

Abstract
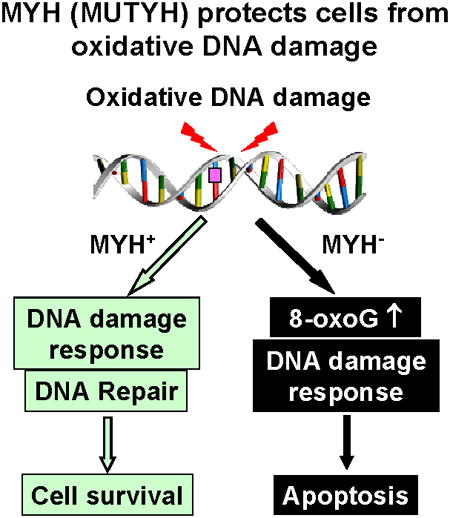
MutY DNA glycosylase homologs (MYH or MUTYH) reduce G:C to T:A mutations by removing misincorporated adenines or 2-hydroxyadenines paired with guanine or 7,8-dihydro-8-oxo-guanine (8-oxoG). Mutations in human the MYH (hMYH) gene are associated with the colorectal cancer predisposition syndrome MYH-associated polyposis. To examine the function of MYH in human cells, we regulated MYH gene expression by knockdown or overproduction. The hMYH knockdown human HeLa cells are more sensitive to the killing effects of H2O2 than the control cells. In addition, hMYH knockdown cells have altered cell morphology, display enhanced susceptibility to apoptosis, and have altered DNA signaling activation in response to oxidative stress. The cell cycle progression of hMYH knockdown cells is also different from that of the control cells following oxidative stress. Moreover, hMYH knockdown cells contain higher levels of 8-oxoG lesions than the control cells following H2O2 treatment. Although MYH does not directly remove 8-oxo-G, MYH may generate favorable substrates for other repair enzymes. Overexpression of mouse MYH (mMYH) in human mismatch repair defective HCT15 cells makes the cells more resistant to killing and refractory to apoptosis by oxidative stress than the cells transfected with vector. In conclusion, MYH is a vital DNA repair enzyme that protects cell from oxidative DNA damage and is critical for proper cellular response to DNA damage.
Keywords: DNA repair, MutY homolog, Base excision repair, Oxidative stress, DNA damage response
Graphical abstract
1. Introduction
The human genome is vulnerable to DNA-damaging agents of both endogenous and environmental origin. Reactive oxygen species (ROS) are commonly generated as by-products during mitochondrial oxidative phosphorylation, in response to inflammation, and following exposure to ionizing radiation and chemicals [1]. ROS-induced DNA damage includes DNA strand breaks and base lesions that are mainly repaired by base excision repair (BER) [2,3]. The first step in BER is carried out by a lesion-specific DNA glycosylase, which cleaves the N-glycosidic bond between the base and deoxyribose sugar [4].
The most abundant form of DNA damage is 8-oxo-7,8-dihydroguanine (8-oxo-G or GO). This lesion is highly mutagenic because it can base-pair with adenine during DNA replication to induce G:C to T:A transversions [5-9]. In human cells, hMTH1 (E. coli MutT homolog), hOGG1 glycosylase (E. coli MutM or Fpg functional homolog), hMYH or MUTYH glycosylase (E. coli MutY homolog), hNEIL1 glycosylase (E. coli Nei homolog), and hMSH2/hMSH6 mismatch repair enzyme complex act to reduce the mutagenic effect associated with GO (reviewed in [10]). Human MYH (also called hMUTYH) can excise adenines or 2-hydroxyadenines misincorporated opposite to GO [10-16]. Thus, MYH protects cells from the mutagenic effect of GO. Germline mutations in the hMYH gene cause autosomal recessive colorectal adenomatous polyposis (MYH-associated polyposis, MAP) [17-21]. Tumors from affected patients contain somatic G:C to T:A mutations in the adenomatous polyposis coli (APC), K-ras, and other genes [17,22,23]. Myh knockout mice exhibit a 5.3-fold increased rate of spontaneous intestinal adenoma/adenocarcinoma [24]. The tumor suppressor function of MYH has been attributed to its function in mutation avoidance [11,25]. Recently, an alterative model has been proposed that MYH suppresses tumorigenesis by inducing cell death due to oxidative stress [26,27].
Genotoxic stress activates DNA damage response that enhances DNA repair, arrests cell-cycle progression, and triggers apoptosis via cell cycle checkpoints [28,29]. We have shown that MYH glycosylase is associated with the checkpoint sensor Rad9/Rad1/Hus1 (9-1-1) complex in both fission yeast Schizosaccharomyces pombe and human cells [30,31]. The 9-1-1 complex has been proposed to provide a platform to coordinate BER because it interacts with and stimulates the activity of almost every enzyme in the long-patch BER pathway [32]. Several lines of evidence support that S. pombe Myh1 (SpMyh1) is an “adaptor” to recruit checkpoint proteins to DNA lesions. (1) DNA damage-induced phosphorylation of S. pombe Hus1 is dependent on SpMyh1 expression [30]. (2) SpMyh1 is required for 9-1-1 localization to telomeres that are highly susceptible to oxidative damage [33]. (3) Disruption of the SpMyh1/Sp9-1-1 interaction in vivo increases the mutation rate in S. pombe and sensitizes the yeast to H2O2 [34]. In human cells, the interaction of MYH and 9-1-1 is enhanced following ionizing radiation; and hMYH and 9-1-1 co-localize in the nucleus following H2O2 treatment [31]. In addition, knockdown (KD) of hMYH decreases phosphorylation of Chk1 induced by hydroxyurea and UV [35]. However, the rationale for this finding is not clear because hMYH is not the major recognition factor for hydroxyurea-induced replication stress and UV damage. Recently, the hMYH Q324H (according to the old nomenclature) variant has been shown to be defective in interaction with the 9-1-1 complex and to affect DNA repair and DNA damage response in vivo [36]. Thus, in the mammalian system, MYH also acts as an adaptor for checkpoint sensors. To examine the function of hMYH in human cells, we have knocked down and overproduced MYH expression and examined the cell's response to oxidative stress. We show that hMYH protects cells from apoptosis and GO damage under oxidative stress. MYH is also a key mediator for checkpoint activation.
2. Materials and methods
2.1. Cell culture and cell extracts
Human HeLa S3 and HCT15 cell lines were purchased from American Type Cell Culture (ATCC). HeLa cells were maintained in DMEM (Cellgro) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin. HCT15 cells were grown in RPMI 1640 medium (Cellgro) supplemented with 10% FBS and penicillin/streptomycin at 37°C in 5% CO2. Cell extracts were prepared from cells grown to late log phase. The cell pellet from one 10 cm dish (∼ 1 × 107 cells) was lysed in 0.3-0.5 ml of RIPA buffer (50 mM Tris-HCl, pH7.4, 150 mM NaCl, 1% NP40, 1 mM EDTA, 0.1% TritonX-100, 1 mM phenylmethylsulfonyl fluoride, 1 mM NaF, and 1 mM Na3VO4) by incubation at 4°C for 30 minutes followed by centrifugation at 14,000 rpm for 10 minute. The supernatant was aliquoted and stored at −80°C. The protein concentration was determined by Bio-Rad protein assay (Bio-Rad).
2.2. Antibodies and Western blotting
Antibodies used for Western blotting include: MYH (custom-raised peptide antibody α344) [12], Chk1 (Bethyl, A300-298A), Ser 317-phosphorylated Chk1 (Bethyl, A300-163A), Cdc25C (BD Pharmingen, 51-80701N), Ser 216-phosphorylated Cdc25C (Cell Signaling Technology, 4901S), β-actin (Sigma/Aldrich, 5441), and horseradish peroxidase-conjugated anti-mouse/anti-rabbit antibodies (BioRad, 1706516/1706515).
