

Abstract
Ultraviolet photodissociation at 193 nm (UVPD) and negative electron transfer dissociation (NETD) were compared to establish their utility for characterizing acidic proteomes with respect to sequence coverage distributions (a measure of product ion signals across the peptide backbone), sequence coverage percentages, backbone cleavage preferences, and fragmentation differences relative to precursor charge state. UVPD yielded significantly more diagnostic information compared to NETD for lower charge states (n ≤ 2), but both methods were comparable for higher charged species. While UVPD often generated a more heterogeneous array of sequence-specific products (b-, y-, c-, z-, Y-, d-, and w-type ions in addition to a- and x- type ions), NETD usually created simpler sets of a/x-type ions. LC-MS/UVPD and LC-MS/NETD analysis of protein digests utilizing high pH mobile phases coupled with automated database searching via modified versions of the MassMatrix algorithm was undertaken. UVPD generally outperformed NETD in stand-alone searches due to its ability to efficiently sequence both lower and higher charge states with rapid activation times. However, when combined with traditional positive mode CID, both methods yielded complementary information with significantly increased sequence coverage percentages and unique peptide identifications over that of just CID alone.
Keywords: tandem mass spectrometry, protein identification, negative electron transfer dissociation, ultraviolet photodissociation
Graphical Abstract
A comprehensive comparison of NETD and UVPD MS/MS methods is undertaken for acidic peptides.
INTRODUCTION
Standard proteomics experiments typically involve a “bottom-up” strategy consisting of protein digestion, liquid chromatographic separation using acidic eluents, ionization in the positive mode, tandem mass spectrometry (MS) via collision induced dissociation (CID),1,2 and automated database searching.3,4 This general protocol consistently works well for the analysis of peptides cations.5 In addition, alternative dissociation methods, such as electron capture/transfer dissociation6–11 as well as infrared photodissociation (IRMPD)12–15 and ultraviolet photodissociation (UVPD16–27 have been developed to supplement CID. While CID remains the most popular MS/MS method for peptide cations, it often falls short for the characterization of more acidic species, such as peptides containing several acidic amino acids and/or acidic post translational modifications (PTM), due to low ion signals and poor MS/MS patterns. Acidic peptides ionize readily in the negative mode via deprotonation;28,29 however, CID of peptide anions generally yields low sequence coverage, high abundances of uninformative neutral losses, and overall unpredictable fragmentation patterns.30–32 It is now well recognized that many important modifications such as phosphorylation, sulfonation, acetylation, and glycosylation render proteins more acidic after translation.33 Moreover, all proteomes exhibit a bimodal distributions of protein isoelectric points (pI).34–37 In other words, every proteome contains acidic and basic subsets of proteins with very few proteins possessing a pI close to cellular pH.34–37 As a result of these two factors, there is a need for improved methods for the characterization of the acidic regions of proteins as well as the acidic subset of proteins, including those approaches specifically adapted for analysis of peptide anions.
Several MS/MS and MSn methods have recently been developed for analysis of peptide anions to combat the downfalls of conventional CID.38–44 The majority of these techniques have utilized electrons or photons to promote primarily Cα-C(O) bond cleavages, resulting in a- and x-type sequence ions. For example, the interaction of peptides with high-energy electrons, termed electron detachment dissociation (EDD), has been shown to be useful for sequencing peptide anions.38,39 Activated-electron photodetachment dissociation (a-EPD), an MS anion activation method, uses UV irradiation at 262 nm in the first step to produce charge-reduced peptide radicals which are subsequently dissociated by collisional activation in the second step.40,45 EDD and a-EPD were compared recently for characterization of peptide anions, and while both techniques yielded mostly the same types of ions (a/x-type), the abundances were significantly different.40 Cleavages next to aromatic and histidine residues were favored for a-EPD, whereas cleavages next to negative charge solvation sites on the backbone were preferential for EDD.40 Recently, a method involving high kinetic energy metastable ions (MAD-MS) was used to dissociate phosphorylated and sulfonated peptide anions; Cα-C(O) bond cleavages were again the main dissociation channels.41 The development of EDD, a-EPD, and MAD-MS all represent promising advances for peptide anion characterization, but these methods generally afford low dissociation efficiencies and thus require long averaging times to generate interpretable spectra.40,41 Also, implementing these methods on liquid chromatographic timescales is problematic because the required activation times often range from half a second up to multiple seconds for peptide dissociation.39,45 Two alternative anion dissociation techniques that require less averaging and lower activation times to produce reasonable spectra, therefore allowing more facile integration with LC-MS/MS workflows, are negative electron transfer dissociation (NETD)42,43 and 193 nm ultraviolet photodissociation (UVPD).44 The former involves the reaction of peptide anions with electron-deficient radical cations, causing electron transfer from the peptide to the reagent that results in backbone dissociation to create mainly a and x ions.42,43 This method has shown great potential in sequencing peptides in high-throughput experiments.46 193 nm UVPD delivers high-energy (6.4 eV) photons to peptides and has been shown to yield diagnostic spectra dominated by a and x ions and utilizing activation times as little as 5 ns (in combination with LC-MS/MS).44 UVPD has more recently been used in conjunction with automated database searching and sequencing of peptide anions for the analysis of complex protein samples.47 Since both NETD and UVPD have been promising for the sensitive and high-throughput analysis of acidic proteomes, we have undertaken a comparison of NETD and UVPD for the characterization of a series of modified (e.g., phosphorylated) and unmodified peptide anions. The sequence coverage distribution (a measure of product ion signals across the peptide backbone), sequence coverage percentages, backbone cleavage preferences, and fragmentation differences with respect to precursor charge state were assessed between the methods. Results from the analysis of a seven protein mixture via automated MassMatrix48–50 interpretation and LC-MS/MS are also presented.
