

ABSTRACT
Organ growth occurs through the integration of external growth signals during the G1 phase of the cell cycle to initiate DNA replication. Although numerous growth factor signals have been shown to be required for the proliferation of cardiomyocytes, genetic studies have only identified a very limited number of transcription factors that act to regulate the entry of cardiomyocytes into S phase. Here, we report that the cardiac para-zinc-finger protein CASZ1 is expressed in murine cardiomyocytes. Genetic fate mapping with an inducible Casz1 allele demonstrates that CASZ1-expressing cells give rise to cardiomyocytes in the first and second heart fields. We show through the generation of a cardiac conditional null mutation that Casz1 is essential for the proliferation of cardiomyocytes in both heart fields and that loss of Casz1 leads to a decrease in cardiomyocyte cell number. We further report that the loss of Casz1 leads to a prolonged or arrested S phase, a decrease in DNA synthesis, an increase in phospho-RB and a concomitant decrease in the cardiac mitotic index. Taken together, these studies establish a role for CASZ1 in mammalian cardiomyocyte cell cycle progression in both the first and second heart fields.
Keywords: Heart development, Proliferation, Cardiomyocyte, Congenital heart disease, First heart field, Second heart field, CASZ1, Mouse
Summary: Cardiac-specific deletion of the para-zinc-finger protein CASZ1 in mouse embryos leads to significant heart defects associated with impaired cardiomyocyte proliferation.
INTRODUCTION
Early development of the heart is governed by hyperplastic growth, in which cardiac cells undergo mitogen-dependent activation during the G1 phase of the cell cycle (Ahuja et al., 2007). During early stages of heart development, cardiomyocytes of the first and second heart fields are highly proliferative, resulting in substantial growth of the embryonic heart. The overall rate of cardiomyocyte proliferation gradually declines concomitant with the onset of cardiomyocyte terminal differentiation (Soonpaa et al., 1996; Christoffels et al., 2000; Pasumarthi and Field, 2002; Sedmera et al., 2003; Ikenishi et al., 2012). After this period, the vertebrate heart continues to grow largely through hypertrophy and by recruitment and proliferation of cells from the neural crest and the epicardium (Li et al., 1996; Creazzo et al., 1998; Manner et al., 2001; Ahuja et al., 2007; Kelly, 2012; Maillet et al., 2013).
Understanding the transcriptional mechanisms of cardiomyocyte proliferation is crucial for uncovering pathologies and treatments for congenital heart disease. Past studies have shown that cardiomyocyte hyperplastic growth occurs in response to input from a large network of growth factor signaling pathways (Lavine et al., 2005; Ahuja et al., 2007; Bersell et al., 2009; Heallen et al., 2011; Porrello et al., 2011; Xin et al., 2011, 2013; Eulalio et al., 2012; von Gise et al., 2012; Wadugu and Kuhn, 2012). However, many basic questions regarding the mechanisms underlying how these growth factors regulate the cardiomyocyte cell cycle remain unanswered. Central to this issue, it is not known which cardiac-specific transcription factors act to drive or commit cardiomyocytes into the next round of division.
CASZ1 is a para-zinc-finger transcription factor that has been shown to be expressed during and required for vertebrate heart development, with depletion of Casz1 in Xenopus embryos leading to the failure of a small subset of progenitor cells to differentiate into cardiomyocytes, resulting in aberrant cardiac morphogenesis and eventual death (Vacalla and Theil, 2002; Liu et al., 2006; Christine and Conlon, 2008; Amin et al., 2014; Sojka et al., 2014). The evolutionary role of Casz1 in heart development is further emphasized by genome-wide association studies showing genetic association of the CASZ1 locus with blood pressure and hypertension (Levy et al., 2009; Takeuchi et al., 2010; Lu et al., 2015). Consistently, it has been demonstrated that CASZ1 has an essential role in blood vessel assembly and lumen formation (Charpentier et al., 2013a,b). Together, these studies implicate a potential link between Casz1 and cardiovascular dysfunction. However, the genetic requirement and endogenous role for Casz1 in mammalian cardiac development remain to be established.
Here we report that Casz1 is expressed in cardiomyocytes during the earliest stages of mammalian heart development, and using genetic fate mapping we show that Casz1-positive cells give rise to derivatives of both the first and second heart field, including cardiomyocytes in the left and right ventricles and the left and right atria. Through the generation of a conditional null allele, we demonstrate that Casz1 is essential for early mammalian heart development and define a role for Casz1 in the proliferation of cardiomyocytes during chamber formation. We go on to demonstrate an essential role for Casz1 in the cardiomyocyte cell cycle, showing that loss of Casz1 leads to a prolonged or arrested G1 phase that is associated with a marked reduction in DNA synthesis, an increase in phospho-RB, and a decrease in the cardiac mitotic index. Taken together, our results demonstrate a role for Casz1 in the G1-to-S phase progression of cardiomyocytes.
RESULTS
Casz1 is expressed in the developing myocardium
To address the role of Casz1 in mammalian heart development, we cloned full-length Casz1 from adult mouse heart tissue and conducted a detailed expression analysis. We found that Casz1 is first expressed in the cardiac crescent (E7.5, Fig. 1A) and continues to be expressed in the heart during cardiac looping (E8, Fig. 1B,F; E8.5-E9.5, Fig. 1C,D,G,H), when we observed Casz1 expression in the future left and right ventricles, in both the compact layer and the trabeculae, and in the primitive atria (E9.5, Fig. 1D,H). Sectioning of heart tissue at these stages further revealed Casz1 expression in both the myocardium and endocardium (E8.5, Fig. 1G). By E11.5 we found expression of Casz1 in the heart but also in other tissue types, including the limb bud, nasal placode, somites, telencephalon, hindbrain and, consistent with recent reports, the eye (Konstantinides et al., 2015; Mattar et al., 2015) (Fig. 1E).
Fig. 1.

Casz1 is expressed in the developing mouse heart. (A-H) Whole-mount in situ hybridization at E7.5 (A), E8.0 (B,F), E8.5 (C,G), E9.5 (D,H) and E11.5 (E). Casz1 is first expressed in myocardial precursor cells beginning at E7.5 (A). At E8.0, Casz1 transcripts are found in the linear heart tube (B) and as the heart undergoes looping (C,D). Casz1 is also expressed in the eye, the nasal placode, somites, telencephalon, hindbrain and the developing limb bud (E). (F-H) Vibratome sections of whole-mount embryos reveal that, at early stages of heart development, although Casz1 is not found in the pharyngeal mesoderm, it is expressed in the differentiating myocardium at the venous pole of the heart (F). Casz1 is expressed in the myocardium and the endocardium (G) and in the trabeculae (H). (I-L) CASZ1 colocalizes with the cardiomyocyte marker tropomyosin (TMY) in the myocardium of an E14.5 left ventricle. The boxed region in K of the left ventricular myocardium is enlarged in L to highlight expression of CASZ1 within the nuclear bodies of the nucleus. (M,N) CASZ1 is not expressed at E14.5 in Tie2-positive endothelial cells (M) nor Wt1-positive epicardial cells (N). Sections of Tie2-Cre;R26R-EGFP reporters were used to mark endothelial cells (M) and a Wt1-Cre;R26R-EGFP reporter to mark epicardial cells (N). (O,P) Immunofluorescent staining for CASZ1 and ISL1 shows coexpression in outflow tract myocardium at E10.5. The boxed region in O of OFT myocardium is enlarged in P. Nuclei in K-O were stained with DAPI (blue). MP, myocardial progenitors; HT, developing heart tube; LH, looped heart; N, neural tube; E, eye; HB, hindbrain; NP, nasal placode; T, telencephalon; LB, limb bud; S, somites; PME, pharyngeal mesoderm; MY, myocardium; EN, endocardium; TR, trabeculae; CAC, common atrial chamber; CVC, common ventricular chamber; CL, compact layer; EPI, epicardium; LV, left ventricle; OFT, outflow tract; FG, foregut. Scale bars: 20 µm in I-K,M,N; 5 µm in L; 10 µm in P.
We observed that CASZ1 protein is expressed in a pattern similar to that of Casz1 mRNA. Our data demonstrate that CASZ1 is expressed in defined subdomains of the nucleus (Fig. 1I-L). Given that it is not technically possible to investigate nuclear domains in cardiac tissue in vivo, we examined the compartmentalization of CASZ1 in human primary endothelial cells (HUVECS), a cell type previously shown to express CASZ1 (Charpentier et al., 2013b). From these studies we demonstrate that CASZ1 colocalizes with promyelocytic leukemia (PML), a defining protein of PML bodies (supplementary material Fig. S1). PML bodies are well-defined nuclear subdomains found to associate with a distinct set of genomic loci; however, these loci are undefined and, moreover, there is no known function for PML in cardiovascular development or disease (Matera et al., 2009).
