

Abstract
Tumor necrosis factor alpha (TNF-α) is known to exacerbate ischemic brain injury; however, the mechanism is unknown. Previous studies have evaluated the effects of TNF-α on neurons with long exposures to high doses of TNF-α, which is not pathophysiologically relevant. We characterized the rapid effects of TNF-α on basal respiration, ATP production, and maximal respiration using pathophysiologically relevant, post-stroke concentrations of TNF-α. We observed a reduction in mitochondrial function as early as 1.5 h after exposure to low doses of TNF-α, followed by a decrease in cell viability in HT-22 cells and primary neurons. Subsequently, we used the HT-22 cell line to determine the mechanism by which TNF-α causes a rapid and profound reduction in mitochondrial function. Pre-treating with TNF-R1 antibody, but not TNF-R2 antibody, ameliorated the neurotoxic effects of TNF-α, indicating that TNF-α exerts its neurotoxic effects through TNF-R1. We observed an increase in caspase 8 activity and a decrease in mitochondrial membrane potential after exposure to TNF-α which resulted in a release of cytochrome c from the mitochondria into the cytosol. These novel findings indicate for the first time that an acute exposure to pathophysiologically relevant concentrations of TNF-α has neurotoxic effects mediated by a rapid impairment of mitochondrial function.
Keywords: cytokines, inflammation, mitochondria, neurotoxicity, stroke, TNF-alpha
Inflammatory mechanisms play a crucial role in the pathophysiologic processes after the onset of ischemic stroke (Vila et al. 2000). Ischemic brain injury is complex, and intracellular signaling that regulates innate and adaptive immunity, inflammation, cell death, angiogenesis, and repair processes plays an important role in the initiation, progression, and resolution of ischemia. Initiation of these critical intracellular regulators occurs through rapid cytokine signaling after ischemia, highlighting cytokines as key regulators of stroke damage (Hallenbeck 2002). Tumor necrosis factor alpha (TNF-α) is one of many pro-inflammatory cytokines associated with worsened clinical outcomes after stroke and exacerbations of infarct size in pre-clinical models (Nawashiro et al. 1997; Ormstad et al. 2011). However, our understanding of the pro-inflammatory effects of TNF-α is based primarily on studies of its peripheral actions.
TNF-α mRNA increases within 1 h in the ischemic injury core, and the expression of its immunoreactive protein increases within 2–6 h after the onset of ischemia in preclinical models (Botchkina et al. 1997). TNF-α is increased in the serum of stroke patients between 6 and 12 h after symptom onset (Liu et al. 1994; Sotgiu et al. 2006). TNF-α signaling occurs through the TNF-α receptor (TNF-R), where ligand binding recruits adaptor proteins to a core signaling complex allowing for differences in signal transduction depending upon the stimulatory pattern (Wallach et al. 2002).
The response to TNF-α is cell type dependent. In bovine and rat oligodendrocytes, TNF-α induces apoptosis, while in astrocytes, TNF-α enhances major histocompatibility complex class II and intracellular adhesion molecule 1 expression (Chao et al. 1995). In primary septo-hippocampal cultures, exposure to 30 ng/mL of TNF-α caused significant cytotoxicity but only with the addition of Actinomycin-D (Zhao et al. 2001). When rat primary cortical neurons are exposed to 10 ng/mL of TNF-α for 24 h, neurite retraction or formation of apoptotic bodies was not observed (Reimann-Philipp et al. 2001). However, in PC12 cells, TNF-α was cytotoxic (Reimann-Philipp et al. 2001). While these studies show neurotoxic effects of TNF-α, cultured embryonic rat hippocampal, septal, and cortical neurons are protected from glucose deprivation-induced injury and excitatory amino acid toxicity by exposure to TNF-α (Cheng et al. 1994). Furthermore, TNF-α can induce the expression of anti-apoptotic proteins B-cell lymphoma 2 and B-cell lymphoma-extra large (Bcl-xl) in hippocampal neurons (Tamatani et al. 1999). These few studies used high concentrations and long periods of exposure to TNF-α. Also, there is a single study of adipocytes that used high concentrations and 4-day exposures to assess mitochondrial function (Chen et al. 2010).
In the present study, we tested the hypothesis that acute exposure to TNF-α concentrations seen in serum of stroke patients (Lambertsen et al. 2012; Nayak et al. 2012) causes neuronal cell death through rapid mitochondrial dysfunction. As such, we characterized the effect of acute exposure to low doses of TNF-α in a mouse hippocampal neuronal cell line (HT-22) and mouse primary cortical neurons. A rapid and profound mitochondrial dysfunction was observed after as little as 1.5 h of exposure to TNF-α which preceded cell death. These findings suggest that the damaging effect of TNF-α may be because of the impairment of neuronal mitochondrial function.
Materials and methods
Cell culture
An immortalized mouse hippocampal cell line, HT-22, was cultured in Hyclone Dulbecco’s modified Eagle’s medium (DMEM)/high glucose (Fisher Scientific, Waltham, MA, USA) with 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA, USA) and 1% penicillin/streptomycin (Fisher Scientific). The HT-22 mouse hippocampal neuronal cell line was provided by The Salk Institute for Biological Research (La Jolla, CA, USA), and cell passages 15–23 were used. When HT-22 cells were at least 80% confluent, the cells were trypsinized and spun down at 325.5 × g for 3 min. The cells were counted with Nexcelom Bioscience Cellometer AutoT4 (Lawrence, MA, USA). Primary mouse cortical neurons were purchased from Life Technologies (Carlsbad, CA, USA) and cultured in Neurobasal® Medium supplemented with GlutaMAX™ – I and B-27® supplement (Life Technologies).
