

Abstract
Many RNA regulatory proteins controlling pre-mRNA splicing contain serine:arginine (SR) repeats. Here we found that these SR domains bound hydrogel droplets composed of fibrous polymers of the low-complexity domain of heterogeneous ribonucleoprotein A2 (hnRNPA2). Hydrogel binding was reversed upon phosphorylation of the SR domain by CDC2-like kinases 1 and 2 (CLK1/2). Mutated variants of the SR domains changing serine to glycine (SR-to-GR variants) also bound to hnRNPA2 hydrogels, but were not affected by CLK1/2. When expressed in mammalian cells, these variants bound nucleoli. The translation products of the sense and antisense transcripts of the expansion repeats associated with the C9ORF72 gene altered in neurodegenerative disease encode GRN and PRN repeat polypeptides. Both peptides bound to hnRNPA2 hydrogels independent of CLK1/2 activity. When applied to cultured cells, both peptides entered cells, migrated to the nucleus, bound nucleoli, and poisoned RNA biogenesis, which caused cell death.
Among familial causes of amyotrophic lateral sclerosis (ALS) and/or frontotemporal dementia (FTD), between 25 and 40% of cases are attributed to a repeat expansion in a gene designated C9ORF72. The hexa-nucleotide repeat sequence GGGGCC normally present in 2 to 23 copies is expanded in affected patients to 700 to 1,600 copies (1, 2). The pattern of genetic inheritance of the C9ORF72 repeat expansion is dominant, and multiple lines of evidence suggest that the repeat expansion causes disease. Two theories have been advanced to explain repeat-generated toxicity. First, in situ hybridization assays have identified nuclear dots containing either sense or anti-sense repeat transcripts (3–5), leading to the idea that the nuclear-retained RNAs might themselves be toxic. More recently, equally clear evidence has been generated showing that both the sense and anti-sense transcripts of the GGGGCC repeats associated with C9ORF72 can be translated in an ATG-independent manner known as repeat associated non-ATG (RAN) translation (6). Depending upon reading frame, the sense transcript of the repeats can be translated into glycine:alanine (GAN), glycine:proline (GPN), or glycine:arginine (GRN) polymers. RAN translation of the anti-sense transcript of the GGGGCC repeats of C9ORF72 lead to the production of proline:alanine (PAN), proline:glycine (PGN) or proline:arginine (PRN) polymers. These repeat-encoded polymers are expressed in disease tissue (5, 7–9). The disordered and hydrophobic nature of these polymers, at least the GAN, GPN, and PAN versions, properly predicted that they would aggregate into distinct foci within affected cells (5, 9). Another plausible explanation for repeat-generated toxicity is the idea that the polymeric aggregates resulting from RAN translation of either the sense or anti-sense repeats are themselves toxic.
Here we investigated a third and distinct interpretation as to the underlying pathophysiology associated with repeat expansion of the hexanucleotide repeats associated with the C9ORF72 gene. We suggest that two of the six RAN translation products, GRN encoded by the sense transcript and PRN encoded by the anti-sense transcript, act to alter information flow from DNA to messenger RNA (mRNA) to protein in a manner that poisons both pre-mRNA splicing and the biogenesis of ribosomal RNA.
SR domains of pre-mRNA splicing factors bind hnRNPA2 hydrogels in a phosphorylation-regulated manner
Our standard method of retrieving proteins enriched in unfolded, low complexity sequences involves the incubation of cellular lysates with a biotinylated isoxazole (b-isox) chemical (10). When incubated on ice in aqueous buffers, the b-isox chemical crystallizes. X-ray diffraction analyses of the b-isox crystals revealed the surface undulation of peaks and valleys separated by 4.7Å. When exposed to cell lysates, it is hypothesized that disordered, random coil sequences can bind to the surface troughs of b-isox crystals and thereby be converted to an extended β-strand conformation. When the crystals are retrieved by centrifugation, they selectively precipitate DNA and RNA regulatory proteins endowed with low complexity sequences. When these methods were employed to query the distribution of nuclear proteins precipitated by b-isox microcrystals, scores of proteins annotated as being involved in the control of pre-mRNA splicing were retrieved (11).
Many splicing factors contain long repeats of the dipeptide sequence serine:arginine (SR). Given the low complexity nature of SR domains, we hypothesized that it was this determinant that facilitated b-isox precipitation. Focusing on a member of the SR protein family that has been studied extensively, serine:arginine splicing factor 2 (SRSF2), we appended its SR domain to green fluorescent protein (GFP) to ask whether the SR domain might be sufficient to mediate b-isox precipitation. GFP is a well-folded protein that, alone, is not precipitated by b-isox crystals (10). When fused to the SR domain of SRSF2, GFP was precipitated efficiently by b-isox crystals (fig. S1, A and B).
When incubated at high concentrations, the low complexity domains of certain RNA regulatory proteins, including FUS, EWS, TAF15 and hnRNPA2, polymerize into amyloid-like fibers. In a time and concentration-dependent manner, these fibers adopt a hydrogel-like state (10). No evidence of polymerization or hydrogel formation was observed upon incubation of the GFP fusion protein containing the SR domain of SRSF2 (designated GFP:SRSF2). We then asked whether the fusion protein might be bound and retained by hydrogel droplets formed from polymers of the low complexity domain of hnRNPA2 (10). Indeed, GFP:SRSF2 bound avidly to hydrogel droplets formed from the LC domain of hnRNPA2 (Fig. 1A).
