

Summary
The heart responds to stress signals by hypertrophic growth, which is accompanied by activation of the MEF2 transcription factor and reprogramming of cardiac gene expression. We show here that class II histone deacetylases (HDACs), which repress MEF2 activity, are substrates for a stress-responsive kinase specific for conserved serines that regulate MEF2-HDAC interactions. Signal-resistant HDAC mutants lacking these phosphorylation sites are refractory to hypertrophic signaling and inhibit cardiomyocyte hypertrophy. Conversely, mutant mice lacking the class II HDAC, HDAC9, are sensitized to hypertrophic signals and exhibit stress-dependent cardiomegaly. Thus, class II HDACs act as signal-responsive suppressors of the transcriptional program governing cardiac hypertrophy and heart failure.
Introduction
The adult myocardium responds to stress by hypertrophic growth, which is accompanied by an increase in size of cardiac myocytes, assembly of sarcomeres, and activation of a fetal cardiac gene program (Chien, 1999). Induction of fetal cardiac genes results in altered contractility and calcium handling and correlates with impaired cardiac function (Lowes et al., 2002). A variety of pathologic stimuli, including myocardial infarction, hypertension, contractile abnormalities, and pressure overload elicit the hypertrophic response. Autocrine and paracrine signaling pathways involving angiotensin II (ANGII), endothelin-1 (ET-1), and activators of the adrenergic system also contribute to myocyte hypertrophy.
Several calcium-dependent signaling molecules, including the calcium/calmodulin-dependent phosphatase calcineurin, calcium/calmodulin-dependent protein kinase (CaMK), and mitogen-activated protein kinases (MAPKs) have been implicated in the transduction of hypertrophic stimuli (Frey et al., 2000). However, relatively little is known of the terminal steps in these pathways that reprogram cardiac gene expression in the nucleus. It is also unclear how different intracellular signaling pathways all result in the hypertrophic phenotype, and whether this reflects the convergence of these pathways on a common downstream target or crosstalk between signaling pathways at more upstream steps.
Histone acetyltransferases (HATs) and histone deacetylases (HDACs) govern gene expression patterns by being recruited to target genes through association with specific transcription factors (Jenuwein and Allis, 2001). HATs promote gene activation by acetylating nucleosomal histones, thereby relaxing chromatin structure. HAT activity is opposed by HDACs, which deacetylate histones, resulting in chromatin condensation and transcriptional repression (Johnson and Turner, 1999).
Vertebrate HDACs are categorized into three classes (Grozinger and Schreiber, 2002). Class I includes HDACs 1, 2, 3, and 8, which are expressed ubiquitously. Class II HDACs (4, 5, 7, and 9) are highly expressed in striated muscles and brain and contain N-terminal extensions that interact with positive and negative transcriptional cofactors. The N-terminal regions of class II HDACs also contain two conserved CaMK phosphorylation sites (McKinsey et al., 2000a, 2002). Phosphorylation of these sites leads to the binding of 14-3-3 proteins, which induces HDAC nuclear export and results in derepression of HDAC target genes (McKinsey et al., 2000b; Grozinger and Schreiber, 2000).
Among the targets of class II HDACs is myocyte enhancer factor-2 (MEF2), a MADS-box transcription factor highly expressed in myocytes (Black and Olson, 1998). Association of class II HDACs with MEF2 results in repression of MEF2 activity (McKinsey et al., 2002). This repressive influence can be relieved by CaMK-dependent phosphorylation of HDACs, which induces their dissociation from MEF2 (McKinsey et al., 2000a, 2000b; Lu et al., 2000a, Zhang et al., 2001b). MEF2-interacting transcriptional repressor (MITR) is a predominant splice variant of HDAC9 expressed in the heart (Sparrow et al., 1999; Zhou et al., 2001). MITR associates with MEF2 in a signal-responsive manner (Zhang et al., 2001b), but lacks a catalytic domain. Nevertheless, MITR efficiently suppresses MEF2 activity by recruiting other corepressors (Sparrow et al., 1999; Zhang et al., 2001a).
Here, we show that diverse hypertrophic signals in vivo lead to the activation of a cardiac HDAC kinase that phosphorylates the signal-responsive sites in class II HDACs and MITR. Mutant proteins lacking these phosphorylation sites act as signal-resistant repressors of cardiomyocyte hypertrophy and fetal cardiac gene expression in vitro. Conversely, HDAC9 knockout mice are super-sensitive to hypertrophic stimuli and spontaneously develop cardiac hypertrophy with advanced age. These findings demonstrate that class II HDACs act as signal-responsive repressors of cardiac hypertrophy.
Results
Activation of a Cardiac HDAC Kinase by Hypertrophic Stimuli
Class II HDACs and MITR possess a common structure with two conserved CaMK phosphorylation sites flanking a nuclear localization sequence (NLS) near their N termini (Figures 1A and 1B). To determine whether a kinase specific for these phosphorylation sites might be activated in the heart in response to hypertrophic stimuli, we developed an in vitro kinase assay using portions of HDAC4, HDAC5, and HDAC9/MITR encompassing the conserved CaMK sites fused to GST as substrates (Figure 1C). Phosphorylation of GST-HDAC proteins was readily detected in this assay (Figure 1C, WT). GST-HDAC fusion proteins in which the CaMK sites (Ser-246 in HDAC4, Ser-259 in HDAC5, and Ser-218 in HDAC9/MITR) were mutated to alanines (Figure 1C, Mut) were not phosphorylated by cardiac extracts, demonstrating that the kinase activity is specific for the CaMK sites.
Figure 1. Elevated HDAC Kinase Activity in Hypertrophic Hearts.

(A) Schematic diagram of class II HDACs. Class II HDACs have a bipartite structure, with a C-terminal catalytic (HDAC) domain and an N-terminal extension with a MEF2 binding domain. Conserved phosphorylation sites flank the nuclear localization sequence (NLS) and a nuclear export sequence (NES) is near the C terminus. MITR is a splice variant of HDAC9 that lacks an HDAC domain.