Cell extracts (about 25 μg of total protein) were separated on SDS-polyacrylamide gels and transferred to nitrocellulose membranes. The membranes were blocked with phosphate-buffered saline (PBS) with 0.1% Tween-20 and 10% nonfat dry milk, reacted with primary antibodies, and then incubated with secondary antibodies with wash between each step [37]. Western blotting was detected by the Enhanced Chemiluminescence (ECL) analysis system (USB Corporation, 72552) or ECL Plex (GE Healthcare) according to the manufacturer's protocols.
2.3. Knockdown and overproduction of MYH
Lentiviruses expressing shRNAs against hMYH (TRCN0000056604, TRCN0000056605) and non-target (NT) shRNA (SHC016V) were obtained from Sigma/Aldrich. Lentivirus particles were transduced into HeLa S3 cells with 8 μg/mL polybrene and cells were selected with 50 μg/ml of puromycin. MYH KD was confirmed by reverse transcription-quantitative PCR (RT-qPCR) and Western blotting.
For overexpression of MYH, vector pcDNA3.1 and pcDNA3.1 expressing mouse MYH (kindly provided by Dr. Yusaku Nakabeppu at Kyushu University, Japan) [38,39] were transfected into HCT15 cells using Fugene6 (Roche, Nutley, NJ, USA). The cells were replanted at 48 hrs after the transfection and stably transfected cells were selected with 50 μg/ml hygromycine. MYH over-expression was confirmed by Western blotting.
2.4. Detection of hMYH mRNA level by RT-qPCR
Total RNA from HeLa cells was isolated by using TRIZOL reagents (Invitrogen) according to the manufacture's protocol. 100 ng RNA was used as a template for RT-qPCR reactions using primers for hMYH and β-actin (see below). Real time PCR was performed using the Light Cycler 480 II Detection System (Roche) with iScript™ SYBR® Green One-Step Kit (BioRad, 1708892). The reactions were carried out with an initial step for RT at 50°C for 10 min followed by PCR cycles (95°C for 30 sec, 60°C for 30 sec, and 70°C for 45 sec). The mRNA level of hMYH was calculated relative to that of β-actin as ΔCt which is the difference between the number of cycles required to go above background in hMYH and β-actin samples. The fold difference of hMYH mRNA levels of control cells (NT) over hMYH shRNA knockdown (KD) cells was calculated according to the formula 2ΔCt(NT)-ΔCt(KD). The reactions were carried out in duplicate and data are averaged from three independent experiments.
Primers for hMYH: 5′-AGCCGGAAGAGGTGGTATTG-3′ and 5′-CTCTGAGACCCACACAGCATA-3′.
Primers for β-actin: 5′-ACCAACTGGGACGACATGGA-3′ and 5′-TACATGGCTGGGGTGTTGAA-3′
2.5. Cell morphological examination and cell viability analysis
Cells were seeded one day before H2O2 treatments. The culture medium was replaced with fresh medium containing H2O2 at indicated concentrations for 1 hr or left untreated. Cells were then washed with PBS, and recovered with fresh medium for 24 hrs. Cell morphology was examined under a light microscope (Nikon Eclipse TS100).
Cell viability was measured using the neutral red uptake assay [40]. Cells were seeded in 96-well flat bottom tissue culture plates. One day post-seeding, the cells were treated with H2O2 as described above. After recovery for 24 hrs, the plates were incubated for 2 h in regular medium containing 40 μg/ml of neutral red (3-amino-7-dimethylamino-2-methyl-phenazine hydrochloride, Sigma). The cells were then washed with PBS twice; the dye was extracted from each well with acidified ethanol solution. Plates were incubated at room temperature with gentle shaking for 15 minutes and the absorbance at 540 nm was read in Multiskan Spectrum microplate spectrometer (Thermo Scientific).
2.6. Colony formation assays
For clonogenic survival assays, HeLa or HCT15 cells were seeded at 1000 cells per well in 6-well plates in regular media for one day. The cells were treated with H2O2 for 1 hr or left untreated. Cells were washed with PBS twice and recovered in fresh media. After 10 days, cells were stained with 0.5% crystal violet in 20% methanol. After 30 minutes, plates were washed with water to remove the background color and allowed to air dry. Images were scanned and analyzed.
2.7. Apoptosis TUNEL assay
The apoptotic cells were detected by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay in accordance with the manufacturer's protocol (Promega). Briefly, cells were cultured in 4-chamber slides in regular medium for 24 hrs. The cells were then treated with H2O2 and recovered for 24 hrs in fresh media as described above. The cells were fixed with 4% paraformaldehyde in PBS for 25 minutes at 4°C, permeabilized with 0.2% Triton X-100 for 5 min at room temperature, and end-labeled with TdT for 1 hr at 37°C using the DeadEnd Fluorometric TUNEL assay reagents. Mounting media containing 4′,6′-diamidineno-a-phenylindole (DAPI) (Vector Laboratories) was added to stain nuclei. Images were captured by Nikon E400 fluorescent microscope with an attached CCD camera.
2.8. Quantification of 8-oxoG
Cells were seeded on a 4-chamber culture slide and treated with H2O2 and recovered for 5 hrs in fresh media as described above. Cells were fixed in 1:1 methanol:acetone for 20 minutes at -20°C followed by 0.05 N HCl treatment for 5 minutes. RNA was removed by 100 μg/ml RNase A in 150 mM NaCl and 15 mM sodium citrate for 1 hr at 37°C and DNA was denatured in situ with 0.15 N NaOH in 70% ethanol for 4 minutes. Cells were then treated with 5 μg/ml proteinase K in 20 mM Tris-HCl (pH 7.5) and 1 mM EDTA for 10 minutes at 37°C and blocked with 5% normal goat serum in PBS at room temperature for 1 hr. Cells then were incubated with anti-8-oxo-dG antibody (Trevigen, 4354-MC-050) at 4°C overnight. Next, the cells were washed three times for 15 min each in PBS and incubated with Alexa Fluro 488 goat anti-mouse antibodies (Molecular Probes) at a 1:250 dilution in PBS for 1 hr at room temperature at dark. The slides were then washed three times in PBS with 0.05% Tween-20. Nuclear DNA was counterstained with 5 μg/ml DAPI. Slides were mounted with cover slip using mounting medium (Vector, H-1200) and images were captured with a Nikon E400 fluorescent microscope with an attached CCD camera.
2.9. Cell cycle analysis
Double thymidine blocked synchronized cells were used to study the effects of MYH on cell cycle progression after H2O2 treatment. Cells were seeded in 10 cm plates (106 cells/dish) and treated with 2 mM thymidine for 18 hours. Cells were washed twice with PBS and fresh medium was added for additional 9 hour to release cell cycle. For the second block, medium containing 2 mM thymidine was then applied for another 17 hour. Cells were washed with PBS twice and then treated with H2O2 for one hour at the indicated concentration. At different time intervals post-treatment, cells were collected for cell cycle analysis. The collected cells were washed twice with PBS and resuspended in 200 μl PBS. Cells were kept in 80% ethanol for longer than 2 hours at -20°C. After all samples were collected and fixed, they were washed twice with PBS and stained with solution containing 50 μg/ml propidium iodide and 100 μg/ml RNase for 30 minutes. After one wash with PBS, cells were resuspended in 0.5 ml PBS and subjected to flow cytometry analyses by FACScan flow cytometer equipped with a 488nm blue laser (BD Biosciences) at University of Maryland Baltimore Greenebaum Cancer Center. Data analysis was performed with FlowJo (Tree Star, Ashland, OR) using the Watson cell cycle algorithm.
