

Significance
Recent surveys of the microbial communities living on and in the human body—the human microbiome—have revealed strong variation in community membership between individuals. Some of this variation is stable over time, leading to speculation that individuals might possess unique microbial “fingerprints” that distinguish them from the population. We rigorously evaluated this idea by combining concepts from microbial ecology and computer science. Our results demonstrated that individuals could be uniquely identified among populations of 100s based on their microbiomes alone. In the case of the gut microbiome, >80% of individuals could still be uniquely identified up to a year later—a result that raises potential privacy concerns for subjects enrolled in human microbiome research projects.
Keywords: forensic genetics, microbial ecology, metagenomics, human microbiome, strain variation
Abstract
Community composition within the human microbiome varies across individuals, but it remains unknown if this variation is sufficient to uniquely identify individuals within large populations or stable enough to identify them over time. We investigated this by developing a hitting set-based coding algorithm and applying it to the Human Microbiome Project population. Our approach defined body site-specific metagenomic codes: sets of microbial taxa or genes prioritized to uniquely and stably identify individuals. Codes capturing strain variation in clade-specific marker genes were able to distinguish among 100s of individuals at an initial sampling time point. In comparisons with follow-up samples collected 30–300 d later, ∼30% of individuals could still be uniquely pinpointed using metagenomic codes from a typical body site; coincidental (false positive) matches were rare. Codes based on the gut microbiome were exceptionally stable and pinpointed >80% of individuals. The failure of a code to match its owner at a later time point was largely explained by the loss of specific microbial strains (at current limits of detection) and was only weakly associated with the length of the sampling interval. In addition to highlighting patterns of temporal variation in the ecology of the human microbiome, this work demonstrates the feasibility of microbiome-based identifiability—a result with important ethical implications for microbiome study design. The datasets and code used in this work are available for download from huttenhower.sph.harvard.edu/idability.
Recent large-scale investigations of the human microbiome have revealed great variability in the body site-specific community structure and function of microbial organisms across healthy individuals (1, 2). In addition, it has been shown that features of the human microbiome might stably associate with individuals over substantial periods of time (3–5). These observations suggest that individuals might be uniquely and stably identified within a population based on their resident microbiota. However, to date there have been no rigorous efforts to quantitatively establish the feasibility of microbiome-based identifiability. To do so requires demonstrating (i) that one can identify a “metagenomic code” that is specific to an individual in a sample population; (ii) that the code can be robustly redetected at a later time; (iii) that the code is unlikely to erroneously match a previously unseen sample; and (iv) that such codes can be constructed for a sizeable fraction of individuals (Fig. 1). These criteria emphasize that human microbiome identifiability is intimately associated with microbiome establishment, structure, personalization, and temporal stability—fundamental topics in ecological approaches to microbiome research.
Fig. 1.
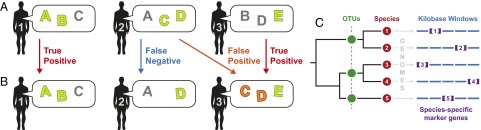
Metagenomic codes (overview). (A) Three individuals and their metagenomic features (represented by capital letters) are shown. For each individual, a subset of features is highlighted that is unique among the three individuals. We refer to these sets as metagenomic codes. (B) The same three individuals reevaluated after weeks to months. Individual 1’s microbiome has remained stable, and his code still uniquely identifies him among the population (a true positive). Individual 2 has lost metagenomic feature C, and his code no longer identifies him (a false negative). Individual 3 has lost feature B and gained feature C. Individual 3 is still a true positive with respect to his own code, but also matches individual 2’s code (a false positive). (C) Illustration of the four metagenomic feature types considered in this work: OTUs, species, kilobase windows from reference genomes (kbwindows), and species-specific marker genes (markers) (see Methods and Table 1 for details).
At the same time, the human microbiome can be viewed as a reservoir of genetic variation extending beyond an individual’s own genome. Hence, the degree to which the human microbiome is identifiable is relevant to forensic genetics and genetic information privacy beyond the ecological significance outlined above. Human genetic information has been applied to differentiate individuals for over a century, beginning with the definition and application of the ABO blood types (6). In more recent decades, the description of higher-resolution genetic variants—notably, short tandem repeats (STRs)—has substantially boosted the identifying power of human genetic information. These technologies are now widely applied in forensics to link suspects to crime scenes, identify disaster victims, and establish familial relationships. Under ideal circumstances, identifying codes based on human genetic markers are expected to be unique among billions of individuals (7), although practical concerns (e.g., sample contamination and relatedness among individuals) can reduce this number considerably (8).
Like STRs, SNPs in the human genome have strong identifying power, with an estimated 30–80 independent SNPs required to uniquely pinpoint each person on Earth (9). Such SNPs can be readily inferred from a variety of nucleotide sequencing methods commonly applied in modern biomedical research (10). These advancements, coupled with a drive to make such data open to a wider audience, have led to increased concerns for subject privacy in genomics research (11, 12). These privacy concerns extend beyond subject identification: human SNPs are increasingly powerful for subject characterization, including prediction of physical traits, disease risk, demography, and family history (10, 11). In part due to these privacy concerns, human DNA sequences are routinely removed from microbiome datasets (where they arise as contaminants) before publication (13). However, the prospect remains of linking these datasets back to their donors based on individualized microbiome sequences alone. Moreover, just as human SNPs can be used to characterize an individual, human microbiome features are increasingly being associated with a variety of subject traits, including diet (14), health status (15), age, and geography (16). Hence, if subjects are routinely identifiable based on their microbiomes, one could potentially discern additional private information about those subjects at the same time.