(B) Amino acid homologies surrounding the regulatory serines (arrowheads) in the class II HDACs.
(C) Schematic diagram of HDAC kinase assay. Kinase assays were performed with GST-HDAC substrates. The region of each HDAC that was used as substrate is underlined in (A). A kinase from heart lysates phosphorylates the wild-type (WT) substrates, but not mutant (Mut) substrates containing alanine in place of the regulatory serine residue.
(D and E) HDAC kinase assays were performed with heart extracts from thoracic aorta-banded (TAB) or sham-operated mice (D) or from wild-type (wt) or αMHC-calcineurin transgenic (Cn-Tg) mice. Values represent the average of at least three independent assays.
(F) HDAC kinase assays were performed with heart extracts derived from wild-type (lane 1) and Cn-Tg (lanes 2–23) mice in the absence (lanes 1 and 2) or presence (lanes 3–23) of diverse inhibitors (upper image). Kinase activity relative to that in extracts from Cn-Tg mice (lane 2) was determined (lower image). Inhibitors were added to reactions at concentrations 20 times the IC50. Combo1 contained AIP and Go6983 and combo2 contained HA1004 and Go6983. Inhibitor experiments were performed at least three times with comparable results. A representative experiment is shown. For (D–F), enzyme activity was quantified as described in Experimental Procedures.
Constriction of the thoracic aorta in mice creates a pressure gradient in excess of 50 mm Hg and results in approximately a 50% increase in cardiac mass within 21 days (Hill et al., 2000). Analysis of cardiac extracts from sham-operated and thoracic aorta-banded (TAB) mice showed that pressure overload increased HDAC kinase activity 2.1- and 1.7-fold, using GST-HDAC9 and GST-HDAC5 substrates, respectively (P < 0.05; Figure 1D). Hypertrophic hearts from transgenic mice expressing activated calcineurin (Molkentin et al., 1998) also showed pronounced elevation of HDAC kinase activity (Figure 1E). Cardiac hypertrophy due to expression of activated forms of CaMK and the MAP kinase MEK5 in the heart also resulted in stimulation of HDAC kinase activity (data not shown).
To further characterize the HDAC kinase, we tested its sensitivity to a panel of inhibitors (Figure 1F and data not shown). The HDAC kinase was inhibited by staurosporin and K252a, which are general serine/threonine kinase inhibitors. The kinase was also partially inhibited by HA1004, which is a broad inhibitor known to inhibit CaMK, PKA, and PKG. However, the kinase was not inhibited by compounds that inhibit CaMK (KN62, KN93, or AIP), PKA (H-89 or 4-cyano-3-methylisoquinoline), PKC (GF109203X, Gö6976, Gö6983, or Ro-31-8425), MEK (PD98059), p38 (SB203580), cdk (olomoucine and roscovitine), or PI-3-kinase (rapamycin and wortmannin). The kinase was also not inhibited by inhibitors of PKG, Raf, or MLCK (data not shown). Combinations of the above inhibitors also failed to inhibit the kinase. Paradoxically, inhibitor combinations that contained HA1004 were less effective than HA1004 alone. We suspect this is due to counteracting effects of one inhibitor on the action of another.
These results demonstrate that diverse hypertrophic cues stimulate the activity of a kinase (or kinases) specific for the phosphorylation sites that inactivate class II HDACs and suggest that the kinase measured in this assay does not correspond to kinases previously implicated in hypertrophic signaling.
Signal-Resistant HDACs Inhibit Hypertrophy and Acetylation of Fetal Cardiac Genes
In light of the finding that cardiac hypertrophy was associated with enhanced HDAC kinase activity, which would be predicted to inactivate HDACs and MITR, we tested whether mutant proteins lacking the regulatory phosphorylation sites were able to prevent hypertrophic gene expression in primary rat cardiomyocytes. As shown in Figure 2A, the hypertrophic marker genes atrial natriuretic factor (ANF) and β-myosin heavy chain (β-MHC) are upregulated in cardiomyocytes stimulated with the adrenergic agonist phenylephrine (PE), a potent inducer of hypertrophy. Infection of cardiomyocytes with adenoviruses that expressed HDAC5 and MITR with serine-to-alanine mutations at positions 259/498 and 218/448, respectively, abrogated the induction of these genes. The HDAC5 and MITR mutant proteins had no effect on expression of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Because MITR was effective in repressing fetal gene expression and is the predominant form of HDAC9 expressed in the heart, we used it in subsequent studies of the mechanisms involved in the blockade to hypertrophic signaling.
Figure 2. Inhibition of Fetal Gene Expression and Acetylation by Signal-Resistant MITR.

(A) Neonatal rat cardiomyocytes were infected with adenoviruses encoding phosphorylation site mutants of HDAC5 (Ad-HDAC5 S259/498A) or MITR (Ad-MITR S218/448A) and were then stimulated with PE for 24 hr. Transcripts for ANF, β-MHC, and GAPDH were detected by dot blot. ANF and β-MHC expression are shown relative to that in untreated, uninfected controls and normalized to the level of GAPDH.
(B) Cardiomyocytes were infected with adenoviruses encoding Myc-tagged MITR or MITR S218/448A at the indicated multiplicity of infection (moi). Cells maintained in serum-free medium were stimulated with PE for 24 hr. The relative levels of β-MHC mRNA in Ad-MITR versus Ad-MITR S218/448A-infected cells were quantified by dot blot and normalized to the level of GAPDH.
(C) The expression of ectopic MITR in cardiomyocyte lysates was determined by Western blotting with an anti-Myc antibody (upper image). The blot was reprobed with an anti-α-tubulin antibody to reveal total protein input (bottom image).
(D) Soluble chromatin was prepared from cardiomyocytes infected with either Ad-GFP or Ad-MITR S218/448A and treated for 24 hr with FBS or PE. Chromatin was immunoprecipitated with an antibody specific for acetylated histone H3 and precipitated genomic DNA was analyzed by PCR using primers for ANF, β-MHC, GAPDH, and α-cardiac actin promoters. Positions of primers (numbers) and the GATA sites (black boxes) relative to the transcription initiation sites of each gene are shown. The lower image shows a DNA input control in which PCR amplification was performed prior to immunoprecipitation.
