

Abstract
Long noncoding RNAs (lncRNAs), which have emerged in recent years as a new and crucial layer of gene regulators, regulate various biological processes such as carcinogenesis and metastasis. HOTAIR (Hox transcript antisense intergenic RNA), a lncRNA overexpressed in most human cancers, has been shown to be an oncogenic lncRNA. Here, we explored the role of HOTAIR in HeLa cells and searched for proteins regulated by HOTAIR. To understand the mechanism of action of HOTAIR from a systems perspective, we employed a quantitative proteomic strategy to systematically identify potential targets of HOTAIR. The expression of 170 proteins was significantly dys-regulated after inhibition of HOTAIR, implying that they could be potential targets of HOTAIR. Analysis of this data at the systems level revealed major changes in proteins involved in diverse cellular components, including the cytoskeleton and the respiratory chain. Further functional studies on vimentin (VIM), a key protein involved in the cytoskeleton, revealed that HOTAIR exerts its effects on migration and invasion of HeLa cells, at least in part, through the regulation of VIM expression. Inhibition of HOTAIR leads to mitochondrial dysfunction and ultrastructural alterations, suggesting a novel role of HOTAIR in maintaining mitochondrial function in cancer cells. Our results provide novel insights into the mechanisms underlying the function of HOTAIR in cancer cells. We expect that the methods used in this study will become an integral part of functional studies of lncRNAs.
Annotation of the human genome has revealed that, although less than 2% of the genome sequence encodes proteins (1), at least 90% is actively transcribed into noncoding RNAs (ncRNAs)1. NcRNAs, once thought to be the “dark matter” of the genome, have attracted widespread attention and are implicated in the regulation of many major biological processes impacting development, differentiation, and metabolism (2). They are divided into two major classes according to size: small ncRNAs, including microRNAs (miRNAs) and other noncoding transcripts of less than 200 nucleotides (nt), and longer regulatory ncRNAs, referred to as long noncoding RNAs (lncRNAs) (3). LncRNAs are in general defined as mRNA-like, nonprotein coding transcripts longer than 200 nucleotides (4, 5). Using the most advanced sequencing platforms and algorithms for assembling transcripts from deep RNA-sequencing reads, it is estimated that there are about 20,000 distinct lncRNAs in humans (6, 7). Although the function of most lncRNAs remains to be elucidated, recent findings suggest their involvement in fundamental cellular processes, including apoptosis and the cell cycle (8–10), chromatin modification (11, 12), genomic reprogramming (13, 14), gene imprinting (15), and RNA processing (16). LncRNAs have recently been discovered to exhibit unique profiles in various human cancers, and aberrant lncRNA expression has been functionally linked to tumorigenesis (6, 17, 18). Identification of cancer-associated lncRNAs and their interplay with target genes are now important areas of research in cancer biology; lncRNAs may be one of the missing pieces in the oncogene network puzzle.
HOTAIR (Hox transcript antisense intergenic RNA), which has a length of 2158 nt and is located within the Homeobox C (HOXC) gene cluster on chromosome 12, is one of the few well-studied lncRNAs (19, 20). It is unique in that it is overexpressed in the vast majority of cancer types analyzed so far and has been recognized as an oncogenic lncRNA (19). Recently, HOTAIR has been shown to induce proliferation and metastasis in a variety of tumors and is a negative prognostic indicator for several cancers (19, 20). Work pioneered by Howard Chang and colleagues uncovered a possible mechanism for HOTAIR in cancer (20–22). HOTAIR interacts with polycomb repressive complex 2 (PRC2), which enhances H3K27 trimethylation to decrease expression of multiple genes, especially metastasis-suppressing genes (20–22). Subsequent studies demonstrated that HOTAIR serves as a molecular scaffold for at least two distinct histone modification complexes, coordinating their functions in transcription repression (12). Several transcriptome-wide studies have detected extensive changes in cellular transcript levels in response to inhibition of HOTAIR, indicating that HOTAIR can regulate hundreds of genes (22, 23), providing insight into mechanisms underlying the function of HOTAIR in cancer cells. Although informative, transcript abundances do not necessarily reflect cellular protein levels because protein activity can be influenced by an array of post-transcriptional regulatory mechanisms and the correlation between protein and mRNA levels is generally modest (24, 25). It is therefore necessary to analyze cellular protein levels after inhibition of HOTAIR at the proteomics level.
In a previous study, we successfully employed a quantitative proteomic approach using SILAC (stable isotope labeling by amino acids in cell culture) methodology to identify targets of miR-21 in cancer cells (26). Here, we carried out global proteomic profiling to identify genes regulated by HOTAIR in HeLa cells. Using SILAC-based quantitative proteomics, we found that the expression of 170 proteins was dysregulated by inhibition of HOTAIR. Many interesting differentially-expressed proteins that potentially play functional roles during HOTAIR inhibition were identified. Analysis of this data at the systems level revealed major changes in proteins involved in diverse cellular components, in particular in the cytoskeleton and respiratory chain. Further functional studies revealed that vimentin (VIM), a key protein involved in the cytoskeleton, contributes to various phenotypic effects observed after inhibition of HOTAIR. By correlating our proteomic data with that from functional studies, novel insights into the mechanism underlying the function of HOTAIR in cancer cells emerge.
EXPERIMENTAL PROCEDURES
Cell Culture and RNA Interference
Human cervical cancer cell line HeLa, Chang Liver, human macrophage-like cell line U937, multiple myeloma cell line U266, lung cancer cell line H1299, hepatocellular carcinoma line HepG2 and gastric cancer cell line SGC7901 were purchased from American Type Culture Collection (Manassas, VA). HeLa, Chang Liver, U937, U266, H1299, HepG2 and SGC7901 were grown in DMEM (Hyclone, Logan, UT) containing 10% fetal bovine serum (FBS) (Gibco, Gaithersburg, MD), 2 mm glutamine, 50 U/ml penicillin and 50 mg/ml streptomycin at 37 °C in a humidified atmosphere with 5% CO2.
For transient transfection, HeLa cells were transfected with 10 nm of siRNA targeting HOTAIR (siHOTAIR-I or siHOTAIR-II) or negative control siRNA (siNC) using Lipofectamine RNAiMAX (Invitrogen, Gaithersburg, MD). RNA expression of HOTAIR was verified at 48 h after transfection by qRT-PCR as described below. For VIM gene knockdown, HeLa cells were transfected with 10 nm of siRNA targeting VIM (siVIM-I, siVIM-II or siVIM-III) and a negative control siRNA (siNC). Cells were harvested 48 h after transfection and VIM gene knockdown was assessed by Western blotting. All the siRNAs were purchased from GenePharma Co. Ltd. (Shanghai, China). siRNA sequences are listed in supplemental Table S1.
A human VIM expression plasmid (pVIM) (Catalogue NO.: EX-D0114-M13) and control plasmid (pEGFP) were purchased from GeneCopoeia, Inc. (Rockville, MD). The VIM and control plasmids were expressed in HeLa cells by transient transfection using Lipofectamine 2000 Reagent (Invitrogen), according to the manufacturer's protocol. Cells were collected 48 h after transfection and overexpression of VIM was confirmed by Western blotting.
For stable transfection, HeLa cells were transfected with a plasmid expressing small interfering RNA molecules targeting HOTAIR. A pGPU6/GFP/Neo siRNA Expression Vector kit was purchased from GenePharma Co., Ltd. The RNAi sequences used were listed in supplemental Table S1 and a scrambled sequence (GTTCTCCGAACGTGTC ACGT) which has no significant homology to human gene sequences was used as a control. HeLa cells were transfected with pGPU6/GFP/Neo/HOTAIR or pGPU6/GFP/Neo/control using Lipofectamine 2000 Reagent (Invitrogen) and were then selected for neomycin resistance for 3 weeks, at which point one clone was selected from the pGPU6/GFP/Neo/control or pGPU6/GFP/Neo/HOTAIR transfected cells. Cells selected from pGPU6/GFP/Neo/control transfected HeLa cells were designated as HeLa-NC cells, and those from the pGPU6/GFP/Neo/HOTAIR transfected HeLa cells as HeLa-KD cells. The expression level of HOTAIR was determined by qRT-PCR.
RNA Isolation and qRT-PCR
Total RNA was extracted from cultured cells using Trizol reagent (Invitrogen) according to the manufacturer's protocol. RNA was reverse transcribed into first strand cDNA using a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA), and quantitative PCR was carried out using SYBR Green PCR Master Mix (Roche Diagnostics Ltd, Mannheim, Germany) and a LightCycler 480 Real-Time PCR system (Roche). The GAPDH gene was used as an endogenous control gene for normalizing the expression of target genes. Each sample was analyzed in triplicate. The thermo cycling program consisted of holding at 95 °C for 5 min, followed by 40 cycles of 10 s at 95 °C, 30 s at 60 °C and 30 s at 72 °C. Melting-curve data were then collected to verify PCR specificity and the absence of primer dimers. Primer sequences are listed in supplemental Table S1.
Detection of Apoptosis
Transiently or stably transfected HeLa cells (1 × 106) were harvested 48 h after transfection, washed three times in phosphate-buffered saline (PBS), and then stained with Annexin V-FITC and PI according to the manufacturer's instructions (Beyotime, Haimen, China). Samples were acquired on a FACScan flow cytometer (Becton Dickinson, San Jose, CA) and analyzed with the BD FACSDiva software 6.0 (Becton Dickinson).
Cell Cycle Progression Assay
Transiently or stably transfected HeLa cells (1 × 106) were harvested, washed three times with ice-cold PBS and fixed with 70% ethanol overnight at 4 °C. Cells were stained with PI (Beyotime), and a cell cycle profile was determined using a BD FACSAria III Cell Sorting System (Becton Dickinson). Ten thousand events were acquired for each sample, and cell cycle distributions were determined using ModFit LT software (Becton Dickinson). Experiments were performed in triplicate. Results are presented as the percentage of cells in a particular phase.