Fig. 1. CLK1/2-mediated release of GFP-fused SR domain from mCherry:hnRNPA2 hydrogel droplets.

Hydrogel droplets composed of mCherry fused to the low complexity domain of hnRNPA2 were incubated with protein solution of GFP-fused to SR domains from either SRSF2 (A) or SRSF2G1/G2 (B). Both GFP proteins bound well to the mCherry:hnRNPA2 hydrogels as revealed by GFP signal trapped at the periphery of hydrogel droplets (22). Upon overnight incubation with either CLK1 or CLK2, pre-bound GFP-fused SR domain of SRSF2 was released from the mCherry:hnRNPA2 hydrogels in the presence of ATP [third and fifth panels of (A)]. The GFP-fused to the SR domain of SRSF2G1/G2 was resistant to CLK1/2-mediated release from hydrogels [third and fifth panels of (B)].
The SR domains of splicing factors can be phosphorylated (12–14). Two related protein kinase enzymes, CDC2-like kinase 1 (CLK1) and CDC2-like kinase 2 (CLK2), phosphorylate serine residues within SR domains (fig. S1C) (15–18). In order to ask whether phosphorylation of SR domains might affect their binding to hydrogel droplets formed from the LC domain of hnRNPA2, we pre-bound the GFP:SRSF2 fusion protein then exposed the droplets to ATP alone, CLK1/2 enzymes alone, or a mix of ATP and enzymes. Release of the GFP:SRSF2 test protein was observed in a time, enzyme and ATP dependent manner (Fig. 1A).
The CLK1/2 protein kinases themselves contain SR domains, presumably helping to guide these enzymes to the proper sub-nuclear locations where they serve to regulate the activities of SR domain-containing splicing factors (17). Hydrogel droplets were co-exposed to GFP:SRSF2 along with a derivative of CLK2 containing an SR domain. In this case, exposure to ATP alone facilitated release of GFP:SRSF2, presumably due to activation of the CLK2 enzyme held by its SR domain in proximity to the GFP:SRSF2 test protein (fig. S1D).
The SRSF2 splicing factor contains two SR domains, one located between residues 117 and 169 of the polypeptide, and another located between residues 177 and 221 (fig. S2). Sixteen out of twenty one serine residues within the former SR domain were mutated to glycine, leading to a variant designated SRSF2G1. Likewise, fourteen out of seventeen serine residues within the latter SR domain were mutated to glycine, leading to the SRSF2G2 variant. These two mutants were recombined to produce the SRSF2G1/G2 variant (fig. S2). The altered SR domain of the SRSF2G1/G2 variant was fused to GFP (GFP:SRSF2G1/G2), expressed in bacteria, purified and exposed to hnRNPA2 hydrogel droplets. Like the native SR domain, the GFP:SRSF2G1/G2 variant bound to the hydrogel droplets. By contrast, when the bound hydrogel droplets were exposed to ATP and either of the CLK1/2 enzymes, no GFP was released (Fig. 1B).
Binding of native and serine-to-glycine variants of SRSF2 to nuclear puncta
SR domain-containing pre-mRNA splicing factors localize to various puncta in the nucleus of eukaryotic cells (19). In interphase nuclei, SR-containing proteins are found in puncta variously termed “interchromatin granule clusters” or nuclear speckles. These puncta are roughly 1–3 μM in diameter and are composed of smaller granules connected by a thin fibril (20). Hypophosphorylated SR domains associate with the periphery of nucleoli in a region termed nucleolar organizing region (NOR) associated patches (NAPs) (21). Knowing that the SR domain of the SRSF2G1/G2 mutant binds to hnRNPA2 hydrogels in a manner immune to CLK1/2-mediated release, we transfected cultured cells with GFP-tagged versions of the four SRSF2 variants (the native protein, the SRSF2G1 mutant, the SRSF2G2 mutant and the SRSF2G1/G2 mutant). Unlike the native SRSF2 protein, which distributed to nuclear speckles, the other three proteins associated with nucleoli (Fig. 2A). When co-transfected with an expression vector encoding CLK1 enzyme, partial release from nucleoli was observed for the SRSF2G1 and SRSF2G2 mutants, yet no release was observed for the SRSF2G1/G2 mutant (Fig. 2B). Thus, it appears that by changing serine residues to glycine in the three mutants of SRSF2, we created mimics of the hypophosphorylated state of SR proteins. Because the SRSF2G1 and SRSF2G2 proteins can only be partially phosphorylated by CLK1, and because the SRSF2G1/G2 variant cannot be phosphorylated, we reason that these proteins become trapped in nucleoli at an early stage of the pathway of nuclear speckle formation and pre-mRNA splicing.
Fig. 2. Native or S-to-G mutated variants of SRSF2 localize to different nuclear puncta.