In this work, we applied insights from computing theory and microbial ecology to construct metagenomic codes from sets of individual-specific and maximally stable metagenomic features. This approach enabled rigorous assessment of human microbiome identifiability in a large cohort. Microbiome features were generally less unique and less stable than features of the human genome, meaning that microbiome-based identifiability did not match the exceptionally high specificity of genomic identifiability outlined above. However, based on a typical body site-specific microbiome, approximately one third of individuals could be precisely pinpointed at later time points among populations of hundreds (with few false positives). Thus, microbiome-based identifiability is possible for a nontrivial fraction of individuals in a typical cohort: a potential genetic information privacy issue not typically considered in microbiome study design.
Results
We considered four types of metagenomic features from which to construct personalized metagenomic codes (Fig. 1 and Table 1). Two were taxon-level feature types: operational taxonomic unit (OTU) abundance derived from 16S ribosomal gene sequencing and bacterial and archaeal species abundance assayed from whole metagenome shotgun (WMS) sequencing. In addition, we considered two gene-level feature types assayed from WMS sequencing: species-specific marker genes (markers) from the MetaPhlAn database (17) and tiled kilobase windows (kbwindows) drawn from a large set of bacterial reference genomes. All feature abundance measurements were based on sequencing data from individuals sampled during the Human Microbiome Project (HMP) (13), with individuals sampled at multiple times serving as the focal population for the evaluation of feature stability and code construction. Our process consisted of (i) adapting a classical algorithm from computer science to the task of metagenomic code construction; (ii) training the algorithm to prioritize optimal metagenomic features; (iii) constructing codes based on multiple body sites and feature types using individuals’ first-visit samples; and (iv) comparing these codes to samples from later time points and independent validation cohorts to quantify metagenomic code stability and specificity. These analyses and results are expanded in the sections below, with additional details provided in Methods.
Table 1.
Properties of metagenomic features and detection thresholds
| Feature description | Short name | Sequencing basis | Units | Confident detection threshold | Relaxed detection threshold | Confident nondetection threshold | Body sites | Paired samples per body site |
| Operational taxonomic units | OTUs | 16S rRNA gene | Relative abundance | >1e−3 | >1e−4 | <1e−5 | 18 | 25–105 |
| Microbial species | Species | Whole metagenome shotgun | Relative abundance | >1e−3 | >1e−4 | <1e−5 | 6 | 14–50 |
| Species-specific marker genes | Markers | Whole metagenome shotgun | RPKM | >5 | >0.5 | <0.05 | 6 | 14–50 |
| Kilobase windows from microbial reference genomes | kbwindows | Whole metagenome shotgun | RPKM | >5 | >0.5 | <0.05 | 6 | 9–45 |
In analyses of per-feature stability, a feature was considered detected if its abundance exceeded the confident detection threshold; a feature was considered acquired if it initially fell below the confident nondetection threshold and then later exceeded the confident detection threshold. When defining a metagenomic code, features with abundance between the confident detection and confident nondetection thresholds were considered ambiguous. When reevaluating a code at a later time point, the relaxed feature detection thresholds were used to add robustness to temporal variation.
Defining Metagenomic Codes Using Hitting Sets.
Our approach for defining metagenomic codes is based on the concept of a hitting set. For a collection of nonempty sets {a1, a2,…, aN}, a hitting set S is a set that has at least one element in common with each ai; the set S is said to “hit” each ai (18). If removing any element from S would cause it to not hit at least one ai, then S is called a minimal hitting set. For example, S = {A, C} is a minimal hitting set for the sets a1 = {A, B}, a2 = {B, C, D}, and a3 = {A, E}. To construct metagenomic codes using hitting sets, we consider a population of N individuals in which each individual i possesses a set of metagenomic features ui. For a given individual i, we define sets aij containing the features present in individual i but absent from each other individual j (aij = ui − uj). For example, in a Venn diagram comparing features found in individuals 1 and 2, the set a12 would represent features that were found exclusively in individual 1. If we can make a new set Si that contains at least one element from each aij set (i.e., a hitting set for the aij sets), then Si will be a metagenomic code that is unique to individual i: each other individual is missing at least one element from Si, and therefore the features in Si collectively distinguish individual i from the rest of the population. This process will only be impossible for individual i if another individual j possesses all of individual i’s features; in that case there are no features that distinguish i from this j, the corresponding aij set is empty, and thus we cannot build a hitting set for all aij sets.
Finding a minimal hitting set for a collection of sets is nondeterministic polynomial-time (NP)–hard (19), and an efficient greedy approximation can be used instead (20). This method iteratively grows a candidate hitting set by adding the most common element among nonhit sets; in the metagenomic code application, this translates to prioritizing features that are rare in the population (SI Appendix, Fig. S1). This greedy approach guarantees a hitting set at most log2(M) times larger than the minimal hitting set, where M is the number of distinct elements across the sets to be hit. We used a similar form of greedy optimization to identify metagenomic codes under the hitting set framework; however, rather than prioritizing rare features to build unique codes of minimal size, we instead prioritized features that would promote code stability and specificity over time.
Determinants of Metagenomic Feature Stability in the Human Microbiome.
This prioritization process required that we first identify which properties of microbes or microbial genes within the human microbiome indeed promoted code stability, which speaks to the ecology of the microbiota in addition to its ability to differentiate among human hosts. We thus considered simple temporal stability of individual microbial features and two ecological properties: (i) a feature’s population prevalence, the fraction of individuals in the population that possess the feature; and (ii) a feature’s per-sample abundance, the relative number of copies of that feature in a particular individual. The human gut microbiome has been found to contain individual-specific strains of bacterial species, with a substantial fraction of this variation remaining stable over 1 or more years (4, 5). We extended these results by quantifying the stability of additional metagenomic feature types at a wider array of body sites. We then identified properties of metagenomic features that promoted mid- to-long-term stability, which we prioritized when building metagenomic codes.