Cell Proliferation Assay
A Cell Counting Kit-8 (Boster, Wuhan, China) was used to determine cell proliferation. Briefly, transiently or stably transfected HeLa cells (1 × 103) were plated in triplicate in 96-well plates. 10 μl cell proliferation reagents were added to each well and cells were incubated for 2 h at 37 °C. Cell numbers were estimated by measuring the optical density (OD) at 450 nm. Absorbance of cell-free wells containing medium was set as zero.
Wound Healing Assay
Transiently or stably transfected HeLa cells were seeded into a six-well plate and allowed to grow to 70% confluence in complete medium. Cell monolayers were wounded using a plastic tip (1 mm) that touched the plate. Cells were then washed with PBS to remove debris, transfected and incubated for 24 h. The number of cells migrating to the wound surface and the average distance cells migrated was determined under an inverted microscope at designated time points.
Cell Invasion Assay
Transwell chambers (Corning, 8.0 μm pore size) coated with Matrigel (BD Biosciences, Bedford, MA) were used to measure the invasiveness of cancer cells. In brief, transiently or stably transfected HeLa cells (2 × 105) were plated in the upper chamber in serum-free media. The bottom chamber was covered with media containing 10% FBS. After incubating for 48 h, cells that migrated to the bottom of the chamber insert was fixed in methanol for 15 min and stained with Giemsa stain. Invading cells were photographed and counted on the stained membrane under a microscope. Each membrane was divided into four quadrants and the average from four quadrants was calculated.
SILAC and Protein Extraction
HeLa cells were grown in SILAC DMEM Medium (Pierce Biotechnology, Rockford, IL) containing 10% FBS, and either the 13C6-l-lysine (heavy) or 12C6-l-lysine (light) for more than seven generations before harvesting, to ensure high labeling efficiency (>99%). Heavy-labeled cells were transfected with 10 nm siHOTAIR-I and light-labeled HeLa cells were transfected with 10 nm siNC. After incubating for 48 h, cells were washed three times with ice-cold PBS, transferred to clean 1.5 ml Eppendorf tubes and lysed with RIPA lysis buffer (50 mm Tris-HCl, 150 mm NaCl, 0.1% SDS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1 mm PMSF, 100 mm leupeptin, and 2 mg/ml aprotinin, pH 8.0) on ice for 15 min. Cellular debris was removed by centrifugation at 13,200 × g for 30 min at 4 °C. Protein concentration was measured using a BCA Protein Assay Kit (TIANGEN, Beijing, China).
Protein Separation and In-gel Digestion
The “Light” and “Heavy” lysates were mixed in a 1:1 ratio based on protein weight (100 μg of each), boiled in SDS-PAGE sample buffer, separated by 12% SDS-PAGE and stained with Coomassie Brilliant Blue. The entire gel lane was cut into 30 sections for in-gel digestion. Excised sections were chopped into small pieces of 1 mm2, washed in deionized water and completely destained using 100 mm ammonium bicarbonate (NH4HCO3) in 50% acetonitrile (ACN). A reduction step was performed in 100 mm dl-Dithiothreitol (DTT) at 37 °C for 1 h. The proteins were alkylated in 50 mm iodoacetamide (IAA) at room temperature for 1 h in the dark. Gel sections were first washed in deionized water, then ACN, and finally dried in a SpeedVac system (Thermo Fisher Scientific). Digestion was carried out using 20 μg/ml sequencing grade modified trypsin (Promega, Madison, WI) in 50 mm NH4HCO3 at 4 °C for 45 min and then incubated at 37 °C overnight. The supernatants were transferred into a 200 μl microcentrifuge tube and the gels were extracted twice with extraction buffer (67% ACN containing 2.5% trifluoroacetic acid). The peptide extracted and the supernatant of the gel slices were combined and then dried in a SpeedVac.
LC-MS/MS Analysis
The dried peptides from each gel slice were reconstituted in 5% ACN/0.1% formic acid and analyzed using a maXis impact UHR-QTOF system (Bruker Daltonics, Bremen, Germanys) coupled to a Dionex Ultimate 3000 nano-flow HPLC (Dionex, Sunnyvale, CA). Peptide mixtures from each gel slice were first desalted online using a C18 PepMap trap column (300 μm i.d., 5 cm long, LC Packings), and eluted to a C18 PepMap analytical column (75 μm i.d., 15 cm long, LC Packings). Peptides were separated using a linear gradient from 10–50% of solvent B (98% ACN, 2% water and 0.1% formic acid) over 45 min at a flow rate of 300 nl/min at room temperature, where solvent A was water containing 2% ACN and 0.1% formic acid. Two biological replicates were analyzed. TOF-MS screening measurements were all performed on a predefined 50–2200 m/z acquisition window at 2500 TOF summations (∼2 Hz). CID MS/MS acquisition was performed over the same 50–2200 m/z window with three intensity-binned precursors of charge +2 to +4, and at least 1000 counts selected for fragmentation. Accumulation times for MS/MS were also intensity-binned from a maximum of 5000 summations (∼1 Hz, if precursor ≤ 1 × 103 ion counts) to a minimum of 2000 summations (∼2.5 Hz, if precursor ≥ 2 × 104 ion counts). To test the effects of increasing the accumulation time, additional experiments were also performed using either a minimal time (2500 summations, ∼2 Hz) or a maximal time (15,000 summations, ∼0.33 Hz). An optimized set of isolation windows was used based on the precursor m/z to achieve at least 90% precursor recovery prior to fragmentation. Selected precursors that had been analyzed >2 times were actively excluded from analysis for 15s. Ion transmission optimization for MS/MS was also performed on four key parameters for the collision cell and the ion cooler cell (RF guide voltages CCRF and ICRF, transfer time ICTT, and prepulse time ICPP).
Data Processing and Protein Quantification
Raw files were processed using LC/MS software DataAnalysis 4.0 SP4 (Bruker Daltonics) and converted into XML files. The XML file of each MS/MS run was imported to Proteinscape v3.0 (Bruker Daltonics). Protein identification was performed by searching the MS/MS data on a local Mascot server v2.4 (Matrix Science, London, UK) against the IPI human 3.87 database (including 91,491 entries). Search parameters used were as follows: enzyme specificity, trypsin/with no proline restriction; maximum missed cleavages, 2; carbamidomethyl (+57.0215 Da, Cys) as fixed modification; oxidation (+15.9949 Da, Met), Lys (+6.0201 Da, SILAC heavy amino acid) as variable modifications; precursor ion mass tolerance, 0.1 Da; and MS/MS mass tolerance, 0.1 Da. The false discovery rate (FDR) was set to 1% using Mascot Percolator (an algorithm that uses semi-supervised machine learning to improve the discrimination between correct and incorrect spectrum identifications). Detection of at least two matching peptides per protein was set as a requirement for unambiguous identification. In each independent technical replicate, the relative quantification of the proteins was performed by WARP-LC 3.0 (Bruker Daltonics) and Proteinscape v3.0 (Bruker Daltonics). The average peak area ratio of the “control”/“treated sample” was calculated for all the peptides by ProteinScape v3.0. Peptide ratios were normalized by dividing by the overall median of all peptides. When ratios for individual peptide matches were combined into ratios for protein hits, the Grubbs' method was used for detecting and removing outliers, and protein ratios were calculated as the geometric mean of the ratios of corresponding peptides. Protein ratios from replicate experiments were averaged. In all subsequent data analyses, we used only those proteins that were identified in both of the two independent experiments. Protein ratios were log2 transformed and the frequency distribution of the quantified proteins was calculated to determine differentially expressed proteins.
Bioinformatics Analysis
Classification of HOTAIR-regulated proteins was performed using PANTHER (Protein Analysis Through Evolutionary Relationships) (http://www.pantherdb.org), which classifies genes and proteins by their functions (27). GO classification of the differentially expressed proteins were also performed using DAVID Bioinformatics Resources 6.7 (28, 29).
The protein–protein interaction (PPI) network of HOTAIR-regulated proteins was built by searching against the STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) database version 9.1(30) with default settings except that organism was set to “human.” PPI networks for “cytoskeleton” and “respiratory chain” associated proteins were respectively extracted from the PPI network of the HOTAIR-regulated proteins by selecting only proteins belong to the “cytoskeleton” or “respiratory chain” category. PPI networks were then visualized by Cytoscape v3.1.0 (31).
Western Blotting
Protein extracts (10 μg) prepared with RIPA lysis buffer were resolved on a 12% SDS-PAGE gel, and transferred to an Immobilon-P PVDF transfer membrane (Millipore, Bedford, MA) by electroblotting. After blocking with 5% nonfat milk, membranes were incubated overnight with a 1:1000 dilution of antibodies at 4 °C. Blots were then incubated with peroxidase-conjugated anti-mouse or anti-rabbit IgG (KPL, Gaithersburg, MD) for 1 h at room temperature at a 1:1000 dilution and then developed using a SuperSignal West Pico kit (Pierce Biotechnology). Immunoblots were scanned using an Image Scanner (GE Healthcare, Waukesha, WI). Blot densitometry analysis was performed using Image J (National Institutes of Health, Bethesda, MD). The following antibodies were used for Western blot analyses: rabbit anti-VIM polyclonal (Abcam, Cambridge, MA), rabbit anti-FBL polyclonal, rabbit anti-MGMT polyclonal, rabbit anti-STAT5B polyclonal, rabbit anti-VASP polyclonal, rabbit anti-PSMD10 polyclonal (Abclonal Technology, Wuhan, China), and mouse anti-GAPDH polyclonal (CWBIO, Beijing, China). All analyses were performed in triplicate.