GFP fusion proteins linked to either the native, full-length SRSF2 or the SRSF2G1, SRSF2G2 or SRSF2G1/G2 mutants were transfected in U2OS cells in the absence (A) or presence (B) of a co-expressed mCherry:CLK1 fusion protein. The native SRSF2 protein localized to nuclear speckles and was dispersed into the nucleoplasm in the presence of co-transfected mCherry:CLK1. The SRSF2G1 and SRSF2G2 mutants localized to nucleoli as deduced by co-staining with antibodies specific to the nucleolar marker, Fibrillian. The SRSF2G1 mutant was partially redistributed from nucleoli to the cytoplasm in the presence of mCherry:CLK1. The SRSFG2 mutant was partially redistributed from nucleoli to the nucleoplasm in the presence of mCherry:CLK1. Co-expression of mCherry:CLK1 had no effect on the nucleolar localization of the SRSF2G1/G2 mutant.
The GRN and PRN RAN translation products of C9ORF72 bind hnRNPA2 hydrogels
The sense and anti-sense transcripts of the GGGGCC repeat expansions associated with familial forms of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) can be translated in an ATG independent manner (5, 7, 9). Depending upon reading frame, the sense repeat transcript encodes GAN, GPN or GRN polymers. Likewise, the anti-sense transcripts of the repeats encode PAN, PGN and PRN polymers. We focused on the GRN translation product of the sense repeat transcript, and the PRN translation product of the anti-sense repeat transcript, for three reasons. First, these polymers are considerably more hydrophilic than the GAN, GPN, PAN and PGN polymers, and less likely to aggregate. Second, the GRN and PRN polymers might, by virtue of the abundance of arginine residues, be self-programmed to return to the nucleus after cytoplasmic translation (owing to the fact that nuclear localization signals tend to be enriched in basic amino acids). Third, these polymers are reminiscent of the SRSF2G1, SRSF2G2 and SRSF2G1/G2 variants that bound to hnRNPA2 hydrogel droplets independent of the effects of the CLK1 enzyme (Fig. 1), and associated tightly with nucleoli in living cells (Fig. 2).
GFP derivatives were prepared to contain 20 repeats of the dipeptide sequence SR, GR or PR (22). After expression in bacterial cells and purification, each fusion protein was incubated with hnRNPA2 hydrogels. Unlike GFP itself, which did not bind to any of the hydrogels used, the GFP:SR20, GFP:GR20 and GFP:PR20 fusion proteins bound avidly to hnRNPA2 hydrogel droplets. When protein-bound hydrogels were exposed to CLK1 or CLK2 in the presence of ATP, GFP:SR20 was liberated, but not GFP:GR20 or GFP:PR20 (Fig. 3). We interpret these results in the same way as observations made with GFP:SRSF2 and its serine to glycine variants (Figs. 1 and 2). CLK1/2-mediated phosphorylation of the serine residues in the GFP:SR20 fusion protein is interpreted to facilitate its release from hnRNPA2 hydrogel droplets. Because the GRN and PRN polymers have no serine residues, they cannot be phosphorylated and released form hydrogels upon exposure to CLK1/2 and ATP.
Fig. 3. Binding of translation products of C9ORF72 hexanucleotide repeat expansion to mCherry:hnRNPA2 hydrogel droplets.

Recombinant fusion proteins linking GFP to 20 repeats of the SR, GR or PR polymers (GFP:SR20, GFP:GR20 or GFP:PR20) were applied to slide chambers containing mCherry:hnRNPA2 hydrogel droplets. After overnight incubation at 4°C, all three proteins were trapped to the periphery of the hydrogels droplets (top panels). When incubated with reaction mixtures containing either the CLK1 or CLK2 protein kinase enzymes, pre-bound GFP:SR20 was released from the hydrogels in an ATP-dependent manner. GFP:GR20 or GFP:PR20 pre-bound to mCherry:hnRNPA2 hydrogel droplets were immune to the release by CLK1 or CLK2, even in the presence of ATP.
The GRN and PRN RAN translation products of C9ORF72 penetrate cells, migrate to the nucleus, bind nucleoli and kill cells
Polymeric versions of the GRN and PRN translation products of C9ORF72 were synthesized to contain 20 dipeptide repeats terminated by an epitope tag (22). The synthetic peptides were solubilized in aqueous buffer and applied to cultured U2OS cells for 30 min at 10 μM. The cells were then fixed and stained with antibodies capable of recognizing the HA epitope tag. Both the GR20 and PR20 polymers entered cells, migrated to the nucleus and bound to nucleoli (Fig. 4A). The morphology of U2OS cells was altered after prolonged exposure to the GR20 and PR20 translation products of C9ORF72 hexanucleotide repeats. Alteration in cell morphology was more pronounced for the PR20 peptide than GR20. Within 24 hours of exposure to 10 μM levels of PR20, U2OS cells began to display a spindle-like phenotype. Upon exposure to 30 μM of the PR20 peptide for 24 hours, almost all cells were detached from the culture substrate and dead (fig. S3A). Similar effects on cell morphology and viability were observed for cultured human astrocytes (fig. S3B).
Fig. 4. Synthetic GR20 and PR20 peptides bind nucleoli and kill cells.

(A) Peptides containing 20 repeats of GR or PR (GR20 or PR20, respectively) were synthesized to contain an HA epitope tag and applied to cultured U2OS cancer cells (left panels) or human astrocytes (right panels). Cells were fixed and stained with either the HA reacting antibody (green signal) or an antibody to the nucleolar protein Fibrillarin (red signal). Both GR20 and PR20 synthetic peptides associated prominently with nucleoli. Measurements of U2OS cell viability revealed toxicity in response to both PR20 (B) and GR20 (C) synthetic peptides. Cell viability was measured at 72 or 12 hours after initial treatment of PR20 or GR20, respectively. In the case of GR20 peptide, the medium was replaced every 2 hours to supplement fresh peptide. The PR20 and GR20 synthetic peptides killed U2OS cells with IC50 levels of 5.9 and 8.4 μM, respectively.