TNF-α treatment
Recombinant Mouse TNF-α was purchased from R&D Systems (Minneapolis, MN, USA) and reconstituted at 50 μg/mL in phosphate-buffered saline (PBS) containing 0.1% bovine serum albumin. Dilutions were made in Hyclone DMEM/high glucose with 10% FBS and 1% penicillin/streptomycin to obtain concentrations of 1, 10, 100, and 1000 pg/mL. The media was removed from each well by gentle aspiration. After removing the media, 100 μL of Hyclone DMEM/high glucose or the various concentrations of TNF-α were added for either 1.5, 3, 6, or 12 h. These time points were chosen based upon the post-stroke increase in TNF-α in serum patients and in the brains of animal models (Liu et al. 1994; Botchkina et al. 1997; Zaremba and Losy 2001; Intiso et al. 2003). For primary mouse cortical neurons, TNF-α at concentrations of 0, 100, and 1000 pg/mL were added for 1.5, 3, 6 and 24 h.
Mitochondrial function assessment
About 15 000 HT-22 cells or 16 000 primary mouse cortical neurons were seeded in a XFe96 cell culture microplate. HT-22 cells were treated with 1–1000 pg/mL of TNF-α (six replicates per treatment condition) 24 h after seeding. Primary mouse cortical neurons were treated with 100 or 1000 pg/mL of TNF-α 3 days after seeding. After treating with TNF-α, mitochondrial function was assessed with the XFe 96 Analyzer (Seahorse Bioscience, North Billerica, MA, USA) using a Mito Stress test kit at 1.5, 3, 6, and 12 h (HT-22 cells) or 1.5, 3, 6, and 24 h (primary neurons). Non-mitochondrial-derived oxygen consumption rate (OCR) was measured using the first measurement after addition of rotenone and antimycin a. To calculate basal respiration, the measurement prior to oligomycin addition was subtracted from non-mitochondrial-derived OCR. Proton leak was measured using the third measurement after oligomycin injection subtracted from non-mitochondrial-derived OCR. ATP production was measured from subtracting proton leak from basal respiration. Maximal respiration was calculated using the first measurement after trifluorocarbonylcyanide phenylhydrazone (FCCP) injection subtracted from non-mitochondria-derived OCR.
Cell viability
Cell viability was assessed using Calcein AM (Life Technologies), and reconstituted at 2 mM in dimethylsulfoxide. About 15 000 HT-22 cells or 16 000 primary mouse cortical neurons were seeded in a black-walled clear bottom 96 well plate. HT-22 cells were exposed to 1–1000 pg/mL of TNF-α and the primary mouse cortical neurons were treated with 100 or 1000 pg/mL of TNF-α. After exposure to TNF-α for 1.5, 3, 6, and 12 h (HT-22 cells) or 1.5, 3, 6, and 24 h (primary neurons), the plate was washed three times with PBS 1X. A total of 100 μLof 1 μM Calcein AM was added to the wells. The plate was incubated at 22°C in the dark for 30 min. The plate was read using a BioTek Synergy H1 Hybrid reader (Winooski, VT, USA).
Flow cytometry
HT-22 cells were grown to 90% confluence, a single cell suspension was made using cell dissociation buffer, and the cells were fixed in 10% formaldehyde for 30 min and subsequently permeabilized in 70% ethanol for 30 min on ice. To reduce non-specific antibody binding, HT-22 cells were blocked in 3% bovine serum albumin in PBS for 15 min and subsequently incubated with 1 μg PE anti-mouse CD120a (TNF R Type I/p55) or PE anti-mouse CD120b (TNF R Type II/p75) from Biolegend (San Diego, CA, USA). Data were acquired by counting 10 000 events and analyzed using FACSCalibur (BD Biosciences, San Jose, CA, USA) to determine the presence or absence of TNF-R1 and/or TNF-R2.
TNF-R1 or TNF-R2 antibody treatment
HT-22 cells were pre-treated for 1 h with 4, 6, or 8 μg/mL of LEAF™ Purified anti-mouse CD120a (TNF R Type I/p55), 4, 6, or 8 μg/mL of LEAF™ Purified anti-mouse CD120b (TNF R Type II/ p75), or 8 μg/mL of LEAF™ Purified Armenian Hamster IgG Isotype Ctrl from Biolegend. Media was removed and cells were then treated with 100 pg/mL of TNF-α for 1.5 h or 3 h. Cell viability was assessed with Calcein AM at both 1.5 and 3 h, and mitochondrial basal respiration was assessed with the XFe 96 Analyzer from Seahorse Bioscience at 1.5 h.
Caspase 8 and caspase 3/7 activity
Caspase 8 and Caspase3/7 activity was assessed with the Caspase-Glo™ 8 and Caspase-Glo™ 3/7 assay kits (Promega, Madison, WI, USA) after treating HT-22 cells with 1, 10, 100, 1000 pg/mL of TNF-α for 1.5 h.
TMRE mitochondrial membrane potential assay
Mitochondrial membrane potential was assayed using a TMRE mitochondrial membrane potential assay (Abcam, Cambridge, MA, USA) by addition of 300 nM TMRE and 20 μM FCCP after treating HT-22 cells with 1, 10, 100, 1000 pg/mL of TNF-α for 1.5 h.
Cytochrome c release from mitochondria to cytosol
After exposure to 100 or 1000 pg/mL of TNF-α for 3, 6, 12, and 24 h, HT-22 cells were fractioned into cytosolic and mitochondrial fractions using the Mitochondrial/Cytosol Fractionation Kit (Biovision, Milpitas, CA, USA). The protein concentration of each fraction was determined using the Pierce 660 nm protein assay (Thermo Scientific, Waltham, MA, USA). Cytochrome c levels in the cytosolic and mitochondrial fractions were determined using a rat/mouse cytochrome c Quantikine ELISA (R&D Systems, Minneapolis, MN, USA). Percentage release of cytochrome c was calculated using the following equation: total cytosolic cytochrome c/(cytosolic + mitochondrial cytochrome c) (Tanaka et al. 2004).
