the tumor-intrinsic effect of loss of NEDD9 [64]. Consistent with this study, Loskutov et al. found that NEDD9 inhibition by an antisense therapy decreased primary tumor growth and metastasis in xenograft models of breast cancer, and showed that NEDD9 is required for MMP14 enzymatic recovery through the late endosomes (Fig. 3). Recovery of MMP14 enabled disengagement of tissue inhibitor of MMPs 2 (TIMP2) and tumor invasion [66]. Depletion of NEDD9 decreased targeting of the MMP14/TIMP2 complex to late endosomes and increases trafficking of MMP14/TIMP2 from early/sorting endosomes back to the surface in a small GTPase ADP ribosylation factor-6 (Arf6)-dependent manner. Re-expression of NEDD9 or a decrease in Arf6 activity restored MMP14 activity and the invasive properties of tumor cells [66]. Subsequent studies showed that NEDD9 function is required not only for the trafficking of MMP14, but also for the trafficking of ligand-bound integrins, likely through the inactivation of tyrosine phosphorylated caveolin-1 (CAV1) (Fig. 3). In the absence of NEDD9, the trafficking of ligand-bound integrins from early to late endosomes was impaired, resulting in a significant decrease in degradation of ligand/integrin complexes and an increase in CAV1-mediated recycling of ligand-bound integrins from early endosomes back to the plasma membrane without ligand disengagement, thus leading to low adhesion and migration of breast cancer cells [65].
NEDD9 also strongly influences TGFβ signaling in breast cancer [61–63]. NEDD9 potentiates the TGFβ signaling pathway by interacting with inhibitory SMADs (SMAD6 and SMAD7), abrogating their recruitment to TGFβ receptor, thereby enhancing signal transduction (Fig. 2) [63]. TGFβ increases breast tumor-initiating cell numbers in claudin-low breast cancer cell lines by orchestrating a specific gene signature enriched in stem cell processes. NEDD9 is necessary to mediate these TGFβ-specific effects through a positive feedback loop that integrates TGFβ/SMAD and Rho-actin-SRF-dependent signals (Fig. 3) [61].
Of considerable relevance to breast cancer biology, Bradshaw et al. found that estrogens regulate NEDD9 phosphorylation and scaffolding activity [74]. As noted above, 105 kDa NEDD9 is both tyrosine and serine phosphorylated and undergoes further modifications to produce a 115-kDa hyper-phosphorylated protein that is serine, threonine, and tyrosine phosphorylated [32, 34, 75], with integrin-mediated adhesion inducing accumulation of the 115-kDa form [33, 75, 76]. Bradshaw at colleagues showed that 105-kDa NEDD9 phospho-form was predominant in ER-positive versus ER-negative breast cancer cell lines. Levels of the 105-kDa NEDD9 phospho-form were significantly increased after 3 days of estrogen exposure due to a slower rate of protein decay. Exogenous expression of NEDD9 failed to induce cell spreading in the presence of estrogen, and this was reversed by tamoxifen treatment. The authors concluded that stabilization of 105-kDa NEDD9 may be inhibitory to NEDD9-dependent cell spreading, providing a link to a clinical phenotype of ER-positive breast cancers associated with better prognosis. Additional phosphorylation events that generate hyper-phosphorylated 115-kDa NEDD9 may be required for NEDD9-dependent cell spreading in ER-positive breast cancers [74].
Studies of NEDD9 expression in primary tumor samples showed frequent NEDD9 overexpression in breast tumors, and association of NEDD9 expression with more aggressive tumor phenotypes [60, 64, 77]. Kong et al. analyzed 20 breast primary tumor samples and demonstrated a statistically significant increase in NEDD9 mRNA expression in tumors, compared with their adjacent normal mammary tissues [60]. This group also evaluated NEDD9 protein expression in paraffin-embedded mammary tissue sections from 84 breast cancer patients in parallel with the surrounding normal breast epithelia. While normal mammary epithelial cells displayed no or weak NEDD9 staining, breast carcinoma cells were positive for NEDD9 staining in the cytoplasm and/or in nucleus. Additionally, NEDD9 expression was associated with adverse prognostic markers, including estrogen receptor (ER) negativity, a triple negative or HER2-positive(+) subtype, and high tumor grade: 31.82% of the triple negative tumors and 24.00% of Her2+ tumors had high levels of NEDD9 expression, whereas only 11.62% of the ER+ tumors showed high expression of NEDD9 protein [60]. NEDD9 staining in tumor tissue microarrays of 200 breast cancer cases positively correlated with disease progression. The lowest intensity was found in normal tissue, followed by a 10-fold increase in DCIS and a 30-fold increase in invasive ductal carcinoma [64]. Intriguingly, only a 10-fold increase in expression over normal tissue was observed in lymph node metastases, suggesting that high level of NEDD9 is selected during the invasion from the primary site, but that NEDD9 is either less important during growth at distant metastatic sites, or actively counter-selected, perhaps to support mesenchymal to epithelial transition (MET) by tumor cells, to establish a colony [60, 64].
Intriguingly, several studies of NEDD9 raise the possibility that under some conditions, loss of NEDD9 rather than its overexpression can induce features associated with tumor promotion in mammary tissue. For example, siRNA depletion of NEDD9 identified this gene as an inhibitor of migration in untransformed MCF10A breast epithelial cells [78]. As previously discussed, Nedd9 knockout was associated with increased number of lung metastases following tail vein injection of the tumor cells in some mouse models [71]. Down-regulation of the Nedd9 mRNA was a part of a transcriptional signature associated with enhanced metastasis to the lung in one TGFβ-associated mammary cancer model [79]. Significant overexpression of NEDD9 in the low invasive potential breast cancer cell line MCF-7 promotes migration and invasion [59], but also induces apoptosis and mitotic defects that trigger cell cycle arrest checkpoints [29, 36, 76, 80]. These data are compatible with the idea that cells must acquire prior genetic lesions that counteract NEDD9-dependent cell cycle arrest before NEDD9 is effective as a metastasis-promoting protein [48].
3.3 Lung Cancer
Somatic mutations of the LKB1 tumor suppressor gene, or loss of the region of chromosome 19p containing LKB1, are present in about one third of lung adenocarcinomas [81]. The tumor suppressor and anti-metastatic effect of normal LKB1 expression is exerted, in part, through down-regulation of NEDD9 expression [81–84]. Upregulation of NEDD9 was identified as a part of the genomic signature of LKB1 loss [83, 84]. LKB1 negatively regulates NEDD9 transcription by promoting cytosolic translocation of the NEDD9 transcriptional co-activator CRTC1 from the nucleus. Ectopic expression of either NEDD9 or CRTC1 partially reversed the inhibitory function of LKB1 on metastasis of lung cancer cells [82]. In mouse models, RNAi-mediated silencing of Nedd9 inhibited tumor progression of Lkb1-deficient lung tumors, whereas ectopic NEDD9 expression accelerated tumor growth.