CASZ1 protein is expressed in the tropomyosin (TMY)-positive cardiomyocytes (Fig. 1I-L) and its expression is mutually exclusive to that of Tie2 (Tek)-positive derived endothelial cells (Fig. 1M) and Wt1-positive derived epicardial cells (Fig. 1N). We further find that CASZ1 is co-expressed in second heart field cells with the second heart field marker ISL1 (Cai et al., 2003; Sun et al., 2007) (Fig. 1O,P). Collectively, these data demonstrate that CASZ1 is expressed in the nucleus of cardiomyocytes and, by inference, in PML bodies of the first and second heart fields during the early stages of cardiogenesis.
Casz1-expressing cells give rise to first and second heart field derivatives
To determine whether cardiomyocytes that express Casz1 contribute to derivatives of the first and second heart field, we performed genetic lineage tracing of Casz1-expressing cells. To permanently label this population of cells and its descendants, we generated a tamoxifen-inducible CreERT2 allele (Casz1CreERT2) by homologous recombination in embryonic stem cells (ESCs) and subsequently passed the Casz1CreERT2 through the germline (supplementary material Fig. S2). Treatment with tamoxifen results in Cre-driven recombination and, in the presence of a tomato reporter (R26RtdT), in the identification of Casz1-expressing cells and their progeny. Co-staining with the cardiomyocyte marker TMY enabled lineage identification of tagged cell populations. From this lineage analysis we identified cardiomyocytes derived from Casz1-expressing cells (supplementary material Fig. S1C,D; Fig. 2A-J) at E12.5 in the left (Fig. 2I,J) and right (Fig. 2A,B,G,H) ventricular walls, the interventricular septum (Fig. 2C,D), the compact layer (Fig. 2G,H) and the trabeculae (Fig. 2E,F). We found no evidence that Casz1-expressing cells can give rise to cardiac fibroblasts or cardiac endothelium; however, we note that the recombination frequency of the Casz1CreERT2 allele with the current tamoxifen regime is low and therefore we cannot formally rule out this possibility. Collectively, our studies show that E8.5 Casz1-expressing cells give rise to cardiomyocytes derived from both the first and second heart fields.
Fig. 2.
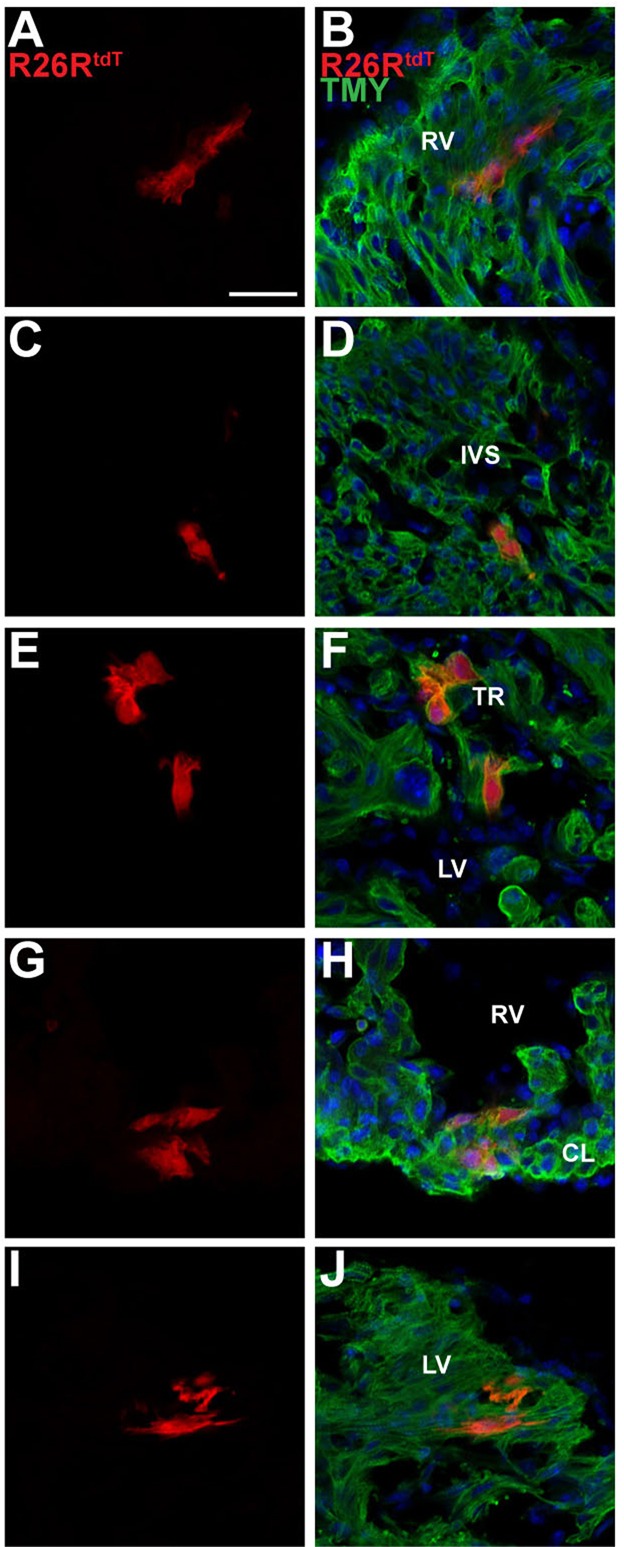
Lineage tracing with Casz1CreERT2-neo labels cardiomyocytes in first and second heart field derivatives. Immunofluorescent staining for cardiac TMY and with DAPI (blue) of the Casz1CreERT2/+;R26RtdT/+ embryo depicted in supplementary material Fig. S2 shows that Casz1-expressing cells give rise to cardiomyocytes located in the walls of the left (I,J) and right (A,B,G,H) ventricles, the interventricular septum (C,D) and the trabeculae (E,F). RV, right ventricle; LV, left ventricle; IVS, interventricular septum; TR, trabeculae; CL, compact layer. Scale bar: 20 µm.
Casz1 is essential for cardiac development
To determine the requirement for Casz1 in cardiac development, we mapped the cardiac transcriptional start site(s) of Casz1 (supplementary material Fig. S3B). Our analysis identified two transcriptional start sites in embryonic heart: one 275 bp upstream and the other 139 bp upstream from the predicted translational start site. Using these data we generated a conditional floxed null allele (Casz1flox-neo) by homologous recombination in ESCs. Successful targeting of the selection cassette generated a Casz1flox-neo allele in which exon 6 is flanked with loxP sites (supplementary material Fig. S3A). Exon 6 was chosen because it is a crucial coding exon located downstream of both Casz1 transcriptional start sites and contains the CASZ1 nuclear localization signal. Furthermore, in the event that cryptic splicing occurs to any of the next three exons it would lead to the introduction of a frameshift and, if the protein were made, it would lack a nuclear localization signal and all zinc- finger domains (supplementary material Fig. S3).
The presence of the Casz1flox-neo allele in ESCs was confirmed by Southern blot, PCR and genomic sequence analysis (supplementary material Fig. S3C,D; data not shown). ESCs harboring Casz1flox-neo were used to generate chimeric mice, which passed Casz1flox-neo through the germline. The PGKneo cassette (Liu et al., 2003) was removed by mating F1 heterozygous mice to mice expressing FlpE recombinase (Rodriguez et al., 2000) and Casz1f/+ progeny were mated to an Nkx2.5-Cre line to generate Casz1f/+;Nkx2.5Cre/+ (Moses et al., 2001). The Casz1f/+;Nkx2.5Cre/+ mice were mated to Casz1f/f mice to generate a cardiac-specific conditional Casz1 mutation: Casz1f/f;Nkx2.5Cre/+. Expression of Cre from the Nkx2.5-Cre allele was confirmed by mating to R26RtdT reporter mice (Madisen et al., 2010) (supplementary material Fig. S4), and loss of Casz1 in Casz1f/f;Nkx2.5Cre/+ in cardiac but not neural tissue (dorsal root ganglia) was confirmed by immunohistochemistry (supplementary material Fig. S3E-H).
We observed that heterozygotes containing the Casz1f/+;Nkx2.5Cre/+ alleles are viable, fertile and display no obvious phenotypic abnormalities. By contrast, no homozygous mice for the floxed allele, Casz1f/f;Nkx2.5Cre/+, were recovered postnatally, indicating that loss of Casz1 in the developing heart is embryonic lethal.
Analysis of timed intercrosses of Casz1f/+;Nkx2.5Cre/+ mice failed to identify viable homozygous Casz1f/f;Nkx2.5Cre/+ embryos subsequent to E14.5 (Fig. 3). Gross examination of Casz1f/f;Nkx2.5Cre/+ embryos demonstrated that E12.5 mutants are indistinguishable from wild-type littermates (Fig. 3A,B). However, at E13.5, Casz1f/f;Nkx2.5Cre/+ embryos exhibit inflated pericardial sacs, severe edema and blood hemorrhaging, which are consistently indicative of circulatory distress (Fig. 3C,D) (Conway et al., 2003).
Fig. 3.