EXPERIMENTAL
Materials and Sample Preparation
All peptides were purchased from AnaSpec (Fremont, CA), and all proteins were purchased from Sigma Aldrich (St. Louis, MO) except for chicken lysozyme C which was purchased from MP Biomedicals, LLC (Solon, OH). All peptides and proteins were used without further purification. HPLC grade methanol and water (Fisher Scientific) were used for preparing peptide working solutions. HPLC grade ammonium hydroxide and argon of ≥99.998% purity were purchased from Sigma Aldrich (St. Louis, MO). All optical components were purchased from Edmund Optics Inc. (Barrington, NJ).
An equimolar mixture of bovine α-casein, β-casein, β-lactoglobulin A, BSA, ribonuclease A and chicken lysozyme C was made in 100 mM sodium bicarbonate. 200 μg of total protein was reduced with dithiothreitol (DTT) for 30 minutes then incubated with iodoacetamide at 40°C for one hour in the dark. Excess iodoacetamide was quenched with additional DTT. 4 μL of 1 μg/μL trypsin in 1 mM HCl was added, giving a protein to enzyme ratio of 50:1. The digestion solution was incubated overnight at 37°C. The digest was desalted using C18 cartridges then dried and reconstituted in water to a concentration of 1 μg/μL.
Instrumentation
A Bruker Daltonics (Billerica, MA) HCT Ultra ETD II ion trap mass spectrometer was modified to allow the introduction of 193 nm photons into the trapping volume. Briefly, two 25 mm CaF2 windows were mounted in the vacuum manifold lid above the ion trap. The laser beam was passed through 2 mm holes in the ring electrode (oriented vertically) of the quadrupole ion trap. Broadband deep UV enhanced 12.5 mm mirrors were placed in 45 degree mounts under and next to the ion trap to reflect the laser beam back out of the mass spectrometer. This configuration allowed facile alignment of the laser through the system by monitoring the intensity and profile of the beam exiting the system. Ultraviolet photons (193 nm, 5 ns pulse, 500 Hz, 8 mJ/pulse max) were generated by an ExciStar XS 500 laser (Coherent Inc., Santa Clara, CA) and used without focusing or collimating the beam. An external trigger supplied through the auxiliary interface port gated a pulse/delay generator (Model 505, Berkely Nucleonics Corporation, San Rafael, CA) which triggered the laser during the fragmentation period of the ion trap scan function (two pulses total). An override in the instrument user interface and replacement of the methane carrier gas with argon allowed efficient production of fluoranthene radical cations by argon chemical ionization and transmission of the radical cations to the ion trap. A reagent crucible temperature of 60 °C, a filament current of 1 μA, and electron energy of 70 eV were used.
Mass Spectrometry and Liquid Chromatography
10 μM peptide working solutions composed of 70/30 (v/v) methanol/water with 0.1% ammonium hydroxide were infused at 1.5 μL/min for electrospray ionization. NETD experiments were performed with a low mass cutoff of m/z 160, 10 to 15 ms of reagent accumulation, and 150 to 250 ms reaction time. UPVD experiments were performed with 4 ms activation periods allowing two 4 mJ laser pulses and a qz-value of 0.110 which allowed detection of low m/z product ions.
LC-MS/MS experiments were performed with a DionexUltiMate 3000 system (Sunnyvale, CA) using a capillary flowsplitter. An Agilent ZORBAX 300 Extend-C18 column (Santa Clara, CA) (150 × 0.3 mm, 3.5 μm particle size) was used for all separations. The column temperature was held at 25°C. For negative ion mode experiments (NETD and UVPD), eluents A and B consisted of 0.05% ammonium hydroxide in water and acetonitrile, respectively. A 120 minute gradient consisting of the first five minutes at 3% eluent B and a linear increase to 35% eluent B over 120 minutes at a flow rate of 4 uL/min was used to separate the protein mixture digest. CID experiments were performed in positive mode and utilized 0.1% formic acid in water and acetonitrile for eluents A and B, respectively. The same gradient were used for positive mode except the gradient ended at 40% eluent B. 5 μL of 1 μg/μL protein mixture digest was injected on column. Data-dependent acquisition consisted of a survey scan of m/z 400 – 2000 followed by five MS/MS scan events for all activation methods. For LC-MS/NETD, the precursor charge state preference was set to exclude singly charged ions. No precursor charge state preference was specified for LC-MS/UVPD. A preference for doubly charged ions was utilized for LC-MS/CID experiment. For all LC-MS/MS experiments, eight averages were used for all MS/MS events and dynamic exclusion was set to one repeat and exclude for 60 seconds.