Multiple Reaction Monitoring (MRM) Analysis
Expression changes in targeted proteins were further verified by MRM using the MIDAS (MRM initiated detection and sequencing) Workflow. In silico determination of best candidate MRM transitions for target proteins was performed based on the mProphet scoring model integrated into the skyline software (AB SCIEX, Foster City, CA). The detectability, retention time and fragmentation pattern of the transition candidates were further validated by an EMS scan, followed by four EPI scans on the QTRAP 6500 System (AB SCIEX). For the quantitation of each peptide, we selected four validated transitions and the MRM experiments were performed in triplicate (technical replicate). Declustering potential (DP) and collision energy (CE) values for each of the validated transitions were also optimized using Skyline software. The LC-MRM run was then performed for predetermined transitions using a QTRAP 6500 System (AB SCIEX) coupled with a nanoLC-Ultra 1D system (Eksigent, Dublin, CA) in MRM mode. Briefly, in-solution digestion of proteins was performed as previously described (32). 3 μg of tryptic peptides from the sample or control group were desalted on a 200 μm × 0.5 mm trap chip and then eluted onto a 75 μm × 150 mm column chip for MS analysis. Both the trap and column chips were ChromXP C18-CL 3 μm 120Å phase (Eksigent). Peptides were separated using a linear gradient formed by solvent A (98% water, 2% ACN and 0.1% formic acid) and solvent B (98% ACN, 2% water and 0.1% formic acid) going from 10–50% solvent B over a 45 min period at a flow rate of 300 nl/min at room temperature.
MS analysis was performed on a QTRAP 6500 System equipped with an IonDrive™ Turbo V Ion Source (AB SCIEX). The source temperature was set to 160 °C, and the ion spray voltage was set to 2300 V. Previously optimized DP and CE values were used for each analyte. Curtain gas was set to 30, and collision gas (CAD) was set to high. Resolutions on quadrupole part 1 (Q1) and quadrupole part 3 (Q3) were both unit resolution.
Data from the intensity chromatograms of the transitions were extracted with the MultiQuant program (AB SCIEX). Peak areas for each transition were extracted and peptide peak areas were calculated by summing the peak areas of the corresponding transitions. Each peptide peak area for individual peptides in the sample group was compared with the corresponding peptide peak areas in the control group to estimate the relative differences between two groups. Comparisons of different groups were performed using the Student's t test, with p < 0.05 and CV < 10% being considered significant.
Immunofluorescence Staining, Imaging, and Computational Analysis
Transiently or stably transfected HeLa cells were grown on a sterile glass bottom culture dish. The culture dishes were washed three times with 1×PBS and cells were fixed in 4% formaldehyde for 30 min. Cells were permeabilized in 1% FBS, 0.2% Triton-X100 on ice for 5 min. After washing in PBS, cells were blocked with 1% BSA for 1 h at room temperature and then incubated with rabbit anti-VIM polyclonal at a dilution of 1:200 for 2–4 h in darkness. Cells were then washed three times with PBS and incubated with Dylight 488-conjugated Goat anti-Rabbit IgG (H+L) (Abbkine, Redlands, CA) at a dilution of 1:100 for 1 h in darkness. Cells were mounted in ProLong Gold antifade reagent with 4′6-diamidino-2-phenylindole (DAPI) after a brief wash with PBS for 10 min at room temperature. Subsequently, cells were washed three times with 1×PBS thoroughly and examined using a LSM 710 laser scanning confocal microscope (Carl Zeiss, Oberkochen, Germany). Whole cells were photographed with the image focused on the vimentin fibers localized closest to the cell periphery as described previously (33, 34) using an OMX V4™ Super-resolution Imaging Systems (GE Healthcare). All analyses and quantifications were performed in a blinded manner. At least 100 cells were quantified for each experiment and condition, seven to eight images (40 nm pixels) being acquired from different cells. Image analysis was performed as previously described (35).
ELISA Analysis of the Ubiquinol-Cytochrome C Reductase (UQCR)
HeLa cells (1 × 106) were harvested 48 h after transfection with 10 nm of siNC or siHOTAIR-II, washed three times with ice-cold PBS and lysed with RIPA lysis buffer on ice for 15 min. Cellular debris was removed by centrifugation at 13,200 × g for 30 min at 4 °C. The concentration of UQCR was determined using an ELISA Kit for UQCR (Cloud-Clone Corp, Houston, TX) according to the manufacturer's protocol.
Glucose Uptake Assay
Transiently or stably transfected HeLa cells were harvested, washed three times with PBS and incubated at 37 °C for 30 min with 100 μm 2-(N-(7-Nitrobenz-2-oxa-1,3-diazol −4-yl)amino)-2-deoxyglucose (2-NBDG) (Invitrogen) dissolved in glucose-free and serum free-media as previously described(34). Cells were then washed with PBS. Intracellular 2-NBDG was examined using a LSM 710 laser scanning confocal microscope (Carl Zeiss). Fluorescence was measured with a fluorescence-activated cell sorter (FACS) instrument (Becton Dickinson).
Reactive Oxygen Species (ROS) Measurement
The intracellular ROS level was measured using Reactive Oxygen Species Assay Kit (Beyotime). In brief, cells were harvested and incubated with 10 μm DCFH-DA for 20 min at 37 °C, protected from light. DCF fluorescence was measured with a BD Accuri™ C6 flow cytometer (Becton Dickinson).
Measurement of Mitochondrial Membrane Potential (Δψ)
The Δψ of HeLa cells after HOTAIR knockdown was measured using a Mitochondrial Membrane Potential Assay Kit with JC-1 (Beyotime). Fluorescence emission was analyzed by flow cytometry (JC-1 monomers: excitation wavelength 488 nm, emission filter 530/30 nm; JC-1 aggregates: excitation wavelength 488 nm, emission filter 585/42 nm) using a BD Accuri™ C6 flow cytometer (Becton Dickinson). The Δψ was also quantified by microscopic examination of cells stained with MitoTracker Deep Red FM. In brief, the cell medium was aspirated after treatment, and MitoTracker Deep Red FM (Invitrogen) was added (500 nm in cell growth medium) for 30 min at 37 °C. After incubation, the cells were fixed in ice-cold, 100% methanol for 15 min at −20 °C and rinsed three times with PBS for 5 min. The fluorescence emission was detected under a LSM 710 laser scanning confocal microscope (Carl Zeiss).
Transmission Electron Microscopy (TEM) Observation
The ultrastructural alterations of HeLa cells after HOTAIR knockdown were observed through TEM. Briefly, HeLa cells were washed three times with PBS, and prefixed in 4% paraformaldehyde for 2 h and postfixed in 1% osmium tetroxide at 4 °C for 1 h. After that, cells were scraped into 1.5 ml Eppendorf tubes and washed three times with 0.1 m sodium cacodylate buffer (pH 7.4), and then dehydrated and embedded in resin. The ultrathin sections were made and stained with uranyl acetate and lead citrate. The samples were analyzed by a H-7000FA transmission electron microscopy (Hitachi, Tokyo, Japan) at an operating voltage of 75 kV.
Xenograft Mouse Model
Four-week-old female athymic BALB/c mice were purchased from Vital River Laboratories (Beijing, China). All animal procedures were performed in accordance to protocols approved by the Institutional Animal Care and Use Committee at the Institute of Hydrobiology. For xenograft models, 2 × 106 HeLa cells transfected with siNC or siHOTAIR, and HeLa cells expressing either control shRNA or shHOTAIR were injected subcutaneously in the right flank of BALB/c nude mice (five mice per group). After 20 days, these mice were sacrificed and tumors were weighed. The tumors were homogenized, and proteins were extracted for Western blotting.
Statistical Analysis
Statistical analysis was carried out to assess differences between experimental groups. Statistical significance was analyzed by Student's t tests and expressed as a p value. p < 0.05 was considered to be statistically significant. One asterisk and two asterisks indicate p < 0.05 and p < 0.001, respectively.
RESULTS
Functional Effects of HOTAIR Inhibition in HeLa Cells
HOTAIR, a negative prognostic factor, has been correlated with cancer cell proliferation, apoptosis, invasion, and metastasis (3, 36–38) and is overexpressed in various cancer cell lines (22, 39–41). Here, we used quantitative real-time PCR (qRT-PCR) to detect the relative abundance of HOTAIR in different cell lines, HeLa cells showing the highest levels of HOTAIR expression (supplemental Fig. S1). To determine the function of HOTAIR in HeLa cells, we used siRNA to knockdown its expression and evaluated its knockdown efficiency by qRT-PCR. The abundance of HOTAIR in HeLa cells was reduced more than 50% at 48 h after transfection (Fig. 1A). Flow cytometry demonstrated that transfection of HeLa cells with siHOTAIR for 48 h resulted in a significant increase in the percentage of apoptotic cells relative to cells transfected with negative control siNC (Fig. 1B). Quantitative analysis indicated that the percentage of apoptotic cells was significantly higher (p < 0.05; Fig. 1C) for siHOTAIR-transfected cells than for siNC-transfected cells. We next examined the effect of HOTAIR knockdown on the cell cycle. The percentage of cells in G1 phase was reduced whereas that in S phase and G2/M phase increased (Fig. 1D). At the same time, cell growth was significantly suppressed relative to the negative control siNC group (Fig. 1E). Furthermore, HOTAIR inhibition significantly decreased HeLa cell invasion as determined using a Boyden chamber assay (Fig. 1F and 1G), and cell migration as determined with a wound-healing assay (Fig. 1H and 1I). These data demonstrate that HOTAIR is involved in the regulation of cell growth, the cell cycle, apoptosis, and migration and invasion of HeLa cells.
Fig. 1.

Functional effects of HOTAIR inhibition on HeLa cells. A, HeLa cells were transfected with siHOTAIR or siNC for 48 h. HOTAIR knockdown efficiency was determined by qRT-PCR. The expression level of HOTAIR was normalized to GAPDH. B, Apoptosis was determined by annexin V staining and flow cytometry. C, Apoptosis rate of HeLa cells at 48 h after transfection with siHOTAIR or siNC. D, Effect of HOTAIR knockdown on cell cycle progress. The percentage of cells in the G1 phase was significantly decreased whereas that in S phase and G2/M phase was increased after HOTAIR inhibition. E, HOTAIR knockdown in HeLa cells significantly inhibits cell growth. F, Effect of HOTAIR knockdown on cell invasion, as determined in a Boyden chamber assay. G, Numbers of cells on the underside of the filter. Significantly enhanced invasion (p < 0.05) is indicated. H, HOTAIR inhibition led to a significant reduction of cell migration as determined by a wound-healing assay. I, Quantification of the wound healing assay. Data are presented as means ± S.D. and represent results from three independent experiments. Statistically significant differences are indicated: *p < 0.05; **p < 0.01.