A metagenomic feature’s abundance at an individual’s first sampling visit was a strong, positive correlate of feature stability, which we quantified as the probability of redetecting the feature at the individual’s second sampling visit, weeks to months later (Fig. 2A). Notably, feature absence at the second sampling time point was best explained by temporal variation, as technical variation in feature detection was low (the median replicate pair was 97–99% similar; Methods and SI Appendix, Fig. S2A). The difference in temporal stability between the most and least abundant taxa was more extreme at skin and vaginal sites than in the oral cavity and gut (mean stability ratio of 3.4:1 vs. 1.8:1). Relative to oral and gut environments, the skin and vaginal body sites are characterized by lower pH (21, 22) and support less diverse microbial communities (1). Moreover, quantitative estimates of human microbial biomass at the skin [102–107 CFU/cm2 (23)] and vaginal [107 CFU/g secretion (24)] sites are orders of magnitude smaller than biomass estimates for oral and gut sites [108–1012 CFU/mL (25)]. Low-abundance taxa at skin and vaginal sites thus represent a smaller amount of overall biomass, which may allow them to be more easily perturbed, or such taxa may reflect transient, and not stably associated, members of the associated microbial communities.
Fig. 2.
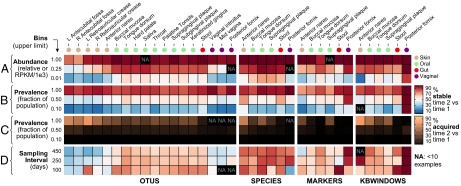
Properties associated with microbiome feature stability. For each (body site, feature type) combination, we counted cases of features confidently detected across subjects’ first sampling visits (time 1). The fraction of these cases that remained confidently detected at subjects’ second sampling visits (time 2; weeks to months later) provided a measure of feature stability. Stability was positively and strongly correlated with (A) feature abundance and (B) feature prevalence. (C) Highly prevalent features that were not detected in subjects’ time 1 samples had a high probability of being acquired by time 2, particularly at more exposed sites (e.g., skin). (D) Sampling time interval had a less marked effect on stability. NA, a (body site, feature type) combination with <10 confident detection events at time 1. Abundance values for OTUs and species reflect relative abundance; abundance values for markers and kbwindows reflect RPKM units.
Feature prevalence was also a strong determinant of stability (Fig. 2B). Low-prevalence features (those that were rare in the population) disappeared much more frequently between subjects’ first and second sampling visits than more common features, suggesting that the former may also reflect transient visitors to the human microbiome or potentially spurious detection events. As mentioned above, naïve prioritization of these rare features might result in a smaller (minimal) metagenomic code, but such minimal codes would be very unstable over time.
Surprisingly, we found that individuals who lacked otherwise highly prevalent features had a high probability of acquiring those features over time (Fig. 2C). Highly prevalent features also tended to be locally abundant (for species, Spearman’s r = 0.68; two-tailed P < 10−58), and the enhanced acquisition of locally abundant, highly prevalent features is consistent with models of microbiome assembly that include neutral processes (26). Such models assume that stochastic loss and dispersal from a common source are primary drivers of microbiome variation. This effect was particularly evident for metagenomic features measured at sites on the skin, which may result from (i) increased rates of disturbance at the skin (leading to stochastic loss of species) combined with (ii) enhanced rates of direct transfer of skin-associated microbes between individuals by skin-to-skin contact (27) or between individuals and the built environment, which supports a disproportionate fraction of skin-associated microbes (28). A fraction of apparent acquisition events may also be explained by undersampling of low-abundance features at the initial time point.
Last, although in practice this information would not be available at an initial time point to prioritize for feature selection, we considered the effect of the time interval between individuals’ first two sampling visits. The average sampling interval was 194 d, ranging from a minimum of weeks (30 d) to a maximum of 1 y (364 d). Compared with the influence of body site, feature abundance, and feature prevalence, sampling time interval appeared to have a remarkably smaller effect on feature stability, although some cases of increased stability over shorter time periods were apparent (e.g., gene-level features in the stool; Fig. 2D).
Biologically Informed Greedy Code Construction.
Per-feature stability results thus indicated that unique metagenomic codes optimized solely for rare features would not be robust to temporal variation (Fig. 2B). Based on the stability and habitat specificity of abundant features (Fig. 2A), we designed a greedy algorithm to construct hitting sets that prioritize features with large “abundance gaps”: the difference between each feature’s abundance in individual i and its next highest abundance in the population. This procedure had the effect of enriching more abundant features within codes while secondarily enriching less prevalent features, the latter of which are (i) more discriminative and (ii) less likely to be acquired by other individuals over time (leading to loss of code uniqueness; Fig. 2C). Our algorithm operated as follows, with criteria for confident detection and confident nondetection defined in Table 1:
-
•
Create a vector of confidently detected features, F, for an individual, i, ranked by descending abundance gap, as defined above. Create an empty code set, S, and a set containing all other individuals in the population, J.
-
•
Remove the highest ranked feature (f) from F. Delete individuals from J for whom f was confidently not detected. If f differentiates at least one additional population member (i.e., at least on individual was deleted from J), add f to S.
-
•
Repeat the previous step, stopping when either F becomes empty (no features remain) or J becomes empty (we have distinguished individual i from the rest of the population). If J is empty, then S is a unique metagenomic code for individual i; else, individual i has no unique code.
-
•
Optionally, after J is empty but before F is empty, continue adding features to S, stopping when S reaches a desired minimum size, d, or when F is empty. This procedure adds robustness to noise and, effectively, error correction to avoid false positives; in our study, d = 7.
-
•
Optionally, after adding f to S, delete remaining features in F with similar presence/absence profiles to f. When using the d option above, this also helps to diversify the features added to an already unique code; we defined similar as Jaccard score >0.8.
A fully documented python implementation of this algorithm is available online at huttenhower.sph.harvard.edu/idability.
Identifiable Microbial Codes in the Human Microbiome.