In clinical specimens, elevated expression of NEDD9 was strongly associated with malignant progression, propensity to form metastases, and with decreased survival [82, 85, 86]. In a cohort of 175 human non-small cell lung carcinomas (NSCLC) high NEDD9 expression was strongly correlated with lymph node metastasis and advanced clinical stage, as well as with history of smoking and high tumor grade. No significant correlations were observed between NEDD9 expression and other clinical features, including NSCLC subtypes and tumor size [82]. In another study, the expression of NEDD9 was analyzed by immunohistochemistry (IHC) in 60 formalin-fixed and paraffin-embedded lung adenocarcinoma tissues. The immunostaining scores revealed a statistically significant upregulation of NEDD9 in metastatic comparing to non-metastatic lung adenocarcinomas (p<0.001) [85].
NEDD9 is thought to act as a trigger of the epithelial-mesenchymal transition (EMT) [69]. Loss of E-cadherin/beta-catenin and up-regulation of N-cadherin are hallmarks of the EMT, and are known to be influenced by NEDD9 [11, 68]. Miao at al. used IHC to evaluate expression of NEDD9 and its correlation with expression of E-cadherin, β-catenin and N-cadherin in 105 cases of non-small cell lung carcinoma and the corresponding normal lung tissues. NEDD9 was overexpressed in 56.2% of the NSCLC samples compared to normal lung tissue, and correlated with reduced membrane expression of E-cadherin and beta-catenin that was highly statistically significant. Additionally, overexpression of NEDD9 correlated positively with lymph node metastases. Notably, the mean overall survival of NSCLC patients overexpressing NEDD9 (39.10 +/− 6.49 months) was markedly shorter than patients with normal NEDD9 expression (56.67 +/− 7.44 months; Log-Rank, P = 0.001). In multivariate analysis, overexpression of NEDD9 (P = 0.013) and TNM stage (P = 0.001) were significant independent prognostic factors for reduced overall survival in NSCLC [86, 87].
3.4 Melanoma
A relationship between NEDD9 overexpression and metastatic potential is well documented in melanoma. Using an inducible mouse model of melanoma, Kim et al. characterized metastatic variants with an acquired focal chromosomal amplification that corresponds to a much larger amplification in human metastatic melanomas [88]. Within the amplified region, Nedd9 was the only gene that was consistently overexpressed (with or without amplification) in mouse metastatic melanoma models driven by either H-RAS or C-MET oncogene. NEDD9 knockdown reduced frequency of metastases in mouse xenografts, as well as reduced proliferation and inhibited invasion in melanoma cell lines. NEDD9 overexpression by itself failed to increase melanoma metastatic potential, but in conjunction with RAS overexpression resulted in enhanced invasion in Boyden Chamber by 3.4-fold, and significantly increased the number of metastases in mouse xenograft models [88]. In concordance with these results, Rozenberg et al. used an in vivo mouse model of metastasis to show that metastatic melanoma cell lines have high NEDD9 expression, but that NEDD9 lentiviral transfection and overexpression did not confer metastatic potential on non-metastatic cells [89]. These data are compatible with the observations in mammary cancer models, discussed above, indicating that NEDD9 overexpression is only pro-tumorigenic in the context of other lesions that remove checkpoints or reduce apoptosis.
In humans, gain of chromosome 6p, containing NEDD9 gene, is a frequent event in metastatic, but not primary melanomas [90]. Tumor microarray analysis demonstrated that NEDD9 expression is significantly higher in human metastatic melanoma samples comparing to primary melanomas, and level of NEDD9 expression in >50% of primary melanomas was higher than in benign nevi, indicating a selection for cells with NEDD9 overexpression in the process of tumorigenesis [88]. In addition to gain of the chromosome 6p, loss of function of LKB1 tumor suppressor leads to upregulation of NEDD9. Germline mutations of LKB1 are implicated in Peutz-Jeghers syndrome, which includes aberrant mucocutaneous pigmentation. Somatic LKB1 mutations occur in 10% of cutaneous melanoma, explaining overexpression of NEDD9 in these tumors [91].
3.5 Other malignances
NEDD9 overexpression is documented to occur and in some cases linked to the process of tumorigenesis of many different malignances, including colon [14, 15, 92, 93], liver, [12], pancreatic [94], ovarian [95, 96], prostate [18, 97], and kidney cancer [19], gastrointestinal stromal tumors [98], glioblastoma [17, 99, 100], and neuroblastoma [9, 10, 101].
High expression of NEDD9 predicts adverse outcomes of colorectal cancer patients. Analysis of NEDD9 expression by IHC in 92 patients with colorectal cancer showed high expression of NEDD9 in 68/92 colorectal cancer samples, compared with 12/92 normal tissues (P<0.01). High level of NEDD9 expression significantly correlated with advanced stage, tumor grade, lymph node metastases, and distant metastases. Patients with a higher NEDD9 expression had a significantly shorter overall survival (P<0.01). NEDD9 expression was an independent predictive factor of overall survival in multivariate analysis [92]. Kim et al. [14] found that NEDD9 mediates hypoxia-induced migration of colorectal carcinoma cells. NEDD9 is highly expressed in cultured colorectal carcinoma cells exposed to hypoxia, and in the hypoxic areas of human colorectal cancer specimens. Hypoxia-inducible factor-1α (HIF-1α) mediates the effects of hypoxia on induction of NEDD9 expression via binding to a hypoxia-responsive element of the NEDD9 promoter (Fig. 1A). Importantly, the induction of NEDD9 expression further enhances HIF-1α transcriptional activity by modulating the interaction between HIF-1α and its transcriptional cofactor p300. Decreased level of NEDD9 reduced the expression of hypoxia-inducible genes, including those that regulate cell motility, and led to dramatically reduced cell migration [14]. Xia et al. showed that in colorectal cancer cells, inflammatory mediator prostaglandin E(2) induces overexpression of NEDD9 and subsequent cell proliferation, cell cycle progression, and tumor growth, which are mediated by an activating interaction of NEDD9 with the mitotic kinase Aurora A (Fig. 4) [93]. Additionally, Li et al. identified NEDD9 as a novel WNT signaling target in colorectal cancer. Misregulation of the canonical WNT/β-catenin pathway and aberrant activation of WNT signaling target genes are common in colorectal cancer, and contribute to cancer progression. NEDD9 expression was upregulated by WNT-3a, β-catenin, and Dvl2 in a dose-dependent manner, and was suppressed following beta-catenin down-regulation by shRNA (Fig. 1A) [15].
