

Abstract
DNA methylation at CpG dinucleotides is an important epigenetic regulator common to virtually all mammalian cell types, but recent evidence indicates that during early postnatal development neuronal genomes also accumulate uniquely high levels of two alternative forms of methylation, non-CpG methylation and hydroxymethylation. Here we discuss the distinct landscape of DNA methylation in neurons, how it is established, and how it might affect the binding and function of protein readers of DNA methylation. We review studies of one critical reader of DNA methylation in the brain, the Rett syndrome protein methyl CpG-binding protein 2 (MeCP2), and discuss how differential binding affinity of MeCP2 for non-CpG and hydroxymethylation may affect the function of this methyl-binding protein in the nervous system.
Keywords: non-CpG methylation, hydroxymethylation, MeCP2, Rett syndrome
Methylation of cytosines at the carbon 5 position (5-methylcytosine, mC) constitutes the most common covalent modification of vertebrate genomic DNA. Traditionally, cytosine methylation in vertebrate genomes has been viewed as largely restricted to CpG dinucleotide (CG) sequences, providing a stable epigenetic mark that mediates long-term transcriptional silencing. Indeed, 60–90% of all CGs are methylated in mammalian genomes, and CG methylation (mCG) has been shown to play critical roles in genomic imprinting, X-chromosome inactivation, cellular differentiation, and development (1). In addition, the disruption of cellular DNA methylation patterns has been linked to human disease, including multiple cancers (2, 3).
Evidence that DNA methylation has a uniquely important role in the brain emerged almost two decades ago with the discovery of the prominent methyl-DNA–binding protein, methyl-CpG-binding protein 2 (MeCP2), and the later identification that mutations in MeCP2 give rise to the X-linked neurological disorder Rett syndrome (RTT) (4–6). Subsequent studies also have identified neurodevelopmental disorders associated with mutations in DNA methyltransferases (7), suggesting that both the enzymatic “writers” of DNA methylation patterns and the “readers” of these marks have important roles in the brain. In this context, new studies from several laboratories have uncovered extensive cytosine modification in the brain beyond mCG. Non-CG methylation (CH methylation or mCH, in which H = A, C, or T) is now appreciated to accumulate in the human and mouse brain postnatally, reaching levels similar to that of mCG in the neuronal genome (8, 9). Moreover, oxidation of mC by the ten-eleven translocation (Tet) family of dioxygenases leads to the selective accumulation of 5-hydroxymethylcytosine (hmC) in the adult brain, together with its more highly oxidized derivatives 5-formylcytosine and 5-carboxylcytosine (10, 11). This finding suggests that hmC may act as an intermediate in an active DNA demethylation pathway, though growing evidence also suggests that hmC may serve as a stable neuronal epigenetic mark in its own right (12).
The discovery of these previously unidentified brain-enriched forms of DNA methylation provides a new perspective on DNA methylation dynamics in the developing CNS. Because proper establishment and maintenance of DNA methylation have been shown to be essential for normal development and function of the mammalian brain (13), and mutation of readers of DNA methylation (e.g., MeCP2) leads to neurological disease, these advances promise significant insights into the mechanisms underlying both neurodevelopment and disease. Here, we review recent findings regarding the deposition, distribution, and regulation of these distinctive forms of DNA methylation in the brain. We also discuss the implications of these findings for the function of neural methyl-DNA–binding proteins, focusing on recent studies that are relevant to MeCP2 as an important reader of neuronal DNA methylation in the brain.
The Distinctive DNA Methylation Landscape of the Brain
DNA methylation in most somatic tissues is established by the de novo methyltransferases DNA (cytosine-5)-methyltransferase 3A (Dnmt3a) and 3B (Dnmt3b) during early embryogenesis (14), with the maintenance methyltransferase DNA (cytosine-5)-methyltransferase 1 (Dnmt1) acting to propagate established methylation patterns during cellular division by recognizing and copying parent strand methylation at symmetrical CG dinucleotides to the newly synthesized daughter strand (Fig. 1) (1). Traditional sodium bisulfite conversion methods, in conjunction with high-throughput sequencing, have recently enabled genome-wide analysis of methylation patterns across multiple tissues in mammals and other organisms (reviewed in ref. 15). These studies indicate that the mCG landscape is generally similar in postnatal neurons and other cell types, so that intergenic regions and repeat DNA contain high levels of mCG, and active regulatory elements (e.g., enhancers and promoters depletion) are depleted of mCG (8, 9, 16–18). Thus, in neurons, as in other cell types, mCG is thought to silence the transcription of repeats across the genome and to regulate gene expression by promoting transcriptional repression (19, 20). However, in recent years it has become apparent that neuronal genomes also harbor a complement of distinct cytosine modifications. Thus, although neuronal-specific mCG certainly exists and has been characterized (8, 18), relative to other cell types, mCH and hmC appear to expand the role of DNA methylation markedly in the neuronal genome relative to other cell types.
Fig. 1.