3. Results
3.1. hMYH knockdown cells are more sensitive to oxidative stress
To examine the function of hMYH in human cells, we knocked down hMYH gene expression using shRNA. The knockdown was performed in HeLa cells because DNA repair has been studied extensively with this cell line. We have studied protein-protein interactions and assayed MYH activity in HeLa cell extracts [12,31,41,42]. HeLa cells were stably transfected with a set of lentiviruses containing shRNA against hMYH or control non-target shRNA from Sigma/Aldrich. Our result indicated that a combined action of TRCN0000056604 and TRCN0000056605 shRNA gave the best knockdown of hMYH. Reverse transcription-quantitative PCR analysis showed that the mRNA of hMYH in HeLa cells was reduced to 12% with hMYH-shRNA as compared to the non-target shRNA (Fig. 1A). Also, the hMYH protein level was reduced to 30% with hMYH-shRNA (Fig. 1B). We then compared the phenotypes between these hMYH KD cells with the control cells. The hMYH KD HeLa cells had the same growth rate (data not shown) but were more sensitive to the killing effects of H2O2 than the control cells at 100-500 μM of H2O2 (Fig. 1C). Thus, hMYH activity is required for proper cellular response to oxidative DNA damage.
Fig. 1.
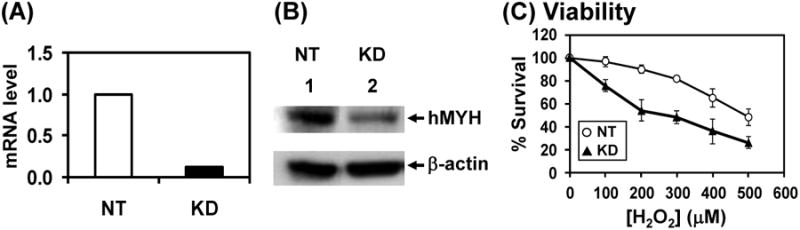
HeLa cells with hMYH-knockdown are more sensitive to H2O2 than control. (A) mRNA level of hMYH is reduced to 12% in knockdown (KD) cells as compared to non-target (NT) control cells. HeLa cells were stably transfected with lentivirus containing shRNA against hMYH or control shRNA. mRNA levels were determined by RT-qPCR. (B) The protein level of hMYH is reduced to 30% in knockdown cells as compared to control cells. Extracts were prepared from control and knockdown cells and Western blotting were analyzed with hMYH and β-actin antibodies. (C) Sensitivity to H2O2 of MYH KD cells. Both non-target and MYH knockdown HeLa cells were treated with H2O2 at indicated concentrations for 1 hr. The survived cells were detected after 24 hrs. Survival differences are statistical significant between two cell lines at 100-500 μM H2O2.
3.2. hMYH knockdown cells have altered cell morphology and display enhanced susceptibility to apoptosis after H2O2 treatment
We also observed that cells with low hMYH expression underwent morphological changes after H2O2 treatment (Fig. 2A). More swelled cells were found in hMYH KD cells when treated with 100 μM or 200 μM of H2O2. In addition, the hMYH KD cells exhibited poor survival rates in colony formation assay following treatments with several doses of H2O2 (Fig. 2B). There are statistically significant differences in colony formation between MYH KD and control cells at 30-150 μM H2O2 (P < 0.05). For example, after 75 μM H2O2 treatment, 60% colony formation was found in non-target cells as compared to untreated cells (Fig. 2B). However, MYH KD cells had only 35% colony formation after 75 μM H2O2 treatment as compared to untreated cells (Fig. 2B). The reduction in MYH KD cells is highly significant (P = 0.001).
Fig. 2.
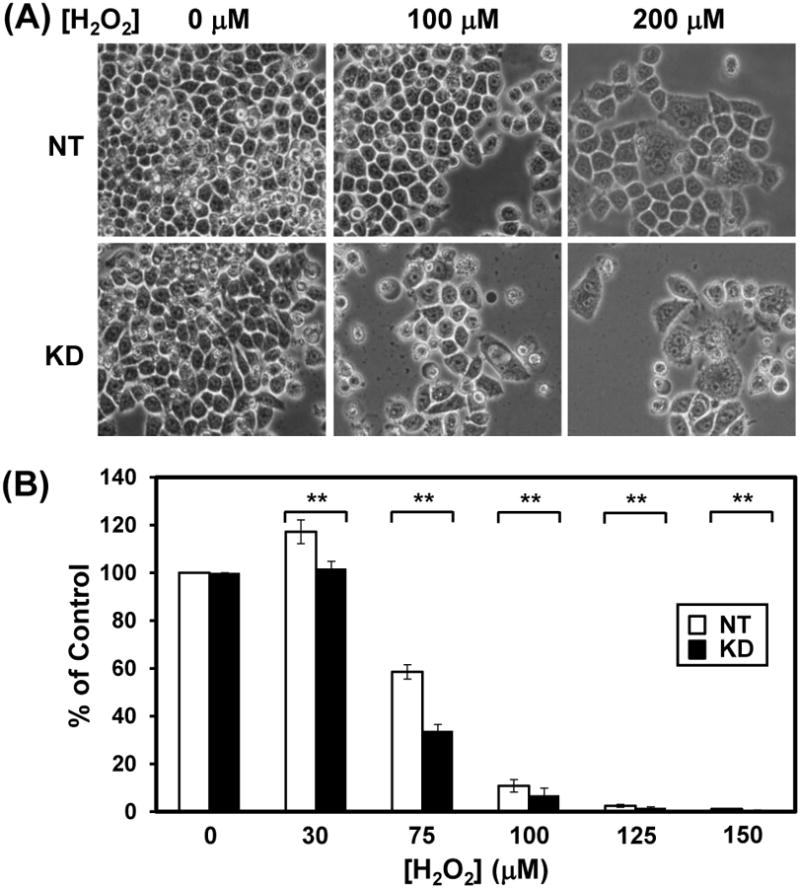
HeLa cells with hMYH-knockdown have altered cell morphology and poor survival rate than control cells after H2O2 treatment. HeLa cells were stably transfected with lentivirus containing shRNA against hMYH or non-target (NT) shRNA. (A) Morphological changes of MYH KD cells after H2O2 treatment. MYH knockdown HeLa cells were treated with 0, 100 and 200 μM H2O2 for 1 hour. Cells were imaged under microscope after 6 days of treatment. Fewer cell number count and more cells undergo morphological changes were observed in H2O2-treated hMYH KD cells as compared to control cells. (B) Colony formation of MYH KD cells after H2O2 treatment. Cells were treated with 30-150 μM H2O2 for 1 h or left untreated (control). Plates were incubated for 10 days. Quantitative analyses of the relative colony formation of NT (white bar) and MYH-ND (black bars) from three experiments. The percentage (%) is calculated from the ratios of H2O2 treated over untreated samples. The error bars reported are the standard deviations of the averages and P-value was calculated by Student's t-test. Two stars indicate P < 0.05.
Following DNA damage, cell cycle checkpoints are activated to arrest cell cycle and allow DNA repair. In case of extreme DNA damage, apoptosis may be triggered. When hMYH is limiting, persistent oxidative damage may result in cell death. In order to confirm that, we checked cell apoptosis by TUNNEL assay. Indeed, over 90% of hMYH KD cells and only 20% of control cells underwent apoptosis (green-stained cells) after 150 μM H2O2 treatment (Fig. 3A) (P < 0.001). However, with 75 μM H2O2, the fractions of cells underwent apoptosis in both cell lines were not significantly different.