If the regulatory phosphorylation sites in MITR and class II HDACs are targets for hypertrophic stimuli, then wild-type HDAC proteins would be expected to be less effective inhibitors of hypertrophy than mutant proteins lacking these sites. To test this possibility, we compared the effects of wild-type MITR and the MITR S218/448A mutant on expression of β-MHC in cardiomyocytes treated with PE. As shown in Figure 2B, at comparable multiplicities of infection (moi’s), Ad-MITR S218/448A was more than twice as effective in suppressing β-MHC expression than wild-type MITR. The activity of Ad-MITR S218/448A in this assay is an underestimate of its relative potency because the mutant protein accumulates to a level about 10-fold lower than the wild-type protein at the same moi (Figure 2C).
To determine whether signal-resistant MITR changed the acetylation state of histones associated with fetal cardiac genes, we performed chromatin immunoprecipitation (ChIP) assays with an antibody specific for acetylated histone H3. Associated genomic DNA was then analyzed by PCR using primers specific for the promoter regions of the ANF and β-MHC genes. As a control, cardiomyocytes were infected with an adenovirus encoding green fluorescent protein (GFP). As shown in Figure 2D, the promoter regions of ANF or β-MHC are highly acetylated in cardiomyocytes infected with Ad-GFP and treated with either fetal bovine serum (FBS) or PE. In contrast, infection with Ad-MITR S218/448A significantly reduced acetylation of these promoters in the presence of hypertrophic stimuli. The GAPDH and α-cardiac actin genes, which are not regulated during hypertrophy, did not show a change in histone acetylation in the presence of Ad-MITR S218/448A. Thus, there was a direct correlation between histone deacetylation and repression of fetal gene expression by signal-resistant MITR, suggesting that MITR repressed fetal genes by reducing the acetylation state of their promoters.
Neonatal cardiomyocytes assemble organized sarcomeres in the presence of hypertrophic agonists. Cardiomyocytes expressing the phosphorylation site mutant of MITR were unable to reorganize sarcomeres or upregulate expression of ANF protein in response to stimulation by PE or FBS (Figures 3A and 3B). Identical results were obtained with adenovirus encoding the HDAC5 S259/498A mutant (data not shown). Similarly, Ad-MITR S218/448A prevented induction of β-MHC and ANF in response to FBS, ET-1, ANGII, and insulin-like growth factor-1 (IGF-1; Figure 3C). These findings suggested that phosphorylation of the regulatory serines in MITR and HDAC5 was necessary for hypertrophy in response to diverse agonists.
Figure 3. Prevention of Hypertrophy by MITR.

(A and B) Cardiomyocytes were cultured either in the absence of virus or were infected with adenoviruses encoding GFP or Myc-tagged MITR S218/448A. After stimulation with PE or FBS for 24 hr, cells were fixed and stained with antibody against sarcomeric α-actinin (A, red signal) or ANF (B, perinuclear red signal). Adenovirus-infected cells were identified by GFP fluorescence or costaining with anti-Myc antibody to reveal ectopic MITR (green signal). Scale bar equals 20 μm.
(C) Cardiomyocytes were cultured and infected with adenoviruses as described above. Cells maintained in serum-free medium were stimulated with FBS, PE, ET-1, ANGII, or IGF-1. After 24 hr, RNA was isolated and the indicated transcripts were measured by dot blot. Numbers indicate the expression level of each transcript in the presence of Ad-MITR S218/448A relative to Ad-GFP and normalized to GAPDH.
Generation of HDAC9 Mutant Mice
To further investigate the potential involvement of class II HDACs as suppressors of cardiac hypertrophy in vivo, we inactivated the mouse HDAC9/MITR gene by homologous recombination in ES cells and generated HDAC9 mutant mice (Figures 4A–4C). We chose to focus on HDAC9 because among the class II HDACs, it is expressed at the highest levels in the heart and because MITR, the primary product of the HDAC9 locus, was highly effective in suppressing hypertrophy in vitro.
Figure 4. Targeting of Mouse HDAC9.

(A and B) A diagram of the HDAC9 and MITR proteins is shown above the mouse HDAC9 locus. Sizes of introns in kilobases (kb) and exons (filled boxes) are indicated. In the targeting vector, a nuclear LacZ (nLacZ) reporter was inserted in-frame with exon 4. Homologous recombination resulted in deletion of exons 4 and 5, which encode the MEF2 binding domain of HDAC9/MITR. Insertion of the nLacZ gene introduced a BamHI site that was used in conjunction with the indicated external 5′ probe to distinguish the wild-type (6.5 kb) and mutant (4.1 kb) alleles by Southern blot (B).
(C) Genomic DNA from mice of the indicated genotypes was analyzed by PCR using primers specific for the 5′ and 3′ regions of the targeted mutation.
(D) The positions of the primers within HDAC9 exons are shown (lower image). Targeting the HDAC9 locus results in an aberrant mRNA splicing event between exons 3 and 6, as evidenced by the 300 base pair (bp) PCR product using primers P1 and P3, which is not present in wild-type controls. Sequencing of this RT-PCR product revealed a frameshift that introduced stop codons that prevent translation of sequences downstream of exon 3. L7 transcripts were amplified as a control for cDNA integrity.
(E) The consequences of HDAC9 gene disruption on the expression of mRNA transcripts for HDACs 1–8 were assessed by semi-quantitative RT-PCR using heart RNA from mice of the indicated HDAC9 genotypes.
The targeted mutation deleted most of exon 4 and all of exon 5, which encode residues 99–176 that encompass the MEF2 binding domain of the protein (Figures 4A–4C). HDAC9 mutant mice were obtained at predicted Mendelian ratios and showed no discernible pathological or histological abnormalities at early age (data not shown).