Casz1 is required for cardiac development. (A-D) Gross morphology of Casz1f/+;Nkx2.5+/+ wild-type (WT) and Casz1f/f;Nkx2.5Cre/+ embryos. By E13.5, Casz1f/f;Nkx2.5Cre/+ embryos are beginning to exhibit edema and have decreased blood flow throughout the vasculature. (E-H) SEM analysis of WT and Casz1f/f;Nkx2.5Cre/+ hearts at E12.5 and E13.5 highlights cardiac defects. At E12.5, Casz1f/f;Nkx2.5Cre/+ embryos have cardiac malformations, including an enlarged right atria and aberrant left ventricle. These defects are more pronounced at E13.5. (I-N) Histological analysis of WT and Casz1f/f;Nkx2.5Cre/+ hearts. Casz1f/f;Nkx2.5Cre/+ embryos exhibit severe cardiac hypoplasia and ventral septal defects. H&E staining of transverse sections shows thinning of the ventricular walls beginning at E12.5, leading to severe cardiac hypoplasia. Boxed areas are enlarged to the right. Arrow (L) highlights the ventricular septal defect. (M,N) Quantification of ventricular wall thickness in WT and Casz1f/f;Nkx2.5Cre/+ embryos at E10.5 (M) and E12.5 (N). Data are shown as mean±s.e.m.; *P<0.05, WT versus Casz1f/f;Nkx2.5Cre/+; NS, not significant. RV, right ventricle; LV, left ventricle; RA, right atria; LA, left atria; OFT, outflow tract; IVS, interventricular septum; CL, compact layer; TR, trabeculae. Scale bars: 60 µm in I,J,K,L; 15 µm in I′,J′,K′,L′.
Ultrastructural analysis using scanning electron microscopy (Hullinger et al. 2012) of Casz1f/f;Nkx2.5Cre/+ hearts at E12.5 revealed cardiac malformations, including an enlarged right atrium and malformed left ventricle (Fig. 3E,F). These defects were more pronounced by E13.5 (Fig. 3G,H), with ballooning of the right atria indicative of blood pooling in the right ventricle (Deng et al., 1996). Collectively, these results demonstrate an essential requirement for Casz1 in mammalian heart development between E10.5 and E12.5.
Casz1 is required for growth of the cardiac chambers
Histological examination showed that Casz1f/f;Nkx2.5Cre/+ and wild-type hearts were indistinguishable at E10.5 (mean ventricular wall thickness of 15.4±0.44 µm versus 16.1±0.70 µm, respectively; n=3, P=0.42) (Fig. 3I-J′,M). However, by E12.5, severe thinning of the myocardium and an associated decrease in wall thickness were observed in Casz1f/f;Nkx2.5Cre/+ embryos as compared with wild-type littermate controls [mean of 22.6±1.2 µm (n=2) versus 14.3±0.91 µm (n=4); P<0.005], indicating that Casz1 is required in the ventricular myocardium. In addition, we observed an underdeveloped interventricular septum and decreased trabeculation (Fig. 3K-L′,N). By E13.5, Casz1f/f;Nkx2.5Cre/+ hearts were hypoplastic, with narrower ventricular lumens and membranous ventricular septal defects (supplementary material Fig. S5). Together, these results demonstrate that Casz1 is essential for development of the cardiac chambers and further imply that Casz1 is essential for cardiomyocyte growth.
Casz1 embryonic nulls phenocopy Casz1f/f;Nkx2.5Cre/+ embryos
To test whether Casz1 is required for embryogenesis prior to E12.5 we generated a complete embryonic null allele of Casz1 by mating Casz1f/f mice to a Sox2-Cre driver (Hayashi et al., 2002) to create Casz1f/f;Sox2Cre/+. Extensive studies have established that Sox2-Cre is expressed and functions in all cells of the epiblast (Fig. 4A,B) (Hayashi et al., 2002; Barrow et al., 2007; Arnold et al., 2009; Delgado-Esteban et al., 2013). We found that Casz1f/f;Sox2Cre/+ mice, as with Casz1f/f;Nkx2.5Cre/+, do not survive beyond E14.5. Histological examination at E12.5 confirmed that Casz1f/f;Sox2Cre/+ homozygotes have a cardiac phenotype indistinguishable from that of Casz1f/f;Nkx2.5Cre/+ mice (mean ventricular wall thickness of 41.2±2.1 µm versus 25.8±1.6 µm, respectively; n=2, P<0.0005) (Fig. 3I-N and Fig. 4E-I). These results demonstrate that an initial and essential requirement for Casz1 in the mouse embryo is in the developing heart and confirm that loss of Casz1 in cardiac tissue leads to hypoplastic growth and embryonic lethality.
Fig. 4.

Casz1 null embryos phenocopy Casz1 cardiac null embryos. (A,B) Whole-mount lacZ staining of Sox2+/+;R26RlacZ/+ and Sox2Cre/+;R26RlacZ/+ E10.5 embryos demonstrates that Sox2-Cre is ubiquitously expressed. (C,D) Gross morphology of E12.5 Casz1f/+;Sox+/+ wild-type (WT) and Casz1f/f;Sox2Cre/+ embryos. (E-H) Histological analysis of WT and Casz1f/f;Sox2Cre/+ hearts. Casz1f/f;Sox2Cre/+ embryos exhibit severe cardiac hypoplasia and ventral septal defects. H&E staining of transverse sections shows cardiac hypoplasia at E12.5 Boxed areas are enlarged to the right. Arrow (G) highlights the ventricular septal defect. (I) Quantification of ventricular wall thickness in WT and Casz1f/f;Sox2Cre/+ embryos at E12.5. Data are shown as mean±s.e.m.; ***P<0.0005, WT versus Casz1f/f;Sox2Cre/+. Labels as in Fig. 3. Scale bars: 60 µm in E,G; 15 µm in F,H.
Casz1 is essential in the second heart field
Cells from the second heart field give rise to the right ventricle, the interventricular septum, the outflow tract, both atria, and the atrial septum (Kelly, 2012). Our data demonstrated that CASZ1 is co-expressed with the second heart field marker ISL1 at E10.5 (Fig. 1O,P), and our fate mapping studies identified descendants of Casz1-expressing cells within second heart field derivatives (Fig. 2A,B,G,H). We further observed that loss of Casz1 in Nkx2.5-positive cells leads to a hypoplastic right ventricle. Based on these observations we hypothesize that Casz1 is required for second heart field development.
To test this hypothesis we generated mice that lack Casz1 in the second heart field by crossing Casz1f/f mice to an Isl1-Cre driver (Srinivas et al., 2001) to create Casz1f/f;Isl1Cre/+. Gross examination of Casz1f/f;Isl1Cre/+ embryos showed that they are viable and indistinguishable from heterozygous and wild-type littermates, at least until E14.5 (Fig. 5G,H). At E14.5, Casz1f/f;Isl1Cre/+ hearts have a normal left ventricle (mean of 82.6±3.7 µm versus 79.7±5.4 µm in ventricular wall thickness of n=2, P=0.66) (Fig. 5J) while displaying reduced thickness in the right ventricle (mean of 64.5±1.7 µm versus 37.9±4.3 µm; n=2, P<0.005) (Fig. 5I). Although there were significant differences in thickness of the ventricular free wall, we did not detect any ventricular septal defects (Fig. 5A-F). Consistently with right ventricular hypoplasia, no Casz1f/f;Isl1Cre/+ mice were recovered postnatally, indicating that Casz1 is required for the growth of the second heart field.
Fig. 5.
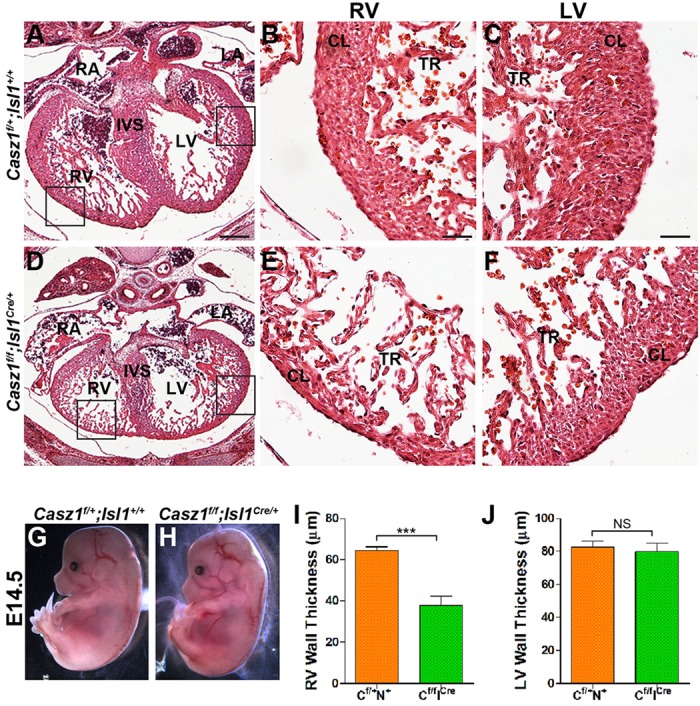
Casz1 is required in the second heart field. Histological analysis of WT and Casz1f/f;Isl1Cre/+ hearts at E14.5 demonstrates that Casz1f/f;Isl1Cre/+ hearts are misshapen and have a thin right ventricle. Boxed areas are enlarged to the right. (G,H) Gross morphology of E14.5 Casz1f/+;Isl1+ wild-type (WT) and Casz1f/f;Isl1Cre/+ embryos. (I,J) Quantification of right (I) and left (J) ventricular wall thickness in WT and Casz1f/f;Isl1Cre/+ embryos at E14.5. Data are shown as mean±s.e.m.; ***P<0.0005, WT versus Casz1f/f;Isl1Cre/+; NS, not significant. Labels as in Fig. 3. Scale bars: 60 µm in A,D; 15 µm in B,C,E,F.