In hepatocellular carcinoma (HCC) cells, NEDD9 expression is regulated by forkhead box C1 (FOXC1), a member of the Fox family of transcription factors (Fig. 1A). FOXC1 promotion of HCC metastasis through the induction of EMT and cell migration may depend on up-regulation of NEDD9 expression [12].
NEDD9 protein and mRNA levels are elevated in pancreatic carcinomas compared to adjacent noncancerous tissues [94]. High NEDD9 expression significantly correlates with clinical stage, lymph node metastasis status, and histologic grade. Pancreatic cancer patients with a higher NEDD9 expression had a significantly shorter survival time than those patients with lower NEDD9 expression, and multivariate analysis revealed that NEDD9 is an independent factor of poor prognosis [94].
In murine models of spontaneously arising ovarian cancer (MISIR-TAg), MISIR-Tag;Nedd9−/− mice exhibited delayed tumor development, decreased tumor burden, and reduced number of lung metastases comparing to MISIR-Tag;Nedd9+/+ mice. Additionally, the number of tumor infiltrating macrophages and NK-cells were decreased in MISIIR-Tag;Nedd9−/− mice [96]. Wang et al. analyzed NEDD9 expression in 129 archived epithelial ovarian cancer specimens by IHC, and in 28 freshly frozen specimens by Western blotting [95]. NEDD9 was overexpressed in ovarian cancer compared with noninvasive epithelial ovarian tumors and normal ovarian specimens. Overexpression of NEDD9 significantly correlated with advanced-stage, high tumor grade, and suboptimal primary cytoreductive surgery. Multivariate analysis indicated that NEDD9 overexpression (P = 0.033), advanced stage (P < 0.001), and high-grade carcinoma (P = 0.01) were independent predictors of poor survival [95].
NEDD9 promotes migration, invasion and EMT of the PC-3 cell line, derived from a prostate cancer bone metastasis [18]. Both NEDD9 and its canonical effectors associated with EMT are strongly induced by TGFβ in prostate cancer cells. Prostate cancer cell lines with stable overexpression of NEDD9 have a mesenchymal phenotype and significantly enhanced cell invasion, while knockdown of endogenous NEDD9 expression diminishes TGFβ-triggered tumor invasion. The level of NEDD9 expression in primary prostate tumors, measured by IHC, positively correlates with Gleason score, serum PSA level, and presence of bone metastases [97]. Studies in prostate cancer, renal cell carcinoma (RCC) and glioblastoma have identified NEDD9 as a target of the micro-RNA miR-145, an important repressor of pluripotency of embryonic stem cells and a tumor suppressor in different cancers, and showed decreased level of miR-145 and reciprocal increase of NEDD9 expression in tumor samples compared to normal tissue (Fig. 1A) [17–19].
NEDD9 overexpression is implicated in the pathogenesis of central and peripheral nervous system malignant tumors. NEDD9 is a necessary and specific downstream effector of FAK that promotes highly invasive behavior of glioblastoma cells [100]. In neuroblastoma cells NEDD9 depletion by siRNAs significantly decreased cellular migration in a 3D migration assay, regardless of whether these cells have undergone a Rac1-dependent conversion to a mesenchymal morphology [101]. Studies in the human neuroblastoma cell line SH-SY5Y identified a putative retinoic acid response element in the 5′ region of the NEDD9 promoter (Fig. 1A). Regulation of NEDD9 may be an important means whereby all-trans retinoic acid promotes cell spreading and neurite outgrowth in SH-SY5Y human neuroblastoma cells [9, 10].
4. NEDD9 in other human pathological conditions
Although at a more nascent stage than studies of NEDD9 in cancer, a number of studies have implicated function of this protein as important in several other disease states. The mechanistic insights yielded in the study of these diseases may ultimately contribute to the understanding of NEDD9 function in cancer. In brief summary, the upregulation of NEDD9 by loss of VHL and by hypoxia detected in colon cancer and kidney tissue has implications for other conditions associated with hypoxia, including notable stroke. Nedd9 is upregulated in the neurons of the cerebral cortex and hippocampus after transient global ischemia in rats. Induced Nedd9 is tyrosine phosphorylated, bound to FAK in dendrite and soma of neurons, and promotes neurite outgrowth, contributing to recovery of neurologic function after cerebral ischemia [102]. A recent study found that NEDD9 expression is elevated in human autosomal dominant polycystic kidney disease (ADPKD) and in mouse ADPKD models. Although genetic ablation of Nedd9 does not independently influence cystogenesis, constitutive absence of Nedd9 strongly promotes cyst formation in a mouse model of ADPKD. ADPKD arises from defective signaling by mutated forms of the ciliary proteins PKD1 and PKD2, resulting in defective intracellular calcium homeostasis, and other signaling defects [103]. The biological activities of NEDD9 in ADPKD reflect of its activation of the Aurora-A kinase, which induces resorption of cilia, limiting this defective signaling (Fig. 4) [104]. In addition, several recent studies implicate NEDD9 and Aurora-A with regulation by and regulation of calcium/calmodulin signaling, not only in ciliary signaling, but also in control of mitosis, which may inform the oncogenic activity of NEDD9 and Aurora-A [105–107].
In addition, NEDD9 participates in diverse processes related to function of the immune system, such as T-cell receptor signaling, autoimmunity, chemotaxis, lymphocyte adhesion, and response to viral infections. Studies of NEDD9 in the immune system provide a useful complement to consideration of NEDD9 function in adherent cell types; these are discussed in greater depth in a review by Seo and colleagues [108]. In brief summary, NEDD9 was independently discovered as a protein highly phosphorylated following ligation of the β1 integrin receptor in T cells {Minegishi, 1996 #22}. Tyrosine phosphorylated NEDD9 interacts with Crk and C3G to mediate signal transduction from β1 integrin receptor {Ohashi, 1998 #192}, resulting in T cell activation and IL-2 production by human peripheral T cells [109]. The HTLV-1 Tax protein interacts with and induces NEDD9, connecting the protein to viral leukemogenesis {Iwata, 2005 #103}. Similar signaling functions have been identified in B cells and other lymphoid lineages {Astier, 1997 #28;Manie, 1997 #24}{Hunter, 1997 #25}. NEDD9 plays an important role in lymphocyte movement, chemotaxis, and cell adhesion, influencing functions of both T and B lymphocytes, with NEDD9 required for lymphocyte trafficking and marginal zone B cell maintenance [38]. Phosphorylation of NEDD9 by ABL kinases and activation of Rap1 are important for these processes {Gu, 2012 #134}, as well as the integrin/Crk signaling pathways mentioned above. Finally, as an intracellular protein, NEDD9 is not a suitable target for cancer immunotherapy; however, given its role in modulating immune system maturation, and antigen response or tolerance, NEDD9 expression may have a function in predicting immune response.