To confirm that the mutation created a null allele, we performed RT-PCR analysis using primers representing exon sequences within and surrounding the targeted deletion region (Figure 4D). Using primer P2 (from deleted exon 4) and P3 (from exon 6), we detected a PCR product in RNA from hearts of wild-type and HDAC9+/− mice, but not of HDAC9 −/− mice. In contrast, using primer P1 (from exon 3) and primer P3, RT-PCR products were detected in RNA samples from mice of all genotypes. In HDAC9+/− mice, products of 549 and 300 bp, corresponding to the wild-type and mutant transcripts, respectively, were detected, whereas in HDAC9 −/− mice, only the mutant transcript was detected (Figure 4D). These results suggest that replacement of exon 4 and exon 5 of HDAC9 with LacZ-Neo induced alternative splicing of the targeted allele, such that exon 3 was spliced to exon 6. This was confirmed by sequence analysis of RT-PCR products. Such splicing caused a frameshift and introduced two stop-codons immediately following exon 3 (data not shown). Thus, no functional HDAC9 or MITR protein is produced in homozygous mutants. Furthermore, due to such alternative splicing, the lacZ gene was not expressed in mutant mice (data not shown).
To determine whether other HDACs might be upregulated in mutant mice to compensate for the lack of HDAC9, we compared the expression of HDACs 1–8 in hearts from wild-type and mutant mice by RT-PCR. Transcripts encoding these other HDACs were expressed at normal levels in mutant hearts (Figure 4E).
HDAC9 Mutant Mice Develop Age-Dependent Cardiac Hypertrophy and Are Hypersensitive to Pressure Overload
Although HDAC9 mutant mice were initially normal, by eight months of age they developed cardiac hypertrophy and showed an average increase of 46% in heart weight/body weight ratios compared to age-matched wild-type controls (P < 0.001; Figures 5A and 5B). In contrast, at one month of age, there was no significant difference in cardiac mass between wild-type and HDAC9−/− mice.
Figure 5. Cardiac Hypertrophy in HDAC9 Mutant Mice.
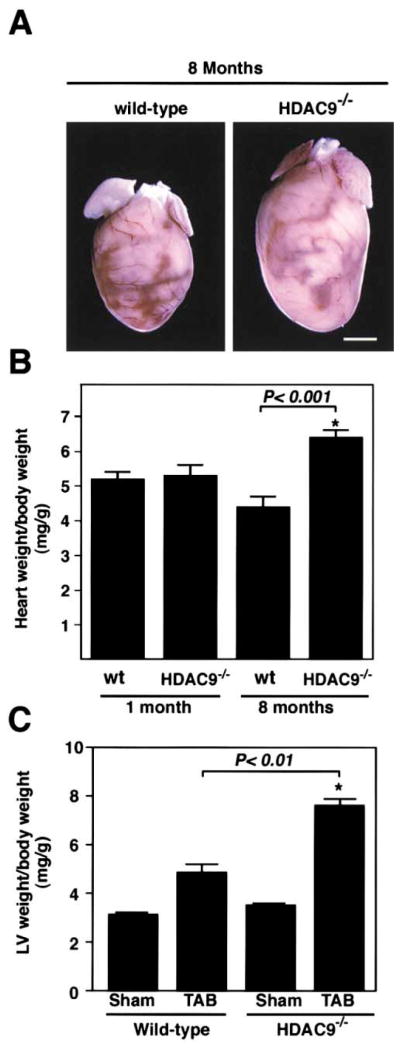
(A and B) HDAC9 mutant mice and wild-type littermates were sacrificed at one and eight months of age and heart weight-to-body weight ratios were determined. Values represent the mean ± standard deviation (SD). N = 5. Scale bar equals 2 mm.
(C) Hypersensitivity to TAB. Six-to-eight-week-old mice were subjected to thoracic aortic banding (TAB) or to sham operation. Twenty-one days later, animals were sacrificed and the ratios of left ventricular (LV) mass-to-body weight were determined. At least five mice of each genotype were analyzed. Values represent the mean ± SD.
Despite the lack of an overt phenotype in young HDAC9−/− mice, we wondered whether they might be sensitized to hypertrophic stimuli, as would be predicted if HDAC9 was a suppressor of stress-induced signaling pathways. Since our earlier finding indicated that pressure overload stimulated a kinase that would inactivate class II HDACs, we compared the responses of wild-type and HDAC9 mutant mice to thoracic aortic banding. Banding of the thoracic aorta of wild-type mice resulted in an average increase in left ventricular (LV) mass of 56% after 21 days (Figure 5C). In contrast, hearts from banded HDAC9−/− mice showed a 105% increase in LV mass over this period (P < 0.01). These findings suggest that HDAC9 acts as a suppressor of the signaling mechanism that conveys a pressure stimulus to hypertrophic growth of the heart.
HDAC9 Mutant Mice Are Sensitized to the Hypertrophic Effects of Activated Calcineurin
To further investigate the potential role of HDAC9 as a suppressor of hypertrophy, we asked whether HDAC9−/− mice were hypersensitive to calcineurin signaling. To address this issue, we crossed HDAC9−/− mice with mice bearing the α-MHC-calcineurin transgene (Cn-Tg). Indeed, hearts from HDAC9 mutants showed an exaggerated response to activated calcineurin (Figures 6A and 6B). Whereas activated calcineurin resulted in a 130% increase in cardiac mass by 4 weeks of age, the HDAC9 mutants showed a 220% increase in response to calcineurin (P < 0.001; Figure 6B). Massive hypertrophy was observed in the right and left ventricular walls and interventricular septum. Histological analysis demonstrated that the increase in cardiac mass was a reflection of the extreme hypertrophy of individual cardiomyocytes in HDAC9 mutant mice (Figure 6A, lower image). HDAC9+/− mice showed an enhanced cardiac growth response to calcineurin that was intermediate between that of wild-type and HDAC9−/− littermates (Figure 6B). The enhanced hypertrophy of HDAC9−/− hearts was mirrored by changes in fetal cardiac gene expression, as Northern blot analysis showed that transcripts for ANF, brain natriuretic peptide (BNP), and β-MHC were super-induced by calcineurin in the absence of HDAC9 (Figure 6C).