Regulation of cell growth by Casz1
The observation that the loss of Casz1 in cardiac tissue leads to a hypoplastic heart and a concomitant decrease in chamber wall thickness led us to hypothesize that Casz1 functions to regulate cardiomyocyte proliferation. To test this hypothesis, we determined the number of cardiomyocytes in wild-type and Casz1f/f;Nkx2.5Cre/+ mice. We found that cardiomyocytes, as marked by expression of TMY, were significantly reduced in Casz1 cardiac null versus wild-type ventricles at E12.5 (mean of 868±69 versus 638±60 cardiomyocytes; n≥3, P<0.05) (Fig. 6A-F; supplementary material Fig. S6). We note that the cardiac hypoplasia observed in Casz1 cardiac null hearts is specific to the cardiomyocytes and does not affect the epicardium or endothelial cells at E12.5 (Fig. 6P,Q). Furthermore, the reduction in cell number is not associated with programmed cell death (supplementary material Fig. S8). Consistently, cardiomyocytes in Casz1 cardiac null hearts continue to express the cardiomyocyte markers TMY and cardiac troponin T (cTNT; also known as TNNT2) and we could detect higher order structures including sarcomeres with Z-disks (Fig. 6R-U). Together, these studies support a role for Casz1 in cardiomyocyte growth.
Fig. 6.

Casz1 regulates cardiomyocyte proliferation. (A-D′) Immunofluorescent staining for TMY and with DAPI (blue) highlights a decrease in the number of differentiated cardiomyocytes at E12.5 in Casz1f/f;Nkx2.5Cre/+ embryos. (E,F) Quantification of cardiomyocyte cell counts per field at E10.5 (E) and E12.5 (F). Data are shown as mean±s.e.m. of three embryos; *P<0.05, WT versus Casz1f/f;Nkx2.5Cre/+; NS, not significant. (G-J) Immunofluorescent staining for EdU and TMY shows a reduction in proliferation in Casz1f/f;Nkx2.5Cre/+ ventricles at E12.5. Boxed areas of left ventricles are magnified beneath. (K,L) Immunofluorescent staining for pHH3 along with TMY demonstrates a decreased cardiomyocyte mitotic index in Casz1f/f;Nkx2.5Cre/+ embryos as compared with WT in sections of E12.5 hearts. (M-O) Quantitative evaluation of reduced cardiomyocyte proliferation in Casz1f/f;Nkx2.5Cre/+ ventricles at E12.5 by EdU incorporation (M), reduced cardiomyocyte mitotic index in Casz1f/f;Nkx2.5Cre/+ ventricles (N) and in Casz1f/f;Isl1Cre/+ right ventricles at E12.5 (O). Data are shown as mean±s.e.m. of three embryos; *P<0.05, ***P<0.0005, WT versus Casz1f/f;Nkx2.5Cre/+ embryos at a given stage. (P,Q) Immunofluorescent staining for cTNT to mark cardiomyocytes and filamin A (FILA) to mark the epicardium and endothelial cells demonstrates a reduction in cardiomyocytes, whereas FILA expression within the epicardium and endothelial cells is comparable to WT at E12.5. (R-U) Immunofluorescent staining for phalloidin demonstrates that actin filaments are intact in Casz1f/f;Nkx2.5Cre/+ E12.5 hearts. Boxed areas are magnified beneath. EN, endothelial cells; S, sarcomere; Z-d, Z-disks (other labels as Figs 1 and 3). Scale bars: 10 µm in A′,B′,C′,D′,K,L,T,U; 20 µm in P-R; 50 µm in I,J; 100 µm in A,B; 200 µm in C,D,G,H.
To validate our findings, we performed a comparative expression analysis of the developing heart using RNA isolated from E10.5 hearts (n=3 cardiac null; n=3 controls), a period before we can detect any cardiac abnormalities in Casz1f/f;Nkx2.5Cre/+ embryos. RNA-seq data were processed for differential expression analysis using Cufflinks (Trapnell et al., 2012); raw data and full analysis are available at the NCBI Gene Expression Omnibus under accession number GSE55394. We used unbiased Gene Ontology and from this analysis determined that the three largest categories of genes downregulated as a result of the loss of Casz1 were ‘regulation of growth’, ‘regulation of systems processes’ and ‘cardiac muscle development’ (supplementary material Fig. S7). Thus, these analyses suggest that Casz1 functions to regulate the growth of the cardiac chambers.
Casz1 is required for cardiomyocyte G1-to-S phase transition
Our phenotypic analysis, cell quantification and transcriptional profiling are all consistent with a role for Casz1 in cardiomyocyte cell division. To gain insight into the mechanisms of Casz1-mediated cardiomyocyte growth, we conducted a detailed analysis of cell proliferation. Our results show a significant reduction in EdU incorporation in myocardial cells in the developing myocardium of Casz1 cardiac null ventricles versus controls (mean of 19.5±0.9% versus 13.6±0.8%; n=3, P<0.0001) (Fig. 6G-J,M). The cardiomyocyte mitotic index in E12.5 Casz1f/f;Nkx2.5Cre/+ ventricles (mean of 19.6±1.1% versus 13.7±0.98%; n=3, P<0.005) and Casz1f/f;Isl1Cre/+ right ventricles (mean of 19.3±1.1% versus 13.5±2.1%; n=2, P<0.05) was also reduced, as assessed by phospho-histone H3 (pHH3) staining (Fig. 6K,L,N,O). Thus, Casz1 acts to control cardiomyocyte proliferation in both the first and second heart fields.
To further define the mechanism by which Casz1 functions in cardiomyocyte cell division, we conducted cell cycle profiling of cardiac nuclei from control and Casz1 cardiac null heart tissue (Fig. 7A). Our results indicate that cardiac nuclei null for Casz1 display a significant increase in cells in G1 phase (52.3% versus 60.1% in n=4 hearts) and a concomitant decrease of cells in S phase (26.1% versus 19.4%), while displaying no alteration in the percentage of cells in G2 phase (21.5% versus 20.5%). Consistently, we observe a significant increase in Casz1 cardiac null cardiomyocytes expressing the phosphorylated form of the tumor suppressor protein retinoblastoma 1 (RB), a highly effective inhibitor of the G1-to-S transition (mean of 3.5±0.3% versus 5.3±0.7%; P<0.05) (Fig. 7B-D) (Mulligan and Jacks, 1998).
Fig. 7.
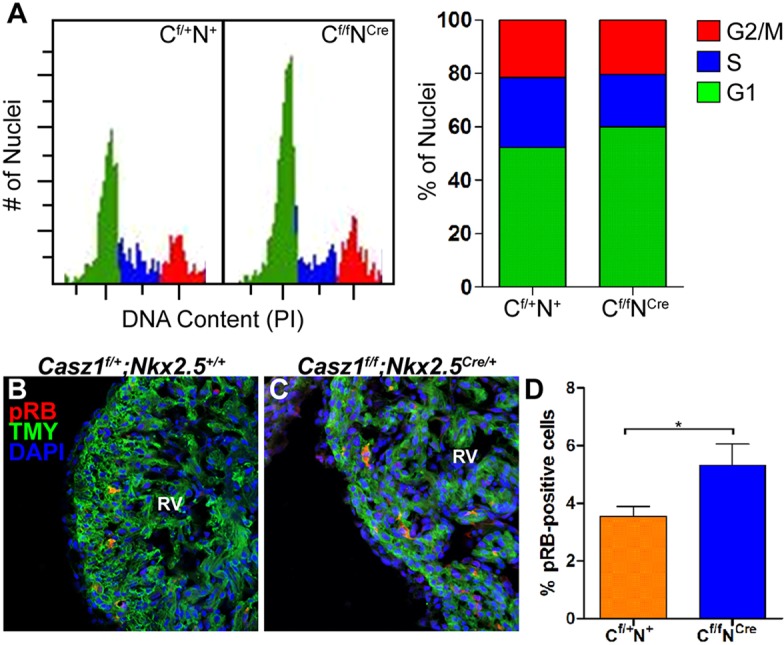
Casz1 is essential for cardiac G1-to-S cell cycle transition. (A) Cell cycle profiles of E12.5 propidium iodide (PI)-stained cardiac nuclei for Casz1f/+;Nkx2.5+/+ and Casz1f/f;Nkx2.5Cre/+ embryos and quantitation of cells with G1, S or G2/M DNA content, showing an increase in G1-phase cells and a decrease in S-phase cells in Casz1 null heart tissue. (B,C) Immunofluorescent staining for phospho-RB (pRB) and TMY demonstrates an increase in the number of pRB-positive cardiomyocytes in Casz1 cardiac null hearts versus controls at E12.5. (D) Quantification of pRB staining results. Data are the mean±s.e.m. of n=2 embryos; *P<0.05, WT versus Casz1f/f;Nkx2.5Cre/+ embryos. RV, right ventricle.