Cytosine methylation and hydroxymethylation. The DNA methyltransferases Dnmt1, Dnmt3a, and Dnmt3b methylate the five-position carbon on cytosine. The maintenance methyltransferase Dnmt1 predominantly methylates hemimethylated CpG dinucleotides (mCG:CG), whereas the de novo methyltransferases Dnmt3a and Dnmt3b catalyze the methylation of cytosines in CN (in which N = G, A, C, or T) dinucleotides (1, 8, 9, 22). Methylcytosine can be modified to hydroxymethylcytosine by the Tet family of dioxygenases, Tet1–3. Current evidence demonstrates that the oxidative conversion of methylcytosine to hydroxymethylcytosine by the Tet enzymes occurs at CG dinucleotides, as is consistent with the overwhelming proportion of hydroxymethylation occurring as hmCG rather than hmCH (8, 37). Additionally, Tet enzymes are capable of catalyzing further oxidation of hmC to formylcytosine and carboxylcytosine to yield substrates for Tdg, ultimately resulting in the generation of a nonmethylated cytosine (10, 11).
mCH.
Although mCH is detected readily by standard bisulfite sequencing methods, appreciation of the functional relevance of this modification likely lagged because the relatively low frequency of methylation at individual CH dinucleotides compared with CG dinucleotides initially rendered this form of methylation difficult to distinguish from the background cytosine nonconversion rate during bisulfite treatment. Indeed, mCH is nearly absent from nonneuronal adult somatic cells (9, 21). However, with the application of whole-genome single-base–resolution DNA methylation profiling methods that use precise controls to distinguish the background nonconverted cytosines that remain after bisulfite treatment from true low-level methyl-cytosines (15), significant levels of mCH were detected, first in ES cells and then in the adult brain (16, 17). Subsequent analysis of NeuN+ neurons has shown that mCH is specifically enriched in neurons relative to other cell types (8, 9). Current data place the global frequency of mCH, within all CH base calls observed in adult human and mouse CNS neurons, at roughly 2–6%, with most identified sites of mCH displaying methylation in 20–25% of sampled genomes (8). Although these numbers are dwarfed by the comparable percentage of methylation at CG dinucleotides (60–90%), it is important to note that, because of the marked depletion of CG dinucleotides from eukaryotic genomes, mCH is estimated to account for up to, or more than half of, the total fraction of methylated cytosines of the adult mouse and human neuronal genomes.
The correlation of mCH levels observed between individuals at both the kilobase and single-site scales suggests that methylation of CH sites is a highly controlled process (8), and functional analysis of mCH is only now beginning. However, at a genome-wide level, it already is clear that neuronal mCH is depleted in expressed genes, with mCH levels throughout the 5′-upstream, gene-body, and 3′-downstream regions inversely correlated with the abundance of the associated transcript (8, 9). Reporter gene studies using in vitro methylated plasmids have confirmed the ability of mCH to repress gene expression (9), although the specific pattern of in vitro methylation tested in these studies did not fully recapitulate endogenous mCH patterns. Thus, mCH appears to serve as a repressive epigenetic mark. Moreover, the discovery of abundant neural mCH in neurons significantly expands the fraction of the neuronal genome under regulation by mC.
Further clues to mCH function can be derived from its method of deposition. Unlike the overall level of mCG, which remains unchanged during development, mCH accumulates significantly in the brain during early postnatal development. Intriguingly, mCH levels increase most rapidly during the primary phase of synaptogenesis in the early postnatal brain (mouse, ∼2–4 wk; human, 0–2 y) (8, 9). This mCH accumulation appears to be driven by a transient, coincident increase in the expression of the de novo DNA methyltransferase Dnmt3a (8). Notably, Dnmt3a has been shown to catalyze mCH in heterologous systems (22), and Dnmt3a knockdown leads to a significant reduction in mCH, but not mCG, in the adult mouse dentate gyrus (9, 23). Analysis of Dnmt3a mutant mice also provides circumstantial evidence corroborating the importance of this postnatal mCH increase for neural function. Indeed, conditional deletion of Dnmt3a from the CNS during late gestation results in shortened lifespan, hypoactivity, and impaired motor coordination (24). These phenotypes likely stem specifically from the loss of mCH in the brain, because we recently have found that mCH is absent from the brain of these Dnmt3a conditional null animals, whereas the level of mCG is largely unaffected (23). Notably, late postnatal, brain-specific conditional knockout of Dnmt3a alone results in no overt physical or behavioral deficits (25), a finding that is consistent with the existence of a critical developmental window in which mCH is deposited in the brain (8).
It is important to note that mCH is not clearly associated with gene repression in all cell types: mCH in ES cells is correlated with gene expression, such that genes containing high levels of mCH are more highly expressed. The distinct roles mCH in different cell types may be related to the unique complement of writers and/or readers of DNA methylation. Dnmt3a and Dnmt3b are both highly expressed in ES cells, whereas Dnmt3a, but not Dnmt3b, is highly expressed in neurons (8, 14). Perhaps as a consequence of the differential expression of these de novo DNA methyltransferases and the mechanisms by which they are targeted to the genome, the predominant form of mCH in ES cells occurs within the context of the CAG trinucleotide, whereas in neurons mCH occurs largely within the CAC trinucleotide (8, 9, 17, 26, 27). The differential distribution and sequence context of mCH may have important implications for readers of DNA methylation and whether they function as activators or repressors of gene expression. Thus, future studies that characterize the function of mCH must consider the unique writers and readers of DNA methylation present in a given cell type.