Figure 6. HDAC9 Mutant Mice Are Hypersensitive to Calcineurin-Mediated Hypertrophy.

(A) HDAC9 mutant mice were bred with mice harboring the αMHC-calcineurin transgene (Cn-Tg). Hearts from one-month-old mice of the indicated genotype were isolated (top images), sectioned, and stained with H and E (middle and bottom images). The bottom image shows high magnification images of H and E-stained cardiomyocytes from the ventricular septum. Scale bar equals 2 mm for top and middle, 20 μm for bottom.
(B) Heart weight-to-body weight ratios of mice of the indicated genotypes were determined at four weeks of age. At least ten mice of each genotype were analyzed. Values represent the mean ± standard deviation.
(C) Total RNA was isolated from hearts of mice of the indicated genotype and expression of transcripts for ANF, brain natriuretic peptide (BNP), β-MHC, and GAPDH was determined by Northern blot analysis. The relative abundance of transcripts was quantified and normalized to GAPDH.
(D) Wild-type or HDAC9 mutant mice were crossed to transgenic mice harboring a MEF2-dependent LacZ reporter gene. Offspring were bred with Cn-Tg mice. At least two littermates of each genotype were sacrificed at 8 weeks of age and cardiac MEF2 activity was detected by staining for β-galactosidase activity. Scale bar equals 2 mm.
We considered the possibility that the exaggerated response of HDAC9−/− hearts to stress signaling might reflect subtle structural or functional cardiac abnormalities that are exacerbated by stress. However, echocardiography failed to reveal any abnormalities in cardiac function in HDAC9 mutant mice up to 8 months of age. We also considered whether noncardiac or systemic abnormalities could contribute to the sensitized phenotype of HDAC9 mutants, but we observed no abnormalities in lung, vascular system, or skeletal muscle of the mutants. Thus, we conclude that the phenotype represents a cell-autonomous abnormality of cardiac myocytes.
The Transcriptional Activity of MEF2 Is Hypersensitive to Calcineurin Signaling in HDAC9−/− Mice
Since MEF2 is activated by stress signals and repressed by class II HDACs, we used a line of transgenic mice, which harbors a LacZ transgene controlled by three tandem copies of the MEF2 consensus binding site, to determine whether HDAC9 regulated the responsiveness of MEF2 to calcineurin signaling in the intact heart in vivo. This MEF2-LacZ transgene provides a read-out of MEF2 transcriptional activity and is only basally active in the postnatal heart (Naya et al., 1999). In the absence of calcineurin signaling, the MEF2-dependent reporter was expressed at low levels in both wild-type and HDAC9 mutant hearts. In contrast, MEF2 activity was elevated in the hearts of α-MHC-calcineurin transgenic mice, and in the HDAC9 mutant, MEF2-LacZ expression was activated to even higher levels in response to calcineurin signaling (Figure 6D). These findings demonstrate that MEF2 responds to signals for pathologic, but not physiologic hypertrophy and that the responsiveness of MEF2 to calcineurin signaling is negatively regulated by HDAC9 in vivo.
Discussion
Stress signals stimulate adult cardiomyocytes to undergo hypertrophy, which is associated with the activation of a fetal cardiac gene program that results in mal-adaptive changes in cardiac contractility and calcium handling. The results of this study show that stress signals stimulate an HDAC kinase that phosphorylates conserved regulatory serine residues in class II HDACs. Mutant forms of class II HDACs that cannot be phosphorylated render cardiac myocytes refractory to hypertrophic stimuli, and mice lacking HDAC9 are sensitized to stress signals that induce hypertrophy. A model consistent with these findings is shown in Figure 7.
Figure 7. Repression of Cardiac Hypertrophy by Class II HDACs.

Class II HDACs (4, 5, 7, and 9) associate with MEF2 and inhibit hypertrophy and the fetal gene program. Stress signals stimulate an HDAC kinase that phosphorylates (P) HDACs at two conserved serine residues. HDAC kinase is also stimulated by calcineurin. When phosphorylated, HDACs bind 14-3-3, dissociate from MEF2, and are exported from the nucleus. Upon release of HDACs, MEF2 is free to associate with histone acetyltransferases (HATs) and to activate downstream target genes that drive a hypertrophic response. MEF2 also interacts with NFAT and GATA transcription factors, which have been implicated in hypertrophic gene expression.
Activation of an HDAC Kinase by Hypertrophic Signals
CaMK activity is elevated in failing human hearts (Kirchhefer et al., 1999), and constitutively activated CaM kinases can induce cardiac hypertrophy in vivo and in vitro (Kato et al., 2000; Passier et al., 2000; Zhang et al., 2002). The ability of CaMK signaling to phosphorylate the conserved regulatory sites in MITR and class II HDACs in transfected cells (McKinsey et al., 2000a, 2000b; Zhang et al., 2001b) suggested that CaMK might be involved in the transduction of hypertrophic stimuli through HDACs in vivo. Indeed, our results demonstrate the existence of a protein kinase activity in cardiac extracts that phosphorylates the CaMK sites in MITR and class II HDACs and point to this enzyme as an effector in hypertrophic signaling pathways. However, the kinase detected in this assay does not appear to correspond to a typical CaMK, based on its insensitivity to all of the CaMK inhibitors we tested (KN62, KN93, and AIP), as well as its failure to bind a calmodulin affinity column and its activity in the presence of EGTA (data not shown). The kinase is also not recognized by an anti-CaMKIV antibody. Although the kinase was partially inhibited by HA1004, which is known to inhibit PKA, PKG, and CaMK, this inhibitor may also inhibit other untested or unknown kinases. Since other inhibitors for PKA, PKG, and CaMK do not inhibit the HDAC kinase, it does not appear that these kinases singly or in combination can account for the activity. Nevertheless, it is possible that the kinase is a CaMK-like kinase, since purified CaMK can phosphorylate the same sites in HDACs. The identity of this stress-responsive kinase remains to be determined.