Statistical analysis
One-way ANOVA and post hoc analyses (Dunnett or Trend for linear analysis) were used to analyze differences among groups using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA, USA). Statistical significance was determined at a p < 0.05.
Results
TNF-α caused a decrease in mitochondrial function at 1.5, 3, 6, and 12 h in HT-22 cells
Figure 1 (a, c, e, g) and Figure S1 show the dose-dependent decrease in basal respiration, ATP production, and maximal respiration at 1.5, 3, 6, and 12 h of TNF-α exposure. At 1.5 h, basal respiration, ATP production, and maximal respiration decreased by approximately 30%, and 40% at 100 and 1000 pg/mL TNF-α, respectively (Fig. 1a and Figure S1a). At 3 and 12 h, but not 6 h, the magnitude of the dose-dependent reduction in basal respiration, ATP production, and maximal respiration induced by TNF-α was increased (Fig. 1 c, e, g and Figure Sb–d).
Fig. 1.
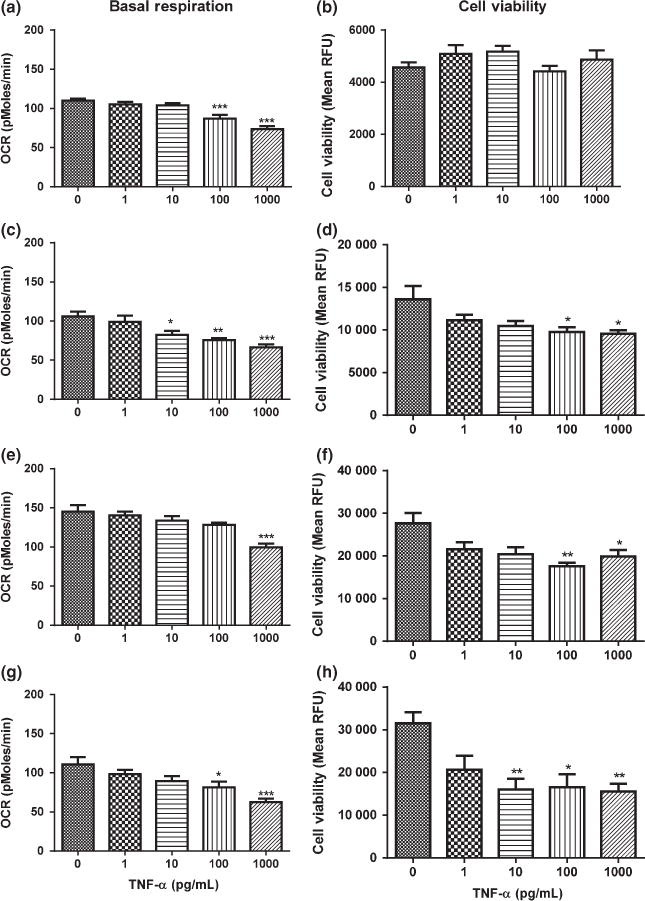
Effects of tumor necrosis factor alpha (TNF-α) on basal respiration and cell viability in HT-22 cells. (a) Basal respiration after exposure to TNF-α for 1.5 h. (b) Cell viability after exposure to TNF-α for 1.5 h. (c) Basal respiration after exposure to TNF-α for 3 h. (d) Cell viability after exposure to TNF-α for 3 h. (e) Basal respiration after exposure to TNF-α for 6 h. (f) Cell viability after exposure to TNF-α for 6 h. (g) Basal respiration after exposure to TNF-α for 12 h. (h) Cell viability after exposure to TNF-α for 12 h. Analysis of variance and Dunnett post hoc tests were used to assess significance. *p < 0.05; **p < 0.01; ***p < 0.001. Depicted are mean + SEM for n = 6/group.
TNF-α caused a delayed decrease in cell viability in HT-22 cells
Based on our observation of decreased mitochondrial function as early as 1.5 h of TNF-α exposure, we determined if the decrease in mitochondrial function preceded cell death. We did not observe a significant decrease in cell viability after 1.5 h of exposure to TNF-α (Fig. 1b). However, cell viability began to decrease after 3 h by approximately 30% at doses of 100 and 1000 pg/mL of TNF-α (Fig. 1d). Additionally, cell viability decreased significantly at 6 and 12 h (Fig. 1f and h). At 6 h, 100 and 1000 pg/mL of TNF-α caused a decrease in cell viability of approximately 37%. At 12 h, 10, 100, and 1000 pg/mL caused a decrease in cell viability of approximately 50%.
TNF-α caused a rapid decrease in mitochondrial function and a delayed decrease in cell viability in mouse primary cortical neurons
To confirm the rapid and profound effects of low doses of TNF-α on mitochondrial function was not specific to a transformed cell line, we exposed mouse primary cortical neurons to low doses of TNF-α for 1.5, 3, 6, and 24 h. After exposure to 100 pg/mL and 1000 pg/mL of TNF-α for 1.5 h, basal respiration was decreased by 48% and 61%, respectively without causing cell death (Fig. 2a and b). Acute exposure to low doses of TNF-α did not decrease cell viability until 24 h after exposure to 1000 pg/mL of TNF-α (Fig. 2h). Since TNF-α rapidly decreases mitochondrial function prior to cell death in both HT-22 cells and mouse primary cortical neurons, we used the HT-22 cell line to determine the mechanism by which TNF-α rapidly affects mitochondrial function.
Fig. 2.