Fig. 3.
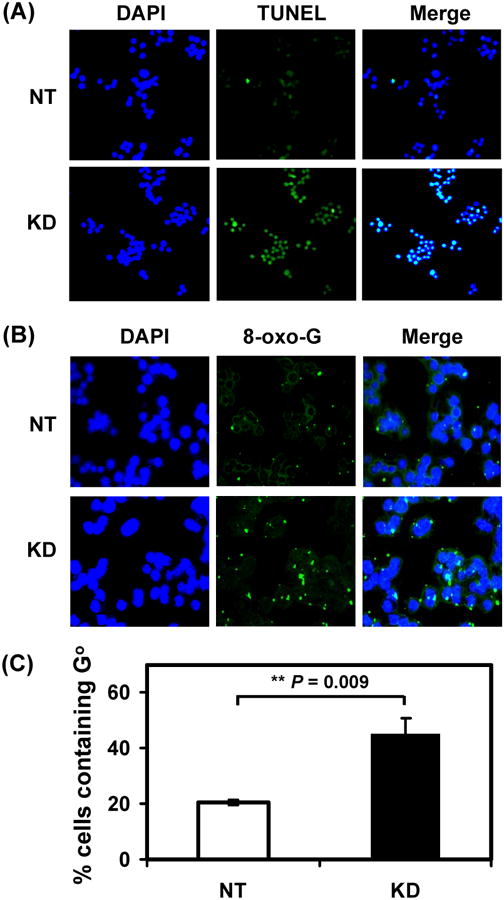
HeLa cells with hMYH knockdown are more sensitive to apoptosis and contain higher level of 8-oxoG than control cells after H2O2 treatment. HeLa cells were stably transfected with lentivirus containing shRNA against hMYH or non-target (NT) shRNA. (A) Cells were treated with 150 μM H2O2 for 1 hr, replaced with fresh media for one day, and subjected to DAPI staining and TUNEL assay. Both were merged in the third column. (B) Cells were treated with 150 μM H2O2 for 1 hr and subjected to immuno-florescent staining with anti-8-oxo-dG antibody. Nuclear DNA was counterstained with DAPI. Upper panel, cells with control NT shRNA. Lower panel was from cells with hMYH knockdown. (C) Quantitative analyses of GO accumulation of NT (white bar) and MYH-ND (black bars) cells from three experiments. The percentage (%) of cells containing GO is presented after H2O2 treatment. The error bars reported are the standard deviations of the averages and P-value shown was calculated by Student's t-test.
3.3. hMYH knockdown cells contain higher amount of 8-oxoG
Repair of GO lesion proceeds via redundant and complimentary mechanisms (reviewed in [10]). hMTH1 (MutT homolog) eliminates 8-oxo-dGTP from the nucleotide pool and hOGG1 (8-oxo-G glycosylase) removes GO adducts paired with cytosines. hMYH glycosylase and the hMSH2/hMSH6-mediated mismatch repair increase replication fidelity by removing the adenine misincorporated opposite GO [25,43,44]. Although MYH does not directly remove GO, MYH may generate favorable substrates for other repair enzymes such as OGG1. To see whether hMYH has a role in GO removal, we measured the GO level in hMYH KD cells. As shown in Fig. 3B (1st panel) and Fig. 3C, 20% of HeLa cells with non-target shRNA contained GO after treatment with 75 μM H2O2. In contrast, 45% of hMYH KD cells contained GO after H2O2 treatment (Fig. 3B, 2nd panel and Fig. 3C) (P = 0.009). Thus, hMYH affects GO content under oxidative stress.
3.4. hMYH knockdown cells have altered DNA signaling activation in response to H2O2 challenge
DNA damage activates ATM or ATR which in turn phosphorylates a large number of proteins including Chk1, Chk2, p53, and BRCA1 [45]. Some of these target proteins are transducers that can phosphorylate additional downstream effectors such as Cdc25. Upon phosphorylation by Chk1 or Chk2, Cdc25 phosphatase is degraded leading to the accumulation of phosphorylated Cdc2 and G1/S or G2/M arrest [29]. The effect of hMYH on the phosphorylation of Chk1 and Cdc25 in response to H2O2 treatment was investigated. Interestingly, the level of hMYH increased after 6 hrs post H2O2 treatment in control cells (Fig. 4, 1st panel, compare lane 3 to lane 1) but not in hMYH KD cells (Fig. 4, 1st panel, compare lane 8 to lane 6). Total Chk1 protein was slightly reduced in untreated hMYH KD cells (Fig. 4, 2nd panel, compare both bands in lane 1 to lane 6). H2O2 treatment did not induce much change in total Chk1 level in both control and KD cells (Fig. 4, 2nd panel). The level of phosphorylated Chk1 increased rapidly in both control and MYH KD cells at one hr post stress and then declined (Fig. 4, 3rd panel). The rarios of phosphorlated Chk1 over total Chk1 protein did not change between the two cell lines. Both the phosphorylated form and total amounts of Cdc25C were reduced about 3-fold in post H2O2-treated hMYH KD cells (Fig. 4, 4th and 5th panels). Phosphorylated Cdc25C increased substantially at 6-24 hrs post-H2O2 treatment in control cells (Fig. 4, 5th panel, lanes 3 and 4). Thus, MYH KD cells have a reduced DNA damage response.
Fig. 4.
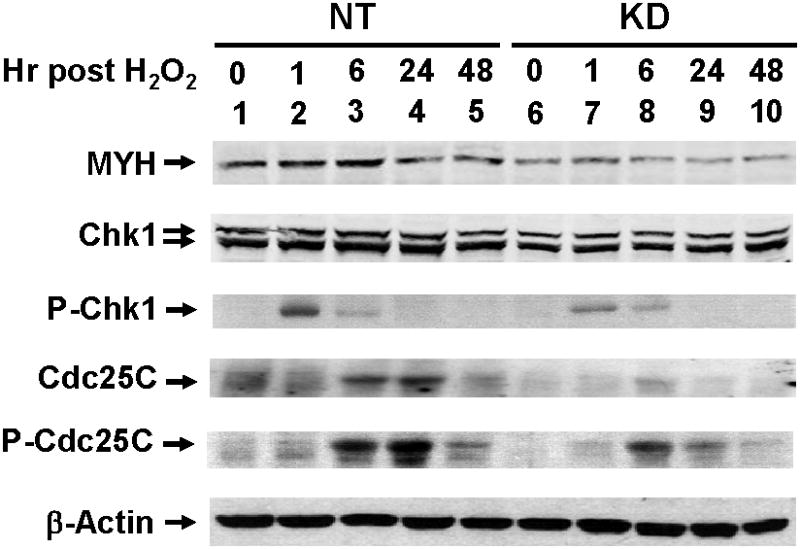
DNA damage signaling in hMYH-knockdown HeLa cells after H2O2 treatment. HeLa cells were stably transfected with lentivirus containing shRNA against hMYH or non-target (NT) shRNA. Cells were treated with 150 μM H2O2 for 1 hr and then recovered for different time intervals as indicated. Cell extracts were prepared for Western blotting analysis with antibodies against different proteins. P-Chk1 and P-Cdc25C are Ser 317-phosphorylated Chk1 and Ser 216-phosphorylated Cdc25C, respectively.