We further investigated the functional effects of HOTAIR knockdown in stably-transfected HeLa cells. HOTAIR knockdown cells (HeLa-KD) showed significantly diminished levels of HOTAIR RNA compared with cells transfected with negative control shRNA (HeLa-NC) (supplemental Fig. S2A). Like the siHOTAIR-transfected cells, HeLa-KD cells showed suppressed cell growth (supplemental Fig. S2B), and decreased invasion (supplemental Fig. S2E and 2F) and migration capacity (supplemental Fig. S2G and 2H). However, there were some phenotypic differences in cell cycle progress and cell apoptosis between siHOTAIR-transfected cells and HeLa-KD cells (supplemental Fig. S2C and 2D), possibly arising from differences between transient and stable transfection of cells.
Identification of HOTAIR-regulated Proteins Using a Quantitative Proteomic Approach
Because HOTAIR is expressed at high levels in HeLa cells, we reasoned that knockdown of HOTAIR would change the expression of its target genes. Knockdown of endogenous HOTAIR rather than overexpression is preferable for investigating ncRNA-dependent gene regulation and function (23). We therefore employed a SILAC-based quantitative proteomics approach to identify proteins dysregulated by HOTAIR knockdown in HeLa cells (Fig. 2A). A total of 3195 proteins were identified using the criteria described in the Materials and Methods at a FDR of 1% (supplemental Table S2), the majority of which had a log2 fold change value (siHOTAIR/siNC) between −0.55 and +0.55 after HOTAIR inhibition (supplemental Fig. S3). One hundred Seventy proteins had a log2 fold change ≥ 0.55 or ≤ −0.55 and were regarded as dysregulated after HOTAIR inhibition. Of these, 118 were up-regulated and 52 were down-regulated (supplemental Table S3). Supplemental Table S4 provides detailed protein quantitation information from the replicate experiments. All raw data has been deposited in the PeptideAtlas database (http://www.peptideatlas.org) under the identifier PASS00496 (http://www.peptideatlas.org/PASS/PASS00496).
Fig. 2.

Quantitative proteomic identification of HOTAIR-regulated proteins in HeLa cells. A, Workflow for the identification of HOTAIR-regulated proteins. HeLa cells were differentially labeled by growing them in medium containing light or heavy amino acids (SILAC). Proteins were extracted from the labeled cells 48 h after transfection with siHOTAIR or siNC, then equal amounts of protein from each sample were combined. The protein mix was separated by 12% SDS-PAGE and the resulting gel was cut into 30 sections. Each of the fractions was in-gel digested and analyzed via LC-MS/MS. B, Heatmap showing the expression of differentially expressed proteins after HOTAIR inhibition. C, PANTHER Protein Class ontology classification of the 170 proteins differentially expressed after HOTAIR expression silencing. HOTAIR-regulated proteins were classified into 22 classes.
Bioinformatic Analysis of HOTAIR-regulated Proteins
To evaluate the biological relevance of the HOTAIR-regulated proteins, we first classified them using PANTHER Protein Class ontology (42). The 127 proteins assigned to Panther Protein Class categories were classified into 22 classes (Fig. 2C and supplemental Table S5), the five largest of which were the nucleic acid binding (36 proteins), cytoskeletal protein (18 proteins), hydrolase (17 proteins), enzyme modulator (17 proteins), and oxidoreductase (16 proteins) classes. We next classified the HOTAIR-regulated proteins by their GO molecular function, biological process and cellular components to get an overview of the GO distribution (supplemental Fig. S4). Of note, GO cellular component classification results also revealed that many HOTAIR-regulated proteins were assigned to the cytoskeleton and mitochondrion (supplemental Fig. S4C). GO biological process analysis revealed that the HOTAIR-regulated proteins are involved in a variety of biological processes, such as oxidation reduction, translation, biological generation of precursor metabolites and energy, and RNA processing (supplemental Fig. S4A). In the GO molecular function category, nucleotide binding, RNA binding and structural molecule activity were the three major categories (supplemental Fig. S4B). Taken together, these bioinformatic results suggest that HOTAIR may be a critical regulator of diverse cellular functions across widespread biological processes.
A HOTAIR-regulated protein interaction network, generated using the STRING(30) database, revealed that 113/170 (66.5%) proteins were involved in protein–protein interactions (Fig. 3A). Cytoskeleton- and mitochondrion-related proteins formed two highly connected clusters, suggesting that physiological interactions between these two clusters might contribute to the cooperation and/or coordination of their functions in mediating the function of HOTAIR. Notably, vimentin (VIM) and Ubiquinol-Cytochrome C Reductase (UQCR) were hubs in the cytoskeleton and respiratory chain network, respectively (Fig. 3B and 3C), suggesting that these two proteins may play an important role in mediating the function of HOTAIR in HeLa cells. Twenty-seven proteins were cytoskeleton proteins including actin, microtubule, intermediate filament cytoskeleton and their related proteins (Fig. 3D). These results confirm that HOTAIR is a critical regulator of diverse cellular functions across widespread biological processes, especially of the cytoskeleton and mitochondrion-related processes.
Fig. 3.

Bioinformatic analysis of HOTAIR-regulated proteins. A, The PPI network for HOTAIR-regulated proteins was constructed by searching against the STRING database v9.0 with default settings except that organism was set to “human.” 113 of the 170 DEPs are involved in this network. The 170 DEPs were classified into several functional groups according to their GO categories. White nodes indicate that these proteins were not involved in the PPI network. Orange nodes indicate cytoskeleton-related proteins, and red nodes indicate mitochondria-related proteins. B, The PPI network of cytoskeleton-related proteins generated by STRING 9.0 and visualized with Cytoscape v3.1.0. C, The PPI network of mitochondrial respiratory chain-related proteins generated by STRING 9.0 and visualized with Cytoscape v3.1.0. D, Heatmap showing the expression of 27 cytoskeleton-related proteins after HOTAIR inhibition. E, Heatmap showing the expression of 32 mitochondrial respiratory chain-related proteins after HOTAIR inhibition.
Validation of the Quantitative Proteomics Data
We then validated some of the proteins identified by SILAC experiments using Western blotting (Fig. 4A), selecting eight proteins based on how well their biological functions and importance are understood. Results from Western blotting and densitometric analysis were consistent with the SILAC experiments.
Fig. 4.

Validation of differential protein expression. A, Western blotting of eight differentially expressed proteins (FBL, STAT5B, MGMT, VIM, VASP, GNAI2, C1QBP and PSMD10) at 48 h after HOTAIR inhibition. Values are expressed as the percent change relative to the controls and Western blots are from one representative experiment. Western blot analysis detected changes in expression consistent with those detected by MS. GAPDH was used as an internal control. The change in the expression of VIM and FBL was further verified by MRM. MS spectrum of target peptides of VIM (B) and FBL (C) selected for MRM validation. Results of the MRM analysis were consistent with SILAC data.
MRM, a highly specific and sensitive mass spectrometry technique based on the selection of a peptide ion and one or more characteristic fragment ions was used to provide further independent validation of SILAC results. MRM can be used to accurately and reproducibly detect and quantify specific molecules in a complex mixture (43, 44). The expression of VIM and FBL was significantly up-regulated after HOTAIR knockdown (Fig. 4B and 4C), in agreement with SILAC results. Details of the dynamic MRM settings, including the Q1 and Q3 masses, retention times, DPs, and CEs of 40 ion pairs are given in supplemental Table S6.
VIM Contributes to the Effects of HOTAIR Knockdown
As mentioned above, and verified by both Western blotting and MRM analysis (Fig. 4A and 4B), VIM was found to be significantly up-regulated after HOTAIR inhibition. In addition, bioinformatic analyses suggested that VIM may play an important role in mediating the functions of HOTAIR in HeLa cells (Fig. 3B). To investigate whether VIM has a role in mediating the functions of HOTAIR and explore the molecular consequences of VIM inhibition/overexpression, we first examined the mRNA and protein expression levels of VIM in HeLa-KD cells; results were consistent with those observed in transiently-transfected cells (Fig. 4A, Fig. 5A and supplemental Fig. S5A). Furthermore, knockdown of HOTAIR enhanced expression of VIM in lung cancer cell line H1299 but not in gastric cancer cell line SGC7901 (supplemental Fig. S5). Consistent with previous reports (23), our results confirm that HOTAIR regulates different sets of genes in different cancer cells.
Fig. 5.

VIM contributes to the effects of HOTAIR knockdown. A, HeLa cells were transfected with siHOTAIR or siNC for 48 h and VIM mRNA expression levels were determined by qRT-PCR. The expression level of VIM was normalized to GAPDH. B, HeLa cells were transfected with siVIM for 48 h, then VIM mRNA expression levels were determined by qRT-PCR. The expression level of VIM was normalized to GAPDH. C, Western blot analysis of VIM protein expression 48 h after transfection with siVIM (siVIM-I, siVIM-II and siVIM-III) or siNC. GAPDH was used as an internal control. D, HeLa cells were transfected with pVIM or pEGFP. VIM expression levels were determined by Western blotting at 24 h and 48 h after transfection. EGFP serves as the negative control and GAPDH as the loading control. E, Effect of VIM knockdown and overexpression on cell invasion as determined with a wound-healing assay. F, Quantification of the wound healing assay. G, Effect of VIM knockdown and overexpression on cell invasion, as determined with a Boyden chamber assay. H, Numbers of cells on the underside of the filter. Significantly enhanced invasion (p < 0.05) is indicated. Data are presented as means ± S.D. and results are from one representative experiment of at least three. *p < 0.05; **p < 0.01. I, HOTAIR suppressed tumor growth and regulated VIM expression in nude mice. HeLa cells transfected with siHOTAIR or siNC and HeLa cells expressing either control shRNA or shHOTAIR were injected subcutaneously into the right flank of nude mice. After 20 days, mice were sacrificed and tumors were dissected and weighed. Representative photographs of xenografts were taken 20 days after injection of HeLa cells transfected with siHOTAIR or HeLa-KD cells. J, Quantification of tumor weight. Data are presented as means ± S.D. (n = 5). K, Western blotting of VIM protein expression in tumors excised from the mice indicated 20 days after injection.