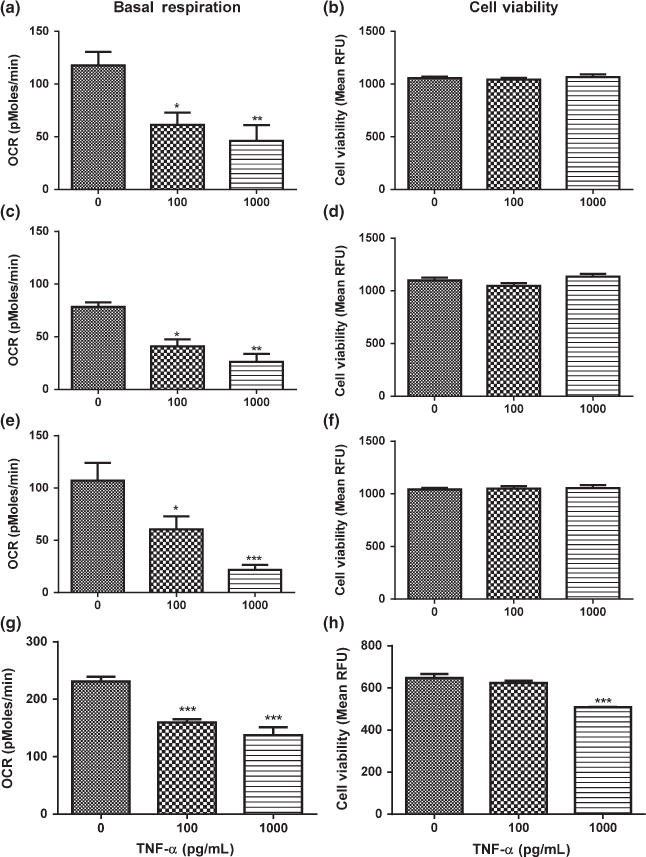
Effects of tumor necrosis factor alpha (TNF-α) on basal respiration and cell viability in mouse primary cortical neurons. (a) Basal respiration after exposure to TNF-α for 1.5 h. (b) Cell viability after exposure to TNF-α for 1.5 h. (c) Basal respiration after exposure to TNF-α for 3 h. (d) Cell viability after exposure to TNF-α for 3 h. (e) Basal respiration after exposure to TNF-α for 6 h. (f) Cell viability after exposure to TNF-α for 6 h. (g) Basal respiration after exposure to TNF-α for 24 h. (h) Cell viability after exposure to TNF-α for 24 h. Analysis of variance and Dunnett post hoc tests were used to assess significance. *p < 0.05; **p<0.01; ***p< 0.001. Depicted are mean + SEM for n = 6/group. When SEM is not depicted, it is too small for representation.
HT-22 cells express TNF-R1 and TNF-R2
It is known that TNF-α mediates its effects through TNF-R1 and TNF-R2. There is expression of TNF-R1 on all cell types, while expression of TNF-R2 is primarily on hemopoietic and endothelial cells (Maddahi et al. 2011). To determine the expression pattern of TNF-α receptors on HT-22 cells, flow cytometry analysis of HT-22 cells with antibodies specific for TNF-R1 and TNF-R2 was performed. HT-22 cells expressed both TNF-R1 (24.56%) and TNF-R2 (87.17%) under normal culture conditions (Figure S2).
TNF-α exerts its neurotoxic effects through TNF-R1
Pre-treating HT-22 cells with 4, 6, or 8 μg/mL of TNF-R1 antibody for 1 h ameliorated the TNF-α induced decrease in mitochondrial basal respiration (Fig. 3a), ATP production, and maximal respiration (data not shown) at 1.5 h. As observed in Fig. 1b, TNF-α did not kill HT-22 cells at 1.5 h (Fig. 3 b and e). The decrease in cell viability caused by 100 pg/mL of TNF-α at 3 h was ameliorated when TNF-R1 was blocked (Fig. 3c). However, when HT-22 cells were pretreated with 4, 6, or 8 μg/mL of TNF-R2 antibody, the neurotoxic effects of TNF-α on mitochondrial basal respiration at 1.5 h (Fig. 3d) or cell viability at 3 h (Fig. 3f) was not affected.
Fig. 3.
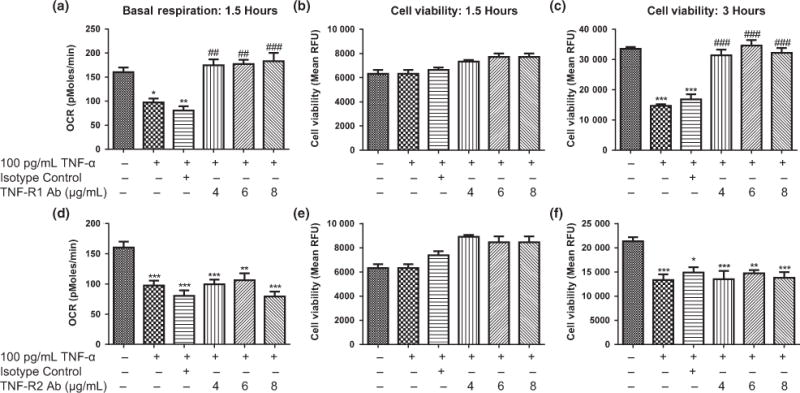
Effects of blocking TNF-α receptor (TNF-R1 or TNF-R2) on TNF-α induced changes in mitochondrial basal respiration and cell viability. (a) Effects of TNF-R1 antibody on TNF-α induced changes in mitochondrial basal respiration at 1.5 h. (b) Effects of TNF-R1 antibody on TNF-α induced changes in cell viability at 1.5 h. (c) Effects of TNF-R1 antibody on TNF-α induced changes in cell viability at 3 h. (d) Effects of TNF-R2 antibody on TNF-α induced changes in mitochondrial basal respiration at 1.5 h. (e) Effects of TNF-R2 antibody on TNF-α induced changes in cell viability at 1.5 h. (f) Effects of TNF-R2 antibody on TNF-α induced changes in cell viability at 3 h. Analysis of variance and Dunnett post hoc tests were used to assess significance, *p < 0.05, **p < 0.01; ***p < 0.001; ##p < 0.01 ; ###p < 0.001; *compared to Vehicle; #compared to 100 pg/mL TNF-α; Depicted are mean + SEM for n = 6/group.