The stability of the GR20 and PR20 peptides was analyzed by immunoblotting. Following the administration of a single dose of each peptide, U2OS cells were incubated for indicated time periods. After retrieval of culture medium, cells were then washed with phosphate buffered saline, lysed and deposited onto nitrocellulose dot blots that were probed with antiserum specific to the HA epitope. These measurements gave evidence of a relatively short half-life for the GR20 peptide (20–30 min), but a much longer half-life for the PR20 peptide (72 hours) (fig. S4, A and B).
Cell viability was then measured for cultures exposed to varying levels of the PR20 (22). An IC50 value of 5.9 μM was observed for the PR20 peptide (Fig. 4B). Similar cellular toxicity was observed for the GR20 peptide, but required that the GR20 peptide be added every two hours (Fig. 4C). Cell death in response to the PR20 peptide was also time dependent. After administration of 10 μM of the PR20 peptide, half-maximal impact on cell viability was observed roughly 36 hours later (fig. S4E). When exposed to a 30 μM dose of the peptide, 50% cell death was observed at 6 hours (fig. S4F).
Exposure of cultured cells to the GR20 and PR20 translation products of C9ORF72 impairs both pre-mRNA splicing and the biogenesis of ribosomal RNA
Having observed that GR20 and PR20 translation products of the C9ORF72 hexanucleotide repeats bound nucleoli and killed cultured cells, we wondered whether this might be the consequence of alterations in RNA biogenesis. To this end, cultured human astrocyte cells were exposed for 6 hours to the synthetic PR20 peptide and used to prepare RNA for deep sequencing. Computational analysis of the RNA-seq data predicted alteration in splicing in a variety of cellular mRNAs (22). Validation of predicted changes in pre-mRNA splicing was conducted by use of strategically designed PCR primers (22). PCR products consistent with predicted alterations in splicing were subjected to DNA sequencing (fig. S5A). In all cases, predicted changes in pre-mRNA splicing were confirmed, with the degrees of effect on splicing ranging from modest in the cases of the NACA and RAN GTPase mRNAs, to severe in the cases of the PTX3 and GADD45A mRNAs. Administration of the PR20 peptide caused exon 2-skipping of the mRNA encoding the RAN GTPase, thus resulting in removal of the first 88 residues of the protein (Fig. 5A and fig. S5B). Furthermore, PR20 administration caused exon 2-skipping of the mRNA encoding the pentraxin-related protein PTX3, thereby predicting an in frame deletion of 135 amino acids (Fig. 5B and fig. S5C). PR20 administration also caused the mRNA encoding nascent polypeptide-associated complex subunit alpha (NACA) to contain a different 5′ UTR (Fig. 5C). Finally, PR20 administration caused the mRNA encoding the growth arrest and DNA damage-inducible (GADD45) protein to include the full intronic sequences on both sides of exon 2 in the mature transcript, thereby altering the open reading frame in a manner expected to inactivate the GADD45 protein if translated from the aberrantly spliced mRNA (Fig. 5D).
Fig. 5. Effect of PR20 peptide on RNA processing.

Aberrant splicing of the RAN GTPase, PTX3, NACA and GADD45A transcripts in PR20-treated cells was validated by RT-PCR (A to D). Schematic diagrams show either normal splicing (black lines) or mis-splicing (red lines). A bold red line in panel (D) indicates retention of intron. (A) RT-PCR analysis of RAN GTPase transcript: arrow indicates normal transcript (252 bp) and arrowhead indicates exon 2-skipped transcript (212 bp). (B) RT-PCR analysis of PTX3 transcript: arrow indicates normal transcript (844 bp) and arrowhead indicates exon 2-skipped transcript (442 bp). (C) RT-PCR analysis of NACA transcript: arrow indicates normal transcript (386 bp) and arrowhead indicates transcript with aberrant 5′ UTR (314 bp). (D) RT-PCR analysis of GDD45A transcript: arrow indicates normal transcript (573 bp) and arrowhead indicates intron-retention transcript (1,283 bp). (E) Scatter plot of RNA abundance measured from RNA-seq data (top panel). X-axis designates RNA abundance (log2(FPKM)) for the control sample, and the Y-axis corresponds to RNA abundance for the PR20 treated sample. Each dot represents a single mRNA species, with green dots representing transcripts of individual ribosomal protein genes. The distribution of RNA abundance fold-change between the PR20 treated sample and the control sample is shown in the bottom panel of E. Black line represents the distribution of all genes, and green line represents the distribution of ribosomal protein genes. Expression of members of the ribosomal protein gene family was significantly up-regulated by PR20 treatment (P < 2.2e-16, Kolmogorov–Smirnov test). (F) Aberrant ribosomal RNA (rRNA) processing in PR20-treated cells as analyzed by qPCR. Data are plotted as normalized fold-change against control and error is represented by standard deviation of triplicate experiments. The Y-axis indicates fold changes relative to untreated control. Black bars on X-axis below histograms indicate approximate locations of qPCR primers for 45S, 18S-5′ junction, 18S, 18S-3′ junction, 5.8S-5′ junction, 5.8S, 5.8S-3′ junction, 28S-5′ junction, and 28S rRNA (from left to right).