We applied the code-building algorithm above to first-visit samples from 120 individuals with multiple visits from the HMP cohort (60 with WMS sequencing data). The algorithm was applied separately for four metagenomic feature types (OTUs, species, markers, and kbwindows) at a variety of body sites (Fig. 3 and Table 1). We identified population-unique metagenomic codes for the majority of individuals and body sites using gene-level features (markers and kbwindows), but not using taxon-level features (OTUs and species; Fig. 3A). This difference was due to the smaller number and nonrandom assortment of taxon-level features, which frequently resulted in individuals whose taxa were a subset of other individuals’ taxa (meaning they had no unique taxon-level code). Although the average difference between individuals’ microbial community composition and ecology is substantial, the pool of organisms populating any one body site habitat is proportionally constrained at the species level (1). The presence and absence of marker genes and kbwindows instead capture strain-level variation within species and thus provided a richer universe of features with which to distinguish individuals (Fig. 4).
Fig. 3.
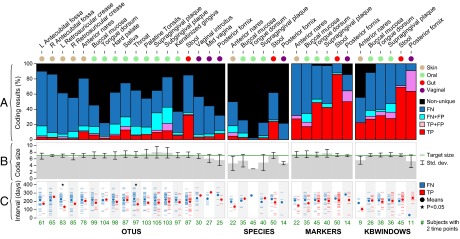
Temporal stability of metagenomic codes. (A) We identified unique metagenomic codes for individuals based on their first sampling visits (time 1); an individual whose microbial features were a subset of a second individual’s features had no unique code (black bars). Red bars represent true positives (TPs): codes that uniquely identified their owners at time 1 and again at the second sampling visit (time 2; weeks to months later). Blue bars represent false negatives (FNs): codes that matched no one at time 2. Pink and cyan bars represent false positives (FPs): codes that matched someone other than their owner at time 2, either in addition to their owner (TP+FP) or instead of their owner (FN+FP). (B) Average and SD of metagenomic code size. A target size (seven features) was imposed to reduce FPs. (C) Distribution of sampling time intervals for TPs and FNs, with each individual represented by a hash mark. FNs were weakly associated with longer sampling time intervals than TPs in a few body sites and very weakly in aggregate (Mann–Whitney u test). Green numbers indicate the number of individuals profiled at time 1 and time 2 for each (body site, feature type) combination (see Methods for an explanation of why kbwindows numbers differ from species and markers numbers).
Fig. 4.
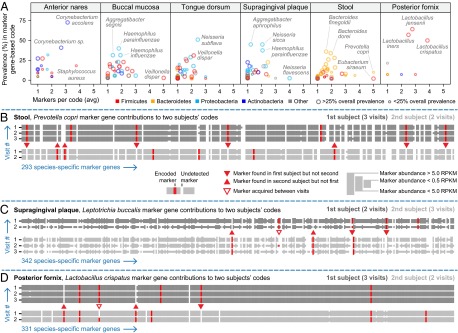
Influence of strain-level variation on marker gene-based codes. (A) Species varied greatly in their likelihood to contribute marker genes to a code (vertical axis) and the numbers of marker genes thus contributed (horizontal axis). Samples from the anterior nares and posterior fornix body sites were typically identified by individual strains (several markers each) of a few dominant taxa, whereas stool and oral sites were instead identified by combinations of species within (e.g., Bacteroides) or across genera, respectively. (B) Each row depicts the abundance of 293 Prevotella copri-specific marker genes in a stool metagenome. The three dark gray rows correspond to three sampling visits from one subject (HMP identifier 158802708) and the two light gray rows correspond to two visits from a second subject (159166850). Certain markers were consistently absent in one subject across visits and consistently present in the other, indicative of stable carriage of subject-specific strains of P. copri. Red markers were included in the subjects’ codes; triangles indicate encoded markers that differentiated the first subject from the second subject (or vice versa). Heights of marker genes within each row vary with gene abundance (binned according to the confident detection, relaxed detection, and confident nondetection thresholds used in the construction and evaluation of metagenomic codes; see inset key). (C) This panel uses the same format as B to explore marker profiles of Leptotrichia buccalis from the supragingival plaque (oral) samples of HMP subjects 159591683 and 159207311. Here, an open triangle represents an encoded marker gene that was acquired between time points (in a potential lateral transfer or strain replacement event), which could contribute to a possible false positive match. (D) This panel uses the format from B and C to explore marker profiles of Lactobacillus crispatus from the posterior fornix (vaginal) samples of HMP subjects 160502038 and 764042746.
Notably, although marker genes were defined to be species-specific based on a catalog of sequenced genomes (17), we found that markers selected for inclusion in codes were significantly enriched for orphaned markers—i.e., marker genes that were confidently detected in the apparent absence of their source species (for all body sites, fold enrichment >4; Fisher’s exact test, two-tailed P < 0.001; SI Appendix, Table S1). Such markers were most likely carried in another genomic background due to mechanisms such as lateral transfer (29), and were of similar stability to other markers included in codes. These orphaned marker genes capture previously unseen and highly individualized genomic variation, granting additional distinguishing power to marker gene-based codes and suggesting that strain-specific, lateral gene transfer events may be a substantial contributor to individual microbiome structure (see SI Appendix, Fig. S3 for an example).
Factors Leading to Loss of Code Robustness over Time (FNs).
After identifying individuals’ metagenomic codes from their first-visit samples, we next compared the codes to individuals’ second-visit samples (taken 30–300 d later) to assess code stability. An individual’s code was considered stable if all of its features were redetected in that individual’s second-visit sample (based on a relaxed threshold; Table 1). Taxon-level codes were very unstable: averaged over body sites, only 15% of OTU-based codes and 13% of species-based codes matched their owners’ second-visit samples [we refer to these as true positives (TPs); Fig. 3A]. For the remaining individuals, at least one code taxon vanished over time, and therefore the code no longer matched its owner at the second time point [we refer to these as false negatives (FNs)]. Gene-level codes were much more stable: 52% of marker- and kbwindow-based codes correctly identified their owners’ second-visit samples (averaged over body sites; Fig. 3A). The gut habitat (as represented by stool) produced the most stable codes across all feature types, with 86% TPs among marker-based codes (see SI Appendix, Fig. S3 for an example). Agreement in coding results for pairs of technical replicates tended to be very strong, particularly for WMS samples (SI Appendix, Fig. S2B). This finding suggests that FNs result primarily from reduced robustness to temporal variation and not technical variation.