MassMatrix was used for all database searching.48–50 This algorithm, which was originally developed for positive mode CID, was modified to allow searching of negative mode MS/MS data by both UVPD and NETD. The methods were optimized individually based on the specific fragmentation behavior associated with each method. For example, NETD searches were performed with several forms of a- and x-type ions (e.g. a/x ± 1 due to hydrogen migration), and therefore searching scores are elevated compared to UVPD, which uses only one form of a- and x- ions. Preferential cleavages at proline, glutamic acid, and aspartic acid that yield specific Y- and y-type ions, and c/z ions are also included in searches for UVPD.44
Using these modified MassMatrix algorithms, LC-MS/MS data sets were searched against the cow International Protein Index (IPI v3.73, Sept 27, 2011) modified to include the chicken lysozyme C sequence using the MassMatrix database search engine v2.4.0. LC-MS/MS were performed in triplicate, and the data from the replicate experiments were merged prior to database searching. Search parameters utilized were trypsin, 2 missed cleavages, phosphorylation of S and T as variable modifications, iodoacetamide/carbamidomethylation of C as a fix modification, precursor ion tolerance of 2 Da and a product ion tolerance of 1 Da. Peptide hits were filtered by pp and pp2 scores less than 5 and pptag scores less than 0.548 for which the pp and pp2 scores are a statistical measure of the number of matched product ions and the total abundance of matched product ions, respectively. All suspect peptide spectral matches (PSMs) were manually verified.
RESULTS AND DISCUSSION
Figure 1 shows NETD (A and C) and UVPD (B and D) spectra of doubly and triply deprotonated KPLLIIAEDVEGEY. NETD of the doubly charged peptide results in production of eight x-ions and two a-ions, corresponding to ten out of thirteen possible backbone cleavage sites. The dominant dissociation pathways include the neutral loss of CO2 and non-dissociative electron transfer (NETnoD), the latter yielding intact charge-reduced precursor ions (labeled as [M-2H]−•). The most abundant sequence-informative products are the x10, x11, x12 and x13 ions. A fluoranthene adduct of the intact precursor is also observed in all NETD spectra and is denoted by the label Δ in Figures 1A and C. The m/z value of the fluoranthene adduct species can be accurately predicted based on the consistent mass shift of 202 Da and thus can be subtracted from the spectra prior to the database searches. UVPD of the doubly charged species yields an extensive array of sequence-informative product ions. Whereas NETD exclusively produced a- and x-type product ions, UVPD yields b-, y-, c-, z-, Y-, d- and w-type product ions in addition to the dominant a- and x-ions. The large array of product ions upon UVPD yielded 100% sequence coverage as well as complementary N- and C-terminal product ions (i.e., a/x product ion pairs) for nine of the thirteen backbone cleavage sites. UVPD also promotes the loss of CO2 from the precursor as well as charge-reduction by photo-induced electron detachment, a pathway noted in our previous UVPD study of peptide anions.44 NETD of the triply charged peptide in Figure 1C provides complete sequence coverage and complementary a- and x-ions for seven of the thirteen peptide backbone cleavage sites. The triply charged peptide yielded five additional a-ions and four x-ions relative to the doubly charged peptide. Although the a/x ion series has higher abundance and affords greater coverage than observed for the corresponding doubly charged peptide, the loss of CO2 and NETnoD remain the most dominant pathways. The fragmentation pattern produced by UVPD of the triply charged species is similar to that of the doubly charged species. Complete sequence coverage is achieved, and complementary a/x ion pairs are observed for nine of the thirteen peptide backbone cleavage sites.
Figure 1.

NETD (A and C) and UVPD (B and D) spectra of doubly and triply charged KPLLIIAEDVEGEY. Δ indicates fluoranthene adduction to the precursor.
For the peptide KPLLIIAEDVEGEY, charge reduction accounts for 59% and 42% of product ions (based on peak area) produced by NETD of the 2- and 3- species, respectively. Similarly, UVPD resulted in 31% and 30% charge reduction for the 2- and 3- species, respectively. Neutral loss of CO2 from the precursor accounts for 24% and 34% of the product ions upon NETD and 15% and 25% upon UVPD for the 2- and 3- species, respectively. UVPD produced less extensive charge reduction and CO2 loss from the precursor than NETD; however small molecule neutral losses (CO2, NH3 and H2O) from sequence ions were more notable upon UPVD. NETD produced only one detectable neutral loss sequence ion (x7 - CO2) for the doubly charged peptide. The loss of CO2 loss was observed from the x7, x9, x10, x11, x12 and x13 ions upon NETD of the triply charged peptide, as well as ammonia loss from a13. CO2, NH3 and H2O losses occurred for a majority of the sequence ions upon UVPD, with even multiple neutral losses in some cases. This outcome is attributed to the energetics of NETD versus UVPD. Polfer and coworkers estimated the recombination energy of electron transfer with fluoranthene radical cations to be 2.4 – 4.5 eV depending on the site of electron abstraction.42 In the same study they also reported that the more energetic electron abstraction by Xe radical cations (estimated recombination energy of 6.7–8.7 eV) vastly increased the number of fragment ions observed.42 However, the additional fragment ions mostly corresponded to sequential neutral losses from a• and x-ions instead of new diagnostic products arising from different fragmentation channels (i.e. b, c, y, z-ions).42 The greater energy deposition upon absorption of a single 193 nm photon (6.4 eV) and wider array of fragmentation channels accessible compared to electron abstraction by fluoranthene radical cations accounts for the larger array of product ion types as well as the greater extent of neutral losses. UVPD product ions may also possess greater internal energy upon formation, thus contributing to more extensive secondary dissociation and thus elevated levels of neutral losses compared to NETD.