3.5. hMYH knockdown perturbs cell cycle progression
Because hMYH KD cells have altered DNA signaling activation in response to H2O2, we investigate cell cycle progression by flow cytometry. Cells were synchronized at S phase by double thymidine block, treated with 75 μM H2O2 for one hour, and then collected for cell cycle analysis. The FACS profiles and their quantitative evaluation are shown in Fig 6. Prior to H2O2 treatment, the MYH KD cells contained more G1, fewer S and fewer G2 cells than the control cells. Specifically, the S3 (late S phase) population was lower in the MYH KD cells (Fig. 5E, 0 hr time points). The cell cycle was progressed slightly slower in the MYH KD cells (6-24 hrs time points in Fig. 5C and 5D). At 24 hrs after release, the control cells had equal population in G1 and S while the MYH KD cells had less S population than G1 population (24 hrs time points in Fig. 5C and 5D). The S3 (late S phase) population was much lower in the MYH KD cells (Fig. 5E, 24 hrs time points). The major difference between control and the MYH KD cells is a pronounced delay of the stressed MYH KD cell population in the transit through the S phase of the cell cycle.
Fig. 6.
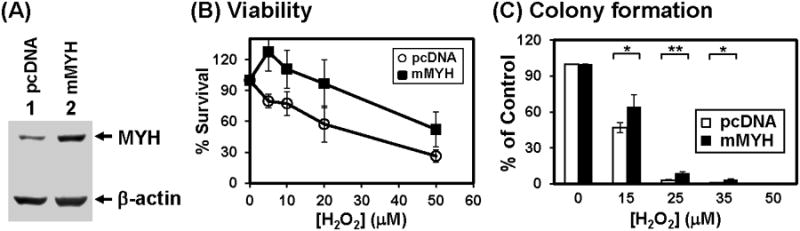
HCT15 (MSH6-defective) human cells expressing exogenous mMYH is more resistant to hydrogen peroxide. (A) Mouse MYH protein was over-expressed in HCT15 cells. Cells were transfected with pcDNA (vector) or mMYH. Extracts were prepared for Western blotting with hMYH and actin antibodies. Both endogenous hMYH and expressed mMYH were detected. (B) Sensitivity to H2O2 of MYH overproducing cells. Both control and MYH overproducing cells were treated with H2O2 at indicated concentrations for 1 hr. The survived cells were detected after 24 hrs. Survival differences are statistical significant between two cell lines at 5, 10, and 20 μM H2O2. (C) Quantitative analyses of the relative colony formation in cells containing vector (white bar) or mMYH (black bar) from three experiments. The percentage (%) is calculated from the ratios of H2O2 treated over untreated samples. The error bars reported are the standard deviations of the averages and P-value was calculated by Student's t-test. One star indicates P < 0.1 and two stars indicate P < 0.05.
Fig. 5.

Cell cycle progression in control and MYH knockdown HeLa cells after exposure to H2O2. HeLa cells were stably transfected with lentivirus containing shRNA against hMYH (KD) or non-target (NT) shRNA. Cells were synchronized at S phase by double thymidine block, treated with 75 μM H2O2 for one hour, and then collected for cell cycle analysis. 0 hr reflects the time of H2O2 treatment and removal of thymidine. (A) The FACS profiles of NT control cells. (B) The FACS profiles of MYH knockdown cells. (C) and (D), Quantitative evaluations of (A) and (B), respectively. The panels show the percentage of population in G1 (black bars), S (grey bars) and G2 (open bars) phases of the cell cycle. (E) Quantitative analyses of late S-phase (S3) population in both NT and MYH KD cells.
3.6. MYH overproduction protects cells from killing by oxidative damage
Adenine misincorporated opposite GO can be repaired by both MYH-mediated base excision repair and MSH2/MSH6-mediated mismatch repair [25,43,44]. We have shown that the DNA binding and glycosylase activities of hMYH are enhanced by hMSH2/hMSH6 complex [41], suggesting that this interaction may be a means by which hMYH repair and mismatch repair cooperate in reducing replication errors caused by oxidized bases. Because mismatch repair can repair oxidative damage [43,44,46], we used human mismatch repair defective HCT15 colon tumor cell line which carries a frameshift mutation in hMSH6 gene [47] to obtain a better effect of hMYH overproduction on DNA repair. We observed that HCT15 cells are more sensitive to H2O2 than HeLa cells (data not shown), therefore lower doses of H2O2 were used in experiments with HCT15 cells. Mouse MYH (mMYH) [48] is 77% identical to hMYH. This high conservation suggests that mMYH will function in human cells. Mouse MYH (mMYH) was overexpressed in HCT15 cells (Fig. 6A). Cells overexpressing mMYH were more resistant to killing by 5-20 μM of H2O2 than cells containing vector alone in viability assay (Fig. 6B) (P < 0.1). In the colony formation assay, mMYH overexpressing cells had better survival after 15-35 μM of H2O2 treatments than control cells (Fig. 6C) (P < 0.1). Moreover, only 2% of HCT15 cells expressing mMYH underwent apoptosis as compared to the control cells where 30% cells underwent apoptosis after treatment with 25 μM of H2O2 (Figs. 7A and 7B) (P = 0.001). The differences between colony formation and apoptosis are due to the time of measurement. Colony formation is measured at 10 days after peroxide treatment while apoptosis is detected at 24 hrs following peroxide treatment. Some cells do not show apoptosis signal at 24 hrs may die after 10 days. Thus, cells overexpressing mMYH are more resistant to peroxide treatment than the control cells with vector.
Fig. 7.
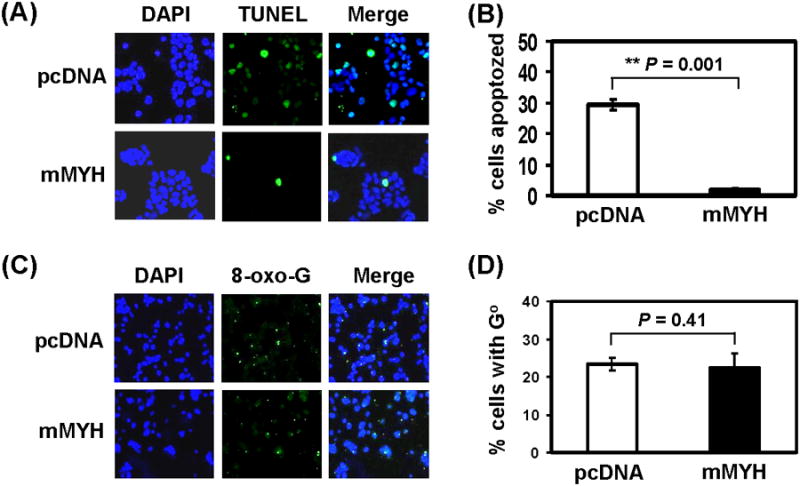
HCT15 (MSH6-defective) human cells expressing exogenous mMYH are more refractory to apoptosis than control cells, but contain the same levels of 8-oxo-G lesions as control cells expressing only endogenous hMYH after peroxide treatment. (A) Cells were stably transfected with pcDNA or mMYH and then treated with 0 or 25 μM H2O2 for 1 hr, recovered in fresh media for one day, and subjected to TUNEL analysis and DAPI staining. (B) Quantitative analyses of apoptosis in cells containing vector (white bar) or mMYH (black bar) from three experiments. The percentage (%) of apoptozed cells is presented after H2O2 treatment. The error bars reported are the standard deviations of the averages and P-value shown was calculated by Student's t-test (C) Cells similar as in (A) were treated with 35 μM H2O2 for 1 hr and subjected to immuno-florescent staining with 8-oxo-dG antibody and DAPI staining. (D) Quantitative analyses of GO accumulation in cells containing vector (white bar) or mMYH (black bar) from three experiments. The percentage (%) of cells containing GO is presented after H2O2 treatment. The error bars reported are the standard deviations of the averages and P-value shown was calculated by Student's t-test.