The role of VIM in HeLa cells was evaluated by silencing/overexpression of VIM using transient transfection of validated siRNAs and cDNAs. mRNA and protein expression levels of VIM decreased dramatically at 48 h after transfection of siRNA targeted against VIM (Fig. 5B and 5C). Overexpression of VIM in HeLa cells was confirmed by Western blotting after transiently transfected with cDNAs encoding VIM or GFP (Fig. 5D).
The effects of VIM on the migration of HeLa cells were evaluated in a wound healing assay. VIM inhibition resulted in a significant increase in cell migration compared with the negative control (Fig. 5E and 5F). Cotransfection of HeLa cells with siHOTAIR and siVIM partially abrogated the effect of HOTAIR knockdown on cell migration (Fig. 5E and 5F). In contrast, overexpression of VIM reduced cell migration significantly (Fig. 5E and 5F).
We next examined the effect of VIM on the invasion of HeLa cells using a Matrigel invasion assay. The number of invaded cells increased significantly in VIM knockdown cells compared with the negative control, whereas overexpression of VIM resulted in reduced invasion capability (Fig. 5G and 5H). Furthermore, cotransfection of HeLa cells with siHOTAIR and siVIM reversed the effect of HOTAIR knockdown on cell invasion.
VIM knockdown in HeLa cells also affected cell growth, cell cycle progress and apoptosis (supplemental Fig. S6). Transfection of HeLa cells with siVIM or cotransfection of HeLa cells with siVIM and siHOTAIR both lead to inhibited cell growth and disturbed cell cycle progress. The apoptosis rate significantly decreased after VIM knockdown, but when cotransfected with siVIM and siHOTAIR, increased apoptosis rate in HeLa cells was observed. However, overexpression of VIM did not cause any significant change in cell growth, cell cycle progress or apoptosis in HeLa cells (supplemental Fig. S6).
A xenograft mouse model was used to further validate the observations obtained from in vitro studies (Fig. 5I). HeLa cells transiently transfected with siHOTAIR and HeLa-KD cells were both tested using the model. Knockdown of HOTAIR suppressed the tumorigenicity of HeLa cells, as measured by tumor mass (Fig. 5J), consistent with previous functional studies in pancreatic cancer cells(23, 45, 46). Western blotting subsequently verified the up-regulation of VIM in HOTAIR knockdown xenografts compared with control xenografts (Fig. 5K), indicating that HOTAIR can regulate the expression of VIM in vivo.
Based on these results, we propose that HOTAIR functions as a negative regulator of VIM and that at least some of the various biological effects of HOTAIR are mediated by regulation of VIM expression in HeLa cells.
Inhibition of HOTAIR or Overexpression of VIM Affects the Organization of the VIM IF Network
VIM IFs are responsible for maintaining cell shape, integrity of the cytoplasm, and stabilizing cytoskeletal interactions(47). To determine whether HOTAIR knockdown can affect the hierarchical structure and distribution of VIM, we compared the distribution of VIM IFs before and after inhibition of HOTAIR. HOTAIR knockdown resulted in a marked increase in the collapse of the VIM IF network around the nucleus (Fig. 6A). The percentage of the VIM IF network which collapsed in siHOTAIR-transfected cells (42.6 ± 3.7%) was significantly higher than that of the siNC-transfected group (16.1 ± 6.6%) (p < 0.01; Fig. 6B). As the VIM IF network is correlated with cell stiffness and shape (35), we next investigated the impact of HOTAIR knockdown on the organization of the VIM IF network. Inhibition of HOTAIR led to increased disorganization and entanglement of the VIM IF network, compared with cells transfected with control siRNA (Fig. 6C). Quantitative analysis also revealed that HOTAIR knockdown increased the diameter of VIM IFs significantly (Fig. 6D). An increase in the diameter of vimentin fibers and the collapse of the VIM network were also observed in HeLa-KD cells (Fig. 6). VIM overexpression has the same effects on the collapse of VIM IF network and the width of VIM fibers as HOTAIR knockdown has (Fig. 6). These results show that inhibition of HOTAIR expression can lead to the reorganization of the VIM IF network, and support the notion that various phenotypic effects observed after HOTAIR inhibition, at least in part, are because of the regulation of VIM expression.
Fig. 6.

Inhibition of HOTAIR or overexpression of vimentin affects organization of the vimentin IF network. A, Representative confocal microscopy images showing the organization of vimentin IF in HeLa cells after HOTAIR knockdown or VIM overexpression, or in HeLa-KD cells B, Quantification of vimentin IF collapse in cells in A. C, Representative cells showing organization of vimentin IF under super-resolution microscopy after HOTAIR knockdown or VIM overexpression, or in HeLa-KD cells. D, Quantification of the diameter of vimentin IFs in the cells in C. (Scale bars: 30 μm in A, 5 μm in C). At least three independent experiments were performed under each condition with at least 100 cells (B) or five 15 × 15 μm images of different cells (D) quantified per experiment. Data are presented as means ± S.D. and represent results from three independent experiments. *p < 0.05; **p < 0.01.
Inhibition of HOTAIR Lead to Mitochondrial Dysfunction
Thirty-two proteins associated with mitochondrial function were dysregulated after inhibition of HOTAIR. Notably, the expression of UQCRQ (Ubiquinol-Cytochrome C Reductase, Complex III Subunit VII), was markedly decreased (> fivefold) (Fig. 3E). ELISA assays confirmed the down-regulation of UQCR after HOTAIR inhibition (Fig. 7A). Increased ROS generation was also observed (Fig. 7B and 7C). These finding suggest that HOTAIR inhibition may cause the deficiency of complex III and impairment of the mitochondrial respiratory chain. Glucose metabolism is largely dependent on mitochondria to generate energy in cells and mitochondrial dysfunction could result in a reduced ability to uptake and utilize glucose (48). Examination of the effect of HOTAIR silencing on glucose uptake indicated that HOTAIR inhibition led to a significant decrease in glucose uptake in both transiently and stably transfected HeLa cells (Fig. 7D–7F and supplemental Fig. S7), suggesting that HOTAIR inhibition may lead to mitochondrial dysfunction. We further examined the ultrastructural changes of HeLa cells after HOTAIR inhibition. Mitochondrial swelling was observed after HOTAIR knockdown, which was followed by loss of cristae and the appearance of intracellular vacuoles that contain electron dense material (Fig. 7G). These changes were progressive, ultimately resulting in large particle-filled vacuoles and disappearance of mitochondria (Fig. 7G). The mitochondrial membrane potential (Δψ) in HOTAIR knockdown HeLa cells was assessed by staining mitochondria with MitoTracker Deep Red and decreased Δψ was shown after HOTAIR inhibition (Fig. 7H). Δψ was also evaluated using JC-1 dye, which has dual emission depending on the mitochondrial membrane potential. JC-1 forms aggregates in cells with a high FL2 fluorescence indicating a normal mitochondrial membrane potential. We observed a loss of Δψ at 48 h post-transfection of siHOTAIR, as reflected by a reduction in FL2 fluorescence with a concurrent gain in FL1 fluorescence as the dye shifts from an aggregate to monomeric state (Fig. 7I and 7J). These results indicated that HOTAIR knockdown in HeLa cells led to depolarization of Δψ. Taken together, these results suggest that HOTAIR is important for maintaining mitochondrial function in cancer cells.
Fig. 7.

Inhibition of HOTAIR leads to mitochondrial dysfunction. A, The concentration of UQCR in HeLa cells decreased after HOTAIR knockdown as measured using ELISA assays. B, Effect of HOTAIR knockdown on cellular ROS production as detected by flow cytometry analysis. C, Quantification of DCF fluorescence in HeLa cells. Data are presented as means ± S.D. and represent results from three independent experiments. *p < 0.05; **p < 0.01. D, Images of cellular glucose photographed under a confocal microscope. E, HOTAIR affects glucose uptake in HeLa cells. Cells had lower cellular glucose levels after HOTAIR knockdown. Glucose uptake was measured by FACS, following 0.5 h exposure to 2-NBDG (100 μm). F, Quantification of the fluorescence of cellular glucose in HeLa cells. Data are presented as means ± S.D. and represent results from three independent experiments. *p < 0.05; **p < 0.01. G, Electron microscopy images of (a) siNC cells and (b) siHOTAIR cells. Labels: M = mitochondria, n = nucleus. H, HOTAIR knockdown affects the mitochondrial membrane potential. Mitochondria were stained with MitoTracker Deep Red and representative images were obtained by confocal microscopy. I, HeLa cells were stained against JC-1 for flow cytometry after HOTAIR knockdown. There was a significant increase in the number of cells with green fluorescence (FL1 (R3)), indicating a decrease in the Δψ. J, The mean fluorescence intensity ratio (FL2/FL1) in HeLa cells after HOTAIR knockdown. CCCP was the positive control. Data are presented as means ± S.D. and represent results from three independent experiments. *p < 0.05; **p < 0.01.