5. NEDD9 and cancer therapy
Because of its roles in cancer, several studies have considered the potential value of NEDD9 as a therapeutic target, or therapeutic guide. Because of lack of a kinase domain, or any defined catalytic domain, and because it is entirely intracellular, NEDD9 is a difficult molecule to target. In pre-clinical models, NEDD9 has typically been inhibited by specific siRNAs or shRNAs; however, delivery of small interfering RNAs in humans is currently problematic (as reviewed in [110]). However, because NEDD9 serves as a scaffolding molecule for other signaling proteins that play significant roles in cancer development, the effects of NEDD9 overexpression in supporting metastasis could in theory be mitigated by inhibition of its upstream regulators or downstream targets. Inhibition of FAK and SRC has been suggested [73], as well as targeting MMPs [64] or removing chondroitin sulfate E by chondroitinase ABC [67]. Conversely, NEDD9 expression levels could influence response to drugs targeting its partners. In one study, deletion of Nedd9 in MMTV-neu mammary tumors increased their sensitivity to inhibitors of FAK and SRC [73]. NEDD9 binding stabilizes Aurora A, and protects it from ubiquitination and proteasome-dependent degradation (Fig. 4) [37]. NEDD9 depletion sensitizes breast tumor cell lines to the Aurora A inhibitor alisertib, decreasing its IC50 from 200 nmol/L to 20 nmol/L. In a breast cancer mouse xenograft model, combined treatment with siRNA to Nedd9 and alisertib was more effective than either treatment alone in reducing tumor burden and the number of lung metastases [37]. As NEDD9 depletion is synergistic with Aurora A inhibition in in vitro and in vivo experiments, NEDD9 overexpression, common in solid tumors, could confer resistance to small molecule inhibitors of Aurora A.
Beyond direct partners, NEDD9 is an intermediary in signaling cascades for integrins, multiple receptor tyrosine kinases (RTKs) and other transmembrane receptors. NEDD9 overexpression is implicated in resistance of gastrointestinal stromal tumors (GISTs) to imatinib, an inhibitor of the RTK KIT [98]. In imatinib-resistant GIST cell lines, DNA microarray analysis showed that resistant cells did not acquire new mutations in KIT, PDGFRA, PKC or JAK2, but expression of NEDD9 in these cells was more than 500 times higher than in parental cells. Silencing of NEDD9 with siRNA in resistant cells restored sensitivity to imatinib [98]. Together, these studies emphasize the value of analyzing NEDD9 expression in consideration of therapeutic strategy.
6. Conclusion
In summary, study of NEDD9 over nearly 2 decades has now elucidated a pleiotropic activity for this protein, that is likely important for maintaining the homeostasis of normal cells and tissues. As is evident from the preclinical data summarized above, there is strong support for the idea that the level of NEDD9 expression influences the ability of tumors to acquire malignant characteristics. The recent emergence of preclinical data linking NEDD9 expression to additional pathological conditions, including stroke, ADPKD, and potentially some neurodegenerative diseases open new areas for exploration. In the future, it will be of considerable interest to extend these analyses into the clinical arena. Although direct therapeutic targeting of NEDD9 remains on the more distant horizon, at the present time ample evidence suggests that measurement of NEDD9 expression levels in tumors will be a valuable addition to genomic biomarkers, to predict resistance versus susceptibility to drugs targeting its partner proteins. While there is already evidence that this is of value for specific cancer therapies (to inhibitors of SRC, FAK, or AURKA), a broader relevance to inhibition of proteins more distantly connected to NEDD9, or to standard cytotoxic therapies, are topics of considerable interest for further investigation.
Highlights.
The NEDD9 scaffold protein assembles protein complexes that drive tumorigenesis
NEDD9 signaling supports invasion, survival, ciliary resorption, and mitosis
NEDD9 expression influences tumor metastasis and therapeutic response
NEDD9 is a promising biomarker for therapeutic resistance
Acknowledgments
This review and a corresponding Gene Wiki article are written as part of the Gene Wiki Review series--a series resulting from a collaboration between the journal GENE and the Gene Wiki Initiative. The Gene Wiki Initiative is supported by National Institutes of Health (GM083924). Additional support for Gene Wiki Reviews is provided by Elsevier, the publisher of GENE. The authors were supported by R21 CA181287 and an award from the PKD Foundation (to E.A.G.); subsidy of the Russian Government to the Program of competitive growth of Kazan Federal University (to R.G); and by NIH Core Grant CA06927 (to Fox Chase Cancer Center).