The NETD (A and C) and UVPD (B and D) spectra of the doubly and triply deprotonated phosphopeptide TSTEPQyQPGNEL are illustrated in Figure 2. The phosphorylation site can be pinpointed to the tyrosine residue based on the NETD and UVPD spectra for both the doubly and triply charged species. However, for the doubly charged species, UVPD affords greater confidence in the identification of the sequence and the localization of the phosphorylation site as a result of the production of peptide backbone cleavages both N-terminal and C-terminal to the phosphorylated residue and complete sequence coverage of the rest of the peptide. NETD did not result in the loss of the phosphate group from the precursor nor from any of the sequence ions. Importantly, UVPD produced H2PO4− and PO3− reporter ions that can be used confirm peptide phosphorylation in bottom-up proteomic experiments. The UPVD spectrum of singly charged TSTEPQyQPGNEL is shown in Supplemental Figure 1. UVPD of the singly charged species resulted in nearly complete series of a and x ions as well as confident assignment of the site of phosphorylation. In this case, the phosphate reporter ions fall below the low mass cutoff and are not detected, but there is an abundant phosphate neutral loss from the precursor. In general the UVPD spectrum for the singly deprotonated phosphopeptide is more congested with internal ions and neutral losses compared to the UVPD mass spectra obtained for the corresponding doubly and triply charged species.
Figure 2.

NETD (A and C) and UVPD (B and D) spectra of doubly and triply charged TSTEPQyQPGENL (y indicates phosphorylated tyrosine). * indicated the precursor ion.
Since activation/dissociation by NETD is more highly dependent on the peptide charge state than UVPD, a comparison of NETD and UVPD was undertaken for six doubly and triply deprotonated peptides. Sequence coverage distributions were calculated for each peptide and charge state based on the summed peak areas of complementary a/x ion pairs normalized to the most abundant a/x ion pair. Only product ions with S/N ≥ 3 were included in the sequence coverage distributions. The results are shown in Figure 3. In general, NETD performs poorly for the doubly deprotonated peptides as witnessed by the rather sparse bar graphs in Figure 3. For these cases, the most dominant pathways produced x-ions corresponding to cleavage near the N-terminus with very few low abundance a-ions. In fact, there are zero complementary a- and x-ions pairs detected for the doubly charged precursors, signifying notable localization of the charge sites upon NETD. NETD of the triply deprotonated species yields greater abundances and broader distributions of backbone cleavages compared to the doubly charged species, although still occurring with notable gaps in coverage. UVPD produced substantially broader distributions of peptide backbone cleavages for both the doubly and triply charged species of all the peptides analyzed. For the doubly charged species, UVPD produced greater product ion abundances for nearly every possible peptide backbone cleavage site compared to NETD. This is partially attributed to the production of complementary a- and x-ion pairs for most backbone cleavage sites upon UVPD, whereas NETD predominantly produced x-ions. Table 1 summarizes the sequence coverages obtained by NETD and UVPD for the same six peptides in the 2- and 3- charge states. UVPD produced 100% sequence coverage for all the peptides (both doubly and triply charged species). NETD produced average sequence coverage of 37% for the doubly charged species and 83% for the triply charged species.
Figure 3.

Sequence coverage distribution comparison between UVPD and NETD of doubly and triply deprotonated peptide anions. The product ion signals were plotted for summed a/x pairs per backbone cleavage. For each peptide, ion signals were normalized to the highest a/x pair between the UVPD and NETD methods.
Table 1.
Sequence coverage comparison of NETD and UVPD of peptide anions. The percentages were calculated from a- and x-type ions with S/N ≥ 3, and represent the number of observed backbone cleavages divided by the total possible backbone cleavages per peptide.
| Peptide Sequence
|
% Sequence Coverage
|
|||
|---|---|---|---|---|
| [M – 2H]2− | [M – 3H]3− | |||
|
|
|
|||
| NETD | UVPD | NETD | UVPD | |
| RQsVELHSPQSLPR | 15 | 100 | 77 | 100 |
| DAEFRHDSGYEVHHEK | 13 | 100 | 60 | 100 |
| TSTEPQyQPGENL | 50 | 100 | 75 | 100 |
| KAESTVAPEEDTDED | 43 | 100 | 93 | 100 |
| KPLLIIAEDVEGEY | 62 | 100 | 100 | 100 |
| ADSGEGDFLAEGGGVR | 40 | 100 | 93 | 100 |
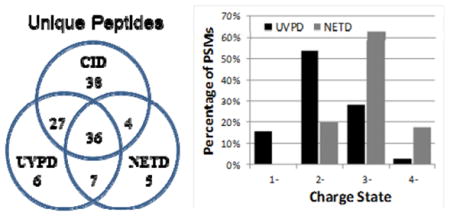
To enable a more extensive comparison of the performance of NETD and UVPD for protemoics-type experiments, as well as benchmarking these MS/MS methods relative to conventional positive mode CID, LC-MS/NETD and LC-MS/UVPD in addition to positive ion mode LC-MS/CID experiments were undertaken for tryptic digests of a model protein mixture containing bovine α-casein, β-casein, β-lactoglobulin A, BSA, ribonuclease A and chicken lysozyme C with automated database searching via MassMatrix. These proteins were chosen for three reasons: they are well characterized model systems, α-casein and β-casein are highly phosphorylated, and the theoretical isoelectric points (based on amino acid content) of these proteins range from ~4.8 for β-lactoglobulin to ~9.3 for lysozyme C and thus should not bias results for either positive or negative mode analyses. Table 2 summarizes the results obtained from the NETD, UVPD and CID data sets. UVPD produced overall higher sequence coverage percentages and greater numbers of unique peptides compared to that of NETD. This outcome can be attributed to the ability of UVPD to efficiently sequence both lower and higher charge states with rapid activation times (4 ms). In contrast, NETD in general yields the best results for higher charged states (n ≥ 3), and uses fairly modest activation times (250 ms), which can limit the number of MS/MS spectra collected. As expected due to higher signal intensities of tryptic peptides (ones that contain at least two protonation sites) in the positive mode, CID generally yielded the most unique peptide identifications and sequence coverage compared to both negative mode MS/MS methods, except for three of the model proteins, αS1-casein, αS2-casein, and β-lactoglobulin A. Both UVPD and NETD, however, often out-performed CID for sequencing and identifying acidic peptide species. UVPD identified a total of nine phosphopeptides corresponding to six unique phosphorylation sites, and NETD also identified nine phosphopeptides corresponding to six unique phosphorylation sites. CID identified seven phosphopeptides corresponding to four unique phosphorylation sites. The Venn diagram in Figure 4 shows the overlap in unique peptides identified in the negative mode by NETD and UVPD and in the positive mode by CID for the protein mixture. A significant overlap in peptide matches was observed between NETD and UVPD with 83% of those identified by NETD also identified by UVPD. UVPD and NETD produced 13 and 12 unique peptides, respectively, that were not identified by CID in positive mode.