The stimulation of HDAC kinase activity by stress signals in vivo, coupled with the ability of signal-resistant HDAC mutants to block hypertrophy of primary cardiomyocytes in the presence of diverse agonists in vitro, suggests that this kinase integrates multiple hypertrophic signaling pathways. Of course, activation of HDAC kinase activity by different hypertrophic stimuli need not be direct and could involve secondary pathways or autocrine signaling loops.
Repression of Cardiac Hypertrophy by MITR and Class II HDACs
The phenotype of HDAC9 mutant mice demonstrates that this HDAC acts in the adult heart to suppress the fetal gene program and hypertrophic growth. In the absence of stress, these mice showed normal cardiac size and function at young age, whereas at old age or when stressed by pressure overload or calcineurin activation, they showed a severely exaggerated hypertrophic response. Remarkably, deletion of only a single HDAC9 allele was sufficient to sensitize animals to hypertrophic stimuli, resulting in a cardiac growth response intermediate between that of wild-type and HDAC9−/− mice. These findings demonstrate that the hypertrophic program is exquisitely sensitive to the repressive influence of HDAC9 and imply that, under conditions of stress, HDACs 4, 5, and 7, which are also expressed in the adult heart, cannot fully compensate for a reduction in HDAC9 expression.
The enhanced hypertrophic phenotype of HDAC9 mutant mice, and the suppression of cardiomyocyte hypertrophy by overexpressed HDACs in vitro, also suggest that in order for stress signals to induce hypertrophy, the stimuli must exceed a threshold of repressive activity that represents the combined actions of class II HDACs. In HDAC9 mutant mice, this threshold is lowered, with resulting exacerbation of the hypertrophic response. This sensitized phenotype also suggests that stress signals do not inactivate all the repressive activity of class II HDACs and that residual nonphosphorylated HDACs dampen the hypertrophic response. Otherwise, removing HDAC9 would not be expected to augment the hypertrophic response.
While HDAC9 mutant mice show an enhanced response to stress signals, they do not show abnormal cardiac growth during postnatal development when the heart enlarges through physiologic hypertrophy. That simply eliminating HDAC activity is insufficient to cause hypertrophy suggests that the hypertrophic program also requires positive signals that are likely to be directed at other targets. (By analogy, releasing the brake of a car is insufficient to make it move without also stepping on the accelerator.) It is notable in this regard that MEF2 is activated by several stress response pathways through mechanisms independent of HDAC derepression (McKinsey et al., 2002).
The lack of enhanced hypertrophy during postnatal cardiac development in HDAC9 mutant mice also suggests that the intracellular pathways that transduce pathologic and physiologic signals leading to hypertrophy are distinct and that HDAC9 responds specifically to stress signals that activate the fetal gene program. The selective activation of fetal cardiac genes by stress signals, but not during normal postnatal growth of the heart, supports the notion that pathologic and physiologic hypertrophy depends on different signaling pathways.
Transcriptional Targets for HDACs in the Hypertrophic Pathway
The super-activation of MEF2 activity in HDAC9 mutant mice in response to calcineurin signaling indicates that MEF2 is a downstream target for repression by HDAC9/MITR in vivo. However, MEF2 need not be the sole transcriptional effector of the stress-response pathways governed by HDACs. It should also be pointed out that MEF2 interacts with GATA and NFAT transcription factors, which also associate with each other (Molkentin et al., 1998; Morin et al., 2000; Youn et al., 2000). These factors have each been shown to be capable of activating the fetal gene program in response to hypertrophic signaling (Liang et al., 2001; Molkentin et al., 1998). Thus, HDAC has the potential to repress hypertrophy-responsive genes by being recruited via MEF2 to gene regulatory regions that do not contain direct binding sites for MEF2 (see Figure 7).
The ability of HDACs to repress cardiac hypertrophy implies that HAT activity is required for hypertrophy and activation of the fetal gene program. Notably, the coactivators p300 and CBP, which possess HAT activity, have been shown to interact with the same region of MEF2 that interacts with class II HDACs and to be activated by the same signals that inactivate class II HDACs (Sartorelli et al., 1997; Impey et al., 2002). The signal-dependent dissociation of phospho-HDACs from MEF2 would therefore allow for the association of MEF2 with p300/CBP and consequent activation of stress-response and fetal cardiac genes.
Therapeutic Implications
Cardiac hypertrophy is associated with an increased risk of morbidity and mortality. Pathologic changes in the hypertrophic heart have been correlated with altered contractility that results from activation of the fetal cardiac gene program (Lowes et al., 2002). The realization that HDACs act as repressors of the hypertrophic program and stress-responsive substrates for an HDAC kinase suggests that therapeutic strategies to inhibit this kinase could be beneficial in the treatment of cardiac hypertrophy. Current strategies for treatment of hypertrophy and heart failure target early steps in hypertrophic signaling pathways, such as cell surface receptors, calcium channels and handling proteins, and components of the β-adrenergic receptor system. Given the plethora of signals that cause hypertrophy and heart failure, an alternative approach would be to target an effector common to many such pathways. Class II HDACs and their regulatory kinase(s) appear to represent such a point of convergence and as such represent potential therapeutic targets.
Experimental Procedures
HDAC Kinase Assays
Adult mouse hearts were homogenized in 1 ml of lysis buffer (PBS containing 1 mM EDTA, 0.1% Triton X-100, 1 mM PMSF, and protease inhibitor cocktail [Roche]). After brief sonication, lysates were clarified by ultra-centrifugation, and protein concentration was determined.