Footnotes
The corresponding Gene Wiki entry for this review can be found here: https://en.wikipedia.org/wiki/NEDD9
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Kumar S, Tomooka Y, Noda M. Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem Biophys Res Commun. 1992;185(3):1155–61. doi: 10.1016/0006-291x(92)91747-e. [DOI] [PubMed] [Google Scholar]
- 2.Law SF, et al. Human enhancer of filamentation 1, a novel p130cas-like docking protein, associates with focal adhesion kinase and induces pseudohyphal growth in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16(7):3327–37. doi: 10.1128/mcb.16.7.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Minegishi M, et al. Structure and function of Cas-L, a 105-kD Crk-associated substrate-related protein that is involved in beta 1 integrin-mediated signaling in lymphocytes. J Exp Med. 1996;184(4):1365–75. doi: 10.1084/jem.184.4.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tikhmyanova N, Little JL, Golemis EA. CAS proteins in normal and pathological cell growth control. Cell Mol Life Sci. 2010;67(7):1025–48. doi: 10.1007/s00018-009-0213-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nikonova AS, et al. CAS proteins in health and disease: an update. IUBMB Life. 2014;66(6):387–95. doi: 10.1002/iub.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Neill GM, Fashena SJ, Golemis EA. Integrin signalling: a new Cas(t) of characters enters the stage. Trends Cell Biol. 2000;10(3):111–9. doi: 10.1016/s0962-8924(99)01714-6. [DOI] [PubMed] [Google Scholar]
- 7.O’Neill GM, et al. A new central scaffold for metastasis: parsing HEF1/Cas-L/NEDD9. Cancer Res. 2007;67(19):8975–9. doi: 10.1158/0008-5472.CAN-07-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knutson DC, Clagett-Dame M. A complex RARE is required for the majority of Nedd9 embryonic expression. Transgenic Res. 2015;24(1):123–34. doi: 10.1007/s11248-014-9825-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Merrill RA, et al. All-trans retinoic acid-responsive genes identified in the human SH-SY5Y neuroblastoma cell line and their regulated expression in the nervous system of early embryos. Biol Chem. 2004;385(7):605–14. doi: 10.1515/BC.2004.075. [DOI] [PubMed] [Google Scholar]
- 10.Merrill RA, et al. Crk-associated substrate (Cas) family member, NEDD9, is regulated in human neuroblastoma cells and in the embryonic hindbrain by all-trans retinoic acid. Dev Dyn. 2004;231(3):564–75. doi: 10.1002/dvdy.20159. [DOI] [PubMed] [Google Scholar]
- 11.Bui LC, et al. Nedd9/Hef1/Cas-L mediates the effects of environmental pollutants on cell migration and plasticity. Oncogene. 2009;28(41):3642–51. doi: 10.1038/onc.2009.224. [DOI] [PubMed] [Google Scholar]
- 12.Xia L, et al. Overexpression of forkhead box C1 promotes tumor metastasis and indicates poor prognosis in hepatocellular carcinoma. Hepatology. 2013;57(2):610–24. doi: 10.1002/hep.26029. [DOI] [PubMed] [Google Scholar]
- 13.Aquino JB, et al. Differential expression and dynamic changes of murine NEDD9 in progenitor cells of diverse tissues. Gene Expr Patterns. 2008;8(4):217–26. doi: 10.1016/j.gep.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 14.Kim SH, et al. Human enhancer of filamentation 1 Is a mediator of hypoxia-inducible factor-1alpha-mediated migration in colorectal carcinoma cells. Cancer Res. 2010;70(10):4054–63. doi: 10.1158/0008-5472.CAN-09-2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, et al. HEF1, a novel target of Wnt signaling, promotes colonic cell migration and cancer progression. Oncogene. 2011;30(23):2633–43. doi: 10.1038/onc.2010.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammerich-Hille S, et al. SAFB1 mediates repression of immune regulators and apoptotic genes in breast cancer cells. J Biol Chem. 2010;285(6):3608–16. doi: 10.1074/jbc.M109.066431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Speranza MC, et al. NEDD9, a novel target of miR-145, increases the invasiveness of glioblastoma. Oncotarget. 2012;3(7):723–34. doi: 10.18632/oncotarget.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo W, et al. HEF1 promotes epithelial mesenchymal transition and bone invasion in prostate cancer under the regulation of microRNA-145. J Cell Biochem. 2013;114(7):1606–15. doi: 10.1002/jcb.24502. [DOI] [PubMed] [Google Scholar]
- 19.Lu R, et al. miR-145 functions as tumor suppressor and targets two oncogenes, ANGPT2 and NEDD9, in renal cell carcinoma. J Cancer Res Clin Oncol. 2014;140(3):387–97. doi: 10.1007/s00432-013-1577-z. [DOI] [PubMed] [Google Scholar]
- 20.Malleter M, et al. A novel large regulator RNA, B2, partially overlaps the HEF1/NEDD9/Cas-L gene. Int J Mol Med. 2010;25(6):897–903. doi: 10.3892/ijmm_00000420. [DOI] [PubMed] [Google Scholar]
- 21.Chapuis J, et al. Association study of the NEDD9 gene with the risk of developing Alzheimer’s and Parkinson’s disease. Hum Mol Genet. 2008;17(18):2863–7. doi: 10.1093/hmg/ddn183. [DOI] [PubMed] [Google Scholar]
- 22.Li Y, et al. Evidence that common variation in NEDD9 is associated with susceptibility to late-onset Alzheimer’s and Parkinson’s disease. Hum Mol Genet. 2008;17(5):759–67. doi: 10.1093/hmg/ddm348. [DOI] [PubMed] [Google Scholar]
- 23.Tedde A, et al. Different implication of NEDD9 genetic variant in early and late-onset Alzheimer’s disease. Neurosci Lett. 2010;477(3):121–3. doi: 10.1016/j.neulet.2010.04.046. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, et al. NEDD9 rs760678 polymorphism and the risk of Alzheimer’s disease: a meta-analysis. Neurosci Lett. 2012;527(2):121–5. doi: 10.1016/j.neulet.2012.08.044. [DOI] [PubMed] [Google Scholar]
- 25.Xing YY, et al. NEDD9 is genetically associated with Alzheimer’s disease in a Han Chinese population. Brain Res. 2011;1369:230–4. doi: 10.1016/j.brainres.2010.10.113. [DOI] [PubMed] [Google Scholar]
- 26.Beck T, Nicolas E, Kopp M, Golemis E. Adaptors for disorders of the brain? The cancer signaling proteins NEDD9, CASS4, and PTK2B in Alzheimer’s disease. Oncoscience. 2014;1(7):486–503. doi: 10.18632/oncoscience.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallez Y, et al. NSP-CAS Protein Complexes: Emerging Signaling Modules in Cancer. Genes Cancer. 2012;3(5–6):382–93. doi: 10.1177/1947601912460050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Briknarova K, et al. The serine-rich domain from Crk-associated substrate (p130cas) is a four-helix bundle. J Biol Chem. 2005;280(23):21908–14. doi: 10.1074/jbc.M501258200. [DOI] [PubMed] [Google Scholar]