Computational analysis of RNA-seq data further revealed changes in the abundance of a subset of cellular RNAs as a function of administration of the PR20 peptide (table S1). A large fraction of the altered RNAs encoded ribosomal proteins (Fig. 5E) or snoRNAs. In both cases, PR20 administration enhanced RNA abundance. Having observed that both the GR20 and PR20 peptides bound to nucleoli, and having observed changes in the abundance of snoRNAs and mRNAs encoding ribosomal proteins, we investigated the central task of nucleoli to synthesize mature ribosomal RNA (rRNA). Nine PCR primer pairs were designed to interrogate the synthesis and processing of rRNA (22). Three monitored the levels of the mature 18S, 5.8S and 28S rRNAs. The other six primers were designed to monitor the 45S rRNA precursor, including pairs that probed: (i) the initial, 5′ end of the precursor that is eliminated along the pathway of rRNA maturation; (ii) the precursor junction at the 5′ end of 18S rRNA; (iii) the precursor junction at the 3′ end of 18S rRNA; (iv) the precursor junction at the 5′ end of 5.8S rRNA; (v) the precursor junction at the 3′ end of 5.8S rRNA; and (vi) the precursor junction at the 5′ end of 28S rRNA (see Fig. 5F). RNA was prepared from human astrocytes exposed for 12 hours to vehicle alone, or 10 μM or 30 μM levels of the PR20 peptide. Slight reductions in 28S rRNA were observed in the samples derived from cells treated with 30 μM of the PR20 peptide. Surprisingly, the level of 5.8S rRNA was reduced by 70% under these conditions (Fig. 5F).
Evidence of impediments in the production of rRNA was confirmed upon evaluation of junctional PCR probes. The qPCR primers specific for the 5′ transcribed spacer at the front end of the rRNA precursor revealed a 20% elevation of the precursor in cells exposed to the lower, 10 μM concentration of the PR20 peptide. The first junctional probe also revealed an elevation in immature rRNA, the second junctional probe revealed normal levels of rRNA precursor, the third probe revealed roughly 20% attenuation of the precursor, the probe monitoring the 3′ junction of 5.8S rRNA revealed 40% attenuation, and the probe monitoring the 5′ junction of 28S rRNA revealed normal precursor levels. Cells exposed to 30 μM levels of the PR20 peptide revealed reductions in the 45S rRNA precursor consistent with a similar, 5′ to 3′ polarity of impediment. Indeed, the qPCR primer pair monitoring processing at the 3′ terminus of 5.8S rRNA revealed a 70% drop. Irrespective of whether these effects result from altered transcription of rRNA genes, altered processing of the 45S rRNA precursor, or both, these assays provide evidence of nucleolar dysfunction in cells treated with the PR20 RAN translation product of the C9ORF72 hexanucleotide repeats.
The GRN and PRN synthetic peptides alter splicing of the EAAT2 transcript in a pattern identical to that observed in ALS patients
Having observed global alterations in pre-mRNA splicing in cells exposed to the PR20 synthetic peptide, we asked whether alterations in pre-mRNA splicing might have been observed in the study of patient-derived tissues. Splicing of the transcript encoding a glutamate transporter designated excitatory amino acid transporter 2 (EAAT2) is altered in ALS patients. Two hallmarks of the altered pattern were the skipping of exon 9 and the inclusion of 1008 residues of intronic sequence downstream from the splice donor site of exon 7 (23).
In order to ask whether a similar pattern of derangement might result from exposure of cells to the PR20 synthetic peptide, we incubated cultured human astrocytes with 10 or 15 μM levels of the polymer for 36 hours, a time point corresponding to roughly 50% reduction in cell viability (fig. S4E). RNA was then extracted and subjected to PCR analysis as a means of resolving the architecture of the EAAT2 transcript (22). PCR products diagnostic of both the exon 9-skipped form of the EAAT2 mRNA, as well as the 1008 residue extension of intron inclusion beyond the splice donor site of exon 7, appeared in a concentration-dependent manner in human astrocytes as a function of exposure to the PR20 polymer (Fig. 6). Both PCR products were sequenced and found to replicate the pattern of aberrant splicing first described in ALS patients precisely (fig. S6A).
Fig. 6. Altered EAAT2 splicing in human astrocytes exposed to PR20 synthetic peptide.