Hydroxymethylation.
Despite a report more than 40 years ago suggesting the existence of hmC in mammalian genomes (28), the presence of significant levels of hmC in CNS neurons remained unexplored until recently. The persistent failure to observe genomic hydroxymethylation likely reflected the inability of traditional bisulfite sequencing methods to distinguish between methylated and hydroxymethylated sites. Kriaucionis and Heintz (29) recently identified the hmC nucleotide in isolated Purkinje and granule cell nuclei using a combination of TLC, HPLC, and mass spectrometry methods. Subsequent development of base-resolution Tet-assisted bisulfite sequencing (TAB-seq) methods, which use β-glucosyltransferase (β-GT)–mediated glucosylation of hmC to protect these sites specifically from oxidation with recombinant Tet enzyme and subsequent conversion to thymine (30), now make it possible to distinguish hmC from C and mC genome-wide.
Current data indicate that hmC is substantially enriched in CNS neurons compared with other cell types, with hmC levels being ∼10-fold greater in the brain than in ES cells (31, 32) and with CG hydroxymethylation (hmCG) accounting for 25% and 40% of all modified CG dinucleotides in the frontal cortex and cerebellum, respectively (8, 29, 33). Brain hmCG levels increase postnatally in parallel with mCH, with an approximately fourfold increase in abundance within the first 6 weeks after birth (8, 34). Although the precise trigger for postnatal hmC accumulation remains unclear, all three Tet family members (Tet1–3) are expressed in the developing CNS (Fig. 1). In addition, Tet1 and Tet3 mRNA levels have been found to respond to neuronal activity (35, 36), raising the possibility that neuronal activity in newly formed circuits helps drive the increase in hmC in the brain and shapes the profile of hmC across the neuronal genome.
It is notable that currently available evidence suggests that most hmC in the brain occurs in the CG context (>99% in mouse adult and fetal cortex) (8). Indeed, given that few individual genomic sites exhibit significant levels of CH hydroxymethylation (hmCH), it remains unclear whether hmCH exists as a genuine species in vivo. Recent characterization of the Tet enzymes has demonstrated a strong preference for oxidation of mCG compared with mCH, providing a possible explanation for the low levels of hmCH (37). However, these steady-state measurements do not rule out the possibility that hmCH is turned over rapidly after conversion from mCH. Another possibility is that hmCH is more abundant in the brain than current methods suggest, and that with new methods of hmCH detection, hmCH may be found to have a more prominent function in the brain.
Genome-wide, base-resolution hmC profiling shows that hmCG is preferentially enriched throughout the gene bodies of highly expressed genes and is depleted from transcriptional start sites (8, 34, 38). Many highly expressed loci thus show depletion of intragenic mCG but still retain relatively high levels of intragenic hmCG. In addition to specific sites of intragenic enrichment for hmCG, an accumulation of hmCG occurs broadly across intergenic regions of the neuronal genome. Thus, in stark contrast to other cell types, such as ES cells, in which hmCG is localized primarily to active regulatory elements, in neurons, even intergenic regions contain substantial levels of hmCG (20–30%) (8, 33).
An important open question regarding neuronal hmC function concerns the extent to which this cytosine modification serves as a stable epigenomic mark that directly regulates genomic function or whether it represents a transient intermediate that marks sites of active CG demethylation. Tet enzymes are capable of catalyzing further oxidation of hmC to 5-formylcytosine and 5-carboxylcytosine to yield substrates for thymine-DNA glycosylase (Tdg), ultimately resulting in the generation of a nonmethylated cytosine (10, 11). In this regard, hmC has been implicated as an intermediate in an active neuronal activity-induced demethylation pathway in the mouse dentate gyrus (39) and has been found to mark sites of subsequent CG demethylation in the frontal cortex (8). Nevertheless, the high steady-state level of hmC in the postnatal CNS and the recently reported long half-life for bulk hmC in the brain (12), combined with the recent identification of cellular factors, such as Uhrf2, that can bind hmCG (40), argue that this cytosine modification also may function as a stable epigenetic mark that directly engages cellular mechanisms to modulate genomic function. It is likely that hmC can function in both capacities, but future studies will be required to tease apart these specific contributions.
Irrespective of the specific molecular role of neuronal hmC, several groups have begun to manipulate Tet activity to define the cellular and organismal consequences of altering neuronal hmC levels. Such studies have implicated hmC regulation in aspects of learning and memory, hippocampal neurogenesis, and neuronal activity-regulated gene expression (35, 41, 42). Coupled with reports that a redistribution of genomic hmC accompanies some forms of neural plasticity (35, 36), these findings suggest that hmC regulation may be intimately involved in CNS function. However, additional research will be required to distinguish more precisely the direct roles for hmC remodeling in neuronal plasticity from the secondary effects related to altered Tet function during neural development.