Taken together, our results show that a loss of Casz1 leads to a prolonged or arrested G1 phase, a decrease in DNA synthesis, an increase in phospho-RB, and a decrease in the cardiac mitotic index. Thus, these results support a role for Casz1 in the cardiomyocyte G1-to-S cell cycle transition.
DISCUSSION
Here we report that Casz1 is expressed in mammalian cardiomyocytes and by genetic fate mapping studies demonstrate that Casz1-positive cells give rise to cardiomyocytes in the first and second heart field. We show through our phenotypic analysis of cardiac conditional null Casz1 embryos that Casz1 is essential for the proliferation of cardiomyocytes in both heart fields and define a role of Casz1 in cardiomyocyte proliferation.
We note that these findings differ from those reported for a Casz1 gene trap line of mice that were generated from an ESC line carrying a βgeo cassette integrated into the ninth intron of Casz1 (Liu et al., 2014). Mice homozygous for the insertion exhibited embryonic arrest at E16.5, a time point in development much later than those reported here for mice in which Casz1 was depleted in both the primary and secondary heart fields (Casz1f/f;Nkx2.5Cre/+), in those depleted of Casz1 in the second heart field (Casz1f/f;Isl1Cre/+) or in a complete embryonic null (Casz1f/f;Sox2Cre/+). Given that the Casz1 conditional allele reported here lacks a nuclear localization signal and all zinc-finger domains, and since the trapped allele and our panel of Casz1 mutants are in the same genetic background, it is unclear why the trapped allele is less penetrant, although it remains formally possible that the allele disrupted by the βgeo insertion occurs in an alternatively spliced form of Casz1 and/or is hypomorphic.
CASZ1 and the G1-to-S phase transition
During early G1, cells respond to mitotic signals to pause or initiate DNA replication (Bertoli et al., 2013). Our analysis of the cardiomyocyte mitotic index coupled with our cell cycle profiling demonstrates a decrease in Casz1 cardiac null cells progressing through S phase. The failure to progress through S phase and the decrease in cardiomyocyte number in Casz1 null hearts are not associated with programmed cell death, and we show that at the period when we observe decreased cardiomyocyte numbers Casz1 null cardiomyocytes maintain their structural integrity and higher order structure (Fig. 6). Given that we observe a decrease in DNA replication in Casz1 null hearts, as shown by EdU, and a concomitant increase in phospho-RB, our data strongly imply that Casz1 acts at the G1-to-S phase transition.
We note in our cell cycle profiling that an appreciable number of cells pass through the G1-to-S phase. Since it is not possible to isolate a pure cardiomyocyte population from our Casz1 null embryos these cells most likely represent other cardiac cell populations, including cardiac fibroblasts and endothelial cells. However, it is formally possible that some of these cells represent cardiomyocytes that have not reached the G1-to-S transition, i.e. a period after which they require Casz1.
One of the major checkpoint controls of the cell cycle is the ‘commitment point’ or ‘restriction point’ that is associated with the G1-to-S phase transition. This point in the cycle, which is also known as ‘start’ in yeast, is the point beyond which a cell will pass through the cycle without external input (Bertoli et al., 2013). Based on our findings, we presently favor a model in which CASZ1 is necessary for cardiomyocytes to pass the restriction point and commit to the next round of division. We note this role of CASZ1 must be stage specific as we fail to see any phenotypic consequences of removing Casz1 during the initial stages of cardiac specification and determination (E6-E8). This observation would imply that this checkpoint control operates under a separate, as yet unidentified mechanism in the early stages of heart development.
In the future, it will be of importance to identify the Casz1-dependent pathway during these early stages of cardiac development and determine what role, if any, CASZ1 plays in adult homoeostasis and injury repair.
MATERIALS AND METHODS
Ethics statement
Research was approved by the Institutional Animal Care and Use Committee at the University of North Carolina and conforms to the Guide for the Care and Use of Laboratory Animals.
Generation of Casz1 mutant mice
BAC clones containing Casz1 genomic DNA from mouse strain 129 were isolated by established methods (Testa et al., 2003). Plasmid pBS-DTRX-Casz1Exon6CKO was constructed that contains the PGKneo expression cassette flanked by two FRT sites and containing a loxP site inserted into intron 5. loxP sites were introduced into the Casz1 locus flanking exon 6 (Liu et al., 2003). Deletion of exon 6 introduces a +2 frameshift into the translational reading frame leading to premature STOP codons, resulting in the elimination of CASZ1 protein expression. Furthermore, alternate splicing to the next three exons would result in proteins that are out of frame. The targeting vector contained 2.2 kb of homologous DNA upstream of the first loxP and 1.8 kb of homologous DNA (right arm) downstream of the PGKneo cassette (Liu et al., 2003). The targeting vector was linearized with NotI and electroporated into 1292/Ola ESCs. G418-resistant colonies were picked, expanded and screened for homologous recombination by Southern blot analysis of HindIII-digested ESC genomic DNA using 5′ and 3′ probes containing sequences outside of those in the targeting vector. Homologous recombinant ESCs were injected into C57BL6/J blastocysts and chimeric male mice were obtained. Male chimeras were mated to C57BL6/J females to establish a mouse line carrying the Casz1flox-neo/+ allele. The frt-flanked PGKneo cassette was removed by mating F1 heterozygous mice to mice expressing the FlpE recombinase (Jackson Labs stock #003800) (Rodriguez et al., 2000). The resulting Casz1flox/flox (Casz1f/f) allele was bred to homozygosity on a C57BL6 background. Casz1 conditional knockout mice and their control littermates were obtained by breeding female Casz1flox/flox mice to male Casz1flox/+ mice expressing Nkx2.5Cre/+. Nkx2.5Cre/+ mice were obtained from Robert Schwartz (Moses et al., 2001). Sox2Cre/+ mice were obtained from Larysa Pevny (Hayashi et al., 2002). Isl1Cre/+ mice were obtained from Li Qian (Srinivas et al., 2001).
In situ hybridization
In situ hybridization was carried out as previously described (Wilkinson, 1992). Specifically, sense and antisense probes were generated using a digoxigenin (DIG) RNA Labeling Kit (Roche). Probes were hybridized overnight at 65°C onto E8.5-E10.5 embryos. DIG-labeled probes were detected by anti-digoxigenin-AP Fab fragments (Roche) and precipitated by BM Purple AP substrate (Roche). Embryos and embryonic hearts were dissected free from surrounding tissues in PBS. Specimens were fixed overnight at 4°C in 4% paraformaldehyde (PFA) in PBS. In situ probes used were Nkx2.5 (gift from Benoit Bruneau; Stennard et al., 2003) and Casz1 (see supplementary material Table S1 for sequences). Tissues were photographed, embedded in 4% low-melt agarose (Promega) and sectioned using a Leica VT1200S vibratome to 30 µm thickness. The sections were mounted on slides and imaged on an Olympus BX61 microscope.
RLM-RACE
5′ RLM-RACE was performed according to the manufacturer's instructions (FirstChoice RLM-RACE Kit, Ambion #AM1700). Mouse adult heart RNA was isolated using standard Trizol (Ambion) extraction.
Histological sectioning and immunohistochemistry
Embryos were fixed in 4% PFA and either paraffin embedded or frozen in OCT (VWR). Paraffin sections (8 µm) were dewaxed, rehydrated and stained with Hematoxylin and Eosin (H&E) using standard methodology. Histology sections were imaged on an Olympus BX61 fluorescence microscope. Digital images were utilized for measurement using ImageJ (NIH) software. Paraffin sections and cryosections (10 µm) were washed in PBS containing 1% Triton X-100 (PBS-T), blocked in PBS-T containing 10% FBS, and incubated overnight at 4°C with the following primary antibodies: rabbit anti-CASZ1 (Santa Cruz, #SC-135453) 1:500; mouse anti-cardiac tropomyosin (TMY) (DSHB, clone CH1) 1:50; mouse anti-Islet1 (DSHB, clone 39.4D5) 1:50; rabbit anti-filamin A (Epitomics, #2242-1) 1:100; mouse anti-GFP (Clontech, #632381) 1:1000; rabbit anti-phospho-histone H3 (Millipore, #06-570) 1:200; mouse anti-cardiac troponin T (DSHB, clone RV-C2) 1:50; mouse anti-sarcomeric myosin (DSHB, clone MF20) 1:50; rabbit anti-caspase 3 (Cell Signaling, #9661) 1:100; rabbit anti-phospho-RB (Cell Signaling, #9308) 1:100; mouse anti-PML (Santa Cruz, #SC-966) 1:500; rabbit anti-cleaved caspase 3 (Asp175; Cell Signaling, #9661) 1:50; or with Alexa Fluor 488-phalloidin (Molecular Probes, #A12379) 1:100. CASZ1 antibody staining required antigen retrieval. Briefly, sections were steamed in 10 mM sodium citrate pH 6.0/0.05% Tween 20 for 20 min, washed in 1× PBS and subjected to processing as detailed above. The following day, sections were washed in PBS-T then incubated for 1 h at room temperature with secondary antibodies: Alexa 488 goat anti-mouse IgG, H+L (Molecular Probes, #A11001) 1:1000; Alexa 488 donkey anti-rabbit IgG (Molecular Probes, #A21206) 1:1000; Alexa 546 goat anti-rabbit IgG (Molecular Probes, #A11010) 1:1000; Alexa 546 goat anti-mouse IgG (Molecular Probes, #A11030) 1:1000. Sections were washed in PBS-T, incubated with 200 ng/ml DAPI (Sigma) and mounted with Permafluor (Thermo Scientific). Stained sections were imaged on Zeiss 700 confocal microscope and ImageJ was used for analysis.