Table 2.
MassMatrix comparison of negative mode NETD and UVPD and positive mode CID LC-MS/MS experiments.
| Protein | % Sequence | Unique Peptides | ||||
|---|---|---|---|---|---|---|
|
|
|
|
||||
| NETD | UVPD | CID | NETD | UVPD | CID | |
| BSA | 51 | 58 | 70 | 26 | 33 | 51 |
| αS1-casein | 35 | 43 | 32 | 9 (4/1)* | 12 (4/1)* | 9 (2/1)* |
| αS2-casein | 38 | 32 | 31 | 6 (4/4)* | 9 (4/4)* | 12 (3/2)* |
| β-lactoglobulin A | 64 | 56 | 48 | 7 | 8 | 11 |
| β-casein | 15 | 24 | 25 | 2 (1/1)* | 6 (1/1)* | 6 (2/1)* |
| Lysozyme C | 24 | 43 | 81 | 2 | 4 | 10 |
| Ribonuclease A | 13 | 42 | 52 | 1 | 3 | 5 |
unique peptides (phosphopeptides/phosphorylation sites)
Figure 4.

Venn diagram comparing the overlap in unique peptides identified in the negative mode by NETD and UVPD and in the positive mode by CID.
Lastly, database searches were undertaken by combining the spectral files for UVPD and positive CID as well as NETD and positive CID to investigate in more detail the overlap between traditional CID and the negative mode methods - these results are illustrated in Table 3. In all cases except for lysozyme C and ribonuclease A, the combined searches significantly increased the sequence coverage percentages and unique peptide identifications over that obtained by CID alone. The combined NETD/CID searches outperformed the UVPD/CID searches for BSA, but underperformed for β-casein. However, even these search outcomes were fairly similar (i.e., the number of unique peptides and sequence coverage percentages were close if not identical). The integrated NETD/CID and UVPD/CID results were similar for most of the other proteins. The variations that were seen between the two different searching strategies are attributed to the differences in optimum ion charge state for each MS/MS method and the corresponding population of charge states generated for the tryptic peptides of each protein (as described in previous sections). To investigate this hypothesis in more quantifiable detail, the charge state distributions of the peptide spectral matches (PSMs) produced by NETD and UVPD are tabulated in Figure 5. For UVPD, the charge state distribution (x̄ = −2.1, s = 0.72, n = 578) is centered about the 2- charge state which corresponded to 54% of the PSMs. In contrast, the charge state distribution for NETD (x̄ = −2.9, s = 0.60, n = 259) is centered about the 3- charge state which corresponded to 63% of the PSMs. These differences in peptide preference highlight the complementary nature of the two MS/MS methods for peptide anions and are responsible for many of the observed variations in the acidic peptide identifications.
Table 3.
MassMatrix comparison of NETD/CID and UVPD/CID combined positive and negative mode data sets.
| Protein | % Sequence | Unique Peptides | ||
|---|---|---|---|---|
|
|
|
|
||
| NETD/CID | UVPD/CID | NETD/CID | UVPD/CID | |
| BSA | 77 | 73 | 54 | 54 |
| αS1-casein | 47 | 46 | 13 (4/1)* | 13 (4/1)* |
| αS2-casein | 46 | 36 | 14 (4/4)* | 15 (5/4)* |
| β-lactoglobulin A | 63 | 63 | 14 | 13 |
| β-casein | 25 | 29 | 6 (2/1)* | 7 (2/1)* |
| Lysozyme C | 81 | 81 | 10 | 10 |
| Ribonuclease A | 52 | 52 | 5 | 5 |
unique peptides (phosphopeptides/phosphorylation sites)
Figure 5.