- 29.Law SF, et al. The docking protein HEF1 is an apoptotic mediator at focal adhesion sites. Mol Cell Biol. 2000;20(14):5184–95. doi: 10.1128/mcb.20.14.5184-5195.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pugacheva EN, Golemis EA. The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nat Cell Biol. 2005;7(10):937–46. doi: 10.1038/ncb1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pugacheva EN, et al. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell. 2007;129(7):1351–63. doi: 10.1016/j.cell.2007.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Law SF, et al. Cell cycle-regulated processing of HEF1 to multiple protein forms differentially targeted to multiple subcellular compartments. Mol Cell Biol. 1998;18(6):3540–51. doi: 10.1128/mcb.18.6.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng M, McKeown-Longo PJ. Cell adhesion regulates Ser/Thr phosphorylation and proteasomal degradation of HEF1. J Cell Sci. 2006;119(Pt 1):96–103. doi: 10.1242/jcs.02712. [DOI] [PubMed] [Google Scholar]
- 34.Liu X, et al. A novel ability of Smad3 to regulate proteasomal degradation of a Cas family member HEF1. EMBO J. 2000;19(24):6759–69. doi: 10.1093/emboj/19.24.6759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nourry C, et al. Direct interaction between Smad3, APC10, CDH1 and HEF1 in proteasomal degradation of HEF1. BMC Cell Biol. 2004;5:20. doi: 10.1186/1471-2121-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dadke D, et al. Deregulation of HEF1 impairs M-phase progression by disrupting the RhoA activation cycle. Mol Biol Cell. 2006;17(3):1204–17. doi: 10.1091/mbc.E05-03-0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ice RJ, et al. NEDD9 depletion destabilizes Aurora A kinase and heightens the efficacy of Aurora A inhibitors: implications for treatment of metastatic solid tumors. Cancer Res. 2013;73(10):3168–80. doi: 10.1158/0008-5472.CAN-12-4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seo S, et al. Crk-associated substrate lymphocyte type is required for lymphocyte trafficking and marginal zone B cell maintenance. J Immunol. 2005;175(6):3492–501. doi: 10.4049/jimmunol.175.6.3492. [DOI] [PubMed] [Google Scholar]
- 39.Ohashi Y, et al. Tyrosine phosphorylation of Crk-associated substrate lymphocyte-type is a critical element in TCR- and beta 1 integrin-induced T lymphocyte migration. J Immunol. 1999;163(7):3727–34. [PubMed] [Google Scholar]
- 40.Manie SN, et al. Involvement of p130(Cas) and p105(HEF1), a novel Cas-like docking protein, in a cytoskeleton-dependent signaling pathway initiated by ligation of integrin or antigen receptor on human B cells. J Biol Chem. 1997;272(7):4230–6. doi: 10.1074/jbc.272.7.4230. [DOI] [PubMed] [Google Scholar]
- 41.Gu JJ, et al. Abl family kinases modulate T cell-mediated inflammation and chemokine-induced migration through the adaptor HEF1 and the GTPase Rap1. Sci Signal. 2012;5(233):ra51. doi: 10.1126/scisignal.2002632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Astier A, et al. Association of the Cas-like molecule HEF1 with CrkL following integrin and antigen receptor signaling in human B-cells: potential relevance to neoplastic lymphohematopoietic cells. Leuk Lymphoma. 1997;28(1–2):65–72. doi: 10.3109/10428199709058332. [DOI] [PubMed] [Google Scholar]
- 43.Ohashi Y, et al. T cell receptor-mediated tyrosine phosphorylation of Cas-L, a 105-kDa Crk-associated substrate-related protein, and its association of Crk and C3G. J Biol Chem. 1998;273(11):6446–51. doi: 10.1074/jbc.273.11.6446. [DOI] [PubMed] [Google Scholar]
- 44.van Seventer GA, et al. Focal adhesion kinase regulates beta1 integrin-dependent T cell migration through an HEF1 effector pathway. Eur J Immunol. 2001;31(5):1417–27. doi: 10.1002/1521-4141(200105)31:5<1417::AID-IMMU1417>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 45.Moore FE, et al. The WW-HECT protein Smurf2 interacts with the Docking Protein NEDD9/HEF1 for Aurora A activation. Cell Div. 2010;5:22. doi: 10.1186/1747-1028-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumar S, et al. Crk at the quarter century mark: perspectives in signaling and cancer. J Cell Biochem. 2014;115(5):819–25. doi: 10.1002/jcb.24749. [DOI] [PubMed] [Google Scholar]
- 47.Feller SM, et al. Physiological signals and oncogenesis mediated through Crk family adapter proteins. J Cell Physiol. 1998;177(4):535–52. doi: 10.1002/(SICI)1097-4652(199812)177:4<535::AID-JCP5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 48.Singh M, et al. Molecular basis for HEF1/NEDD9/Cas-L action as a multifunctional co-ordinator of invasion, apoptosis and cell cycle. Cell Biochem Biophys. 2007;48(1):54–72. doi: 10.1007/s12013-007-0036-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Jong R, et al. BCR/ABL-induced leukemogenesis causes phosphorylation of Hef1 and its association with Crkl. J Biol Chem. 1997;272(51):32649–55. doi: 10.1074/jbc.272.51.32649. [DOI] [PubMed] [Google Scholar]
- 50.Titz B, et al. The proximal signaling network of the BCR-ABL1 oncogene shows a modular organization. Oncogene. 2010;29(44):5895–910. doi: 10.1038/onc.2010.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sattler M, et al. Differential signaling after beta1 integrin ligation is mediated through binding of CRKL to p120(CBL) and p110(HEF1) J Biol Chem. 1997;272(22):14320–6. doi: 10.1074/jbc.272.22.14320. [DOI] [PubMed] [Google Scholar]
- 52.Ota J, et al. Association of CrkL with STAT5 in hematopoietic cells stimulated by granulocyte-macrophage colony-stimulating factor or erythropoietin. Biochem Biophys Res Commun. 1998;252(3):779–86. doi: 10.1006/bbrc.1998.9445. [DOI] [PubMed] [Google Scholar]
- 53.Oda A, et al. CrkL is an adapter for Wiskott-Aldrich syndrome protein and Syk. Blood. 2001;97(9):2633–9. doi: 10.1182/blood.v97.9.2633. [DOI] [PubMed] [Google Scholar]
- 54.Guerrero MS, Parsons JT, Bouton AH. Cas and NEDD9 Contribute to Tumor Progression through Dynamic Regulation of the Cytoskeleton. Genes Cancer. 2012;3(5–6):371–81. doi: 10.1177/1947601912458585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seo S, et al. Crk-associated substrate lymphocyte type regulates myeloid cell motility and suppresses the progression of leukemia induced by p210Bcr/Abl. Cancer Sci. 2011;102(12):2109–17. doi: 10.1111/j.1349-7006.2011.02066.x. [DOI] [PubMed] [Google Scholar]
- 56.Izumchenko E, et al. NEDD9 promotes oncogenic signaling in mammary tumor development. Cancer Res. 2009;69(18):7198–206. doi: 10.1158/0008-5472.CAN-09-0795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iwata S, et al. HTLV-I Tax induces and associates with Crk-associated substrate lymphocyte type (Cas-L) Oncogene. 2005;24(7):1262–71. doi: 10.1038/sj.onc.1208261. [DOI] [PubMed] [Google Scholar]