In summary, current genome-wide profiling studies of mC and hmC in the brain provide an emerging picture of the neuronal methylome and its unique complement of methyl marks. Although the great majority of CG dinucleotides are highly methylated in a similar manner across cell types, the dramatic postnatal build-up of mCH and hmCG in neurons establishes a landscape of cytosine methylation that is markedly distinct from other terminally differentiated cell types. The profile of mCH largely parallels that of mCG across genomic elements, including the build-up of mCH in intergenic regions as well as at regulatory elements of inactive genes, repeat DNA sequences, and the promoters and gene bodies of lowly expressed genes. These mCH sites, which are similar in number to mCG sites, provide a large number of additional sites of action for DNA methylation in the neuronal genome. For hmCG, demarcation of active regulatory elements can result in demethylation. However, the accumulation of hmCG in actively transcribed genes and across intergenic regions of the genome also may reflect the steady-state hydroxymethylation of neuronal DNA. These dramatic transformations also have important implications for our understanding of methyl- and hydroxymethyl-specific readers expressed in the brain.
Binding of MeCP2 to the Brain-Specific Methylome
Protein factors that preferentially bind to methylated DNA are thought to be important mediators of the biological effects of DNA methylation. Indeed, it now is recognized that several structurally unrelated protein domains have the ability to recognize mCG sequences, including protein domains that are present in the methyl-binding domain (MBD) protein family, Kaiso and Kaiso-like proteins, and SRA-domain proteins (43). The identification of variant cytosine modifications calls for a reassessment of the role of methyl-cytosine–binding proteins as specific readers of mCG in neurons, and recent studies suggest specificity among readers for the various oxidized cytosine bases (40). Thus, bulk conversion of mCG to hmCG sites in the brain is likely to create new binding sites for proteins with preferential binding for hmCG while eliminating binding sites for proteins that have specific affinity for mCG. In addition, the appearance of large numbers of mCH dinucleotides in the neuronal genome has the potential to provide new binding sites for readers of DNA methylation. Because of the unique make-up of the neuronal methylome, it is tempting to speculate that methyl-DNA–binding proteins are likely to have distinct distributions and activities in the brain. Below, we focus on the relevance of mCH and hmC to our understanding of the function of one of the best-characterized neuronal methyl-binding proteins, the MBD family member, MeCP2.
MeCP2 was one of two initial mCG-binding factors discovered by Bird and colleagues in a series of pioneering studies that identified proteins with high affinity for methylated DNA (6, 44). MeCP2 exhibits a strong affinity for single, symmetrically methylated CG sequences, and MeCP2 binding to mCG is largely independent of adjacent sequence elements. Study of this protein was revolutionized by the subsequent finding that mutations in the MECP2 gene give rise to RTT, an X-linked progressive autism spectrum disorder that is one of the most common causes of neurological impairment in girls (4). It is now appreciated that MECP2 mutations also result in a constellation of neuropsychiatric abnormalities that extend beyond classic and variant forms of RTT (5). Although present in most somatic cells, MeCP2 is most highly expressed in the brain, especially in neurons, where levels of the protein are five- to 10-fold higher than in other cell types, approaching levels of histones (45, 46). Furthermore, mouse strains selectively lacking MeCP2 in neural tissues have been shown to recapitulate various Rett-like phenotypes faithfully (47, 48). Since the discovery of MeCP2 as a methyl-binding protein, in vivo and in vitro studies have shown that MeCP2 interacts with the NCoR/HDAC3 and Sin3a transcriptional corepressor complexes, suggesting a role for MeCP2 as a transcriptional repressor (49, 50). However, as discussed below, MeCP2 also has been proposed to function as a transcriptional activator (51), and the exact mechanism by which MeCP2 regulates transcription is an area of intensive research. Intriguingly, the buildup of MeCP2 in neurons closely parallels the observed rise in mCH and hmC levels (46), suggesting that MeCP2 may bind these marks to regulate gene expression. Although there have been disparate reports concerning the ability of MeCP2 to recognize mCH and hmC, close consideration of the various studies allows the reconciliation of many of these apparently conflicting findings (see below) and provides new insight into role of MeCP2 in the developing nervous system.
Binding to mCH.
To date, relatively few studies have addressed the ability of MeCP2 to recognize mCH in neurons. Notably, a recent study by Guo et al. (9) reported that the MBD of MeCP2 exhibited clear binding to CH-methylated oligonucleotides in an EMSA (Fig. 2). By reanalyzing previously published MeCP2 ChIP data, the authors report that CH-methylated genomic regions depleted of CGs are enriched in MeCP2-bound chromatin, suggesting a possible in vivo interaction between MeCP2 and mCH in neurons. This finding differs from early characterizations of MeCP2, which reported a strong preference of MeCP2 for DNA methylated in the CG rather than the CH context (Fig. 2) (6, 52). In the earlier experiments, the EMSA analysis used a probe that was methylated at all cytosines outside the CG context. This probe yielded substantially weaker MeCP2 binding than a sequence-matched probe harboring a single mCG site. Here, the results of base-resolution methylation mapping from the brain provide a clue to explain these apparently incongruous findings. These recent methylome analyses report that strong bias for mCH occurs in the CA context [∼60% of mCH appears as CA methylation (mCA)], with several studies identifying a longer preferred sequence than just mCA (8, 9, 17). In this regard, the oligonucleotide probe used in the recent study by Guo et al. (9) included mCH with the preferred sequence context, suggesting that MeCP2 binding to mCH might display sequence preferences.
Fig. 2.