DISCUSSION
HOTAIR is one of the few well-studied lncRNAs and has emerged as a key regulator of carcinogenesis and metastasis and a potential prognostic marker (20, 22, 49). Considerable attention has thus been given to determining its functions as well as to identifying its target genes. Previous studies have shown that HOTAIR can regulate diverse biological processes by modulating levels of hundreds of genes (22, 23). By comparison our proteomic data with previous transcriptomic results (3, 21–23, 41, 50, 51), among 170 proteins identified in the current study, only a few of them were also found to be altered at the mRNA level (supplemental Table S3). The poor correlation between proteomic and genomic results was not unexpected, as previous studies showed that there exists an ∼60 to 80% discordance between mRNA and protein abundances (24, 25). This lack of correlations could be the result of an array of posttranscriptional regulatory mechanisms. Therefore, the current proteomic study represents a complementary strategy for studies of mechanisms that underlie the functions of HOTAIR. In addition, the overlap between these transcriptomic data sets is also limited (Table S3). For example, in pancreatic cancer (23), the altered genes after HOTAIR dysregulation were very different from that in gastrointestinal stromal tumors (41) or small-cell lung cancer (51). Thus, we deduced that HOTAIR may exert its function in different cancer cells through different pathways. To the best of our knowledge, this study is the first to employ proteomic techniques to globally search for lncRNA regulated proteins in cancer cells. We performed quantitative SILAC experiments followed by bioinformatic analyses, and functional assays to identify proteins and pathways regulated by HOTAIR in HeLa cells.
Bioinformatic analyses suggest that many cytoskeletal and cytoskeleton related proteins are found in the 170 DEPs (Fig. 3A), and VIM, a key cytoskeletal protein, may play an important role in mediating the functions of HOTAIR in HeLa cells (Fig. 3B). VIM, an important member of the intermediate filament (IF) family, functions as an organizer of a number of critical proteins involved in attachment, the mitochondria, cell mobility and signaling transduction (52–55). There is growing evidence of an association between the expression of VIM and tumor invasiveness and aggressiveness (56). VIM is thus emerging as an indicator of poor prognosis for cancer patients and an attractive potential target for cancer therapy (47). The precise role of VIM in various fundamental processes remains elusive and the regulation of VIM is highly complex (57). For example, increased invasiveness and migration capacity in a variety of cancer cell types were observed when the expression of VIM is up-regulated (54, 58–60). By contrast, several clinical studies indicated that VIM expression was not associated with metastases in prostate cancer and breast cancer (61–63). In vitro studies also revealed that overexpression of VIM induced by stable transfection of expression constructs in tumoral cells was unable to enhance invasiveness in poorly invasive prostatic tumor (56). Consistent with our results, Li et al. reported that overexpression of vimentin suppresses the proliferative and invasive abilities of HepG2 cells and decrease malignant phenotype of tumor cells in vitro (64). It is likely that the impact of VIM disturbance may vary in different cancer types and cell types. It is thus of great interest that our findings show that HOTAIR functions as a regulator of VIM and that at least some of the various biological effects of HOTAIR are mediated by the regulation of VIM expression in HeLa cells. In agreement with published data, we found that HOTAIR knockdown leads to decreased invasiveness and migration and increased apoptosis of cancer cells, and that tumor growth is significantly inhibited in mice injected with HOTAIR-deficient cells. By investigating the impact of HOTAIR inhibition and VIM knockdown/overexpression in HeLa cells, we have shown that the effects of HOTAIR knockdown can be mimicked by manipulation of VIM expression. We have further shown that HOTAIR knockdown can lead to increases in the diameter and collapse of VIM fibers and VIM overexpression in HeLa cells has the similar effects (Fig. 6). Given that increased collapse of the VIM IF network leads to increased cellular stiffness (65) and decreased migration and invasion potential (47, 66–73), it is likely that the collapse of VIM IFs induced by HOTAIR knockdown results in increased cytoplasmic stiffness and decreased migration and invasion of HeLa cells. Together, these data suggest that VIM is part of a network of HOTAIR-regulated proteins that help cancer cells maintain homeostasis.
Our proteomic and functional studies have also demonstrated that HOTAIR regulates mitochondrial respiratory chain complex activities in HeLa cells. Notably, UQCRQ, a subunit of complex III, was significantly down-regulated and ELISA assays also detected a significant deficiency of complex III after inhibition of HOTAIR. It is well established that the main function of mitochondrial complex III is to accept electrons from ubiquinol, to transfer these to cytochrome c, and to translocate protons across the inner mitochondrial membrane; complex III deficiency thus causes mitochondrial dysfunction and ROS imbalance (74). Consistent with this notion, we found that HOTAIR inhibition leads to depolarization of mitochondrial membrane potential, decreased glucose uptake, mitochondrial ultrastructural abnormalities and enhanced intracellular ROS production (Fig. 7). Therefore, we speculate that HOTAIR inhibition may perturb mitochondrial function in HeLa cells. Mitochondria are highly dynamic and functionally versatile organelles that continuously fission and fuse in response to different physiological needs of the cell (75). Interestingly, it has been long recognized that the cytoskeletal network is fundamental in the positioning and movement of mitochondria (76–79) and VIM co-localizes and interacts with mitochondria to a greater extent than the other cytoskeletal components known to support mitochondria(80). VIM IFs can bind to mitochondria and anchor them with the cytoplasm, and subsequently change the shape, motility and potential of mitochondria (80–82). Modulation of mitochondrial morphology and dynamics has recently been shown to play an important role in cancer migration and invasion (83, 84). To meet the changing energy demands of HeLa cells after inhibition of HOTAIR, mitochondria require an efficient transport and tethering system. However, collapse of the VIM IF network combined with other cytoskeletal disturbances caused by inhibition of HOTAIR may impair mitochondria, and thus disrupt the dynamic equilibrium of mitochondrial energy metabolism. Mitochondrial dysfunction can lead to decreased migration and invasion of cancer cells (83, 84).
Based on our data, we propose a model depicting the molecular mechanism of HOTAIR in regulating migration and invasion of HeLa cells (Fig. 8). We suggest that HOTAIR promotes cell migration and invasion in cancer cells in at least three ways. Firstly, as reported by Tsai et al., HOTAIR may serve as a molecular scaffold linking the PRC2 and LSD1/CoREST/REST protein complexes to regulate hundreds of genes (12). Secondly, HOTAIR may regulate the expression and organization of VIM. As it is a central organizer of the cytoskeletal system, the collapse of the VIM IF network caused by inhibition of HOTAIR may disrupt the dynamic equilibrium of the mitochondrial redox balance. Thirdly, mitochondrial dysfunction could be a mechanism underlying decreased cell migration and invasion. The combination of all three mechanisms regulates the expression of hundreds of proteins and promotes cell migration and invasion of cancer cells and VIM may be a key molecule in HOTAIR-mediated oncogenic signaling. However, there are some phenotypic differences between HOTAIR inhibition and VIM knockdown/overexpression in cells with regard to their growth properties and levels of apoptosis. Thus, the complexity of the mechanism underlying HOTAIR action may be much greater than presently envisaged and some of the other proteins listed in supplemental Table S3 may be as important as VIM in the network of HOTAIR-regulated proteins.
Fig. 8.
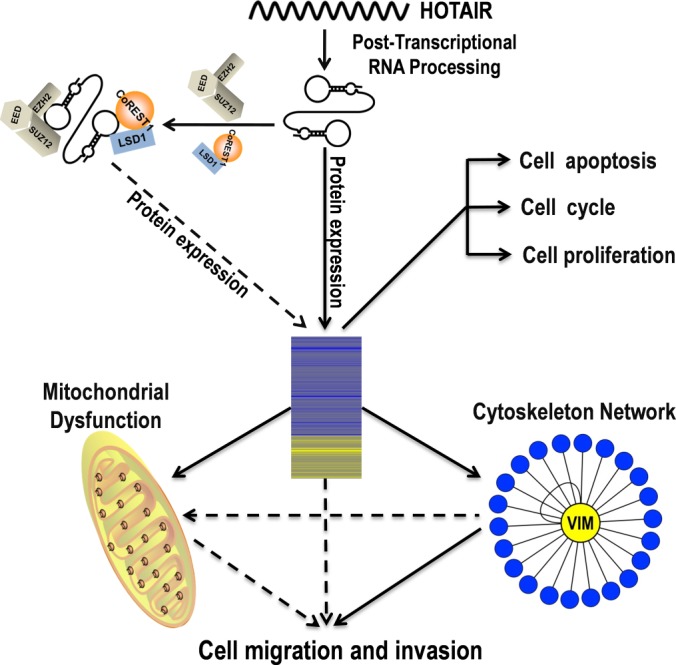
Proposed model depicting the molecular mechanism of HOTAIR in regulating migration and invasion of HeLa cells. HOTAIR promotes cell migration and invasion in HeLa cells via different mechanisms. HOTAIR may serve as a molecular scaffold linking two distinct histone modification complexes to regulate hundreds of genes (12). HOTAIR regulates the expression and organization of vimentin. Our functional study demonstrated that vimentin contributes to the decreased migration and invasion capability of HeLa cells caused by inhibition of HOTAIR. The mitochondrial dysfunction caused by inhibition of HOTAIR may be another cause of the decreased migration and invasion capability of HeLa cells. The combination of all these mechanisms regulates the expression of hundreds of proteins and promotes cell migration and invasion. Vimentin may be a key molecule in HOTAIR-mediated oncogenic signaling.
In conclusion, we have performed the first proteomic analysis of lncRNA regulated proteins in cancer cells and demonstrated that SILAC is an efficient and reliable method for functionally identifying lncRNA regulated proteins. We expect that the methods used in this study will become an integral part of functional studies of lncRNAs. Using this proteomic approach we identified proteins regulated by HOTAIR and showed by further experiments that HOTAIR exerts its effects on migration and invasion of cancer cells, at least in part, through the regulation of VIM. The elucidation of the role of VIM in the HOTAIR-regulated protein network may shed light on the molecular basis of HOTAIR induced-tumor proliferation and metastasis. It is now important to further characterize the interactions between HOTAIR and individual target proteins and to determine the full significance of gene regulation by HOTAIR in cancer cells.
Supplementary Material
Footnotes
Author contributions: L.B. and F.G. designed research; P.Z., Q.X., Y.W., Y.C., and D.G. performed research; P.Z., Q.X., Y.W., Y.C., Z.C., J.F., D.G., and F.G. analyzed data; P.Z., Q.X., J.F., L.B., and F.G. wrote the paper.