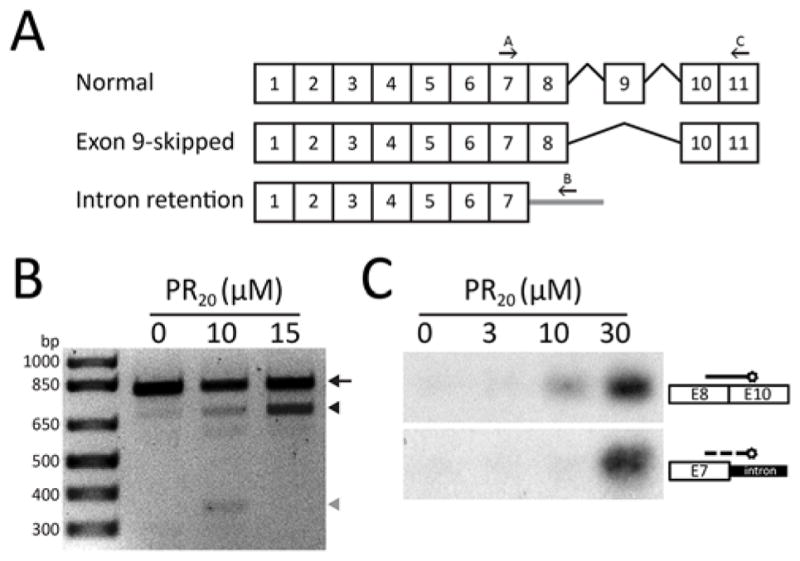
(A) Normal and aberrantly spliced EAAT2 transcripts reveal locations of exon 9-skipping and intron 7 retention. Arrows indicate the primers used for RT-PCR of EAAT2 transcripts (22). (B) RNA prepared from human astrocytes exposed to 0, 10 μM or 15 μM of the synthetic PR20 peptide was interrogated with PCR primers diagnostic of the normal EAAT2 transcript (arrow), the exon 9-skipped variant (black arrowhead) or the intron 7 retention variant (grey arrowhead). No evidence of aberrant EAAT2 transcripts was observed in control cells. Cells exposed for 36 hours to 10 μM of the synthetic PR20 peptide showed equal amounts of the exon 9-skipped and intron 7 retention aberrant transcripts. Cells exposed for 36 hours to 15 μM of the synthetic PR20 peptide showed a significant increase in the amount of the exon 9-skipped EAAT2 transcript. (C) Southern blot probes specific to the exon 9-skipped and intron 7 retention aberrant EAAT2 transcripts revealed exclusive labeling of the PCR products specific to each mRNA isoform. Human astrocytes were exposed to zero, 3 μM, 10 μM or 30 μM levels of the synthetic PR20 peptide for 6 hours. Following PCR amplification and gel electrophoresis, PCR products were blotted onto nitrocellulose and hybridized with isoform-specific probes (22).
It is possible that generalized cell toxicity commonly causes an idiosyncratic pattern of aberrant splicing of the EAAT2 mRNA, thus explaining why cells poisoned by the PR20 peptide replicate the same pattern of improper splicing observed in patient samples. To test this hypothesis, human astrocytes were individually exposed to four other toxins, doxorubicin, taxol, staurosporin and cytochalasin D. RNA was prepared from cells exhibiting clear evidence of toxicity and analyzed by PCR as a means of testing for the patterns of aberrant EAAT2 pre-mRNA splicing commonly observed in astrocytes treated with the PR20 peptide, and brain samples derived from patients suffering the GGGGCC repeat expansion in the C9ORF72 gene. None of the four toxins gave evidence of aberrant splicing of the EAAT2 mRNA (fig. S6B).
Discussion
Here we report several findings that may be relevant to both the basic science of gene expression and the pathophysiology of a specific type of neurodegenerative disease. First, alternative splicing factors containing SR domains interact with fibrous polymers of low complexity domains in a manner reversible by phosphorylation. When appended to a GFP reporter, these SR domains bind to hydrogel droplets formed from polymeric fibers derived from the low complexity domain of hnRNPA2. This binding is reversed upon phosphorylation of serine residues in the SR domains by either of two CDC2-like kinases, CLK1 and CLK2, that are known to phosphorylate SR domains in living cells (16, 17). Mutational change of the serine residues of the SR domain in the SRSF2 alternative splicing factor to glycine resulted in repetitive GRN sequences that retain binding to hnRNPA2 hydrogels, but are not affected by CLK1/CLK2-mediated phosphorylation.
Moving from test tubes to cells, cells expressing variants of SRSF2 wherein serine residues of its two SR domains were uniformly changed to glycine revealed association of the altered splicing factor with nucleoli. Co-transfection of an expression vector encoding the CLK1 kinase failed to liberate the SRSF2G1/G2 variant from its nucleolar localization. Together, these data give evidence of a pathway in which SR domain containing splicing factors first enter a nucleolar compartment in a hypophosphorylated state (21), then migrate to nuclear speckles as a function of phosphorylation by the CLK1/2 family of protein kinase enzymes.
We do not know the identity of the nucleolar target of hypophosphorylated SR domains. Many aspects of the behavior of SR domains in cells can be mimicked by their attachment to hydrogel droplets composed of polymeric fibers of the low complexity domain of hnRNPA2, including that the interaction can be readily reversed by phosphorylation of serine residues by the CLK1/2 protein kinase enzymes. We speculate that the nucleolar target of hypophosphorylated SR domains will also represent a polymeric fiber not unlike the hnRNPA2 fibers described herein.