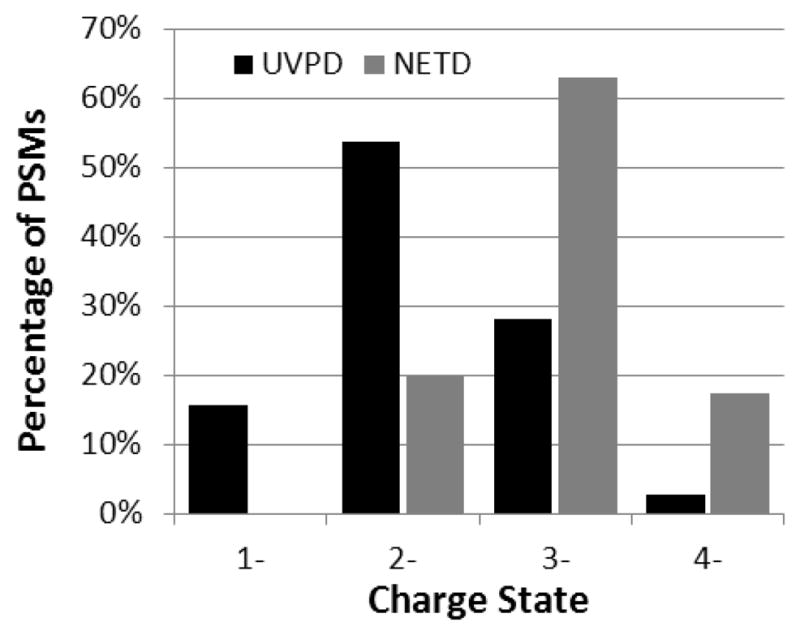
Charge state distribution of peptide spectral matches (PSMs) produced by NETD and UVPD for a protein mixture. The total number of PSMs for NETD and UVPD are 259 and 578, respectively.
CONCLUSION
UVPD and NETD were compared with respect to sequence coverage distributions (a measure of product ion signals across the peptide backbone), sequence coverage percentages, backbone cleavage specificity, and both LC-MS/MS and automated database searching performance metrics for analysis of peptide anions. The substantially shorter activation times and compatibility with a wider range of charge states allowed the collection of a greater number of MS/MS spectra during a typical a LC gradient for UVPD compared to NETD. However, the spectra that were collected for NETD were generally from peptide species that contained several acidic residues and/or acidic post translational modifications – ones that are more difficult for traditional positive mode CID to efficiently and accurately elucidate. Importantly, when combined with positive mode CID, both UVPD and NETD significantly increased the sequence coverage percentages and unique peptide identifications over that of just CID alone, demonstrating their utility and potential for characterizing acidic proteomes.
Supplementary Material
Acknowledgments
J.S.B. acknowledges funding from the NIH (R21GM099028) and the Welch Foundation (F-1155). H.X. acknowledges funding from the NIH (UL1 RR-024989, PI: Dr. Pamela B. Davis).
References
- 1.Hunt DF, Buko AM, Ballard JM, Shabanowitz J, Giordani AB. Sequence analysis of polypeptides by collision activated dissociation on a triple quadrupole mass spectrometer. Biol Mass Spectrom. 1981;8:397–408. doi: 10.1002/bms.1200080909. [DOI] [PubMed] [Google Scholar]
- 2.McLuckey S. Principles of collisional activation in analytical mass spectrometry. J Am Soc Mass Spectrom. 1992;3:599–614. doi: 10.1016/1044-0305(92)85001-Z. [DOI] [PubMed] [Google Scholar]
- 3.Eng JK, McCormack AL, Yates JR., III An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 4.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. ELECTROPHORESIS. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 5.de Godoy LMF, Olsen JV, Cox J, Nielsen ML, Hubner NC, Frohlich F, Walther TC, Mann M. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature. 2008;455:1251–1254. doi: 10.1038/nature07341. [DOI] [PubMed] [Google Scholar]
- 6.Zubarev RA, Kelleher NL, McLafferty FW. Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- 7.Kelleher NL, Zubarev RA, Bush K, Furie B, Furie BC, McLafferty FW, Walsh CT. Localization of Labile Posttranslational Modifications by Electron Capture Dissociation: The Case of γ-Carboxyglutamic Acid. Anal Chem. 1999;71:4250–4253. doi: 10.1021/ac990684x. [DOI] [PubMed] [Google Scholar]
- 8.Zubarev RA, Horn DM, Fridriksson EK, Kelleher NL, Kruger NA, Lewis MA, Carpenter BK, McLafferty FW. Electron Capture Dissociation for Structural Characterization of Multiply Charged Protein Cations. Anal Chem. 2000;72:563–573. doi: 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- 9.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. PNAS. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coon JJ, Syka JEP, Schwartz JC, Shabanowitz J, Hunt DF. Anion dependence in the partitioning between proton and electron transfer in ion/ion reactions. International Journal of Mass Spectrometry. 2004;236:33–42. [Google Scholar]
- 11.Good DM, Wirtala M, McAlister GC, Coon JJ. Performance Characteristics of Electron Transfer Dissociation Mass Spectrometry. Molecular & Cellular Proteomics. 2007;6:1942–1951. doi: 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Bowers WD, Delbert SS, Hunter RL, McIver RT. Fragmentation of oligopeptide ions using ultraviolet laser radiation and Fourier transform mass spectrometry. Journal of the American Chemical Society. 1984;106:7288–7289. [Google Scholar]
- 13.Little DP, Speir JP, Senko MW, O’Connor PB, McLafferty FW. Infrared Multiphoton Dissociation of Large Multiply Charged Ions for Biomolecule Sequencing. Anal Chem. 1994;66:2809–2815. doi: 10.1021/ac00090a004. [DOI] [PubMed] [Google Scholar]
- 14.Hofstadler SA, Sannes-Lowery KA, Griffey RH. Infrared Multiphoton Dissociation in an External Ion Reservoir. Anal Chem. 1999;71:2067–2070. doi: 10.1021/ac990176n. [DOI] [PubMed] [Google Scholar]