* This work was supported by the National Natural Science Foundation of China (Grant No. 31370746), the National Basic Research Program of China (973 Program, 2012CB518700), and the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB14030202).
 This article contains supplemental Figs. S1 to S7 and Tables S1 to S6.
This article contains supplemental Figs. S1 to S7 and Tables S1 to S6.
1 The abbreviations used are:
- ncRNA
- noncoding RNA
- lncRNA
- long noncoding RNA
- miRNA
- microRNA
- HOTAIR
- hoxtranscript antisense intergenicRNA
- nt
- nucleotide
- VIM
- vimentin
- PPI
- protein–protein interaction
- MRM
- multiple reaction monitoring.
REFERENCES
- 1. Esteller M. (2011) Non-coding RNAs in human disease. Nat. Rev. Genet. 12, 861–874 [DOI] [PubMed] [Google Scholar]
- 2. Kugel J. F., Goodrich J. A. (2012) Non-coding RNAs: key regulators of mammalian transcription. Trends Biochem. Sci. 37, 144–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Geisler S., Coller J. (2013) RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 14, 699–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ernst C., Morton C. C. (2013) Identification and function of long non-coding RNA. Fronti. Cell Neurosci. 7, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ponting C. P., Oliver P. L., Reik W. (2009) Evolution and functions of long noncoding RNAs. Cell 136, 629–641 [DOI] [PubMed] [Google Scholar]
- 6. Moran V. A., Perera R. J., Khalil A. M. (2012) Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res. 40, 6391–6400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jia H., Osak M., Bogu G. K., Stanton L. W., Johnson R., Lipovich L. (2010) Genome-wide computational identification and manual annotation of human long noncoding RNA genes. RNA 16, 1478–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kino T., Hurt D. E., Ichijo T., Nader N., Chrousos G. P. (2010) Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci. Signal. 3, ra8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hu W., Yuan B., Flygare J., Lodish H. F. (2011) Long noncoding RNA-mediated anti-apoptotic activity in murine erythroid terminal differentiation. Gene. Dev. 25, 2573–2578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meola N., Pizzo M., Alfano G., Surace E. M., Banfi S. (2012) The long noncoding RNA Vax2os1 controls the cell cycle progression of photoreceptor progenitors in the mouse retina. RNA 18, 111–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang K. C., Yang Y. W., Liu B., Sanyal A., Corces-Zimmerman R., Chen Y., Lajoie B. R., Protacio A., Flynn R. A., Gupta R. A., Wysocka J., Lei M., Dekker J., Helms J. A., Chang H. Y. (2011) A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 472, 120–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tsai M. C., Manor O., Wan Y., Mosammaparast N., Wang J. K., Lan F., Shi Y., Segal E., Chang H. Y. (2010) Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Loewer S., Cabili M. N., Guttman M., Loh Y. H., Thomas K., Park I. H., Garber M., Curran M., Onder T., Agarwal S., Manos P. D., Datta S., Lander E. S., Schlaeger T. M., Daley G. Q., Rinn J. L. (2010) Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat. Genet. 42, 1113–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guttman M., Donaghey J., Carey B. W., Garber M., Grenier J. K., Munson G., Young G., Lucas A. B., Ach R., Bruhn L., Yang X., Amit I., Meissner A., Regev A., Rinn J. L., Root D. E., Lander E. S. (2011) lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 477, 295–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gregg C., Zhang J., Weissbourd B., Luo S., Schroth G. P., Haig D., Dulac C. (2010) High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science 329, 643–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tripathi V., Ellis J. D., Shen Z., Song D. Y., Pan Q., Watt A. T., Freier S. M., Bennett C. F., Sharma A., Bubulya P. A., Blencowe B. J., Prasanth S. G., Prasanth K. V. (2010) The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 39, 925–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Spizzo R., Almeida M. I., Colombatti A., Calin G. A. (2012) Long non-coding RNAs and cancer: a new frontier of translational research? Oncogene 31, 4577–4587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gibb E. A., Vucic E. A., Enfield K. S., Stewart G. L., Lonergan K. M., Kennett J. Y., Becker-Santos D. D., MacAulay C. E., Lam S., Brown C. J., Lam W. L. (2011) Human cancer long non-coding RNA transcriptomes. PloS One 6, e25915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang J., Zhang P., Wang L., Piao H. L., Ma L. (2014) Long non-coding RNA HOTAIR in carcinogenesis and metastasis. Acta Bioch. Bioph. Sin. 46, 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wan Y., Chang H. Y. (2010) HOTAIR: Flight of noncoding RNAs in cancer metastasis. Cell Cycle 9, 3391–3392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li L., Liu B., Wapinski O. L., Tsai M. C., Qu K., Zhang J., Carlson J. C., Lin M., Fang F., Gupta R. A., Helms J. A., Chang H. Y. (2013) Targeted disruption of Hotair leads to homeotic transformation and gene derepression. Cell Rep. 5, 3–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gupta R. A., Shah N., Wang K. C., Kim J., Horlings H. M., Wong D. J., Tsai M. C., Hung T., Argani P., Rinn J. L., Wang Y., Brzoska P., Kong B., Li R., West R. B., van de Vijver M. J., Sukumar S., Chang H. Y. (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim K., Jutooru I., Chadalapaka G., Johnson G., Frank J., Burghardt R., Kim S., Safe S. (2012) HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene 32, 1616–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schwanhausser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 25. Wu L., Candille S. I., Choi Y., Xie D., Jiang L., Li-Pook-Than J., Tang H., Snyder M. (2013) Variation and genetic control of protein abundance in humans. Nature 499, 79–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xiong Q., Zhong Q., Zhang J., Yang M., Li C., Zheng P., Bi L. J., Ge F. (2012) Identification of novel miR-21 target proteins in multiple myeloma cells by quantitative proteomics. J. Proteome Res. 11, 2078–2090 [DOI] [PubMed] [Google Scholar]
- 27. Mi H., Dong Q., Muruganujan A., Gaudet P., Lewis S., Thomas P. D. (2010) PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 38, D204–D210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huang D. W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 29. Huang D. W., Sherman B. T., Lempicki R. A. (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., von Mering C., Jensen L. J. (2013) STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808–D815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T. (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Medzihradszky K. F. (2005) In-solution digestion of proteins for mass spectrometry. Method Enzymol. 405, 50–65 [DOI] [PubMed] [Google Scholar]
- 33. Icenogle L. M., Hengel S. M., Coye L. H., Streifel A., Collins C. M., Goodlett D. R., Moseley S. L. (2012) Molecular and biological characterization of Streptococcal SpyA-mediated ADP-ribosylation of intermediate filament protein vimentin. J. Biol. Chem. 287, 21481–21491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rajaram N., Frees A. E., Fontanella A. N., Zhong J., Hansen K., Dewhirst M. W., Ramanujam N. (2013) Delivery Rate Affects Uptake of a Fluorescent Glucose Analog in Murine Metastatic Breast Cancer. PloS One 8, e76524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rathje L. S. Z., Nordgren N., Pettersson T., Ronnlund D., Widengren J., Aspenstrom P., Gad A. K. B. (2014) Oncogenes induce a vimentin filament collapse mediated by HDAC6 that is linked to cell stiffness. Proc. Natl. Acad. Sci. U.S.A. 111, 1515–1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gibb E. A., Brown C. J., Lam W. L. (2011) The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 10, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gutschner T., Diederichs S. (2012) The hallmarks of cancer: a long non-coding RNA point of view. RNA Bio. 9, 703–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wahlestedt C. (2013) Targeting long non-coding RNA to therapeutically upregulate gene expression. Nat. Rev. Drug Discov. 12, 433–446 [DOI] [PubMed] [Google Scholar]
- 39. Geng Y. J., Xie S. L., Li Q., Ma J., Wang G. Y. (2011) Large Intervening Non-coding RNA HOTAIR is Associated with Hepatocellular Carcinoma Progression. J. Int. Med. Res. 39, 2119–2128 [DOI] [PubMed] [Google Scholar]
- 40. Kogo R., Shimamura T., Mimori K., Kawahara K., Imoto S., Sudo T. (2012) Long Noncoding RNA HOTAIR Regulates Polycomb-Dependent Chromatin Modification and Is Associated with Poor Prognosis in Colorectal Cancers (vol 71, pg 6320, 2011). Cancer Res. 72, 1039–1039 [DOI] [PubMed] [Google Scholar]
- 41. Niinuma T., Suzuki H., Nojima M., Nosho K., Yamamoto H., Takamaru H., Yamamoto E., Maruyama R., Nobuoka T., Miyazaki Y., Nishida T., Bamba T., Kanda T., Ajioka Y., Taguchi T., Okahara S., Takahashi H., Nishida Y., Hosokawa M., Hasegawa T., Tokino T., Hirata K., Imai K., Toyota M., Shinomura Y. (2012) Upregulation of miR-196a and HOTAIR Drive Malignant Character in Gastrointestinal Stromal Tumors. Cancer Res. 72, 1126–1136 [DOI] [PubMed] [Google Scholar]
- 42. Mi H., Guo N., Kejariwal A., Thomas P. D. (2007) PANTHER version 6: protein sequence and function evolution data with expanded representation of biological pathways. Nucleic Acids Res. 35, D247–D252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim K., Kim Y. (2009) Preparing multiple-reaction monitoring for quantitative clinical proteomics. Expert Rev. Proteomic. 6, 225–229 [DOI] [PubMed] [Google Scholar]