Two of the RAN translation products of the hexanucleotide repeats associated with disease variants of the C9ORF72 gene behaved as cytotoxins that impeded pre-mRNA splicing and the biogenesis of ribosomal RNA. The relevant peptides are polymers of one of two di-peptide sequences, GRN or PRN. The density of arginine residues favorable for solubility might also facilitate nuclear import and cell penetrability. Repetitive arginine residues might account for nuclear entry by mimicking the positive charge prototypical of nuclear localization signals (24, 25). Likewise, arginine-rich peptides, such as the HIV TAT peptide, are readily able to penetrate cells (26). Here both of the GR20 and PR20 peptides entered cells, migrated to the nucleus and associated with the periphery of nucleoli. We hypothesize that the binding of GRN and PRN polymers to nuclear puncta suspected to represent an early stage in the complex process of pre-mRNA splicing (21) may clog the pathway. This concept of peptide-induced toxicity differs from earlier studies that gave evidence of cytoplasmic aggregates observed in HeLa cells expressing a fusion protein linking GFP to five repeats of the GR dipeptide (7), immunostaining assays of ALS disease tissue showing cytoplasmic aggregates of the GR RAN translation product (9), and immunostaining of ALS disease tissue showing cytoplasmic aggregates of the PR RAN translation product (5). Our concept of nucleolar binding of the PRN and GRN RAN translation products and consequential impediments to RNA biogenesis is not necessarily mutually exclusive to earlier concepts of aggregate-mediated toxicity generated by any or all of the five RAN translation products of the sense or anti-sense transcripts of the C9ORF72 repeats.
We offer three reasons to believe that the toxicities driven by the GRN and PRN RAN translation products may account for the pathophysiological deficits observed in nerve cell dysfunction in patients carrying repeat expansions in the C9ORF72 gene. First, very specific alterations in splicing of the EAAT2 mRNA have been described from brain tissue derived from C9ORF72 patients, including the skipping of exon 9 and the inclusion of 1008 residues of intronic sequence distal to the splice donor site of exon 7. Administration of the PR20 peptide to human astrocytes derived from normal subjects led to the same changes in EAAT2 pre-mRNA splicing. Second, RNA-seq studies of cells exposed to the GR20 and PR20 peptides revealed changes in the expression of snoRNA known to be important for maturation of ribosomal RNA. These data properly predicted that peptide-treated cells would suffer deficits in ribosomal RNA maturation. Third, recent studies of brain tissue derived from patients carrying repeat expansions in the C9ORF72 gene have given evidence of nucleolar disorder, including impediments in the processing of the 45S ribosomal RNA precursor (27). Thus, we conclude that administration of the GR20 and PR20 peptides to normal human astrocytes leads to pathophysiological deficits that mimic those observed in disease tissue.
In the context of disease progression of both ALS and FTD patients carrying a repeat expansion in the C9ORF72 gene, nerve cell degeneration only begins after 40 or more years of age (28). How is it that RAN translation products appear with such delayed kinetics? Perhaps RAN translation of the hexanucleotide repeats may take place at very low levels in pre-symptomatic decades. Stochastically, the heavy burden of RNA biogenesis demanded of neurons may eventually lead to sufficient expression of the GRN and/or PRN peptides to begin to mildly impede nucleolar function and pre-mRNA splicing. This impediment might favor the generation of damaged ribosomes that could themselves favor RAN translation relative to normal protein synthesis. Alternatively, improperly spliced mRNAs might affect events such as the control of nuclear import/export as regulated by the RAN GTPase, perhaps favoring export of sense or anti-sense transcripts of the expanded hexanucleotide repeats in the C9ORF72 gene for cytoplasmic translation. If either or both of these alterations slightly favored production of the GRN and/or PRN peptides, the process could eventually snowball to the point of nucleolar catastrophy and nerve cell death.
We close with two final considerations. First, we do not know what pathway of cell death results from the toxic activities of the GRN or PRN polymers. If the death pathway were messy, spilling out cellular contents, it is possible that the GRN and PRN polymers of a dead neuron could be taken up by neighboring cells just as we have observed for cultured cells, thereby facilitating the pathological spread of toxicity. Second, we offer the idea that what has been witnessed in this work could reflect the failed birth of a gene. Whereas the repeat expansion in C9ORF72 can be considered to have generated a new protein-coding gene, it is ultimately toxic to the organism. Could it be that the scores of low complexity sequences associated with DNA and RNA regulatory proteins in eukaryotic cells evolved in this same manner?
Supplementary Material
Acknowledgments
We thank M. Brown for suggesting to SLM that the RAN translation products of the hexanucleotide repeats expanded in the C9ORF72 gene might be involved in RNA biogenesis as understood in our solid state conceptualization of information transfer from gene to message to protein. We also thank B. Tu, D. Nijhawan, T. Han, L. Avery and M. Rosen for stimulating discussion, and J. Steitz, C. Emerson, B. Alberts and A. Horwich for helpful comments on the composition of our manuscript. This work was supported by unrestricted endowment funds provided to SLM by an anonymous donor.