GST-HDAC substrates contained amino acids 208–310 of HDAC4, 218–328 of HDAC5, or 180–283 of HDAC9 fused to glutathione S-transferase (GST). GST-fusion proteins were also made with serine-to-alanine mutations at positions 246, 259, and 218 in HDACs 4, 5, and 9, respectively. GST-HDAC protein (1 μg) was conjugated to glutathione-agarose beads. GST-HDAC-bound beads were washed with PBS and subsequently incubated with heart protein lysate (100 μg) in lysis buffer for 2 hr at 4°C. Beads were washed twice with the same buffer and equilibrated with kinase reaction buffer (25 mM HEPES [pH 7.6], 10 mM MgCl2, and 0.1 mM CaCl2). Beads were then resuspended in kinase reaction buffer (30 μl) containing 12.5 μM ATP and 5 μCi [γ-32P]-ATP and reactions were allowed to proceed for 30 min at room temperature. Reactions were then boiled, and phosphoproteins were resolved by SDS-PAGE, visualized by autoradiography, and quantified using a phosphorimager.
Transgenic Mice and β-Galactosidase Staining
Mice bearing transgenes encoding a constitutively active form of calcineurin under control of the α-MHC promoter, and mice carrying the desMEF2-LacZ transgene (MEF2 indicator mice) have been described (Molkentin et al., 1998; Naya et al., 1999). LacZ gene expression was determined as described (Naya et al., 1999).
Cardiomyocyte Cell Culture and Adenovirus Infections
Primary rat cardiomyocytes were prepared as described (Molkentin et al., 1998). Eighteen hours after plating, cells were infected with adenovirus for 2 hr and subsequently cultured in serum-free medium for 24 hr before stimulation for another 24 hr with PE (100 μM; Sigma), ET-1 (100 nM; American Peptide Company, Inc), IGF-1 (100 ng/ml; American Peptide Company, Inc), ANGII (100 nM; American Peptide Company, Inc), or FBS (2.5%; Sigma).
Myc- and FLAG-tagged derivatives of MITR and HDAC5 and their signal-resistant counterparts (S218/448A and S259/498A, respectively) have been described (McKinsey et al., 2000a; Zhang et al., 2001b). For adenovirus production, cDNAs were cloned into the pAC-CMV vector and the resultant constructs were cotransfected into 293 cells with pJM17 using Fugene 6 (Roche). Primary lysates were used to reinfect 293 cells and viral plaques were obtained using the agar overlay method. Clonal populations of adenoviruses were amplified by reinfecting 293 cells and titered.
Immunocytochemistry
Primary cardiomyocytes plated on laminin-coated glass coverslips were fixed and stained as described (Zhang et al., 2001b). Fluorescent images were collected on a LSM 410 Zeiss confocal microscope and were processed with Adobe Photoshop. The following primary and secondary antibodies were used at 1:200 dilution: anti-α-actinin (EA-53; Sigma), anti-Myc (9E10 and A-14; Santa Cruz), anti-rat-ANF (IHC9103; Peninsula Laboratories, Inc.), Texas Red-conjugated anti-mouse or anti-rabbit IgG (Vector Labs), and fluorescein-conjugated anti-mouse or anti-rabbit IgG (Vector Labs).
RNA Isolation and Analysis
Total RNA was purified from cultured cells or intact hearts with Trizol reagent (Invitrogen) according to manufacturer’s instructions. Northern and RNA dot blot analyses were performed with 20 μg and 2 μg, respectively, of total RNA. For RT-PCR, total RNA was used as a template for reverse transcriptase and random hexamer primers (Invitrogen). Sequences of oligonucleotide probes and primers are available upon request.
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assays were performed as described (Lu et al., 2000b), using soluble chromatin prepared from cardiomyocytes. Equal amounts of chromatin from each sample (normalized by ethidium bromide staining of DNA) were immunoprecipitated with an anti-acetylated histone H3 antibody (Upstate Biotechnology). Ten percent of the precipitated DNA was subjected to 28 cycles of PCR in the presence of [32P]-dCTP with primers specific for the ANF, β-MHC, α-cardiac actin, or GAPDH promoters. As a control for DNA content, PCR reactions were also performed on chromatin samples prior to immunoprecipitation. Twenty percent of each reaction mixture was resolved through a 5% native polyacrylamide gel, visualized, and quantified using a phosphorimager (Molecular Dynamics). Primer sequences are available upon request.
Generation of HDAC9 Targeting Construct and Mutant Mice
The HDAC9 targeting construct was generated using the pN-Z-TK2 vector, which contains a nuclear LacZ (nLacZ) cassette and a neomycin-resistance gene (kindly provided by R. Palmiter). Following electroporation into the R1 ES cell line and positive-negative selection, 780 individual ES cell clones were isolated and analyzed by Southern blotting for homologous recombination. Four clones with a disrupted HDAC9 gene were injected into 3.5-day mouse C57BL/6 blastocysts, and the resulting chimeric male mice were bred to C57BL/6 females to achieve germline transmission of the mutant allele.
Thoracic Aorta Banding
Six-to-eight-week-old male wild-type and HDAC9−/− littermates underwent either a sham operation or were subjected to pressure overload induced by thoracic aorta banding as described (Hill et al., 2000).
Acknowledgments
We are grateful to Yin-Chai Cheah for blastocyst injections, Hartmut Weiler for assistance with gene targeting, Robert Gerard for assistance with viruses, and Stuart Schreiber for providing HDAC plasmids. We thank James Richardson, Jeff Starks, and John Shelton for histological sections, and Alisha Tizenor for graphics. This work was supported by grants from the National Institutes of Health, the D.W. Reynolds Clinical Cardiovascular Research Center, the Texas Advanced Technology Program, and the Robert A. Welch Foundation to E.N.O. T.A.M. is a Pfizer Fellow of the Life Sciences Research Foundation.