- 44. Kitteringham N. R., Jenkins R. E., Lane C. S., Elliott V. L., Park B. K. (2009) Multiple reaction monitoring for quantitative biomarker analysis in proteomics and metabolomics. J. Chromatogr. B 877, 1229–1239 [DOI] [PubMed] [Google Scholar]
- 45. Li D., Feng J., Wu T., Wang Y., Sun Y., Ren J., Liu M. (2013) Long intergenic noncoding RNA HOTAIR is overexpressed and regulates PTEN methylation in laryngeal squamous cell carcinoma. Am. J. Pathol. 182, 64–70 [DOI] [PubMed] [Google Scholar]
- 46. Huang J., Ke P., Guo L., Wang W., Tan H., Liang Y., Yao S. (2014) Lentivirus-mediated RNA interference targeting the long noncoding RNA HOTAIR inhibits proliferation and invasion of endometrial carcinoma cells in vitro and in vivo. Int. J. Gynecol. Cancer 24, 635–642 [DOI] [PubMed] [Google Scholar]
- 47. Satelli A., Li S. (2011) Vimentin in cancer and its potential as a molecular target for cancer therapy. CMLS- Cell Mol. Life S. 68, 3033–3046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim J. A., Wei Y., Sowers J. R. (2008) Role of mitochondrial dysfunction in insulin resistance. Circ. Res. 102, 401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lv D. W., Ge P., Zhang M., Cheng Z. W., Li X. H., Yan Y. M. (2014) Integrative network analysis of the signaling cascades in seedling leaves of bread wheat by large-scale phosphoproteomic profiling. J. Proteome Res. 13, 2381–2395 [DOI] [PubMed] [Google Scholar]
- 50. Ge X. S., Ma H. J., Zheng X. H., Ruan H. L., Liao X. Y., Xue W. Q., Chen Y. B., Zhang Y., Jia W. H. (2013) HOTAIR, a prognostic factor in esophageal squamous cell carcinoma, inhibits WIF-1 expression and activates Wnt pathway. Cancer Sci. 104, 1675–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ono H., Motoi N., Nagano H., Miyauchi E., Ushijima M., Matsuura M., Okumura S., Nishio M., Hirose T., Inase N. (2014) Long noncoding RNA HOTAIR is relevant to cellular proliferation, invasiveness, and clinical relapse in small-cell lung cancer. Cancer Medicine 3, 632–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tzivion G., Luo Z. J., Avruch J. (2000) Calyculin A-induced vimentin phosphorylation sequesters 14-3-3 and displaces other 14-3-3 partners in vivo. J. Biol. Chem. 275, 29772–29778 [DOI] [PubMed] [Google Scholar]
- 53. Hendrix MJ, S. E., Seftor RE, Trevor KT. (1997) Experimental coexpression of vimentin and keratin intermediate filaments in human breast cancer cells results in phenotypic interconversion and increased invasive behavior. Am. J. Pathol. 150, 483–495 [PMC free article] [PubMed] [Google Scholar]
- 54. Gilles C., Polette M., Zahm J. M., Tournier J. M., Volders L., Foidart J. M., Birembaut P. (1999) Vimentin contributes to human mammary epithelial cell migration. J. Cell Sci. 112, 4615–4625 [DOI] [PubMed] [Google Scholar]
- 55. Katsumoto T., Mitsushima A., Kurimura T. (1990) The role of the vimentin intermediate filaments in rat 3Y1 cells elucidated by immunoelectron microscopy and computer-graphic reconstruction. Biol. Cell 68, 139–146 [DOI] [PubMed] [Google Scholar]
- 56. Yamasaki T., Seki N., Yamada Y., Yoshino H., Hidaka H., Chiyomaru T., Nohata N., Kinoshita T., Nakagawa M., Enokida H. (2012) Tumor suppressive microRNA138 contributes to cell migration and invasion through its targeting of vimentin in renal cell carcinoma. Int. J. Oncol. 41, 805–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ivaska J., Pallari H. M., Nevo J., Eriksson J. E. (2007) Novel functions of vimentin in cell adhesion, migration, and signaling. Exp. Cell Res. 313, 2050–2062 [DOI] [PubMed] [Google Scholar]
- 58. Lang SH, H. C., Reid IN, Hitchcock IS, Hart CA, Bryden AA, Villette JM, Stower MJ, Maitland NJ. (2002) September 1) Enhanced expression of vimentin in motile prostate cell lines and in poorly differentiated and metastatic prostate carcinoma. Prostate. 52, 253–263 [DOI] [PubMed] [Google Scholar]
- 59. Chu YW, S. E., Romer LH, Hendrix MJ. (1996) Experimental coexpression of vimentin and keratin intermediate filaments in human melanoma cells augments motility. Am. J. Pathol. 148, 63–69 [PMC free article] [PubMed] [Google Scholar]
- 60. Trog D, Y. K., Schild HH, Golubnitschaja O. (2008) May) Up-regulation of vimentin expression in low-density malignant glioma cells as immediate and late effects under irradiation and temozolomide treatment. Amino Acids. 34, 539–545 [DOI] [PubMed] [Google Scholar]
- 61. Heatley M., Maxwell P., Whiteside C., Toner P. (1995) Vimentin and cytokeratin expression in nodular hyperplasia and carcinoma of the prostate. J. Clin. Pathol. 48, 1031–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Seshadri R., Raymond W. A., Leong A. S. Y., Horsfall D. J., McCaul K. (1996) Vimentin expression is not associated with poor prognosis in breast cancer. Int. J. Cancer 67, 353–356 [DOI] [PubMed] [Google Scholar]
- 63. Raymond W. A., Leong A. S. (1989) Vimentin–a new prognostic parameter in breast carcinoma? J. Pathol. 158, 107–114 [DOI] [PubMed] [Google Scholar]
- 64. Li ZM, W. Y., Yang HB, Qin G, Tian L, Deng HX, Wen B. (2008) June) Enhanced expression of human vimentin intermediate filaments in hepatocellular carcinoma cells decreases their proliferative and invasive abilities in vitro. Zhonghua Zhong Liu Za Zhi. 30, 408–412 [PubMed] [Google Scholar]
- 65. Plodinec M., Loparic M., Suetterlin R., Herrmann H., Aebi U., Schoenenberger C. A. (2011) The nanomechanical properties of rat fibroblasts are modulated by interfering with the vimentin intermediate filament system. J. Struct. Bio. 174, 476–484 [DOI] [PubMed] [Google Scholar]
- 66. Cross S. E., Jin Y. S., Tondre J., Wong R., Rao J., Gimzewski J. K. AFM-based analysis of human metastatic cancer cells. Nanotechnology 19:384003, 2008 [DOI] [PubMed] [Google Scholar]
- 67. Lekka M., Laidler P. (2009) Applicability of AFM in cancer detection. Nat. Nanotechnol. 4, 72–72 [DOI] [PubMed] [Google Scholar]
- 68. Xu W., Mezencev R., Kim B., Wang L., McDonald J., Sulchek T. (2012) Cell stiffness is a biomarker of the metastatic potential of ovarian cancer cells. PloS One 7, e46609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ochalek T., Nordt F. J., Tullberg K., Burger M. M. (1988) Correlation between cell deformability and metastatic potential in B16-F1 melanoma cell variants. Cancer Res. 48, 5124–5128 [PubMed] [Google Scholar]
- 70. Watanabe T., Kuramochi H., Takahashi A., Imai K., Katsuta N., Nakayama T., Fujiki H., Suganuma M. (2012) Higher cell stiffness indicating lower metastatic potential in B16 melanoma cell variants and in (-)-epigallocatechin gallate-treated cells. J. Cancer Res. Clin. 138, 859–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Swaminathan V., Mythreye K., O'Brien E. T., Berchuck A., Blobe G. C., Superfine R. (2011) Mechanical stiffness grades metastatic potential in patient tumor cells and in cancer cell lines. Cancer Res. 71, 5075–5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Remmerbach T. W., Wottawah F., Dietrich J., Lincoln B., Wittekind C., Guck J. (2009) Oral Cancer Diagnosis by Mechanical Phenotyping. Cancer Res. 69, 1728–1732 [DOI] [PubMed] [Google Scholar]
- 73. Ward K. A., Li W. I., Zimmer S., Davis T. (1991) Viscoelastic properties of transformed cells: role in tumor cell progression and metastasis formation. Biorheology 28, 301–313 [DOI] [PubMed] [Google Scholar]
- 74. Klimova T., Chandel N. S. (2008) Mitochondrial complex III regulates hypoxic activation of HIF. Cell Death Differ. 15, 660–666 [DOI] [PubMed] [Google Scholar]
- 75. Otera H., Ishihara N., Mihara K. (2013) New insights into the function and regulation of mitochondrial fission. BBA-Mol. Cell Res. 1833, 1256–1268 [DOI] [PubMed] [Google Scholar]
- 76. Heggeness M. H., Simon M., Singer S. J. (1978) Association of mitochondria with microtubules in cultured cells. Proc. Natl. Acad. Sci. U.S.A. 75, 3863–3866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. van der Bliek A. M., Shen Q., Kawajiri S. Mechanisms of mitochondrial fission and fusion. CSH Perspect Biol. 5:a011072, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wu M., Kalyanasundaram A., Zhu J. (2013) Structural and biomechanical basis of mitochondrial movement in eukaryotic cells. Int. J. Nanomed. 8, 4033–4042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kidd M. E., Shumaker D. K., Ridge K. M. (2014) The Role of Vimentin Intermediate Filaments in the Progression of Lung Cancer. Am. J. Resp. Cell Mol. 50, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tang H. L., Lung H. L., Wu K. C., Le A. H. P., Tang H. M., Fung M. C. (2008) Vimentin supports mitochondrial morphology and organization. Biochem. J. 410, 141–146 [DOI] [PubMed] [Google Scholar]
- 81. Matveeva E. A., Chernoivanenko I. S., Minin A. A. (2010) Vimentin intermediate filaments protect mitochondria from oxidative stress. Biol. Membrany 27, 471–481 [Google Scholar]
- 82. Nekrasova O. E., Mendez M. G., Chernoivanenko I. S., Tyurin-Kuzmin P. A., Kuczmarski E. R., Gelfand V. I., Goldman R. D., Minin A. A. (2011) Vimentin intermediate filaments modulate the motility of mitochondria. Mol. Biol. Cell 22, 2282–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Desai S. P., Bhatia S. N., Toner M., Irimia D. (2013) Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophy. J. 104, 2077–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhao J., Zhang J., Yu M., Xie Y., Huang Y., Wolff D. W., Abel P. W., Tu Y. (2013) Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 32, 4814–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.