Footnotes
www.sciencemag.org/cgi/content/full/science.1254917/DC1
Materials and Methods
References and Notes
- 1.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Hölttä-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chiò A, Restagno G, Borghero G, Sabatelli M, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ ITALSGEN Consortium. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M, Qiu J, Sun Y, Ling SC, Zhu Q, Polymenidou M, Drenner K, Artates JW, McAlonis-Downes M, Markmiller S, Hutt KR, Pizzo DP, Cady J, Harms MB, Baloh RH, Vandenberg SR, Yeo GW, Fu XD, Bennett CF, Cleveland DW, Ravits J. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci USA. 2013;110:E4530–E4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, Isaacs AM. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol. 2013;126:845–857. doi: 10.1007/s00401-013-1200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zu T, Liu Y, Bañez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, Ostrow LW, Rothstein JD, Troncoso JC, Ranum LP. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci USA. 2013;110:E4968–E4977. doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LP. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci USA. 2011;108:260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW, 3rd, Rademakers R, Boylan KB, Dickson DW, Petrucelli L. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donnelly CJ, Zhang PW, Pham JT, Heusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 10.Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, Grishin NV, Frantz DE, Schneider JW, Chen S, Li L, Sawaya MR, Eisenberg D, Tycko R, McKnight SL. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–767. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kwon I, Kato M, Xiang S, Wu L, Theodoropoulos P, Mirzaei H, Han T, Xie S, Corden JL, McKnight SL. Phosphorylation-regulated binding of RNA polymerase II to fibrous polymers of low-complexity domains. Cell. 2013;155:1049–1060. doi: 10.1016/j.cell.2013.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roth MB, Murphy C, Gall JG. A monoclonal antibody that recognizes a phosphorylated epitope stains lampbrush chromosome loops and small granules in the amphibian germinal vesicle. J Cell Biol. 1990;111:2217–2223. doi: 10.1083/jcb.111.6.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neugebauer KM, Stolk JA, Roth MB. A conserved epitope on a subset of SR proteins defines a larger family of Pre-mRNA splicing factors. J Cell Biol. 1995;129:899–908. doi: 10.1083/jcb.129.4.899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zahler AM, Lane WS, Stolk JA, Roth MB. SR proteins: A conserved family of pre-mRNA splicing factors. Genes Dev. 1992;6:837–847. doi: 10.1101/gad.6.5.837. [DOI] [PubMed] [Google Scholar]
- 15.Colwill K, Pawson T, Andrews B, Prasad J, Manley JL, Bell JC, Duncan PI. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. EMBO J. 1996;15:265–275. [PMC free article] [PubMed] [Google Scholar]
- 16.Duncan PI, Stojdl DF, Marius RM, Scheit KH, Bell JC. The Clk2 and Clk3 dual-specificity protein kinases regulate the intranuclear distribution of SR proteins and influence pre-mRNA splicing. Exp Cell Res. 1998;241:300–308. doi: 10.1006/excr.1998.4083. [DOI] [PubMed] [Google Scholar]
- 17.Menegay HJ, Myers MP, Moeslein FM, Landreth GE. Biochemical characterization and localization of the dual specificity kinase CLK1. J Cell Sci. 2000;113:3241–3253. doi: 10.1242/jcs.113.18.3241. [DOI] [PubMed] [Google Scholar]
- 18.Aubol BE, Plocinik RM, Hagopian JC, Ma CT, McGlone ML, Bandyopadhyay R, Fu XD, Adams JA. Partitioning RS domain phosphorylation in an SR protein through the CLK and SRPK protein kinases. J Mol Biol. 2013;425:2894–2909. doi: 10.1016/j.jmb.2013.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lamond AI, Spector DL. Nuclear speckles: A model for nuclear organelles. Nat Rev Mol Cell Biol. 2003;4:605–612. doi: 10.1038/nrm1172. [DOI] [PubMed] [Google Scholar]
- 20.Thiry M. Behavior of interchromatin granules during the cell cycle. Eur J Cell Biol. 1995;68:14–24. [PubMed] [Google Scholar]
- 21.Bubulya PA, Prasanth KV, Deerinck TJ, Gerlich D, Beaudouin J, Ellisman MH, Ellenberg J, Spector DL. Hypophosphorylated SR splicing factors transiently localize around active nucleolar organizing regions in telophase daughter nuclei. J Cell Biol. 2004;167:51–63. doi: 10.1083/jcb.200404120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Materials and methods are available as supplementary materials on Science Online.
- 23.Lin CL, Bristol LA, Jin L, Dykes-Hoberg M, Crawford T, Clawson L, Rothstein JD. Aberrant RNA processing in a neurodegenerative disease: The cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron. 1998;20:589–602. doi: 10.1016/S0896-6273(00)80997-6. [DOI] [PubMed] [Google Scholar]
- 24.Kalderon D, Roberts BL, Richardson WD, Smith AE. A short amino acid sequence able to specify nuclear location. Cell. 1984;39:499–509. doi: 10.1016/0092-8674(84)90457-4. [DOI] [PubMed] [Google Scholar]
- 25.Dingwall C, Robbins J, Dilworth SM, Roberts B, Richardson WD. The nucleoplasmin nuclear location sequence is larger and more complex than that of SV-40 large T antigen. J Cell Biol. 1988;107:841–849. doi: 10.1083/jcb.107.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frankel AD, Pabo CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell. 1988;55:1189–1193. doi: 10.1016/0092-8674(88)90263-2. [DOI] [PubMed] [Google Scholar]
- 27.Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, Rothstein JD, Wang J. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- 29.Sheffield P, Garrard S, Derewenda Z. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr Purif. 1999;15:34–39. doi: 10.1006/prep.1998.1003. [DOI] [PubMed] [Google Scholar]
- 30.Ball HL, Mascagni P. Chemical synthesis and purification of proteins: A methodology. Int J Pept Protein Res. 1996;48:31–47. doi: 10.1111/j.1399-3011.1996.tb01104.x. [DOI] [PubMed] [Google Scholar]
- 31.Trapnell C, Pachter L, Salzberg SL. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stephens MA. EDF statistics for goodness of fit and some comparisons. J Am Stat Assoc. 1974;69:730–737. doi: 10.1080/01621459.1974.10480196. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.