References
- Black BL, Olson EN. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol. 1998;14:167–196. doi: 10.1146/annurev.cellbio.14.1.167. [DOI] [PubMed] [Google Scholar]
- Chien KR. Stress pathways and heart failure. Cell. 1999;98:555–558. doi: 10.1016/s0092-8674(00)80043-4. [DOI] [PubMed] [Google Scholar]
- Frey N, McKinsey TA, Olson EN. Decoding calcium signals involved in cardiac growth and function. Nat Med. 2000;6:1221–1227. doi: 10.1038/81321. [DOI] [PubMed] [Google Scholar]
- Grozinger CM, Schreiber SL. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14–3-3-dependent cellular localization. Proc Natl Acad Sci USA. 2000;97:7835–7840. doi: 10.1073/pnas.140199597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozinger CM, Schreiber SL. Deacetylase enzymes. Biological functions and the use of small-molecule inhibitors. Chem Biol. 2002;9:3–16. doi: 10.1016/s1074-5521(02)00092-3. [DOI] [PubMed] [Google Scholar]
- Hill JA, Karimi M, Kutschke W, Davisson RL, Zimmerman K, Wang Z, Kerber RE, Weiss RM. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2863–2869. doi: 10.1161/01.cir.101.24.2863. [DOI] [PubMed] [Google Scholar]
- Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, Goodman RH. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34:235–244. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Johnson CA, Turner BM. Histone deacetylases: complex transducers of nuclear signals. Semin Cell Dev Biol. 1999;10:179–188. doi: 10.1006/scdb.1999.0299. [DOI] [PubMed] [Google Scholar]
- Kato T, Sano M, Miyoshi S, Sato T, Hakuno D, Ishida H, Kinoshita-Nakazawa H, Fukuda K, Ogawa S. Calmodulin kinases II and IV and calcineurin are involved in leukemia inhibitory factor-induced cardiac hypertrophy in rats. Circ Res. 2000;87:937–945. doi: 10.1161/01.res.87.10.937. [DOI] [PubMed] [Google Scholar]
- Kirchhefer U, Schmitz W, Scholz H, Neumann J. Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc Res. 1999;42:254–261. doi: 10.1016/s0008-6363(98)00296-x. [DOI] [PubMed] [Google Scholar]
- Liang Q, De Windt LJ, Witt SA, Kimball TR, Markham BE, Molkentin JD. The transcription factors GATA4 and GATA6 regulate cardiomyocyte hypertrophy in vitro and in vivo. J Biol Chem. 2001;276:30245–30253. doi: 10.1074/jbc.M102174200. [DOI] [PubMed] [Google Scholar]
- Lowes BD, Gilbert EM, Abraham WT, Minobe WA, Larrabee P, Ferguson D, Wolfel EE, Lindenfeld J, Tsvetkova T, Robertson AD, et al. Myocardial gene expression in dilated cardiomyopathy treated with beta-blocking agents. N Engl J Med. 2002;346:1357–1365. doi: 10.1056/NEJMoa012630. [DOI] [PubMed] [Google Scholar]
- Lu J, McKinsey TA, Nicol RL, Olson EN. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci USA. 2000a;97:4070–4075. doi: 10.1073/pnas.080064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, McKinsey TA, Zhang CL, Olson EN. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell. 2000b;6:233–244. doi: 10.1016/s1097-2765(00)00025-3. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Lu J, Olson EN. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. 2000a;408:106–111. doi: 10.1038/35040593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN. Activation of the myocyte enhancer factor-2 transcription factor by calcium/calmodulin-dependent protein kinase-stimulated binding of 14–3-3 to histone deacetylase 5. Proc Natl Acad Sci USA. 2000b;97:14400–14405. doi: 10.1073/pnas.260501497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci. 2002;27:40–47. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin S, Charron F, Robitaille L, Nemer M. GATA-dependent recruitment of MEF2 proteins to target promoters. EMBO J. 2000;19:2046–2055. doi: 10.1093/emboj/19.9.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naya FJ, Wu C, Richardson JA, Overbeek P, Olson EN. Transcriptional activity of MEF2 during mouse embryogenesis monitored with a MEF2-dependent transgene. Development. 1999;126:2045–2052. doi: 10.1242/dev.126.10.2045. [DOI] [PubMed] [Google Scholar]
- Passier R, Zeng H, Frey N, Naya FJ, Nicol RL, McKinsey TA, Overbeek P, Richardson JA, Grant SR, Olson EN. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Invest. 2000;105:1395–1406. doi: 10.1172/JCI8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartorelli V, Huang J, Hamamori Y, Kedes L. Molecular mechanisms of myogenic coactivation by p300: direct interaction with the activation domain of MyoD and with the MADS box of MEF2C. Mol Cell Biol. 1997;17:1010–1026. doi: 10.1128/mcb.17.2.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow DB, Miska EA, Langley E, Reynaud-Deonauth S, Kotecha S, Towers N, Spohr G, Kouzarides T, Mohun TJ. MEF-2 function is modified by a novel co-repressor, MITR. EMBO J. 1999;18:5085–5098. doi: 10.1093/emboj/18.18.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn HD, Chatila TA, Liu JO. Integration of calcineurin and MEF2 signals by the coactivator p300 during T-cell apoptosis. EMBO J. 2000;19:4323–4331. doi: 10.1093/emboj/19.16.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, McKinsey TA, Lu JR, Olson EN. Association of COOH-terminal-binding protein (CtBP) and MEF2-interacting transcription repressor (MITR) contributes to transcriptional repression of the MEF2 transcription factor. J Biol Chem. 2001a;276:35–39. doi: 10.1074/jbc.M007364200. [DOI] [PubMed] [Google Scholar]
- Zhang CL, McKinsey TA, Olson EN. The transcriptional corepressor MITR is a signal-responsive inhibitor of myogenesis. Proc Natl Acad Sci USA. 2001b;98:7354–7359. doi: 10.1073/pnas.131198498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Johnson EN, Gu Y, Morissette MR, Sah VP, Gigena MS, Belke DD, Dillmann WH, Rogers TB, Schulman H, et al. The cardiac-specific nuclear δ(B) isoform of Ca2+/calmodulin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J Biol Chem. 2002;277:1261–1267. doi: 10.1074/jbc.M108525200. [DOI] [PubMed] [Google Scholar]
- Zhou X, Marks PA, Rifkind RA, Richon VM. Cloning and characterization of a histone deacetylase, HDAC9. Proc Natl Acad Sci USA. 2001;98:10572–10577. doi: 10.1073/pnas.191375098. [DOI] [PMC free article] [PubMed] [Google Scholar]