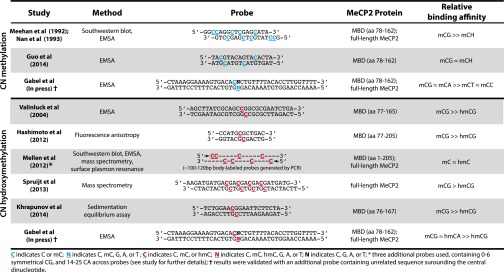
Summary of MeCP2 mC and hmC binding-affinity studies.
To compare the relative affinity of MeCP2 for CG, CA, CC, and CT methylation directly, we recently performed an EMSA (23) using competitor oligonucleotides that differ only in the composition of a single, centrally positioned dinucleotide, *CN (in which *C = C, mC, or hmC, and N = G, A, C, or T). We found that the relative affinity of full-length MeCP2 for mCA, as well as MeCP2 MBD fragments, is similar to that of symmetrically methylated CG. In contrast, MeCP2 exhibited a markedly lower affinity for cytosine methylation occurring in the context of CT methylation (mCT) or CC methylation (mCC) (Fig. 2). These unanticipated sequence preferences for MeCP2 recognition of mCH likely account for early reports that MeCP2 binding was restricted to mCG. Notably, because Meehan and colleagues (6) were unaware of the prevalence of mCA dinucleotides in vivo, the probe they used was largely depleted of mCA, and steric interference from adjacent cytosine modifications and/or proximity to end of the probe sequence may have hindered MeCP2 binding to the two mCA sites present. Our recent findings, together with those of Guo et al. (9), establish mCA as a high-affinity ligand for neuronal MeCP2. This selective, tight binding of MeCP2 to mCA, the most common form of mCH in the brain, suggests a lock-and-key relationship for MeCP2 and mCA that supports a model in which MeCP2 functions with mCA to regulate gene expression in vivo. Future studies will be required to determine if additional sequence features outside the immediate dinucleotide context influence MeCP2 binding and to define the full significance of mCA binding for MeCP2 function in vivo.
Binding to hmC.
The ability of MeCP2 to recognize cytosine hydroxymethylation also has generated conflicting conclusions in the literature. Several groups have reported a reduced affinity of full-length MeCP2 or the isolated MeCP2 MBD domain for hmC-containing DNA using a variety of in vitro approaches, including EMSA, fluorescence polarization, and mass spectroscopy-based proteomics analysis (40, 53–55). In contrast, a recent study by Mellén et al. (38), observed high-affinity MeCP2 binding to both mC- and hmC-containing oligonucleotides by EMSA, Southwestern blot, and surface plasmon resonance analysis using several probe sequences (Fig. 2).
Using the previously described EMSA approach, we recently found that mCG conversion to hmCG substantially decreases the affinity of MeCP2 for the corresponding probe (Fig. 2) (23). Indeed, oligonucleotide sequences that contain either symmetrical or asymmetrical cytosine hydroxymethylation in the CG context compete for MeCP2 binding as poorly as does an unmethylated oligonucleotide, indicating a low affinity for these marks. Likewise, MeCP2 was found to have a low affinity for probes containing CC hydroxymethylation (hmCC) and CT hydroxymethylation (hmCT). Strikingly, however, oligonucleotides containing cytosine hydroxymethylation in the CA context bind to MeCP2 with high affinity, suggesting that oxidative conversion of CA methylation (mCA) to CA hydroxymethylation (hmCA) does not alter the high-affinity binding of MeCP2. These surprising results clarify the seemingly conflicting findings from earlier reports: previous studies that reported low affinity of MeCP2 for hmC used probes in which cytosine hydroxymethylation occurred only in select CG dinucleotides, whereas Mellén et al. (38) generated their oligonucleotide probes via PCR amplification of particular genomic loci in a manner that incorporated hmC throughout the DNA sequence, including hmCG and hmCA. Because CG was underrepresented in their probes, the mC and hmC modifications in the Mellén et al. study were biased toward analysis of the CA dinucleotide. Thus, the high-affinity binding of MeCP2 for both mC and hmC observed by Mellén et al. could have resulted from the presence of hmCA in the probe sequences.
The observation that conversion of methyl-cytosine to hydroxymethyl-cytosine reduces the binding affinity of MeCP2 at CG, but not CA, dinucleotides has important implications for how MeCP2 may bind and function with hmC marks in the genome. Although hmCG is present at appreciable levels in neurons, TLC and genome-wide bisulfite sequencing analyses conducted to date suggest that hmCA, if it exists, is present in the neuronal genome at low levels (8, 29, 33). Thus, oxidative conversion of mC to hmC may primarily influence the distribution of MeCP2 molecules in neuronal chromatin by decreasing the binding affinity of MeCP2 at preexisting mCG sites. In light of the history of the field, however, it would be rash to dismiss the possibility of detection-method bias with respect to hmCH. For example, the TAB-seq method used to measure hmC employs recombinant Tet enzymes, and the extent to which it can detect hmCH is not entirely clear. In this regard, it also is important to note that if hmCA is present in neurons at functionally relevant levels (i.e., if hmCH does not represent background from unoxidized mCH in TAB-seq analysis), current data suggest that, similar to mCH, hmCH likely functions as a repressive mark in the brain. Specifically, Lister and colleagues (8) observed that, unlike hmCG, the limited genic hmCH signal that can be detected in the brain is anticorrelated with gene-expression levels. Given the current ambiguity created by these low levels of detectable hmCH, definitive conclusions about the existence of hmCH and its function in vivo will require additional future studies.