Cell culture
HUVECS (Lonza) were maintained and immunostained as previously described (Charpentier et al., 2013b).
Reporter crosses
Tie2-Cre (Kisanuki et al., 2001) mice were crossed to R26ReGFP reporter mice (Muzumdar et al., 2007). Wt1/IRES/GFP-Cre mice were previously reported (Wessels et al., 2012). Time-mated females were harvested at E14.5 and processed for immunostaining with anti-GFP as detailed above. Nkx2.5-Cre mice (Moses et al., 2001) were crossed to R26RtdT reporter mice (Madisen et al., 2010). Time-mated females were harvested at E7.5, E9.5, E12.5 and E14.5. Sox2-Cre mice (Hayashi et al., 2002) were crossed to R26RlacZ reporter mice (Soriano, 1999) and time-mated females were harvested at E10.5. All embryos were imaged with a Leica MZ 16F dissection microscope.
β-galactosidase staining
Embryos were fixed in X-Gal fixative (0.2% glutaraldehyde, 2 mM MgCl2, 5 mM EGTA, 0.1 M phosphate buffer pH 7.3) for 40 min on ice, washed in detergent (0.1 M phosphate buffer, 2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40, pH 7.3), and incubated in X-Gal staining solution (1 mg/ml X-Gal in dimethylformamide, 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 0.1 M phosphate buffer, 2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40, 20 mM Tris, pH 7.4) at 37°C overnight.
Tamoxifen injections
Tamoxifen (Sigma #T5648) was dissolved in 1:9 ethanol (200 proof):sunflower oil to make a 0.03 mg/µl solution. Time-mated females were subject to intraperitoneal (IP) injection with 3 mg of tamoxifen at E8.5 and embryos were harvested at E12.5. No reporter activity was observed in the absence of tamoxifen. Embryos were fixed in 4% PFA, incubated in Scale U2 solution (Hama et al., 2011) at 4°C for 3 weeks, imaged with a Leica MZ 16F dissection microscope, washed in 1× PBS, immersed in 30% sucrose and frozen in OCT. Cryosections were processed and immunostained as detailed above.
Imaging and statistical analysis
A Leica MZ 16F dissection microscope with a Retiga 4000RV camera was used for whole-mount imaging and an Olympus BX61 with a Retiga 4000R camera was used for color imaging. Higher magnification images were captured using a Zeiss 700 laser scanning confocal microscope. All images and figures were edited and created in either ImageJ or Photoshop CS4 (Adobe). All statistical calculations were performed using Prism 5 (GraphPad). P-values for statistical significance were obtained using a Student's t-test for single variable between control and test samples.
Cardiomyocyte counting assay
Embryos from time-mated females were recovered at E10.5 and E12.5, fixed in 4% PFA and embedded for cryosection. Sections were stained for TMY and with DAPI. For statistical analysis, nine transverse sections were analyzed comprising three sections (representing the anterior, mid and posterior heart) from three different embryos per genotype. The number of TMY-positive and DAPI-stained cardiomyocytes were counted from six 40× fields per section. Counts of total cells positive for TMY and DAPI were averaged from 54 sections. Data are shown as mean±s.e.m. ImageJ was used for analysis and cell counts. Data were compared for statistical significance by Student's t-test.
Proliferation assays
For 5-ethynyl-2′deoxyuridine (EdU) analysis, time-mated females were subject to IP injection with 250 µg EdU (Click-iT EdU Imaging Kit, Invitrogen C10338) 3 h prior to sacrifice. Embryos were recovered at E12.5, fixed with PFA and embedded for cryosectioning (n=3/3). EdU staining was performed according to the manufacturer's instructions. For statistical analysis, nine transverse sections were analyzed comprising three sections (representing the anterior, mid and posterior heart) from three different wild-type embryos and two Casz1f/f;Nkx2.5Cre/+ embryos. The number of EdU-positive cardiomyocytes among total cardiomyocytes (TMY-positive cells) was counted from four 40× fields per section (two fields per ventricle).
For mitotic index analysis, counts of total cells positive for TMY and total cells positive for pHH3 were performed on transverse sections using the same statistical analysis of wild-type and Casz1 mutant hearts at E12.5 (n=3/3) from four 63× fields per section (two fields per ventricle). Mitotic index was calculated by dividing total cells positive for TMY and pHH3 by total cells positive for TMY. Sections were processed for immunostaining as detailed above. Data are shown as mean±s.e.m. ImageJ was used for analysis and cell counts. Data were compared for statistical significance using a Student's t-test.
Scanning electron microscopy (SEM)
A standardized procedure for SEM (Hullinger et al., 2012) of the heart was utilized (Pexieder, 1986). Briefly, the pericardial cavity membrane was excised before the embryos were fixed in 2.5% glutaraldehyde (EM grade, Electron Microscopy Sciences), as previously reported (Tandon et al., 2013), in 1× PBS at 4°C overnight. Embryos were washed in 1× PBS, dehydrated into 100% ethanol and subject to critical point drying. Dried specimens were mounted ventral side up and ion sputtered with gold palladium to ∼40 nm thickness before being scanned with a Zeiss Supra 25 FESEM microscope. SEM photomicrographs were taken in standard orientations and magnifications.
RNA-seq
E10.5 hearts were collected from three Casz1f/f;Nkx2.5Cre/+ and three Casz1f/+;Nkx2.5+/+ embryos. RNA was isolated using standard Trizol extraction. RNA-seq libraries were generated with TruSeq adaptor barcodes from these six samples using standard library preparation protocols (Illumina) by the Vanderbilt Genomic Core. Samples were run over three lanes of a HiSeq2500 (Illumina) to generate on average 103.7 million 50 bp single-end reads per sample. Samples were assessed for quality using Fastqc 0.10.1 and then mapped to the mouse genome (build mm10) using TopHat version 2.0.8 with default parameters (Trapnell et al., 2009, 2012). Aligned reads were then tallied per annotated gene (RefSeq genes downloaded from www.genome.ucsc.edu July 24, 2013) and then normalized using RPKM values and analyzed for differential expression via Cufflinks version 2.1.1 (Trapnell et al., 2012). Raw data and a full analysis are available at GEO under accession number GSE55394.
For Gene Ontology (GO) analysis, genes tested for differential expression with P<0.05 were considered differentially expressed. These genes were split into two classes: upregulated or downregulated in Casz1 mutant hearts. These genes were assessed for GO biological process enrichment using Gorilla (Eden et al., 2009), with the annotated gene list provided as a background. GO terms enriched in either class with a false discovery rate q-value of less than 0.05 were consolidated based on similar processes to generate a more manageable set of GO terms for visualization using REVIGO (Supek et al., 2011).
Embryonic cardiac nuclei isolation and flow cytometry
Embryos from time-mated females were recovered at E12.5. Hearts were dissected in PBS and atria were removed. Ventricles were placed in liquid nitrogen and stored at −80°C. Similar genotypes were pooled (n=4 wild type, n=4 cardiac nulls) and homogenized in liquid nitrogen following a modified cardiac nuclei extraction protocol (Franklin et al., 2011). Briefly, ventricle powder was resuspended in homogenization buffer [10 mM Tris pH 7.4, 250 mM sucrose, 1 mM EDTA, 0.1% NP-40, 10 mM sodium butyrate, 0.1 mM PMSF, 1× protease inhibitors (Roche)] and subjected to ten strokes with a Dounce homogenizer on ice. Nuclei were passed through a cell strainer, centrifuged at 1000 g for 10 min at 4°C and pellets resuspended in PBS, and then fixed with 70% ethanol for 30 min on ice. Nuclei were washed and resuspended in FACS buffer (PBS, 7.5% BSA, 2 mM EDTA, pH 8.0). Nuclei were treated with 100 µg/ml RNase A and stained with 50 µg/ml propidium iodide (Sigma #P4170) and analyzed on an Accuri flow cytometer using CFlow Plus analysis software (BD Biosciences). DNA content was determined using at least 100,000 cardiac nuclei.