Gene-level codes were more robust to temporal variation than taxon-level codes in part because they required fewer, more stable taxa to differentiate individual hosts. A single organism often contributed multiple gene-level features to an individual’s marker- or kbwindow-based code, representing both the organism’s presence as well as individual-specific strain variation (Fig. 4). At the less ecologically diverse skin and vaginal sites, code markers often derived from only a small number of dominant species. Relative to gene-level codes, taxon-level codes depended not only on more taxa, but also required the inclusion of less abundant taxa to achieve uniqueness (which tended to be less stable; Fig. 2A). On the other hand, gene-level codes were able to incorporate multiple distinguishing features from an individual’s most abundant taxa, and they were therefore more robust to temporal variation. Unlike taxon-level codes, gene-level codes were not always robust against interchanges between distinct strains of the same organism over time. Indeed, 17 of 67 marker gene-based codes (25%) that failed to match their owner at the second time point involved the loss of an encoded marker gene, whereas the gene’s parent species remained confidently detected (SI Appendix, Table S2 and Fig. S4).
We considered length of sampling time interval and antibiotic use as two additional factors that might have influenced the likelihood of a code failing to match its owner. FNs tended to be associated with very slightly longer times between sampling visits than TPs, but the differences were only marginally significant (Fig. 3C), suggesting that many of individuals’ unique strains do not drop below the limit of detection solely due to temporal effects on a scale of weeks to months (5). This result was confirmed by logistic regression, which failed to find a statistically significant fit between the odds of a FN and sampling time interval. Although individuals were initially excluded from the HMP cohort if they had previously used antibiotics, a small number of individuals did receive antibiotics between their first and subsequent visits; these individuals were not significantly associated with FNs (Fisher’s exact test; SI Appendix, Table S3).
Factors Leading to Loss of Code Uniqueness (FPs).
The code-building algorithm was guaranteed to produce codes that were unique relative to the sample population on which it was run (in this case, first-visit samples from multivisit HMP individuals), limited only by the dimensionality of available features. However, the algorithm cannot guarantee that codes will remain unique relative to unseen sets of samples, even those derived from the same individuals at later time points. For example, if an individual acquired new metagenomic features between their first and second sampling visits (as seen in Fig. 2C), then that individual’s second-visit sample might match another individual’s first-visit code. We refer to this as a false positive (FP).
In comparisons between individuals’ first- and second-visit samples, FPs occurred for 17% of OTU-based codes, 11% of species-based codes, 8% for marker-based codes, and 12% of kbwindow-based codes (based on the relaxed detection thresholds and averaged over body sites; Fig. 3A). Note that the probability of observing a FP increases as we compare a code with more samples; the FP rate for OTU-based codes is therefore elevated due to the larger number of individuals with 16S samples available for comparison (120 as opposed to 60 with metagenomes). Regardless, marker gene-based codes for stool proved highly robust against loss of uniqueness over time, with only a single FP out of 50 codes (2%). Hence, the general stability of the stool microbiome not only promoted the maintenance of features over time (as observed above), but also appeared to limit feature acquisition events associated with loss of code uniqueness.
We further evaluated code uniqueness in the independent validation subcohort of single visit HMP individuals, i.e., those for whom only first-visit samples were available. Such subjects were not considered in the earlier analyses of feature stability or in the code construction process. For each set of codes, we computed a single value, p, representing the probability of a FP hit per code against one of these previously unseen subjects (Methods and SI Appendix, Table S4). 1/p then represents the estimated population size for which we expect a particular code type to be unique. These population sizes varied among feature types and body sites, averaging on the order of 100s of individuals. Modeling the probability of a spurious hit as an exponential function of population size arrived at similar conclusions (Methods and SI Appendix, Table S5). Marker-based codes for paired HMP stool metagenomes were additionally evaluated against an independent set of 85 stool metagenomes from healthy Danish subjects enrolled in the MetaHIT cohort (30) (SI Appendix, SI Methods). Whether comparing HMP codes to MetaHIT samples or vice versa, codes were predicted to be unique to within ∼700 individuals, consistent with results from the intra-HMP comparison.
Although OTU-based codes were low confidence for most individuals as described above, they were expected to remain unique in populations of ∼500 individuals in the minority of cases where they were stable. More broadly applicable gene-based codes tended to be unique among ∼300 individuals. This difference appears to follow from the fact that gene-based codes draw on a smaller number of taxa than OTU-based codes and hence are more likely to recur in larger populations (SI Appendix, Fig. S5). FPs were particularly common (1/p was small) for codes based on the posterior fornix microbiome, which may reflect the lower level of between-individual microbial diversity at that site (1). These results demonstrate that marker gene-based codes, in addition to being stable over time, tend to remain unique in comparisons with 100s of previously unseen individuals.
Evaluating the Influence of Feature Detection Thresholds on Code Performance.
Defining and evaluating metagenomic codes relies on definitions of confident detection and confident nondetection for individual features (Table 1). We repeated the code definition and evaluation process (Fig. 3A) using alternative detection thresholds (SI Appendix, Fig. S6). Using a relaxed detection threshold during code definition expanded the list of potential code features, thus making it easier to construct unique codes for a given population. However, because lower-abundance features proved less robust to temporal variation (Fig. 2A), codes that incorporated them were progressively less likely to match their owner at subsequent visits. These effects were more noticeable when constructing and evaluating taxon-level codes; gene-level codes typically had large pools of high-abundance features to draw on.