The recent characterization of MeCP2’s binding affinity toward cytosine methylation- and hydroxymethylation-containing DNA provides new in vitro evidence that neuronal-enriched DNA modifications may be important modulators of MeCP2 function. An important next step from these findings is to examine the binding profile of MeCP2 in vivo and to assess the extent to which DNA methylation can explain MeCP2’s distribution across the genome. Several independent studies have reported that MeCP2 binds broadly to neuronal chromatin as detected by ChIP followed by high-throughput sequencing (ChIP-seq) analysis (46, 56, 57). The finding that MeCP2 is expressed at near-histone levels (46), when combined with the largely uniform binding profile detected by ChIP, suggests that MeCP2 interacts with a significant portion of the methylated sites across the genome and in fact may bind unmethylated DNA as well. In the context of this broad binding, however, the MeCP2 ChIP signal has been shown to track with DNA methylation as assessed by affinity-based methods and locus-specific bisulfite sequencing in the brain (46). These findings support a role for DNA methylation in recruiting MeCP2 to the genome in vivo. However, with the realization that DNA methylation occurs at both CA and CG sites in the neuronal genome and that hmCG occurs at high levels in the brain, in vivo MeCP2 binding must be compared with genome-wide base pair-resolution DNA methylation and hydroxymethylation profiles to determine the extent to which MeCP2 binds to each of these marks.
To this end, we recently compared MeCP2 ChIP-seq profiles with base pair-resolution DNA methylation and hydroxymethylation data generated in the brain by whole-genome bisulfite sequencing and the related technique TAB-seq (8, 9, 23). This analysis allowed assessment of the role of mCA marks in establishing the MeCP2 binding profile in vivo that previous, affinity-based approaches may not have detected because of the dual recognition of mC in the mCG or mCA context. Notably, we found that, in the context of broad binding across the genome, the density of the MeCP2 ChIP signal across genes is correlated with mCA density and is inversely correlated with hmCG density. This in vivo binding profile supports in vitro findings regarding the affinity of MeCP2 for these marks, suggesting that mCA may be a high-affinity site for MeCP2 on the neuronal genome, whereas hmCG sites may fail to recruit MeCP2 effectively (23).
In summary, in vitro and in vivo binding analyses suggest that brain-specific distribution of MeCP2 is established, in part, through the build up of mCH and hmCG in the brain. The deposition of mCH (primarily as mCA) during postnatal development provides numerous additional sites in the neuronal genome that may be engaged by the MeCP2 protein. Simultaneously, large-scale conversion of mCG to hmCG may have the effect of removing high-affinity sites for MeCP2 binding. The exact functional consequence of this redistribution of the available high-affinity sites for MeCP2 is not yet clear, but, because CA dinucleotides are distributed across the genome more uniformly than CG dinucleotides, the shift of available high-affinity sites for MeCP2 from mCG to mCA may create a more uniform distribution of methyl-bound MeCP2 molecules across the genome than would be possible if only mCG were present in neurons. It is notable that MeCP2 builds up to exceedingly high levels in neurons postnatally, so that the number of MeCP2 molecules approaches that of histones (46) at the same time that mCA and hmCG approach maximal levels. This high number of MeCP2 molecules, along with the broad distribution of high-affinity mC sites for MeCP2, likely facilitates the nearly ubiquitous binding of MeCP2 across the genome that is observed by ChIP and suggests that MeCP2 functions to modulate transcription, not as a site-specific binding factor in neurons, but as a broadly integrated, core component of neuronal chromatin (46, 56, 57).
Toward a Model of Gene Regulation by mCH, hmC, and MeCP2
The full implications of this unique postnatal remodeling of the neuronal methylome for our understanding of the neural function of specific protein readers of DNA methylation still remain to be explored. Major gaps remain in our understanding of MeCP2 at the molecular level despite clear genetic evidence for the importance of the MeCP2 MBD in neural development (5, 49, 50). With recent insights into the affinity of MeCP2 for mCA and hmCG, a major next step is to integrate this knowledge of MeCP2 binding specificity with in vitro and in vivo biochemical data and gene-expression analysis in MeCP2 mutants to build models of how MeCP2 functions to modulate transcription in vivo. A few studies already have begun to probe how MeCP2 functions with DNA methylation to regulate transcription by comparing the alterations in gene expression that occur in the brain of Mecp2 knockout mice with profiles of hydroxymethylation in the brain (38, 58).