- 15.Wilson JJ, Brodbelt JS. Infrared Multiphoton Dissociation for Enhanced de Novo Sequence Interpretation of N-Terminal Sulfonated Peptides in a Quadrupole Ion Trap. Anal Chem. 2006;78:6855–6862. doi: 10.1021/ac060760d. [DOI] [PubMed] [Google Scholar]
- 16.Morgan JW, Hettick JM, Russell DH. Biological Mass Spectrometry. Vol. 402. Academic Press; 2005. pp. 186–209. [DOI] [PubMed] [Google Scholar]
- 17.Kim TY, Thompson MS, Reilly JP. Peptide photodissociation at 157 nm in a linear ion trap mass spectrometer. Rapid Commun Mass Spectrom. 2005;19:1657–1665. doi: 10.1002/rcm.1969. [DOI] [PubMed] [Google Scholar]
- 18.Ly T, Julian RR. Ultraviolet Photodissociation: Developments towards Applications for Mass-Spectrometry-Based Proteomics. Angewandte Chemie International Edition. 2009;48:7130–7137. doi: 10.1002/anie.200900613. [DOI] [PubMed] [Google Scholar]
- 19.Reilly JP. Ultraviolet photofragmentation of biomolecular ions. Mass Spectrom Rev. 2009;28:425–447. doi: 10.1002/mas.20214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ly T, Julian RR. Residue-Specific Radical-Directed Dissociation of Whole Proteins in the Gas Phase. J Am Chem Soc. 2008;130:351–358. doi: 10.1021/ja076535a. [DOI] [PubMed] [Google Scholar]
- 21.Morgan JW, Russell DH. Comparative Studies of 193-nm Photodissociation and TOF-TOFMS Analysis of Bradykinin Analogues: The Effects of Charge Site(s) and Fragmentation Timescales. J Am Soc Mass Spectrom. 2006;17:721–729. doi: 10.1016/j.jasms.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Thompson MS, Cui W, Reilly JP. Factors That Impact the Vacuum Ultraviolet Photofragmentation of Peptide Ions. Journal of the American Society for Mass Spectrometry. 2007;18:1439–1452. doi: 10.1016/j.jasms.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 23.Kim TY, Reilly JP. Time-Resolved Observation of Product Ions Generated by 157 nm Photodissociation of Singly Protonated Phosphopeptides. J Am Soc Mass Spectrom. 2009;20:2334–2341. doi: 10.1016/j.jasms.2009.08.021. [DOI] [PubMed] [Google Scholar]
- 24.Madsen JA, Boutz DR, Brodbelt JS. Ultrafast Ultraviolet Photodissociation at 193 nm and its Applicability to Proteomic Workflows. Journal of Proteome Research. 2010;9:4205–4214. doi: 10.1021/pr100515x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shin YS, Moon JH, Kim MS. Observation of Phosphorylation Site-Specific Dissociation of Singly Protonated Phosphopeptides. J Am Soc Mass Spectrom. 2010;21:53–59. doi: 10.1016/j.jasms.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 26.Brodbelt JS. Shedding Light on the Frontier of Photodissociation. J Am Soc Mass Spectrom. 2001;22:197–206. doi: 10.1007/s13361-010-0023-6. [DOI] [PubMed] [Google Scholar]
- 27.Time-resolved photodissociation of singly protonated peptides with an arginine at the N-terminus: A statistical interpretation. J Am Soc Mass Spectrom. 2008;19:645–655. doi: 10.1016/j.jasms.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 28.Janek K, Wenschuh H, Bienert M, Krause E. Phosphopeptide analysis by positive and negative ion matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun Mass Spectrom. 2001;15:1593–1599. doi: 10.1002/rcm.417. [DOI] [PubMed] [Google Scholar]
- 29.Gunawardena HP, Emory JF, McLuckey SA. Phosphopeptide Anion Characterization via Sequential Charge Inversion and Electron-Transfer Dissociation. Anal Chem. 2006;78:3788–3793. doi: 10.1021/ac060164j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ewing N, Cassady C. Dissociation of multiply charged negative ions for hirudin (54–65), fibrinopeptide B, and insulin A (oxidized) J Am Soc Mass Spectrom. 2001;12:105–116. doi: 10.1016/S1044-0305(00)00195-1. [DOI] [PubMed] [Google Scholar]
- 31.Bowie JH, Brinkworth CS, Dua S. Collision-induced fragmentations of the (M-H)– parent anions of underivatized peptides: An aid to structure determination and some unusual negative ion cleavages. Mass Spectrom Rev. 2002;21:87–107. doi: 10.1002/mas.10022. [DOI] [PubMed] [Google Scholar]
- 32.Steinborner ST, Bowie JH. The Negative Ion Mass Spectra of [M–H]– Ions Derived From Caeridin and Dynastin Peptides. Internal Backbone Cleavages Directed Through Asp and Asn Residues. Rapid Commun Mass Spectrom. 1997;11:253–258. [Google Scholar]
- 33.Jensen ON. Interpreting the protein language using proteomics. Nat Rev Mol Cell Biol. 2006;7:391–403. doi: 10.1038/nrm1939. [DOI] [PubMed] [Google Scholar]
- 34.Schwartz R, Ting CS, King J. Whole Proteome pI Values Correlate with Subcellular Localizations of Proteins for Organisms within the Three Domains of Life. Genome Research. 2001;11:703–709. doi: 10.1101/gr.gr-1587r. [DOI] [PubMed] [Google Scholar]