Using a more stringent feature redetection threshold during code evaluation decreased the frequency of coincidental matches (SI Appendix, Fig. S6). This result may indicate that erroneous detection events at higher abundance are less frequent, or simply that newly acquired features tend to arise with low abundance. At the same time, true positive rates also tended to decrease when using a more stringent feature redetection threshold. This trend is most likely explained by temporal variation in individual microbiomes: code features that were near the confident detection threshold at time 1 may have dropped below this threshold over time, resulting in a failure to match their respective time 1 codes at time 2 (FNs).
Spurious Matches Are Common with Ecological Distance-Based Identification.
Microbiome samples are frequently compared using ecological measures of distance. These measures establish the degree of similarity between underlying microbial communities, taking into account differences in feature presence, abundance, and phylogenetic distribution. The same measures can be applied to demonstrate that, over time, repeated samples from the same individual are more similar to one another than to samples from other individuals (3), which can serve as an alternative basis for microbiome identifiability.
We evaluated the performance of distance-based microbiome identifiability using the HMP datasets applied above for metagenomic code evaluation. For each metagenomic feature type, we compared each second-visit sample to the collection of first-visit samples from the same body site using the Bray–Curtis and Canberra distance measures. Bray–Curtis distance combines absolute differences between features (making it more sensitive to a few large changes), whereas Canberra distance weights all differences equally (making it more sensitive to many small changes). Under this scheme, there are only two possible results for a given second-visit sample: it is closest to the first-visit sample from the same individual or it is closest to the first-visit sample from another individual. The first scenario represents a true positive, whereas the second scenario represents both a FN (as the sample failed to match the correct individual) and a FP (as the sample spuriously matched another individual).
Ecological distance-based microbiome identification was marked by high FP rates. Focusing on Bray–Curtis distance and averaging over body sites, second-visit OTU-based profiles spuriously matched another individual’s first-visit sample in 81% of cases, species-based profiles matched spuriously in 64% of cases, marker-based profiles in 61% of cases, and kbwindow-based profiles in 59% of cases (SI Appendix, Fig. S7A). Results were surprisingly similar using Canberra distance, which suggests a degree of robustness to the choice of distance measure (SI Appendix, Fig. S7B). True positive rates were reasonably strong and varied from ∼20 to 40% across feature types and body sites: this was comparable to performance of gene-level metagenomic codes at a typical body site and exceeded the performance of taxon-level codes (Fig. 3A). However, high FP rates limit the value of these successful identifications: distance-based identification always produces some closest match, and the match will be incorrect more than half of the time. Hence, unlike metagenomic codes, ecological distance-based approaches are not a feasible strategy for achieving microbiome identifiability in reasonably large populations.
Discussion
Our results indicate that human-associated microbial communities contain sufficient strain-level variation to distinguish individuals relative to a fixed population and robustly over time. Although notions of microbiome personalization have been explored previously using ecological distance measures, we demonstrated that an approach based on discrete metagenomic codes is required for true microbiome-driven identifiability. Although the populations considered here were small relative to real-world human communities (20–50 individuals), we estimate that this lower bound on microbiome-driven identifiability scales to at least hundreds of individuals, and this population size is representative of the cohorts currently used in microbiome research. This finding has important ethical ramifications for microbiome study design, particular those involving stool, as we have shown conclusively that metagenomic samples from a variety of body sites can be linked to individuals without additional identifying information.
Comparing metagenomic codes with previously unseen populations suggested that codes are unique within subpopulations of order-of-magnitude hundreds of people. Beyond this point, we expect to see a FP match between a given code and some unrelated individual. Notably, this coincidental match probability for microbiome-based codes is considerably larger than rates associated with human genomic DNA, which—under ideal conditions—can be vanishingly small (6). This discrepancy between metagenomic and genomic codes is due to at least three factors: (i) the bounded but nontrivial variation in the microbiome over time (relative to an essentially constant human genome); (ii) current limits of detection for rare features (e.g., individual SNPs or low abundance microbes) in the microbiome; and (iii) the nonindependence of microbial features due to ecological covariation. As our results provide only a lower bound on microbially driven identifiability, it is possible that technological advances, such as single cell sequencing and isolation of rare strains, will result in superior identifying power.
Previous research regarding the host-specific stability of the human microbiome has focused largely on stool, which we demonstrated to be exceptionally stable and not representative of other human body sites: the vast majority (86%) of marker gene-based codes from stool uniquely matched their owners after 30–300 d. However, even at the typical body site, ∼30% of codes uniquely identified their owners at later time points with few spurious matches (i.e., codes tended to pinpoint their owners or no one). As a result, it is not safe to assume that microbiome data can be completely anonymized, as a nontrivial fraction of samples can be accurately traced back to their original sources, along with potentially sensitive metadata. Even in the absence of such metadata, the prospect of associating the identified individuals with sensitive phenotypes [e.g., health status, cohabitation (31), or sexually transmitted infections] based on microbiome data alone becomes increasingly real as the list of associations between microbiome features and subject environment, history, and lifestyle expands.
At the same time, the variation in identifying power we observed between body sites underscores the fact that human-associated microbial habitats, in addition to being highly variable between individuals, are dynamical systems that vary nonrandomly within individuals over time. Improving our understanding of this balance between interindividual and temporal variation is critical not only to microbiome-based identifiability, but also to determining the ecological and molecular rules governing the human microbiome.
Methods
Data Collection, Quality Control, and Preprocessing.
Raw microbiome data used in the main analyses of this study were produced through the HMP (13) and are publicly available through the HMP’s data repository (www.hmpdacc.org). Eighty-five additional, publicly available stool metagenomes derived from the MetaHIT project (30) were used in a validation analysis (SRA BioProject PRJEB2054; SI Appendix, SI Methods). Outside of these two published resources, no additional human subject sequencing data were collected or analyzed in this work. OTU abundances were calculated from HMP 16S sequencing data using Mothur (32). Microbial species and species-specific marker gene abundance were quantified from HMP and MetaHIT WMS data using MetaPhlAn in the “rel_ab” and “clade_profiles” analysis modes, respectively (17). By default, MetaPhlAn reports marker gene abundance in units of “reads per kilobase of marker gene (RPK).” RPK values were converted to RPKM units (reads per kilobase per million sample reads) to facilitate comparisons between samples.