Although potentially powerful, such studies will need to be guided by our evolving understanding of the binding specificity of MeCP2 to distinguish primary and secondary effects of MeCP2 loss. In this regard, the observation that hmC is enriched in active genes, together with reports that MeCP2 can bind to hmC in vitro, has been cited as evidence that hmC-bound MeCP2 might function as a transcriptional activator to facilitate gene expression (38, 58). Although independent evidence for both an activating and repressing role for MeCP2 exists (51), the idea that MeCP2 activates gene expression by directly binding hmC within active genes may need to be reassessed in light of the accumulating in vitro and in vivo data demonstrating MeCP2’s reduced affinity for hmCG and the apparent near-absence of neuronal hmCA in neurons (described above). It will be important to develop a model of MeCP2 action in neurons that accounts for the current in vitro and in vivo data showing that oxidation of mCG to hmCG at active genes likely results in reduced MeCP2 affinity at these loci. Future studies that decrease neural hmCG levels through conditional inactivation of the Tet enzymes and then assess the resulting effects on MeCP2 binding and gene expression will be needed to clarify further the precise functional relationship between MeCP2 and neuronal hmC.
Studies of the repressor function of MeCP2 also will need to take into account the potential role that the high affinity of MeCP2 for mCA plays in neuronal gene regulation by MeCP2. The large number of new binding sites provided by mCA as neurons mature may facilitate repression by MeCP2 that either functions similarly to or has a function distinct from mCG. Investigation of the interactions of MeCP2 with its known corepressor binding partners in the context of mCA or mCG binding may uncover specific or common functions for these modified dinucleotides at sites across the genome. As described above, conditional inactivation of Dnmt3a early in postnatal development provides an avenue for eliminating mCA from the genome while leaving mCG largely intact, thereby facilitating studies of mCA-specific aspects of MeCP2 neuronal function. To this end, an initial study from the M.E.G. laboratory examining the effect of disrupting Dnmt3a or MeCP2 function selectively in the brain has uncovered evidence that MeCP2 represses gene expression in neurons by binding to mCA across the length of genes (23). Future work will be needed to explore the mechanism of this regulation by MeCP2 by determining which MeCP2-associated corepressor complexes are required for this repression and how they work together with MeCP2 and mCA to alter gene transcription.
Although recent studies are an important step forward, our understanding of the nature and function of the postnatal program of neural epigenomic remodeling is still in its infancy. Given the pronounced nature of these global, dynamic methylation changes, future study of these epigenetic marks holds the promise of uncovering important new insights into the contribution of epigenetic mechanisms to genomic regulation and neural function. Moreover, these findings will continue to shape our understanding of the role of MeCP2 and other readers of methyl-DNA in aspects of neuronal gene expression, neural development, plasticity, and disease.
Acknowledgments
We thank A. Bird, G. Mandel, M. Coenraads, and members of the M.E.G. laboratory for discussions and for critical reading of the manuscript. This work was supported by a grant from the Rett Syndrome Research Trust and by National Institutes of Health (NIH) Grant 1R01NS048276 (to M.E.G.), a fellowship from the William Randolph Hearst fund (to H.W.G.), and by NIH Grant T32GM007753 and a Howard Hughes Medical Institute Gilliam Fellowship (both to B.K.).
Footnotes
The authors declare no conflict of interest.
This paper results from the Arthur M. Sackler Colloquium of the National Academy of Sciences, “Epigenetic Changes in the Developing Brain: Effects on Behavior,” held March 28–29, 2014, at the National Academy of Sciences in Washington, DC. The complete program and video recordings of most presentations are available on the NAS website at www.nasonline.org/Epigenetic_changes.
This article is a PNAS Direct Submission.
References
- 1.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 2.Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. 2010;70:27–56. doi: 10.1016/B978-0-12-380866-0.60002-2. [DOI] [PubMed] [Google Scholar]
- 3.Huang Y, Rao A. 2014. Connections between TET proteins and aberrant DNA modification in cancer. Trends in Genetics 30(10):464–474.
- 4.Amir RE, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 5.Chahrour M, Zoghbi HY. The story of Rett syndrome: From clinic to neurobiology. Neuron. 2007;56(3):422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Meehan RR, Lewis JD, Bird AP. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 1992;20(19):5085–5092. doi: 10.1093/nar/20.19.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tatton-Brown K, et al. Childhood Overgrowth Consortium Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet. 2014;46(4):385–388. doi: 10.1038/ng.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lister R, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341(6146):1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo JU, et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci. 2014;17(2):215–222. doi: 10.1038/nn.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He YF, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333(6047):1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: Potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286(41):35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bachman M, et al. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat Chem. 2014;6(12):1049–1055. doi: 10.1038/nchem.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tucker KL. Methylated cytosine and the brain: A new base for neuroscience. Neuron. 2001;30(3):649–652. doi: 10.1016/s0896-6273(01)00325-7. [DOI] [PubMed] [Google Scholar]
- 14.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 15.Lister R, Ecker JR. Finding the fifth base: Genome-wide sequencing of cytosine methylation. Genome Res. 2009;19(6):959–966. doi: 10.1101/gr.083451.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie W, et al. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012;148(4):816–831. doi: 10.1016/j.cell.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ziller MJ, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500(7463):477–481. doi: 10.1038/nature12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith ZD, Meissner A. DNA methylation: Roles in mammalian development. Nat Rev Genet. 2013;14(3):204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki MM, Bird A. DNA methylation landscapes: Provocative insights from epigenomics. Nat Rev Genet. 2008;9(6):465–476. doi: 10.1038/nrg2341. [DOI] [PubMed] [Google Scholar]