TNF-α caused an increase in caspase 8 activity but not caspase 3/7 activity in HT-22 cells
After exposure to 1, 10, 100, 1000 pg/mL of TNF-α for 1.5 h, caspase 8 and caspase 3/7 activity was assessed with caspase 8 and caspase 3/7 luciferase assays. TNF-α caused a dose-dependent increase in caspase 8 activity (Fig. 4a); however, caspase 3/7 activity was not affected by TNF-α exposure at this early time (Figure S3). At the highest TNF-α concentration of 1000 pg/mL, caspase 8 activity was increased by 46%.
Fig. 4.
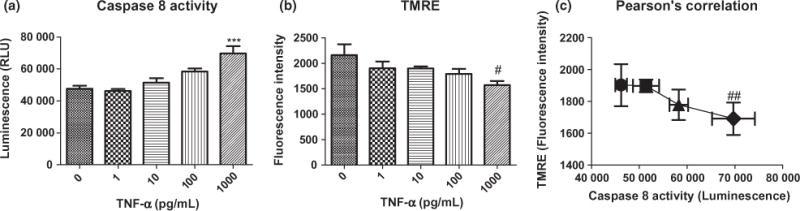
(a) Effects of 1.5 h of exposure to tumor necrosis factor alpha (TNF-α) on caspase 8 activity. Analysis of variance and Dunnett post hoc tests were used to assess significance, ***p < 0.001 (b) Effects of 1.5 h of treatment with TNF-α on mitochondrial membrane potential. #Indicates analysis of variance with post-test for linear trend found a significant dose-dependent decrease in mitochondrial membrane potential, R square = 0.3571, p = 0.0163 (c) ##Indicates a significant Pearson’s correlation between caspase 8 activity and mitochondrial membrane potential. R square = 0.9437, p = 0.086. Depicted are mean + SEM for n = 6/group.
TNF-α caused a decrease in mitochondrial membrane potential in HT-22 cells
Mitochondrial membrane potential dissipation results from the activation of caspases; thus, the increase in caspase 8 activity after 1.5 h of exposure to TNF-α led us to hypothesize that TNF-α causes mitochondrial membrane potential to decrease as a result of the activation of caspase 8. TNF-α exposure for 1.5 h resulted in a significant dose-dependent decrease in mitochondrial membrane potential (Fig. 4b), and there was a significant correlation between the increase in caspase 8 activity and decrease in mitochondrial membrane potential (Fig. 4c).
TNF-α induced cytochrome c release from mitochondria in HT-22 cells
After exposure to 100 and 1000 pg/mL of TNF-α for 3, 6, 12, and 24 h, there was a dose-dependent increase in cytochrome c release from mitochondria (Fig. 5). Exposure to TNF-α for 3, 12, and 24 h with 100 and 1000 pg/mL TNF-α resulted in a significant increase in cytochrome c release, and after exposure for 6 h only 1000 pg/mL of TNF-α resulted in a significant increase in cytochrome c release (p < 0.05).
Fig. 5.
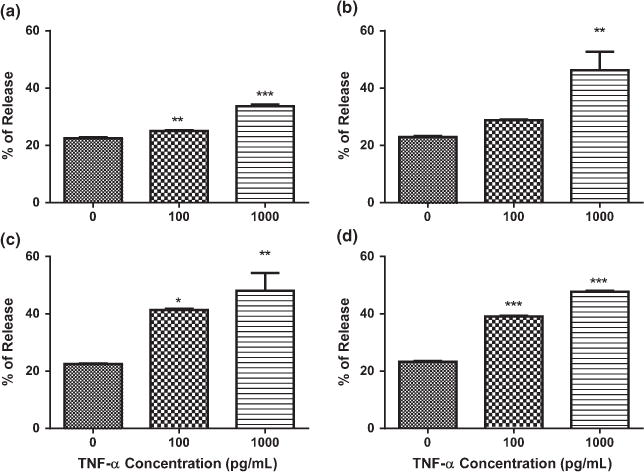
Effects of tumor necrosis factor alpha (TNF-α) on cytochrome c release. (a) Exposure to TNF-α for 3 h. (b) Exposure to TNF-α for 6 h. (c) Exposure to TNF-α for 12 h. (d) Exposure to TNF-α for 24 h. Analysis of variance and Dunnett post hoc tests were used to assess significance. *p < 0.05; **p < 0.01; ***p < 0.001. Depicted are mean + SEM for n = 6/group. When SEM is not depicted, it is too small for representation.
Discussion
The present study shows, for the first time, that TNF-α rapidly and profoundly reduces neuronal cell mitochondrial function. It is widely accepted that mitochondria play a role in neuro-inflammation and neurodegenerative CNS disorders such as multiple sclerosis, Alzheimer’s disease, and Parkinson’s disease (Di Filippo et al. 2010). With an insult like ischemic stroke, microglia activation occurs, leading to the release of TNF-α along with other pro-inflammatory cytokines (Davalos et al. 2005). Pro-inflammatory cytokines can activate mitochondria-induced apoptosis leading to cell death (Huang et al. 2005). Our findings suggest that TNF-α exposure caused neuronal mitochondrial dysfunction as expressed by decreased mitochondrial respiration as early as 1.5 h of exposure. The magnitude of the dose-dependent decrease in mitochondrial respiration was increased at 3 and 12 h but not at 6 h in HT-22 cells. After 6 h of exposure to TNF-α, HT-22 cells appear to activate compensatory mechanisms to respond to the mito-toxic effects of TNF-α. However, this compensation is not effective in preventing neuronal cell death.