- 58.Jeang KT, et al. Life, death, and tax: role of HTLV-I oncoprotein in genetic instability and cellular transformation. J Biol Chem. 2004;279(31):31991–4. doi: 10.1074/jbc.R400009200. [DOI] [PubMed] [Google Scholar]
- 59.Fashena SJ, et al. Dissection of HEF1-dependent functions in motility and transcriptional regulation. J Cell Sci. 2002;115(Pt 1):99–111. doi: 10.1242/jcs.115.1.99. [DOI] [PubMed] [Google Scholar]
- 60.Kong C, et al. NEDD9 is a positive regulator of epithelial-mesenchymal transition and promotes invasion in aggressive breast cancer. PLoS One. 2011;6(7):e22666. doi: 10.1371/journal.pone.0022666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bruna A, et al. TGFbeta induces the formation of tumour-initiating cells in claudinlow breast cancer. Nat Commun. 2012;3:1055. doi: 10.1038/ncomms2039. [DOI] [PubMed] [Google Scholar]
- 62.Giampieri S, et al. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol. 2009;11(11):1287–96. doi: 10.1038/ncb1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Inamoto S, et al. Crk-associated substrate lymphocyte type regulates transforming growth factor-beta signaling by inhibiting Smad6 and Smad7. Oncogene. 2007;26(6):893–904. doi: 10.1038/sj.onc.1209848. [DOI] [PubMed] [Google Scholar]
- 64.McLaughlin SL, et al. NEDD9 depletion leads to MMP14 inactivation by TIMP2 and prevents invasion and metastasis. Mol Cancer Res. 2014;12(1):69–81. doi: 10.1158/1541-7786.MCR-13-0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kozyulina PY, et al. Pro-metastatic NEDD9 regulates individual cell migration via caveolin-1-dependent trafficking of integrins. Mol Cancer Res. 2014 doi: 10.1158/1541-7786.MCR-14-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Loskutov YV, et al. NEDD9/Arf6-dependent endocytic trafficking of matrix metalloproteinase 14: a novel mechanism for blocking mesenchymal cell invasion and metastasis of breast cancer. Oncogene. 2014 doi: 10.1038/onc.2014.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Iida J, et al. Role for chondroitin sulfate glycosaminoglycan in NEDD9-mediated breast cancer cell growth. Exp Cell Res. 2014 doi: 10.1016/j.yexcr.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 68.Tikhmyanova N, Golemis EA. NEDD9 and BCAR1 negatively regulate E-cadherin membrane localization, and promote E-cadherin degradation. PLoS One. 2011;6(7):e22102. doi: 10.1371/journal.pone.0022102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sanz-Moreno V, et al. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135(3):510–23. doi: 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 70.Dilworth SM. Polyoma virus middle T antigen and its role in identifying cancer-related molecules. Nat Rev Cancer. 2002;2(12):951–6. doi: 10.1038/nrc946. [DOI] [PubMed] [Google Scholar]
- 71.Singh MK, et al. Enhanced genetic instability and dasatinib sensitivity in mammary tumor cells lacking NEDD9. Cancer Res. 2010;70(21):8907–16. doi: 10.1158/0008-5472.CAN-10-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bradbury P, Fabry B, O’Neill GM. Occupy tissue: the movement in cancer metastasis. Cell Adh Migr. 2012;6(5):424–32. doi: 10.4161/cam.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Little JL, et al. A requirement for Nedd9 in luminal progenitor cells prior to mammary tumorigenesis in MMTV-HER2/ErbB2 mice. Oncogene. 2014;33(4):411–20. doi: 10.1038/onc.2012.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bradshaw LN, et al. Estradiol stabilizes the 105-kDa phospho-form of the adhesion docking protein NEDD9 and suppresses NEDD9-dependent cell spreading in breast cancer cells. Biochim Biophys Acta. 2011;1813(2):340–5. doi: 10.1016/j.bbamcr.2010.11.018. [DOI] [PubMed] [Google Scholar]
- 75.Zheng M, McKeown-Longo PJ. Regulation of HEF1 expression and phosphorylation by TGF-beta 1 and cell adhesion. J Biol Chem. 2002;277(42):39599–608. doi: 10.1074/jbc.M202263200. [DOI] [PubMed] [Google Scholar]
- 76.O’Neill GM, Golemis EA. Proteolysis of the docking protein HEF1 and implications for focal adhesion dynamics. Mol Cell Biol. 2001;21(15):5094–108. doi: 10.1128/MCB.21.15.5094-5108.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stajduhar E, et al. Expression of growth hormone receptor, plakoglobin and NEDD9 protein in association with tumour progression and metastasis in human breast cancer. Tumour Biol. 2014;35(7):6425–34. doi: 10.1007/s13277-014-1827-y. [DOI] [PubMed] [Google Scholar]
- 78.Simpson KJ, et al. Identification of genes that regulate epithelial cell migration using an siRNA screening approach. Nat Cell Biol. 2008;10(9):1027–38. doi: 10.1038/ncb1762. [DOI] [PubMed] [Google Scholar]
- 79.Minn AJ, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436(7050):518–24. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pugacheva EN, Golemis EA. HEF1-aurora A interactions: points of dialog between the cell cycle and cell attachment signaling networks. Cell Cycle. 2006;5(4):384–91. doi: 10.4161/cc.5.4.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sanchez-Cespedes M, et al. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002;62(13):3659–62. [PubMed] [Google Scholar]
- 82.Feng Y, et al. The CRTC1-NEDD9 signaling axis mediates lung cancer progression caused by LKB1 loss. Cancer Res. 2012;72(24):6502–11. doi: 10.1158/0008-5472.CAN-12-1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Carretero J, et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell. 2010;17(6):547–59. doi: 10.1016/j.ccr.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li L, et al. Synergistic effects of eukaryotic coexpression plasmid carrying LKB1 and FUS1 genes on lung cancer in vitro and in vivo. J Cancer Res Clin Oncol. 2014;140(6):895–907. doi: 10.1007/s00432-014-1607-5. [DOI] [PubMed] [Google Scholar]
- 85.Chang JX, et al. Role of NEDD9 in invasion and metastasis of lung adenocarcinoma. Exp Ther Med. 2012;4(5):795–800. doi: 10.3892/etm.2012.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Miao Y, et al. Overexpression of NEDD9 is associated with altered expression of E-Cadherin, beta-Catenin and N-Cadherin and predictive of poor prognosis in non-small cell lung cancer. Pathol Oncol Res. 2013;19(2):281–6. doi: 10.1007/s12253-012-9580-2. [DOI] [PubMed] [Google Scholar]
- 87.Jin Y, et al. NEDD9 promotes lung cancer metastasis through epithelial-mesenchymal transition. Int J Cancer. 2014;134(10):2294–304. doi: 10.1002/ijc.28568. [DOI] [PubMed] [Google Scholar]