3.6. HCT15 cells overexpressing exogenous mMYH contained the same levels of 8-oxo-G as cells expressing only endogenous hMYH
As shown above, hMYH knockdown cells contain higher amounts of GO. To test whether MYH overexpression has any effect on 8-oxoG level, we compared the GO level in these HCT15 cells following treatment with 35 μM H2O2. The GO level is about the same in mMYH overexpressed cells and the control cells containing vector (Fig. 7C and 7D) (P = 0.41). Thus, overproduction of MYH does not facilitate GO removal.
3.7. MYH overproduction alters DNA damage signaling after oxidative stress
From above, we have detected abnormal signal response of DNA damage in hMYH KD cells. Thus, we examined the DNA damage response in MYH overproduction cells. As shown in Fig. 8 (panel 1, lanes 1-5), the hMYH level was increased after H2O2 treatment in HCT15 cells containing vector. The high level of MYH (hMYH and mMYH) remained almost unchanged in the overproduction cells (Fig. 8, panel 1, lanes 6-10). Total Chk1 protein levels were higher in untreated HCT15 cells overproducing mMYH (Fig. 8, 2nd panel, compare lane 6 to lane 1). H2O2 treatment did not induce significant change in total Chk1 levels in control cells (Fig. 8, 2nd panel, lanes 1-5). Total Chk1 protein levels increased in HCT15 cells overproducing mMYH after H2O2 treatment (Fig. 8, 2nd panel, lanes 6-10). The level of phosphorylated Chk1 is high in mMYH overproducing cells (Fig. 8, panel 3, compare lane 6 to lane 1). The level of phosphorylated Chk1 increased in control cells at six hour post stress and then declined (Fig. 8, 3rd panel, lanes 1-5). However, the level of phosphorylated Chk1 was induced at one hour post stress and then declined in mMYH overproducing cells (Fig. 8, 3rd panel, lanes 6-10).
Fig. 8.
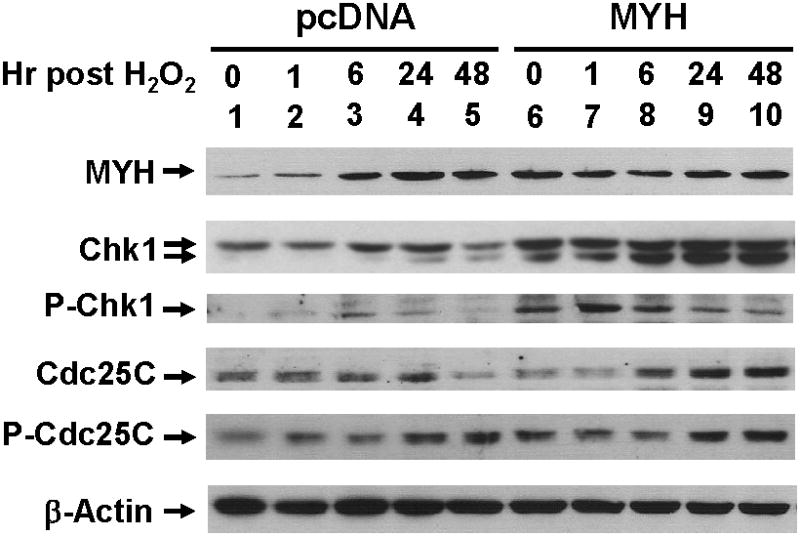
DNA damage signaling in mMYH-overproducing HCT15 cells after H2O2 treatment. Cells transfected with pcDNA or mMYH were treated with 25 μM H2O2 for 1 hr, and then recovered for different time intervals as indicated. Cell extracts were subjected to Western blotting analysis. P-Chk1 and P-Cdc25C are Ser 317-phosphorylated Chk1 and Ser 216-phosphorylated Cdc25C, respectively.
The total amounts of Cdc25C increased at 24 hrs and 6 hrs following treatment with H2O2 in control cells and mMYH overproducing cells, respectively (Fig. 8, 4th panel). The basal levels of phosphorylated Cdc25C were higher in mMYH overproducing cells as compared to pcDNA control (Fig. 8, 5th panel, compare lane 6 to lane 1). The level of phosphorylated Cdc25C increased in control cells at 24-48 hrs post stress (Fig. 8, 5th panel, lanes 1-5). However, the level of phosphorylated Cdc25C declined in 1-6 hrs and then increased after 24 hrs post stress in mMYH overproducing cells (Fig. 8, 5th panel, lanes 6-10). Thus, cells overexpressing MYH have an altered DNA damage response after oxidative stress.
3.8. Cell cycle progression in MYH overproducing cells
Because hMYH overproducing cells have altered DNA signaling activation in response to H2O2, we also investigate cell cycle progression by flow cytometry. Cells were synchronized at S phase by double thymidine block, treated with 25 μM H2O2 for one hour, and then collected for cell cycle analysis. The FACS profiles and their quantitative evaluation are shown in Fig 9. The overall cell cycle profiles were very similar in both control and hMYH overproducing cells (Fig. 9A-9D). When the S-phase was subdivided into four stages, we observed there are more S3 cells at double-thymidine blocked MYH-overexpressing cells at 0-3 hrs. We suggest that MYH-overexpressing cells, but not the control cells, have completely reached the late S phase after synchronization by double thymidine block. In addition, about double amounts of cells at the greater than G2 were observed in MYH-overexpressing cells at all time points. These represent cell aggregation. Thus, there are only subtle differences in cell cycle progression in response to oxidative stress between control and the MYH KD cells.
Fig. 9.

Cell cycle progression in control and MYH overproducing HCT15 cells after exposure to H2O2. Cells stably transfected with pcDNA or mMYH were synchronized at S phase by double thymidine block, treated with 25 μM H2O2 for one hour, and then collected for cell cycle analysis. (A) The FACS profiles of control cells. 0 hr reflects the time of H2O2 treatment and removal of thymidine. (B) The FACS profiles of MYH overproducing cells. (C) and (D), Quantitative evaluations of (A) and (B), respectively. The panels show the percentage of population in G1 (black bars), S (grey bars) and G2 (open bars) phases of the cell cycle. (E) Quantitative analyses of late S-phase (S3) population in both control and MYH overproducing cells.
4. Discussion
In this study, we have investigated the effects of under- and over-expression of MYH on cell survival and damage response to oxidative stress. We show that MYH is a vital DNA repair enzyme that protects human cell from oxidative DNA damage and is critical for proper cellular response to DNA damage. The hMYH knockdown HeLa cells display enhanced susceptibility to apoptosis, contain higher 8-oxoG lesion, and have altered checkpoint signaling activation and cell cycle progression in response to oxidative stress. Overexpression of mouse MYH in human mismatch repair defective cells causes the cells more resistant to killing and more refractory to apoptosis by oxidative stress than the control cells. However, overexpression of MYH does not alter 8-oxoG accumulation and only causes minor change in cell cycle progression in response to oxidative stress.