Neurons have a high ATP demand (Zhu et al. 2012), and a temporary reduction in ATP production results in a profound decrease in neuronal viability (Simpkins et al. 2010). Fiskum et al. (1999) hypothesized that neuronal death that occurs at the core of an infarct after ischemia results from the decrease in mitochondrial and glycolytic ATP production. TNF-α is known to exacerbate infarct size in pre-clinical models of stroke, and this may be because of the profound and rapid decrease in ATP production, caused by TNF-α release in the evolving ischemic core.
Although TNF-R2 is primarily expressed on immune cells, and the expression of TNF-R1 is constitutive on all cell types, HT-22 cells express both receptors (Maddahi et al. 2011). TNF-R2 plays a major role in the lymphoid system and requires membrane-bound TNF-α for full activation; thus, we hypothesized that TNF-α affects mitochondrial function through TNF-R1 because soluble TNF-α signaling is primarily through TNF-R1 (Wajant et al. 2003; Maddahi et al. 2011). Furthermore, there is no correlation between the number of receptors present on a cell and the magnitude of the TNF-induced response (Beyaert and Fiers 1994). Blocking TNF-R1 ameliorated the neurotoxic effects of TNF-α on mitochondrial respiration and cell viability, confirming that TNF-α exerts neurotoxic effects through TNF-R1 not TNF-R2. To further understand the neurotoxic mechanisms of TNF-α, we assessed caspase 8 and caspase 3/7 activity because TNF-α can signal through TNF-R1 and activate caspase 8 or caspase 3/7 (Baud and Karin 2001; McCoy and Tansey 2008). The activation of caspase 8 causes mitochondrial-induced apoptosis through the cleavage of Bcl-2-associated X protein (BAX) and BH3 interacting-domain death agonist (BID) (Aggarwal 2003; McCoy and Tansey 2008). Caspase 8 activity was increased in a dose-dependent manner after exposure to TNF-α for 1.5 h; however, caspase 3/7 activity was not affected at this early time. The increase in caspase 8 activity led us to hypothesize that mitochondrial membrane potential should decrease if caspase 8 is initiating mitochondrial-induced apoptosis (Green and Kroemer 2004). The TMRE mitochondrial membrane potential assay showed a significant dose-dependent decrease in mitochondrial membrane potential after 1.5 h of TNF-α exposure. Furthermore, with a collapse of mitochondrial membrane potential there should be an increase in cytochrome c release from the mitochondria, because of the cleavage of BAX and BID (Baud and Karin 2001; Aggarwal 2003; McCoy and Tansey 2008). After exposure to 100 pg/mL of TNF-α for 3 and 24 h, there was an 11% and 68% increase in cytochrome c release, respectively, from mitochondria. Our observation of a 19.7% decrease in mitochondrial membrane potential and the 30% decrease in ATP production at 1.5 h of treatment with 100 pg/mL TNF-α indicates that sufficient cytochrome c is released to induce apoptosis (Sas et al. 2007). Once cytochrome c is released, cytochrome c interacts with apoptotic protease-activating factor-1, procaspase-9, and dATP forming an apoptosome (Sas et al. 2007). The formation of the apoptosome allows for the activation of caspase-9, which activates procaspase-3, and results in the induction of apoptosis (Sas et al. 2007).
HT-22 cells and primary mouse cortical neurons both showed a rapid mitochondrial impairment and subsequent cell death in response to low concentrations of TNF-α. However, there was a notable difference between the two cell types in the timing of the TNF-α-induced cell death. Whereas HT-22 cells died within 3 h of exposure, primary neurons showed cell loss only at 24 h. The reason for this delayed response of primary neurons is not clear, but is consistent with the comparative resistance of primary neurons to TNF-α in other studies (Reimann-Philipp et al. 2001; Zhao et al. 2001). We suspect, but do not have evidence to support, that primary neurons may have inherent defense mechanisms that resist the neurotoxic effects of a single cytokine, like TNF-α (Tamatani et al. 1999).
Numerous studies reported an association between increased levels of TNF-α mRNA and protein and an exacerbation of ischemic injury (Di Filippo et al. 2010). Administration of TNF-α-blocking antibodies or soluble TNF-R1 prior to inducing stroke reduces infarct volume (Murakami et al. 2005; Di Filippo et al. 2010). Our findings may provide the mechanism of TNF-α exacerbation of stroke damage and shows that TNF-α elicits neurotoxic effects by inducing a rapid mitochondrial dysfunction mediated through TNF-R1 signaling, subsequently activating caspase 8 and leading to the release of cytochrome c.
Current clinical treatment of ischemic stroke concentrates on dissolving and/or removing the clot, without much focus on the secondary damage that occurs as part of the inflammatory ischemic cascade (Works et al. 2013). Inflammation is known to play a major role in the pathophysiology of stroke and progression of damage post-stroke (Vila et al. 2000; Iadecola and Anrather 2011). The identification of a new therapeutic to reduce brain damage because of inflammation post-stroke would be clinically useful. However, TNF-α has both pro-apoptotic and anti-apoptotic activities, and both roles are important physiologically (Aggarwal 2003). When the expression of TNF-α is profound and prolonged, such as after stroke, it has harmful effects (Aggarwal 2003). Thus, understanding the neurotoxic mechanism of TNF-α is important to further advance its potential as a therapeutic target.