- 88.Kim M, et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006;125(7):1269–81. doi: 10.1016/j.cell.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 89.Rozenberg GI, et al. Metastasis in an orthotopic murine model of melanoma is independent of RAS/RAF mutation. Melanoma Res. 2010;20(5):361–71. doi: 10.1097/CMR.0b013e328336ee17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bastian BC, et al. Chromosomal gains and losses in primary cutaneous melanomas detected by comparative genomic hybridization. Cancer Res. 1998;58(10):2170–5. [PubMed] [Google Scholar]
- 91.Liu W, et al. LKB1/STK11 inactivation leads to expansion of a prometastatic tumor subpopulation in melanoma. Cancer Cell. 2012;21(6):751–64. doi: 10.1016/j.ccr.2012.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li P, et al. High expression of NEDD9 predicts adverse outcomes of colorectal cancer patients. Int J Clin Exp Pathol. 2014;7(5):2565–70. [PMC free article] [PubMed] [Google Scholar]
- 93.Xia D, et al. HEF1 is a crucial mediator of the proliferative effects of prostaglandin E(2) on colon cancer cells. Cancer Res. 2010;70(2):824–31. doi: 10.1158/0008-5472.CAN-09-2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xue YZ, et al. Expression of NEDD9 in pancreatic ductal adenocarcinoma and its clinical significance. Tumour Biol. 2013;34(2):895–9. doi: 10.1007/s13277-012-0624-8. [DOI] [PubMed] [Google Scholar]
- 95.Wang H, et al. NEDD9 overexpression is associated with the progression of and an unfavorable prognosis in epithelial ovarian cancer. Hum Pathol. 2014;45(2):401–8. doi: 10.1016/j.humpath.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 96.Gabbasov R, Bickel L, O’Brien S, Litwin S, Seo S, Golemis E, Connolly D. NEDD9 expression promotes epithelial ovarian cancer growth and dissemination. Proceedings of the 105th Annual Meeting of the American Association for Cancer Research. 2014;74(19 Suppl) [Google Scholar]
- 97.Morimoto K, et al. NEDD9 crucially regulates TGF-beta-triggered epithelial-mesenchymal transition and cell invasion in prostate cancer cells: involvement in cancer progressiveness. Prostate. 2014;74(8):901–10. doi: 10.1002/pros.22809. [DOI] [PubMed] [Google Scholar]
- 98.Thao le B, et al. Cas-L was overexpressed in imatinib-resistant gastrointestinal stromal tumor cells. Cancer Biol Ther. 2009;8(8):683–8. doi: 10.4161/cbt.8.8.7779. [DOI] [PubMed] [Google Scholar]
- 99.Ismail HM. Overexpression of s6 kinase 1 in brain tumours is associated with induction of hypoxia-responsive genes and predicts patients’ survival. J Oncol. 2012;2012:416927. doi: 10.1155/2012/416927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Natarajan M, et al. HEF1 is a necessary and specific downstream effector of FAK that promotes the migration of glioblastoma cells. Oncogene. 2006;25(12):1721–32. doi: 10.1038/sj.onc.1209199. [DOI] [PubMed] [Google Scholar]
- 101.Zhong J, et al. NEDD9 regulates 3D migratory activity independent of the Rac1 morphology switch in glioma and neuroblastoma. Mol Cancer Res. 2014;12(2):264–73. doi: 10.1158/1541-7786.MCR-13-0513. [DOI] [PubMed] [Google Scholar]
- 102.Sasaki T, et al. Nedd9 protein, a Cas-L homologue, is upregulated after transient global ischemia in rats: possible involvement of Nedd9 in the differentiation of neurons after ischemia. Stroke. 2005;36(11):2457–62. doi: 10.1161/01.STR.0000185672.10390.30. [DOI] [PubMed] [Google Scholar]
- 103.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76(2):149–68. doi: 10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nikonova AS, et al. Nedd9 restrains renal cystogenesis in Pkd1−/− mice. Proc Natl Acad Sci U S A. 2014;111(35):12859–64. doi: 10.1073/pnas.1405362111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Plotnikova OV, Pugacheva EN, Golemis EA. Aurora A kinase activity influences calcium signaling in kidney cells. J Cell Biol. 2011;193(6):1021–32. doi: 10.1083/jcb.201012061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Plotnikova OV, et al. Rapid calcium-dependent activation of Aurora-A kinase. Nat Commun. 2010;1:64. doi: 10.1038/ncomms1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Plotnikova OV, et al. Calmodulin activation of Aurora-A kinase (AURKA) is required during ciliary disassembly and in mitosis. Mol Biol Cell. 2012;23(14):2658–70. doi: 10.1091/mbc.E11-12-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Seo S, Ichikawa M, Kurokawa M. Structure and function of cas-L and integrin-mediated signaling. Crit Rev Immunol. 2006;26(5):391–406. doi: 10.1615/critrevimmunol.v26.i5.20. [DOI] [PubMed] [Google Scholar]
- 109.Kamiguchi K, et al. Cas-L is required for beta 1 integrin-mediated costimulation in human Tcells. J Immunol. 1999;163(2):563–8. [PubMed] [Google Scholar]
- 110.Fujita Y, et al. A novel platform to enable inhaled naked RNAi medicine for lung cancer. Sci Rep. 2013;3:3325. doi: 10.1038/srep03325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Cai D, et al. The GDP exchange factor AND-34 is expressed in B cells, associates with HEF1, and activates Cdc42. J Immunol. 2003;170(2):969–78. doi: 10.4049/jimmunol.170.2.969. [DOI] [PubMed] [Google Scholar]
- 112.Sakakibara A, Hattori S. Chat, a Cas/HEF1-associated adaptor protein that integrates multiple signaling pathways. J Biol Chem. 2000;275(9):6404–10. doi: 10.1074/jbc.275.9.6404. [DOI] [PubMed] [Google Scholar]
- 113.Cabodi S, et al. Integrin signalling adaptors: not only figurants in the cancer story. Nat Rev Cancer. 2010;10(12):858–70. doi: 10.1038/nrc2967. [DOI] [PubMed] [Google Scholar]
- 114.Baquiran JB, Bradbury P, O’Neill GM. Tyrosine Y189 in the substrate domain of the adhesion docking protein NEDD9 is conserved with p130Cas Y253 and regulates NEDD9-mediated migration and focal adhesion dynamics. PLoS One. 2013;8(7):e69304. doi: 10.1371/journal.pone.0069304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hivert V, Pierre J, Raingeaud J. Phosphorylation of human enhancer of filamentation (HEF1) on serine 369 induces its proteasomal degradation. Biochem Pharmacol. 2009;78(8):1017–25. doi: 10.1016/j.bcp.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 116.Bradbury P, et al. PP2A phosphatase suppresses function of the mesenchymal invasion regulator NEDD9. Biochim Biophys Acta. 2012;1823(2):290–7. doi: 10.1016/j.bbamcr.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 117.Chen L, et al. SYK inhibition modulates distinct PI3K/AKT- dependent survival pathways and cholesterol biosynthesis in diffuse large B cell lymphomas. Cancer Cell. 2013;23(6):826–38. doi: 10.1016/j.ccr.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]