Supplementary Material
Acknowledgements
We are extremely grateful to the Microscopy Services Lab at UNC for microscopy assistance; UNC Animal Models Core for mouse generation; Ross Carroll for H&E staining of histological sections; Benoit Bruneau, Josh Wythe and Patrick Devine for tamoxifen protocols and advice; and Panna Tandon, Christopher Slagle, Lauren Waldron and Michelle Villasmil for critical reading of the manuscript. The CH1, 39.4D5, RV-C2, MF20 monoclonal antibodies developed by Dr Jim Lin, Dr Tom Jessell, Dr Steffano Schiaffino and Dr Donald Fischman, respectively, were obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242, USA.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Funding
This work was supported by grants to F.L.C. from National Institutes of Health (NIH)/NHLBI [RO1 DE018825 and RO1 HL089641]. N.M.A. was supported by an American Heart Association (AHA) award. Deposited in PMC for release after 12 months.
Author contributions
As primary author, K.M.D. performed the majority of experiments and manuscript preparation. N.M.A., L.M.K., H.L. and M.S.C. contributed to experiments and manuscript preparation and editing. A.W. contributed the Wt1-Cre and Tie2-Cre slides and to manuscript preparation and editing. L.H.P. contributed reagents and to experimental design. F.L.C. contributed reagents and to experiments, manuscript editing and preparation.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.119107/-/DC1
References
- Ahuja P., Sdek P. and MacLellan W. R. (2007). Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol. Rev. 87, 521-544. 10.1152/physrev.00032.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin N. M., Gibbs D. and Conlon F. L. (2014). Differential regulation of CASZ1 protein expression during cardiac and skeletal muscle development. Dev. Dyn. 243, 948-956. 10.1002/dvdy.24126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold S. J., Sugnaseelan J., Groszer M., Srinivas S. and Robertson E. J. (2009). Generation and analysis of a mouse line harboring GFP in the Eomes/Tbr2 locus. Genesis 47, 775-781. 10.1002/dvg.20562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrow J. R., Howell W. D., Rule M., Hayashi S., Thomas K. R., Capecchi M. R. and McMahon A. P. (2007). Wnt3 signaling in the epiblast is required for proper orientation of the anteroposterior axis. Dev. Biol. 312, 312-320. 10.1016/j.ydbio.2007.09.030 [DOI] [PubMed] [Google Scholar]
- Bergmann O., Bhardwaj R. D., Bernard S., Zdunek S., Barnabe-Heider F., Walsh S., Zupicich J., Alkass K., Buchholz B. A., Druid H. et al. (2009). Evidence for cardiomyocyte renewal in humans. Science 324, 98-102. 10.1126/science.1164680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersell K., Arab S., Haring B. and Kühn B. (2009). Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138, 257-270. 10.1016/j.cell.2009.04.060 [DOI] [PubMed] [Google Scholar]
- Bertoli C., Skotheim J. M. and de Bruin R. A. (2013). Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 14, 518-528. 10.1038/nrm3629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C.-L., Liang X., Shi Y., Chu P.-H., Pfaff S. L., Chen J. and Evans S. (2003). Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 5, 877-889. 10.1016/S1534-5807(03)00363-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier M. S., Dorr K. M. and Conlon F. L. (2013a). Transcriptional regulation of blood vessel formation: the role of the CASZ1/Egfl7/RhoA pathway. Cell Cycle 12, 2165-2166. 10.4161/cc.25539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier M. S., Christine K. S., Amin N. M., Dorr K. M., Kushner E. J., Bautch V. L., Taylor J. M. and Conlon F. L. (2013b). CASZ1 promotes vascular assembly and morphogenesis through the direct regulation of an EGFL7/RhoA-mediated pathway. Dev. Cell 25, 132-143. 10.1016/j.devcel.2013.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christine K. S. and Conlon F. L. (2008). Vertebrate CASTOR is required for differentiation of cardiac precursor cells at the ventral midline. Dev. Cell 14, 616-623. 10.1016/j.devcel.2008.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffels V. M., Habets P. E. M. H., Franco D., Campione M., de Jong F., Lamers W. H., Bao Z.-Z., Palmer S., Biben C., Harvey R. P. et al. (2000). Chamber formation and morphogenesis in the developing mammalian heart. Dev. Biol. 223, 266-278. 10.1006/dbio.2000.9753 [DOI] [PubMed] [Google Scholar]
- Conway S. J., Kruzynska-Frejtag A., Kneer P. L., Machnicki M. and Koushik S. V. (2003). What cardiovascular defect does my prenatal mouse mutant have, and why? Genesis 35, 1-21. 10.1002/gene.10152 [DOI] [PubMed] [Google Scholar]
- Creazzo T. L., Godt R. E., Leatherbury L., Conway S. J. and Kirby M. L. (1998). Role of cardiac neural crest cells in cardiovascular development. Annu. Rev. Physiol. 60, 267-286. 10.1146/annurev.physiol.60.1.267 [DOI] [PubMed] [Google Scholar]
- Delgado-Esteban M., García-Higuera I., Maestre C., Moreno S. and Almeida A. (2013). APC/C-Cdh1 coordinates neurogenesis and cortical size during development. Nat. Commun. 4, 2879 10.1038/ncomms3879 [DOI] [PubMed] [Google Scholar]
- Deng M. C., Dasch B., Erren M., Möllhoff T. and Scheld H. H. (1996). Impact of left ventricular dysfunction on cytokines, hemodynamics, and outcome in bypass grafting. Ann. Thorac. Surg. 62, 184-190. 10.1016/0003-4975(96)00230-5 [DOI] [PubMed] [Google Scholar]
- Eden E., Navon R., Steinfeld I., Lipson D. and Yakhini Z. (2009). GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48 10.1186/1471-2105-10-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulalio A., Mano M., Dal Ferro M., Zentilin L., Sinagra G., Zacchigna S. and Giacca M. (2012). Functional screening identifies miRNAs inducing cardiac regeneration. Nature 492, 376-381. 10.1038/nature11739 [DOI] [PubMed] [Google Scholar]
- Franklin S., Zhang M. J., Chen H., Paulsson A. K., Mitchell-Jordan S. A., Li Y., Ping P. and Vondriska T. M. (2011). Specialized compartments of cardiac nuclei exhibit distinct proteomic anatomy. Mol. Cell. Proteomics 10, M110.000703 10.1074/mcp.M110.000703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hama H., Kurokawa H., Kawano H., Ando R., Shimogori T., Noda H., Fukami K., Sakaue-Sawano A. and Miyawaki A. (2011). Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat. Neurosci. 14, 1481-1488. 10.1038/nn.2928 [DOI] [PubMed] [Google Scholar]
- Hayashi S., Lewis P., Pevny L. and McMahon A. P. (2002). Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech. Dev. 119 Suppl. 1, S97-S101. 10.1016/S0925-4773(03)00099-6 [DOI] [PubMed] [Google Scholar]
- Heallen T., Zhang M., Wang J., Bonilla-Claudio M., Klysik E., Johnson R. L. and Martin J. F. (2011). Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332, 458-461. 10.1126/science.1199010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hullinger T. G., Montgomery R. L., Seto A. G., Dickinson B. A., Semus H. M., Lynch J. M., Dalby C. M., Robinson K., Stack C., Latimer P. A. et al. (2012). Inhibition of miR-15 protects against cardiac ischemic injury. Circ. Res. 110, 71-81. 10.1161/CIRCRESAHA.111.244442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikenishi A., Okayama H., Iwamoto N., Yoshitome S., Tane S., Nakamura K., Obayashi T., Hayashi T. and Takeuchi T. (2012). Cell cycle regulation in mouse heart during embryonic and postnatal stages. Dev. Growth Differ. 54, 731-738. 10.1111/j.1440-169X.2012.01373.x [DOI] [PubMed] [Google Scholar]
- Kelly R. G. (2012). The second heart field. Curr. Top. Dev. Biol. 100, 33-65. 10.1016/B978-0-12-387786-4.00002-6 [DOI] [PubMed] [Google Scholar]
- Kisanuki Y. Y., Hammer R. E., Miyazaki J.-i., Williams S. C., Richardson J. A. and Yanagisawa M. (2001). Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 230, 230-242. 10.1006/dbio.2000.0106 [DOI] [PubMed] [Google Scholar]
- Konstantinides N., Rossi A. M. and Desplan C. (2015). Common temporal identity factors regulate neuronal diversity in fly ventral nerve cord and mouse retina. Neuron 85, 447-449. 10.1016/j.neuron.2015.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavine K. J., Yu K., White A. C., Zhang X., Smith C., Partanen J. and Ornitz D. M. (2005). Endocardial and epicardial derived FGF signals regulate myocardial proliferation and differentiation in vivo. Dev. Cell 8, 85-95. 10.1016/j.devcel.2004.12.002 [DOI] [PubMed] [Google Scholar]