- 35.Knight CG, Kassen R, Hebestreit H, Rainey PB. Global analysis of predicted proteomes: Functional adaptation of physical properties. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8390–8395. doi: 10.1073/pnas.0307270101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weiller GF, Caraux G, Sylvester N. The modal distribution of protein isoelectric points reflects amino acid properties rather than sequence evolution. Proteomics. 2004;4:943–949. doi: 10.1002/pmic.200200648. [DOI] [PubMed] [Google Scholar]
- 37.Kiraga J, Mackiewicz P, Mackiewicz D, Kowalczuk M, Biecek P, Polak N, Smolarczyk K, Dudek M, Cebrat S. The relationships between the isoelectric point and: length of proteins, taxonomy and ecology of organisms. BMC Genomics. 2007;8:163. doi: 10.1186/1471-2164-8-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kjeldsen F, Silivra OA, Ivonin IA, Haselmann KF, Gorshkov M, Zubarev RA. Cα-C Backbone Fragmentation Dominates in Electron Detachment Dissociation of Gas-Phase Polypeptide Polyanions. Chem Eur J. 2005;11:1803–1812. doi: 10.1002/chem.200400806. [DOI] [PubMed] [Google Scholar]
- 39.Kjeldsen F, Horning OB, Jensen SS, Giessing AMB, Jensen ON. Towards Liquid Chromatography Time-Scale Peptide Sequencing and Characterization of Post-Translational Modifications in the Negative-Ion Mode Using Electron Detachment Dissociation Tandem Mass Spectrometry. J Am Soc Mass Spectrom. 2008;19:1156–1162. doi: 10.1016/j.jasms.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 40.Larraillet V, Vorobyev A, Brunet C, Lemoine J, Tsybin YO, Antoine R, Dugourd P. Comparative Dissociation of Peptide Polyanions by Electron Impact and Photo-Induced Electron Detachment. J Am Soc Mass Spectrom. 2010;21:670–680. doi: 10.1016/j.jasms.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 41.Cook S, Jackson G. Metastable Atom-Activated Dissociation Mass Spectrometry of Phosphorylated and Sulfonated Peptides in Negative Ion Mode. J Am Soc Mass Spectrom. 2011;22:1088–1099. doi: 10.1007/s13361-011-0123-y. [DOI] [PubMed] [Google Scholar]
- 42.Huzarska M, Ugalde I, Kaplan DA, Hartmer R, Easterling ML, Polfer NC. Negative Electron Transfer Dissociation of Deprotonated Phosphopeptide Anions: Choice of Radical Cation Reagent and Competition between Electron and Proton Transfer. Anal Chem. 2010;82:2873–2878. doi: 10.1021/ac9028592. [DOI] [PubMed] [Google Scholar]
- 43.Coon JJ, Shabanowitz J, Hunt DF, Syka JEP. Electron Transfer Dissociation of Peptide Anions. J Am Soc Mass Spectrom. 2005;16:880–882. doi: 10.1016/j.jasms.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 44.Madsen JA, Kaoud TS, Dalby KN, Brodbelt JS. 193-nm photodissociation of singly and multiply charged peptide anions for acidic proteome characterization. Proteomics. 2011;11:1329–1334. doi: 10.1002/pmic.201000565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Antoine R, Joly L, Tabarin T, Broyer M, Dugourd P, Lemoine J. Photo-induced formation of radical anion peptides. Electron photodetachment dissociation experiments. Rapid Commun Mass Spectrom. 2007;21:265–268. doi: 10.1002/rcm.2810. [DOI] [PubMed] [Google Scholar]
- 46.McAlister GC, Russell JD, Rumachik NG, Hebert AS, Syka JEP, Geer LY, Westphall MS, Pagliarini DJ, Coon JJ. Analysis of the Acidic Proteome with Negative Electron-Transfer Dissociation Mass Spectrometry. Anal Chem. 2012;84:2875–2882. doi: 10.1021/ac203430u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Madsen JA, Xu H, Horton AP, Boutz DR, Kaoud TS, Dalby KN, Brodbelt JS. A Flexible Searching Algorithm for Acidic Proteome Characterization by UVPD of Peptide Anions. submitted. [Google Scholar]
- 48.Xu H, Freitas M. A mass accuracy sensitive probability based scoring algorithm for database searching of tandem mass spectrometry data. BMC Bioinformatics. 2007;8:133. doi: 10.1186/1471-2105-8-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu H, Zhang L, Freitas MA. Identification and Characterization of Disulfide Bonds in Proteins and Peptides from Tandem MS Data by Use of the MassMatrix MS/MS Search Engine. Journal of Proteome Research. 2008;7:138–144. doi: 10.1021/pr070363z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu H, Hsu PH, Zhang L, Tsai MD, Freitas MA. Database Search Algorithm for Identification of Intact Cross-Links in Proteins and Peptides Using Tandem Mass Spectrometry. Journal of Proteome Research. 2010;9:3384–3393. doi: 10.1021/pr100369y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.