As with all in vitro studies, our studies have some limitations, and we interpreted our results with caution. First, TNF-α was administered at discrete doses (1–1000 pg/mL) for 1.5–24 h. Post-stroke concentrations change over time and are elevated in patients within 6 h of symptom onset and stay elevated for up to 10 days (Zaremba and Losy 2001; Intiso et al. 2003). Furthermore, TNF-α mRNA is increased in the ischemic core within 1 h, and TNF-α immunoreactive protein is seen within 2–6 h in the brain after middle cerebral artery occlusion in preclinical models (Liu et al. 1994; Botchkina et al. 1997). We observed mitochondrial impairment with clinically relevant concentrations of TNF-α over a critical time window post-stroke. An additional limitation is the use of cell culture models such as a transformed cell line and a primary mouse neuronal culture. Both cell types are models for neurons, but do not have astrocytes, microglia, or endothelial cells; thus, the results should be taken with caution. In view of the similarity between the two models, we believe we have modeled the effects of TNF-α on nerve cells. Finally, we did not evaluate cleavage of BAX and BID. We assumed the cleavage occurred because caspase 8 cleaves BAX and BID, resulting in cytochrome c release. Our results support that exposure to TNF-α at 1.5 h results in an increase in caspase 8 activity, and by 3 h we observed a significant release of cytochrome c from the mitochondria into the cytosol.
This study shows, for the first time, a rapid and profound mitochondrial dysfunction in neurons with acute exposure to low doses of TNF-α. This neurotoxic effect of TNF-α appears to be mediated by TNF-R1, leading to caspase activation, mitochondrial membrane potential collapse, and mitochondrial release of cytochrome c. Collectively, these data help to elucidate the neurotoxic mechanisms of TNF-α, thereby providing potential targets for cytokine-directed therapy for neuroprotection.
Supplementary Material
Figure S1. Effects of TNF-α on ATP production and maximal respiration.
Figure S2. Flow cytometry analysis of HT-22 cell expression of TNF-R1 and TNF-R2.
Figure S3. Effects of 1.5 h of exposure to TNF-α on caspase 3/7 activity.
Acknowledgments
NIH P01 AG022550 and NIH P01 AG027956 to JWS and IDeA CTR support–NIH/NIGMS Award Number U54GM104942 to WVU. TLB is a 2012 Robert Wood Johnson Foundation Nurse Faculty Scholar Grant ID: 70319. DND is supported by training grant T32 GM081741. Both JWS and TLB are supported by NIH grant P20 GM109098. Flow cytometry experiments were performed in the West Virginia University Flow Cytometry Core Facility, which is supported in part by the National Institutes of Health equipment grant number RR020866 and the Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant numbers P30GM103488 (CoBRE) and P20GM103434 (INBRE).
Abbreviations used
- Apaf-1
apoptotic protease-activating factor-1
- BAX
Bcl-2-associated X protein
- Bcl-2
B-cell lymphoma 2
- Bcl-xl
B-cell lymphoma-extra large
- BID
BH3 interacting-domain death agonist
- ICAM-1
intracellular adhesion molecule 1
- MCAO
middle cerebral artery occlusion
- MHC
major histocompatibility complex
- OCR
oxygen consumption rate
- TNF- α
tumor necrosis factor alpha
- TNF-R1
TNF- α receptor 1
- TNF-R2
TNF- α receptor 2
- TNF-R
TNF- α receptor
Footnotes
Supporting information
Additional supporting information may be found in the online version of this article at the publisher’s web-site:
Conflict of interest disclosure
All experiments were conducted in compliance with the ARRIVE guidelines. The authors do not have any conflicts of interest.
References
- Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol. 2003;3:745–756. doi: 10.1038/nri1184. [DOI] [PubMed] [Google Scholar]
- Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11:372–377. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- Beyaert R, Fiers W. Molecular mechanisms of tumor necrosis factor-induced cytotoxicity. What we do understand and what we do not. FEBS Lett. 1994;340:9–16. doi: 10.1016/0014-5793(94)80163-0. [DOI] [PubMed] [Google Scholar]
- Botchkina GI, Meistrell ME, 3rd, Botchkina IL, Tracey KJ. Expression of TNF and TNF receptors (p55 and p75) in the rat brain after focal cerebral ischemia. Mol Med. 1997;3:765–781. [PMC free article] [PubMed] [Google Scholar]
- Chao CC, Hu S, Peterson PK. Glia, cytokines, and neurotoxicity. Crit Rev Neurobiol. 1995;9:189–205. [PubMed] [Google Scholar]
- Chen XH, Zhao YP, Xue M, et al. TNF-alpha induces mitochondrial dysfunction in 3T3-L1 adipocytes. Mol Cell Endocrinol. 2010;328:63–69. doi: 10.1016/j.mce.2010.07.005. [DOI] [PubMed] [Google Scholar]
- Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Di Filippo M, Chiasserini D, Tozzi A, Picconi B, Calabresi P. Mitochondria and the link between neuroinflammation and neurodegeneration. J Alzheimers Dis. 2010;20(Suppl 2):S369–S379. doi: 10.3233/JAD-2010-100543. [DOI] [PubMed] [Google Scholar]
- Fiskum G, Murphy AN, Beal MF. Mitochondria in neurodegeneration: acute ischemia and chronic neurodegenerative diseases. J Cereb Blood Flow Metab. 1999;19:351–369. doi: 10.1097/00004647-199904000-00001. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Hallenbeck JM. The many faces of tumor necrosis factor in stroke. Nat Med. 2002;8:1363–1368. doi: 10.1038/nm1202-1363. [DOI] [PubMed] [Google Scholar]
- Huang Y, Erdmann N, Peng H, Zhao Y, Zheng J. The role of TNF related apoptosis-inducing ligand in neurodegenerative diseases. Cell Mol Immunol. 2005;2:113–122. [PubMed] [Google Scholar]