We considered 5,516 16S samples and 880 WMS samples derived from the 242 individuals enrolled in the HMP, including 106 metagenomes sequenced subsequent to initial publication. Before downstream sample selection and analysis, 16S and WMS samples were evaluated based on median genus-level Bray–Curtis dissimilarity score relative to other samples collected from the same body site. If a sample’s median dissimilarity score exceeded the upper inner fence for all median scores from its body site, then the sample was treated as an outlier and discarded (the upper inner fence is a point 1.5 times the interquartile range above the third quartile). This process removed 222 (4.0%) candidate 16S samples and 49 (5.6%) candidate whole metagenome shotgun samples that were highly atypical for their respective body sites. Following this quality control step, we averaged values derived from pairs of technical replicates (independent sets of measurements for the same subject, body site, and time point).
After performing the quality control steps outlined above, we isolated pairs of samples derived from the same subject and body site at two sampling visits (Table 1). Subjects sampled only once at a particular body site were used as independent validation groups (SI Appendix, Table S4). Paired samples form the basis for all analyses of temporal stability described in the text. Although we refer to subjects’ first and second sampling visits as time 1 and time 2, respectively, for convenience, sampling events were not synchronized across subjects. In addition, we note that subsets of the 242 total HMP subjects with both first- and second-visit samples differed from one body site to another and between the two sequencing methods (16S vs. WMS). For example, a subject could have both first- and second-visit 16S stool samples, but only a first-visit anterior nares WMS sample: this subject would then contribute to analyses of OTU stability in stool but not to analyses of marker gene stability in the nares. Processed paired sample data containing HMP-issued subject identifiers are available for download from huttenhower.sph.harvard.edu/idability.
WMS reads were additionally mapped to a database of 649 microbial reference genomes using the Burrows-Wheeler aligner (33), as described previously (34). The goal of this analysis was to identify strain-level variation in microbial genomic elements outside of the predefined MetaPhlAn marker genes. All genomes were divided into nonoverlapping kilobase-long windows, starting from the 5′ end of each scaffold within the genome. If a genome recruited reads from a WMS sample at >4× depth over >50% of its length, then the genome was considered to have been detected in the sample. In this case, individual abundance values of the genome’s kilobase windows (in RPKM units) were then added to a table of “kbwindows” features for the sample. For some WMS samples, no genomes met the criteria for detection outlined above, and so these samples were excluded from downstream analysis involving kbwindows features. For this reason, sample counts in some kbwindows-based analyses are smaller than those reported in the corresponding species- and marker gene-based analyses of WMS data (Fig. 3).
Numerical Details of Coincidental Match Analysis.
We used two approaches to study loss of code uniqueness (FPs) in comparisons with previously unseen validation subjects. First, we considered all possible pairings between N codes and M validation subjects. Any match between a code and a validation subject was counted as a hit (using the relaxed feature detection thresholds); the total number of such hits was H. Based on this, we estimated the probability p of a match between a code and a random individual to be H/MN; for cases where H = 0 (no FP events were observed), we estimated p as (H + 1)/(MN + 1) using Witten–Bell smoothing (35). 1/p estimates of the size of a previously unseen population in which we would have expected to see one FP; codes would be expected to remain unique in populations below that size. The results of this analysis are reported in SI Appendix, Table S4.
As an alternative approach, we modeled the probability of a code spuriously matching any member of a previously unseen population of size N [FP rate (FPR)] as an exponential function of N
This model assumes that FPR approaches 1 asymptotically as population size (N) goes to infinity. We sampled (with replacement) populations of increasing N from M independent validation subjects and computed an average FPR for each N (for n ≤ M). Fitting the above model to these data yielded estimates of the parameter k (which describes the rate of increase in FPR for increasing N) for each body site + feature type combination. Finally, for each combination we solved
for N. The solution, which we called N50, estimates of the number of previously unseen individuals with whom we could compare a code of a particular type before having a 50% chance of seeing at least one spurious match. Codes have a strong probability of remaining unique in comparisons with new populations of size <N50. Values of k and N50 are reported in SI Appendix, Table S5.
Analysis of Technical Variation.
Although technical replicates were averaged in the main analyses of this work, we performed two analyses on isolated replicate pairs to estimate the effects of technical variation on robust redetection of metagenomic features and codes. We considered 325 replicate pairs for 16S-based metagenomes and 26 replicate pairs for shotgun-based metagenomes; for each pair, both samples were required to pass the quality control procedures outlined above. In the first analysis, we then calculated the probability of a feature being detected in one replicate given that it was confidently detected in the other (SI Appendix, Fig. S2A). In the second analysis, we evaluated the consistency of technical replicates in the construction and evaluation of metagenomic codes (SI Appendix, Fig. S2B). Specifically, for technical replicates A and B corresponding to (body site X, subject Y, and time 1), we separately constructed codes for all site X samples including only replicate A and again including only replicate B. We then compared the code derived for replicate A and the code derived for replicate B to all time 2 samples. If the results were precisely the same (e.g., both the A code and the B code only matched subject Y at time 2), then we scored the replicates as consistent; if there was any deviation, then we scored the replicates as inconsistent. An analogous procedure was repeated for technical replicates of time 2 samples.