Germline mutations in the hMYH gene is associated with human colorectal adenomatous polyposis in MAP patients [17-21] and down-regulation of MYH expression has been shown in human gastric cancer [49]. Cells harboring certain mutations in the hMYH gene are prone to G:C to T:A mutations in APC, K-ras, and other genes [17,22,23]. The protective role of hMYH in cancer formation was thought to stem from reduction of GO-induced mutagenesis. However, MYH may have other functions besides mutation avoidance such as involvement in checkpoint activation and apoptosis upon DNA damage. Oka et al. [26,27] have used Ogg1 knockout mouse cells selectively expressing a nuclear or mitochondrial form of hOGG1 to identify two distinct pathways of cell death induced by oxidative damage. The authors observed that KD of mMYH reduces the cell death triggered by 8-oxoG in both pathways. The authors suggested that MYH glycosylase activity generates strand breaks that lead to cell death and thus preventing cancer formation. Our results are different from the findings of Oka et al. [26,27]. We found that human HeLa cells with hMYH knockdown increase apoptosis and contain higher levels of 8-oxoG lesions while mismatch repair defective human cells overexpressing MYH are more resistant to oxidation stress and reduce apoptosis by oxidative stress. Our results is in agreement of the findings of Molatore et al. [50] and Turco et al. [36] that MYH knockout mouse cells contain higher GO and more sensitive to oxidation stress than the same cells expressing hMYH. Although further studies are necessary to evaluate the discrepancy, the role of MYH in apoptosis in response to oxidative stress may be dependent on cell-type (human vs. mouse cells), the presence of other repair systems, type of DNA damage, and dose and type of DNA damaging agents. It has been demonstrated that human and mouse develop different cancer types when MYH gene is mutated or knocked out [17-21,24]. In response to DNA damage, DNA repair is enhanced and cell cycle is arrested to allow time for DNA repair. Apoptosis is triggered only in the case of extreme DNA damage [28,29]. The question is when and how apoptosis is induced. In the presence of 9-1-1, APE1, and other factors, MYH-mediated BER is enhanced and coordinated to completion. We have observed that MYH protects against apoptosis in two different model cell systems when those cells are subjected to oxidative stress. MYH protects against apoptosis under those conditions because DNA damage response is activated to minimize cytotoxic intermediates such as AP sites and strand breaks.
The 8-oxoG levels are reduced by the combined actions of MYH and other repair pathways. Because MYH does not directly remove 8-oxo-G, our model is MYH may convert A/GO to C/GO which is then repaired by OGG1 or other repair pathways to C/G. In MYH KD but OGG1 positive human cells as in our system, OGG1 can remove GO from GO/AP, GO/C, GO/T, and GO/G mismatches but not from Go/A mispairs. Apoptosis may be triggered following oxidative stress because cells accumulate GO or uncontrolled OGG1 excision activity on parental DNA strands. We have shown that there is an interaction between MutY and MutM in E. coli. The tight binding of MutY with GO mispaired with T, G, and abasic site may block MutM glycosylase activity [51] from generating double-strand breaks or unfavorable repair on parental DNA strands. This similar situation may occur in the mammalian system [52]. In MYH proficient but nuclear or mitochondrial OGG1-depleted cells as in the systems used by Oka et al. [26], cells may be prone to apoptosis because MYH-mediated BER is not coordinated to completion.
We have proposed a model that MYH first recognized a lesion or a mismatch and then recruits the 9-1-1 complex to activate DNA damage response [30,31,33]. Most lines of evidence to support this model are derived from the studies of S. pombe Myh1. Recent findings in the mammalian systems also indicate that MYH acts as an adaptor for checkpoint sensors. An hMYH mutant with reduced interaction with the 9-1-1 complex cannot complement the repair-defective phenotype of myh-defective cells [36]. According to this model, the checkpoint activation such as Chk1 phosphorylation should be dependent on MYH expression. It has been shown that hMYH-disrupted HEK293 cells decrease Chk1 phosphorylation, increase Cdk2 phosphorylation, and increase the amount of Cdc25A upon HU or UV treatment [35]. The authors did not offer any mechanism for MYH-dependent DNA damage response by UV damage or replication stress. In S. pombe, myh1 knockout cells confers a very mild UV and HU sensitivity [53,54]; only myh1 rad1 double mutant display enhanced sensitivity to UV and HU as compared to single mutants [54].
Here, we show that hMYH KD HeLa cells have altered DNA signaling activation following H2O2 treatment. Cells treated with H2O2 generate several types of base lesions including GO whose mutation potential is prevented by MYH [25]. The levels of phosphorylated Chk1 and Cdc25C induced by oxidative stress are lower in hMYH KD HeLa cells than in control cells. In agreement with this, the levels of phosphorylated Chk1 and Cdc25C induced by oxidative stress are elevated in mMYH overproducing cells. Cells overexpressing mMYH contain higher amount of Chk1. The induction of phosphorylation of Chk1 is faster in mMYH overproducing cells as compared to control cells. Thus, MYH is a key regulator for checkpoint activation in both unstressed and oxidatively stressed cells. However, the mechanism by which MYH affects CHK1 and Cdc25C expression and phosphorylation is not clear and requires further investigation.
The altered DNA damage response following H2O2 treatment may perturb cell cycle progression in hMYH KD cells. In the unstressed synchronized MYH KD cell population, we observed more G1, fewer S (particularly the S3 cells) and fewer G2 cells than the control cells. At 24 hs after release, the control cells had equal population in G1 and S while the MYH KD cells had less S population than G1 population. This S phase arrest phenotype in MYH KD cells is consistent with the finding of Turco et al. [36]. These authors reported that sustained S phase arrest in oxidatively stressed cells expressing an hMYH mutant defective in interaction with the 9-1-1 complex [36]. From the cell cycle progression profiles, we observed there are more S3 cells at double-thymidine blocked MYH-overexpressing cells at 0-3 hrs. However, the overall cell cycle profiles were very similar in both control and hMYH overproducing cells. We have shown that the physical association between hMYH and the 9-1-1 complex is enhanced following treatment with ionization radiation and that hMYH colocalizes with hRad9 to foci after stress in human cells [31]. These data support that MYH is an “adaptor” to recruit the 9-1-1 complex to the site of damage. This recruitment enables crosstalk between the two pathways in which the 9-1-1 complex enhances MYH repair activity and simultaneously activates the DNA damage response [30,31]. It has been suggested that the coordination of MYH repair with DNA damage response is essential cell survival following oxidative stress.
Highlights.
In contrast to previous studies using mouse knockout cells, we investigate MYH overproduction in human HeLa cells and MYH knockdown in mismatch repair defective cells.
The hMYH knockdown human HeLa cells are more sensitive to the killing effects of H2O2 than the control cells.
hMYH knockdown cells display enhanced susceptibility to apoptosis, and have altered DNA signaling activation in response to oxidative stress.
hMYH knockdown cells contain higher levels of 8-oxoG lesions than the control cells following H2O2 treatment.
Overexpression of mouse MYH (mMYH) in human mismatch repair defective HCT15 cells makes the cells more resistant to killing and refractory to apoptosis by oxidative stress than the cells transfected with vector.
MYH is a vital DNA repair enzyme that protects cell from oxidative DNA damage and is critical for proper cellular response to DNA damage.
Acknowledgments
The authors thank Dr. Yusaku Nakabeppu (Kyushu University, Japan) for kindly providing the mouse MYH clone. We acknowledge the use of the Shared Flow Cytometry Facility at the University of Maryland Greenbaum Cancer Center. We appreciate the critical reading by Drs. Eric Toth and Amrita Madabushi. This work was supported by National Institutes of Health Grant R01-CA78391.
Footnotes
Conflict of interest statement: The authors declare that there is no conflict of interest.