- 21.Ziller MJ, et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011;7(12):e1002389. doi: 10.1371/journal.pgen.1002389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramsahoye BH, et al. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci USA. 2000;97(10):5237–5242. doi: 10.1073/pnas.97.10.5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gabel HW, et al. Disruption of methyl-CA-dependent long gene repression in Rett syndrome. Nature. 2015 doi: 10.1038/nature14319. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nguyen S, Meletis K, Fu D, Jhaveri S, Jaenisch R. Ablation of de novo DNA methyltransferase Dnmt3a in the nervous system leads to neuromuscular defects and shortened lifespan. Dev Dyn. 2007;236(6):1663–1676. doi: 10.1002/dvdy.21176. [DOI] [PubMed] [Google Scholar]
- 25.Feng J, et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13(4):423–430. doi: 10.1038/nn.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baubec T, et al. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 2015 doi: 10.1038/nature14176. , 10.1038/nature14176. [DOI] [PubMed] [Google Scholar]
- 27.Chen PY, Feng S, Joo JW, Jacobsen SE, Pellegrini M. A comparative analysis of DNA methylation across human embryonic stem cell lines. Genome Biol. 2011;12(7):R62. doi: 10.1186/gb-2011-12-7-r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Penn NW, Suwalski R, O’Riley C, Bojanowski K, Yura R. The presence of 5-hydroxymethylcytosine in animal deoxyribonucleic acid. Biochem J. 1972;126(4):781–790. doi: 10.1042/bj1260781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324(5929):929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu M, et al. Tet-assisted bisulfite sequencing of 5-hydroxymethylcytosine. Nat Protoc. 2012;7(12):2159–2170. doi: 10.1038/nprot.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Globisch D, et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE. 2010;5(12):e15367. doi: 10.1371/journal.pone.0015367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Münzel M, Globisch D, Trindler C, Carell T. Efficient synthesis of 5-hydroxymethylcytosine containing DNA. Org Lett. 2010;12(24):5671–5673. doi: 10.1021/ol102408t. [DOI] [PubMed] [Google Scholar]
- 33.Wen L, et al. Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol. 2014;15(3):R49. doi: 10.1186/gb-2014-15-3-r49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szulwach KE, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci. 2011;14(12):1607–1616. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaas GA, et al. TET1 controls CNS 5-methylcytosine hydroxylation, active DNA demethylation, gene transcription, and memory formation. Neuron. 2013;79(6):1086–1093. doi: 10.1016/j.neuron.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, et al. Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc Natl Acad Sci USA. 2014;111(19):7120–7125. doi: 10.1073/pnas.1318906111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu L, et al. Crystal structure of TET2-DNA complex: Insight into TET-mediated 5mC oxidation. Cell. 2013;155(7):1545–1555. doi: 10.1016/j.cell.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 38.Mellén M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151(7):1417–1430. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145(3):423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spruijt CG, et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152(5):1146–1159. doi: 10.1016/j.cell.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 41.Rudenko A, et al. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron. 2013;79(6):1109–1122. doi: 10.1016/j.neuron.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang RR, et al. Tet1 regulates adult hippocampal neurogenesis and cognition. Cell Stem Cell. 2013;13(2):237–245. doi: 10.1016/j.stem.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Defossez PA, Stancheva I. Biological functions of methyl-CpG-binding proteins. Prog Mol Biol Transl Sci. 2011;101:377–398. doi: 10.1016/B978-0-12-387685-0.00012-3. [DOI] [PubMed] [Google Scholar]
- 44.Lewis JD, et al. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69(6):905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- 45.Kishi N, Macklis JD. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol Cell Neurosci. 2004;27(3):306–321. doi: 10.1016/j.mcn.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 46.Skene PJ, et al. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37(4):457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27(3):327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- 48.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27(3):322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 49.Lyst MJ, et al. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat Neurosci. 2013;16(7):898–902. doi: 10.1038/nn.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guy J, Cheval H, Selfridge J, Bird A. The role of MeCP2 in the brain. Annu Rev Cell Dev Biol. 2011;27:631–652. doi: 10.1146/annurev-cellbio-092910-154121. [DOI] [PubMed] [Google Scholar]
- 51.Chahrour M, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320(5880):1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nan X, Meehan RR, Bird A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993;21(21):4886–4892. doi: 10.1093/nar/21.21.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Valinluck V, et al. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2) Nucleic Acids Res. 2004;32(14):4100–4108. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hashimoto H, et al. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 2012;40(11):4841–4849. doi: 10.1093/nar/gks155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khrapunov S, et al. Unusual characteristics of the DNA binding domain of epigenetic regulatory protein MeCP2 determine its binding specificity. Biochemistry. 2014;53(21):3379–3391. doi: 10.1021/bi500424z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cohen S, et al. Genome-wide activity-dependent MeCP2 phosphorylation regulates nervous system development and function. Neuron. 2011;72(1):72–85. doi: 10.1016/j.neuron.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Baker SA, et al. An AT-hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell. 2013;152(5):984–996. doi: 10.1016/j.cell.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li Y, et al. Global transcriptional and translational repression in human-embryonic-stem-cell-derived Rett syndrome neurons. Cell Stem Cell. 2013;13(4):446–458. doi: 10.1016/j.stem.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]