Supplementary Material
Acknowledgments
We thank N. Segata for assistance profiling MetaPhlAn marker genes, L. J. McIver and A. Shafquat for reviewing online materials, and T. L. Tickle, R. D. Franzosa, and the three anonymous reviewers for helpful comments. This work was funded in part by National Institutes of Health (NIH) National Institute of Allergy and Infectious Diseases (NIAID) Contract HHSN272200900018C (to D.G.), NIH National Human Genome Research Institute (NHGRI) Grant U54HG004969 (to the Broad Institute and D.G.), NIH National Institute of General Medical Sciences Grant P50GM098911 (to B.J.M.B.), NIH NIAID Grant R01 AI101018 (to K.P.L.), Danone Research Grant PLF-5972-GD (to Wendy Garrett and C.H.), NIH NHGRI Grant R01HG005969 (to C.H.), Army Research Office Grant W911NF-11-1-0473 (to C.H.), and National Science Foundation Faculty Early Career Development Grant DBI-1053486 (to C.H.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 6778.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1423854112/-/DCSupplemental.
References
- 1.Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qin J, et al. MetaHIT Consortium A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fierer N, et al. Forensic identification using skin bacterial communities. Proc Natl Acad Sci USA. 2010;107(14):6477–6481. doi: 10.1073/pnas.1000162107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schloissnig S, et al. Genomic variation landscape of the human gut microbiome. Nature. 2013;493(7430):45–50. doi: 10.1038/nature11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Faith JJ, et al. The long-term stability of the human gut microbiota. Science. 2013;341(6141):1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jobling MA, Gill P. Encoded evidence: DNA in forensic analysis. Nat Rev Genet. 2004;5(10):739–751. doi: 10.1038/nrg1455. [DOI] [PubMed] [Google Scholar]
- 7.Cotton EA, et al. Validation of the AMPFlSTR SGM plus system for use in forensic casework. Forensic Sci Int. 2000;112(2-3):151–161. doi: 10.1016/s0379-0738(00)00182-1. [DOI] [PubMed] [Google Scholar]
- 8.Thompson WC, Taroni F, Aitken CG. How the probability of a false positive affects the value of DNA evidence. J Forensic Sci. 2003;48(1):47–54. [PubMed] [Google Scholar]
- 9.Lin Z, Owen AB, Altman RB. Genetics. Genomic research and human subject privacy. Science. 2004;305(5681):183. doi: 10.1126/science.1095019. [DOI] [PubMed] [Google Scholar]
- 10.Greenbaum D, Sboner A, Mu XJ, Gerstein M. Genomics and privacy: Implications of the new reality of closed data for the field. PLOS Comput Biol. 2011;7(12):e1002278. doi: 10.1371/journal.pcbi.1002278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lowrance WW, Collins FS. Ethics. Identifiability in genomic research. Science. 2007;317(5838):600–602. doi: 10.1126/science.1147699. [DOI] [PubMed] [Google Scholar]
- 12.Lunshof JE, Chadwick R, Vorhaus DB, Church GM. From genetic privacy to open consent. Nat Rev Genet. 2008;9(5):406–411. doi: 10.1038/nrg2360. [DOI] [PubMed] [Google Scholar]
- 13.Human Microbiome Project Consortium A framework for human microbiome research. Nature. 2012;486(7402):215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu GD, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenblum S, Turnbaugh PJ, Borenstein E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci USA. 2012;109(2):594–599. doi: 10.1073/pnas.1116053109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yatsunenko T, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Segata N, et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods. 2012;9(8):811–814. doi: 10.1038/nmeth.2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Selman B. Computational science: A hard statistical view. Nature. 2008;451(7179):639–640. doi: 10.1038/451639a. [DOI] [PubMed] [Google Scholar]
- 19.Karp RM. Reducibility Among Combinatorial Problems. Springer; New York: 1972. [Google Scholar]
- 20.Chandrasekaran K, Karp R, Moreno-Centeno E, Vempala S. Proceedings of the Twenty-Second Annual ACM-SIAM Symposium on Discrete Algorithms. Society for Industrial and Applied Mathematics; Philadelphia, PA: 2011. Algorithms for implicit hitting set problems; pp. 614–629. [Google Scholar]
- 21.Grice EA, et al. NISC Comparative Sequencing Program Topographical and temporal diversity of the human skin microbiome. Science. 2009;324(5931):1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravel J, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fredricks DN. Microbial ecology of human skin in health and disease. J Investig Dermatol Symp Proc. 2001;6(3):167–169. doi: 10.1046/j.0022-202x.2001.00039.x. [DOI] [PubMed] [Google Scholar]
- 24.Larsen B, Monif GR. Understanding the bacterial flora of the female genital tract. Clin Infect Dis. 2001;32(4):e69–e77. doi: 10.1086/318710. [DOI] [PubMed] [Google Scholar]
- 25.Evaldson G, Heimdahl A, Kager L, Nord CE. The normal human anaerobic microflora. Scand J Infect Dis Suppl. 1982;35:9–15. [PubMed] [Google Scholar]
- 26.Sloan WT, et al. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ Microbiol. 2006;8(4):732–740. doi: 10.1111/j.1462-2920.2005.00956.x. [DOI] [PubMed] [Google Scholar]
- 27.Meadow JF, Bateman AC, Herkert KM, O’Connor TK, Green JL. Significant changes in the skin microbiome mediated by the sport of roller derby. PeerJ. 2013;1:e53. doi: 10.7717/peerj.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flores GE, et al. Microbial biogeography of public restroom surfaces. PLoS ONE. 2011;6(11):e28132. doi: 10.1371/journal.pone.0028132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smillie CS, et al. Ecology drives a global network of gene exchange connecting the hman microbiome. Nature. 2011;480(7376):241–244. doi: 10.1038/nature10571. [DOI] [PubMed] [Google Scholar]
- 30.Arumugam M, et al. MetaHIT Consortium Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lax S, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science. 2014;345(6200):1048–1052. doi: 10.1126/science.1254529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schloss PD, et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giannoukos G, et al. Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes. Genome Biol. 2012;13(3):R23. doi: 10.1186/gb-2012-13-3-r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Witten IH, Bell TC. The zero-frequency problem: Estimating the probabilities of novel events in adaptive text compression. IEEE Trans Inf Theory. 1991;37(4):1085–1094. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.