- Levy D., Ehret G. B., Rice K., Verwoert G. C., Launer L. J., Dehghan A., Glazer N. L., Morrison A. C., Johnson A. D., Aspelund T. et al. (2009). Genome-wide association study of blood pressure and hypertension. Nat. Genet. 41, 677-687. 10.1038/ng.384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Wang X., Capasso J. M. and Gerdes A. M. (1996). Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J. Mol. Cell. Cardiol. 28, 1737-1746. 10.1006/jmcc.1996.0163 [DOI] [PubMed] [Google Scholar]
- Liu P., Jenkins N. A. and Copeland N. G. (2003). A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 13, 476-484. 10.1101/gr.749203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Yang X., Tan F., Cullion K. and Thiele C. J. (2006). Molecular cloning and characterization of human Castor, a novel human gene upregulated during cell differentiation. Biochem. Biophys. Res. Commun. 344, 834-844. 10.1016/j.bbrc.2006.03.207 [DOI] [PubMed] [Google Scholar]
- Liu Z., Li W., Ma X., Ding N., Spallotta F., Southon E., Tessarollo L., Gaetano C., Mukoyama Y.-S. and Thiele C. J. (2014). Essential role of the zinc finger transcription factor Casz1 for mammalian cardiac morphogenesis and development. J. Biol. Chem. 289, 29801-29816. 10.1074/jbc.M114.570416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X., Wang L., Lin X., Huang J., Charles Gu C., He M., Shen H., He J., Zhu J., Li H. et al. (2015). Genome-wide association study in Chinese identifies novel loci for blood pressure and hypertension. Hum. Mol. Genet. 24, 865-874. 10.1093/hmg/ddu478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L., Zwingman T. A., Sunkin S. M., Oh S. W., Zariwala H. A., Gu H., Ng L. L., Palmiter R. D., Hawrylycz M. J., Jones A. R. et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133-140. 10.1038/nn.2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillet M., van Berlo J. H. and Molkentin J. D. (2013). Molecular basis of physiological heart growth: fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 14, 38-48. 10.1038/nrm3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manner J., Perez-Pomares J. M., Macias D. and Munoz-Chapuli R. (2001). The origin, formation and developmental significance of the epicardium: a review. Cells Tissues Organs 169, 89-103. 10.1159/000047867 [DOI] [PubMed] [Google Scholar]
- Matera A. G., Izaguire-Sierra M., Praveen K. and Rajendra T. K. (2009). Nuclear bodies: random aggregates of sticky proteins or crucibles of macromolecular assembly? Dev. Cell 17, 639-647. 10.1016/j.devcel.2009.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattar P., Ericson J., Blackshaw S. and Cayouette M. (2015). A conserved regulatory logic controls temporal identity in mouse neural progenitors. Neuron 85, 497-504. 10.1016/j.neuron.2014.12.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses K. A., DeMayo F., Braun R. M., Reecy J. L. and Schwartz R. J. (2001). Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis 31, 176-180. 10.1002/gene.10022 [DOI] [PubMed] [Google Scholar]
- Mulligan G. and Jacks T. (1998). The retinoblastoma gene family: cousins with overlapping interests. Trends Genet. 14, 223-229. 10.1016/S0168-9525(98)01470-X [DOI] [PubMed] [Google Scholar]
- Muzumdar M. D., Tasic B., Miyamichi K., Li L. and Luo L. (2007). A global double-fluorescent Cre reporter mouse. Genesis 45, 593-605. 10.1002/dvg.20335 [DOI] [PubMed] [Google Scholar]
- Pasumarthi K. B. S. and Field L. J. (2002). Cardiomyocyte cell cycle regulation. Circ. Res. 90, 1044-1054. 10.1161/01.RES.0000020201.44772.67 [DOI] [PubMed] [Google Scholar]
- Porrello E. R., Johnson B. A., Aurora A. B., Simpson E., Nam Y.-J., Matkovich S. J., Dorn G. W. II, van Rooij E. and Olson E. N. (2011). MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ. Res. 109, 670-679. 10.1161/CIRCRESAHA.111.248880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez C. I., Buchholz F., Galloway J., Sequerra R., Kasper J., Ayala R., Stewart A. F. and Dymecki S. M. (2000). High-efficiency deleter mice show that FLPe is an alternative to Cre-loxP. Nat. Genet. 25, 139-140. 10.1038/75973 [DOI] [PubMed] [Google Scholar]
- Sedmera D., Reckova M., DeAlmeida A., Coppen S. R., Kubalak S. W., Gourdie R. G. and Thompson R. P. (2003). Spatiotemporal pattern of commitment to slowed proliferation in the embryonic mouse heart indicates progressive differentiation of the cardiac conduction system. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 274A, 773-777. 10.1002/ar.a.10085 [DOI] [PubMed] [Google Scholar]
- Sojka S., Amin N. M., Gibbs D., Christine K. S., Charpentier M. S. and Conlon F. L. (2014). Congenital heart disease protein 5 associates with CASZ1 to maintain myocardial tissue integrity. Development 141, 3040-3049. 10.1242/dev.106518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soonpaa M. H., Kim K. K., Pajak L., Franklin M. and Field L. J. (1996). Cardiomyocyte DNA synthesis and binucleation during murine development. Am. J. Physiol. 271, H2183-H2189. [DOI] [PubMed] [Google Scholar]
- Soriano P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70-71. 10.1038/5007 [DOI] [PubMed] [Google Scholar]
- Srinivas S., Watanabe T., Lin C.-S., William C. M., Tanabe Y., Jessell T. M. and Costantini F. (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4 10.1186/1471-213X-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stennard F. A., Costa M. W., Elliott D. A., Rankin S., Haast S. J., Lai D., McDonald L. P., Niederreither K., Dolle P., Bruneau B. G. et al. (2003). Cardiac T-box factor Tbx20 directly interacts with Nkx2-5, GATA4, and GATA5 in regulation of gene expression in the developing heart. Dev. Biol. 262, 206-224. 10.1016/S0012-1606(03)00385-3 [DOI] [PubMed] [Google Scholar]
- Sun Y., Liang X., Najafi N., Cass M., Lin L., Cai C.-L., Chen J. and Evans S. M. (2007). Islet 1 is expressed in distinct cardiovascular lineages, including pacemaker and coronary vascular cells. Dev. Biol. 304, 286-296. 10.1016/j.ydbio.2006.12.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F., Bošnjak M., Škunca N. and Šmuc T. (2011). REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6, e21800 10.1371/journal.pone.0021800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi F., Isono M., Katsuya T., Yamamoto K., Yokota M., Sugiyama T., Nabika T., Fujioka A., Ohnaka K., Asano H. et al. (2010). Blood pressure and hypertension are associated with 7 loci in the Japanese population. Circulation 121, 2302-2309. 10.1161/CIRCULATIONAHA.109.904664 [DOI] [PubMed] [Google Scholar]
- Testa G., Zhang Y., Vintersten K., Benes V., Pijnappel W. W. M. P., Chambers I., Smith A. J. H., Smith A. G. and Stewart A. F. (2003). Engineering the mouse genome with bacterial artificial chromosomes to create multipurpose alleles. Nat. Biotechnol. 21, 443-447. 10.1038/nbt804 [DOI] [PubMed] [Google Scholar]
- Trapnell C., Pachter L. and Salzberg S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105-1111. 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D. R., Pimentel H., Salzberg S. L., Rinn J. L. and Pachter L. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562-578. 10.1038/nprot.2012.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacalla C. M. H. and Theil T. (2002). Cst, a novel mouse gene related to Drosophila Castor, exhibits dynamic expression patterns during neurogenesis and heart development. Mech. Dev. 118, 265-268. 10.1016/S0925-4773(02)00282-4 [DOI] [PubMed] [Google Scholar]
- von Gise A., Lin Z., Schlegelmilch K., Honor L. B., Pan G. M., Buck J. N., Ma Q., Ishiwata T., Zhou B., Camargo F. D. et al. (2012). YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc. Natl. Acad. Sci. USA 109, 2394-2399. 10.1073/pnas.1116136109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadugu B. and Kuhn B. (2012). The role of neuregulin/ErbB2/ErbB4 signaling in the heart with special focus on effects on cardiomyocyte proliferation. Am. J. Physiol. Heart Circ. Physiol. 302, H2139-H2147. 10.1152/ajpheart.00063.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessels A., van den Hoff M. J. B., Adamo R. F., Phelps A. L., Lockhart M. M., Sauls K., Briggs L. E., Norris R. A., van Wijk B., Perez-Pomares J. M. et al. (2012). Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 366, 111-124. 10.1016/j.ydbio.2012.04.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M., Kim Y., Sutherland L. B., Qi X., McAnally J., Schwartz R. J., Richardson J. A., Bassel-Duby R. and Olson E. N. (2011). Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci. Signal. 4, ra70 10.1126/scisignal.2002278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin M., Olson E. N. and Bassel-Duby R. (2013). Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat. Rev. Mol. Cell Biol. 14, 529-541. 10.1038/nrm3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.