- Iadecola C, Anrather J. The immunology of stroke: from mechanisms to translation. Nat Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intiso D, Stampatore P, Zarrelli MM, Guerra GL, Arpaia G, Simone P, Tonali P, Beghi E. Incidence of first-ever ischemic and hemorrhagic stroke in a well-defined community of southern Italy, 1993–1995. Eur J Neurol. 2003;10:559–565. doi: 10.1046/j.1468-1331.2003.00648.x. [DOI] [PubMed] [Google Scholar]
- Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab. 2012;32:1677–1698. doi: 10.1038/jcbfm.2012.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Clark RK, McDonnell PC, Young PR, White RF, Barone FC, Feuerstein GZ. Tumor necrosis factor-alpha expression in ischemic neurons. Stroke. 1994;25:1481–1488. doi: 10.1161/01.str.25.7.1481. [DOI] [PubMed] [Google Scholar]
- Maddahi A, Kruse LS, Chen QW, Edvinsson L. The role of tumor necrosis factor-alpha and TNF-alpha receptors in cerebral arteries following cerebral ischemia in rat. J Neuroinflammation. 2011;8:107. doi: 10.1186/1742-2094-8-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation. 2008;5:45. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y, Saito K, Hara A, et al. Increases in tumor necrosis factor-alpha following transient global cerebral ischemia do not contribute to neuron death in mouse hippocampus. J Neurochem. 2005;93:1616–1622. doi: 10.1111/j.1471-4159.2005.03163.x. [DOI] [PubMed] [Google Scholar]
- Nawashiro H, Tasaki K, Ruetzler CA, Hallenbeck JM. TNF-alpha pretreatment induces protective effects against focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 1997;17:483–490. doi: 10.1097/00004647-199705000-00001. [DOI] [PubMed] [Google Scholar]
- Nayak AR, Kashyap RS, Kabra D, Purohit HJ, Taori GM, Daginawala HF. Time course of inflammatory cytokines in acute ischemic stroke patients and their relation to inter-alfa trypsin inhibitor heavy chain 4 and outcome. Ann Indian Acad Neurol. 2012;15:181–185. doi: 10.4103/0972-2327.99707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormstad H, Aass HC, Amthor KF, Lund-Sorensen N, Sandvik L. Serum cytokine and glucose levels as predictors of poststroke fatigue in acute ischemic stroke patients. J Neurol. 2011;258:670–676. doi: 10.1007/s00415-011-5962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann-Philipp U, Ovase R, Weigel PH, Grammas P. Mechanisms of cell death in primary cortical neurons and PC12 cells. J Neurosci Res. 2001;64:654–660. doi: 10.1002/jnr.1119. [DOI] [PubMed] [Google Scholar]
- Sas K, Robotka H, Toldi J, Vecsei L. Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J Neurol Sci. 2007;257:221–239. doi: 10.1016/j.jns.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Simpkins JW, Yi KD, Yang SH, Dykens JA. Mitochondrial mechanisms of estrogen neuroprotection. Biochim Biophys Acta. 2010;1800:1113–1120. doi: 10.1016/j.bbagen.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotgiu S, Zanda B, Marchetti B, et al. Inflammatory biomarkers in blood of patients with acute brain ischemia. Eur J Neurol. 2006;13:505–513. doi: 10.1111/j.1468-1331.2006.01280.x. [DOI] [PubMed] [Google Scholar]
- Tamatani M, Che YH, Matsuzaki H, Ogawa S, Okado H, Miyake S, Mizuno T, Tohyama M. Tumor necrosis factor induces Bcl-2 and Bcl-x expression through NFkappaB activation in primary hippocampal neurons. J Biol Chem. 1999;274:8531–8538. doi: 10.1074/jbc.274.13.8531. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Nakae S, Terry RD, et al. Cardiomyocyte-specific Bcl-2 overexpression attenuates ischemia-reperfusion injury, immune response during acute rejection, and graft coronary artery disease. Blood. 2004;104:3789–3796. doi: 10.1182/blood-2004-02-0666. [DOI] [PubMed] [Google Scholar]
- Vila N, Castillo J, Davalos A, Chamorro A. Proinflammatory cytokines and early neurological worsening in ischemic stroke. Stroke. 2000;31:2325–2329. doi: 10.1161/01.str.31.10.2325. [DOI] [PubMed] [Google Scholar]
- Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- Wallach D, Arumugam TU, Boldin MP, et al. How are the regulators regulated? The search for mechanisms that impose specificity on induction of cell death and NF-kappaB activation by members of the TNF/NGF receptor family. Arthritis Res. 2002;4(Suppl 3):S189–S196. doi: 10.1186/ar585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Works MG, Koenig JB, Sapolsky RM. Soluble TNF receptor 1-secreting ex vivo-derived dendritic cells reduce injury after stroke. J Cereb Blood Flow Metab. 2013;33:1376–1385. doi: 10.1038/jcbfm.2013.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaremba J, Losy J. Early TNF-alpha levels correlate with ischaemic stroke severity. Acta Neurol Scand. 2001;104:288–295. doi: 10.1034/j.1600-0404.2001.00053.x. [DOI] [PubMed] [Google Scholar]
- Zhao X, Bausano B, Pike BR, et al. TNF-alpha stimulates caspase-3 activation and apoptotic cell death in primary septo-hippocampal cultures. J Neurosci Res. 2001;64:121–131. doi: 10.1002/jnr.1059. [DOI] [PubMed] [Google Scholar]
- Zhu XH, Qiao H, Du F, Xiong Q, Liu X, Zhang X, Ugurbil K, Chen W. Quantitative imaging of energy expenditure in human brain. NeuroImage. 2012;60:2107–2117. doi: 10.1016/j.neuroimage.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Effects of TNF-α on ATP production and maximal respiration.
Figure S2. Flow cytometry analysis of HT-22 cell expression of TNF-R1 and TNF-R2.
Figure S3. Effects of 1.5 h of exposure to TNF-α on caspase 3/7 activity.