References
- 1.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. ASM Press; Washington, D.C.: 2005. [Google Scholar]
- 2.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 3.Krokan HE, Nilsen H, Skorpen F, Otterlei M, Slupphaug G. Base excision repair of DNA in mammalian cells. FEBS Lett. 2000;476:73–77. doi: 10.1016/s0014-5793(00)01674-4. [DOI] [PubMed] [Google Scholar]
- 4.Mol CD, Parikh SS, Putnam CD, Lo TP, Tainer JA. DNA repair mechanisms for the recognition and removal of damaged DNA bases. Annu REv Biophys Biomol Struct. 1999;28:101–128. doi: 10.1146/annurev.biophys.28.1.101. [DOI] [PubMed] [Google Scholar]
- 5.Avkin S, Livneh Z. Efficiency, specificity and DNA polymerase-dependence of translesion replication across the oxidative DNA lesion 8-oxoguanine in human cells. Mutat Res. 2002;510:81–90. doi: 10.1016/s0027-5107(02)00254-3. [DOI] [PubMed] [Google Scholar]
- 6.Maga G, Hubscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci. 2003;116:3051–3060. doi: 10.1242/jcs.00653. [DOI] [PubMed] [Google Scholar]
- 7.Maga G, Villani G, Crespan E, Wimmer U, Ferrari E, Bertocci B, Hubscher U. 8-oxo-guanine bypass by human DNA polymerases in the presence of auxiliary proteins. Nature. 2007;447:606–608. doi: 10.1038/nature05843. [DOI] [PubMed] [Google Scholar]
- 8.Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431–434. doi: 10.1038/349431a0. [DOI] [PubMed] [Google Scholar]
- 9.Wood ML, Dizdaroglu M, Gajewski E, Essigmann JM. Mechanistic studies of ionizing radiation and oxidative mutagenesis: Genetic effects of single 8-hydroxyguanine (7-hydro-8-oxoguanine) residue inserted at a unique site in a viral genome. Biochemistry. 1990;29:7024–7032. doi: 10.1021/bi00482a011. [DOI] [PubMed] [Google Scholar]
- 10.Lu AL, Li X, Gu Y, Wright PM, Chang DY. Repair of oxidative DNA damage. Cell Biochem Biophy. 2001;35:141–170. doi: 10.1385/CBB:35:2:141. [DOI] [PubMed] [Google Scholar]
- 11.David SS, O'Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gu Y, Lu AL. Differential DNA recognition and glycosylase activity of the native human MutY homolog (hMYH) and recombinant hMYH expressed in bacteria. Nucl Acids Res. 2001;29:2666–2674. doi: 10.1093/nar/29.12.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Markkanen E, Dorn J, Hubscher U. MUTYH DNA glycosylase: the rationale for removing undamaged bases from the DNA. Front Genet. 2013;4:18. doi: 10.3389/fgene.2013.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohtsubo T, Nishioka K, Imaiso Y, Iwai S, Shimokawa H, Oda H, Fujiwara T, Nakabeppu Y. Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucl Acids Res. 2000;28:1355–1364. doi: 10.1093/nar/28.6.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slupska MM, Luther WM, Chiang JH, Yang H, Miller JH. Functional expression of hMYH, a human homolog of the Escherichia coli MutY protein. J Bacteriol. 1999;181:6210–6213. doi: 10.1128/jb.181.19.6210-6213.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takao M, Zhang QM, Yonei S, Yasui A. Differential subcellular localization of human MutY homolog (hMYH) and the functional activity of adenine:8-oxoguanine DNA glycosylase. Nucl Acids Res. 1999;27:3638–3644. doi: 10.1093/nar/27.18.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Al Tassan N, Chmiel NH, Maynard J, Fleming N, Livingston AL, Williams GT, Hodges AK, Davies DR, David SS, Sampson JR, Cheadle JP. Inherited variants of MYH associated with somatic G:C to T:A mutations in colorectal tumors. Nat Genet. 2002;30:227–232. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- 18.Halford SE, Rowan AJ, Lipton L, Sieber OM, Pack K, Thomas HJ, Hodgson SV, Bodmer WF, Tomlinson IP. Germline mutations but not somatic changes at the MYH locus contribute to the pathogenesis of unselected colorectal cancers. Am J Pathol. 2003;162:1545–1548. doi: 10.1016/S0002-9440(10)64288-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones S, Emmerson P, Maynard J, Best JM, Jordan S, Williams GT, Sampson JR, Cheadle JP. Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and somatic G:C→T:A mutations. Hum Mol Genet. 2002;11:2961–2967. doi: 10.1093/hmg/11.23.2961. [DOI] [PubMed] [Google Scholar]
- 20.Sieber OM, Lipton L, Crabtree M, Heinimann K, Fidalgo P, Phillips RK, Bisgaard ML, Orntoft TF, Aaltonen LA, Hodgson SV, Thomas HJ, Tomlinson IP. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med. 2003;348:791–799. doi: 10.1056/NEJMoa025283. [DOI] [PubMed] [Google Scholar]
- 21.Sampson JR, Dolwani S, Jones S, Eccles D, Ellis A, Evans DG, Frayling I, Jordan S, Maher ER, Mak T, Maynard J, Pigatto F, Shaw J, Cheadle JP. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362:39–41. doi: 10.1016/S0140-6736(03)13805-6. [DOI] [PubMed] [Google Scholar]
- 22.Jones S, Lambert S, Williams GT, Best JM, Sampson JR, Cheadle JP. Increased frequency of the k-ras G12C mutation in MYH polyposis colorectal adenomas. Br J Cancer. 2004;90:1591–1593. doi: 10.1038/sj.bjc.6601747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lipton L, Halford SE, Johnson V, Novelli MR, Jones A, Cummings C, Barclay E, Sieber O, Sadat A, Bisgaard ML, Hodgson SV, Aaltonen LA, Thomas HJ, Tomlinson IP. Carcinogenesis in MYH-associated polyposis follows a distinct genetic pathway. Cancer Res. 2003;63:7595–7599. [PubMed] [Google Scholar]
- 24.Sakamoto K, Tominaga Y, Yamauchi K, Nakatsu Y, Sakumi K, Yoshiyama K, Egashira A, Kura S, Yao T, Tsuneyoshi M, Maki H, Nakabeppu Y, Tsuzuki T. MUTYH-null mice are susceptible to spontaneous and oxidative stress induced intestinal tumorigenesis. Cancer Res. 2007;67:6599–6604. doi: 10.1158/0008-5472.CAN-06-4802. [DOI] [PubMed] [Google Scholar]
- 25.Lu AL, Bai H, Shi G, Chang DY. MutY and MutY homologs (MYH) in genome maintenance. Front Biosci. 2006;11:3062–3080. doi: 10.2741/2033. [DOI] [PubMed] [Google Scholar]
- 26.Oka S, Ohno M, Tsuchimoto D, Sakumi K, Furuichi M, Nakabeppu Y. Two distinct pathways of cell death triggered by oxidative damage to nuclear and mitochondrial DNAs. EMBO J. 2008 doi: 10.1038/sj.emboj.7601975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oka S, Nakabeppu Y. DNA glycosylase encoded by MUTYH functions as a molecular switch for programmed cell death under oxidative stress to suppress tumorigenesis. Cancer Sci. 2011;102:677–682. doi: 10.1111/j.1349-7006.2011.01869.x. [DOI] [PubMed] [Google Scholar]
- 28.Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol. 2004;5:792–804. doi: 10.1038/nrm1493. [DOI] [PubMed] [Google Scholar]
- 29.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 30.Chang DY, Lu AL. Interaction of checkpoint proteins Hus1/Rad1/Rad9 with DNA base excision repair enzyme MutY homolog in fission yeast, Schizosaccharomyces pombe. J Biol Chem. 2005;280:408–417. doi: 10.1074/jbc.M406800200. [DOI] [PubMed] [Google Scholar]
- 31.Shi G, Chang DY, Cheng CC, Guan X, Venclovas C, Lu AL. Physical and functional interactions between MutY homolog (MYH) and checkpoint proteins Rad9-Rad1-Hus1. Biochem J. 2006;400:53–62. doi: 10.1042/BJ20060774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madabushi A, Lu AL. The novel role of cell cycle checkpoint clamp Rad9-Hus1-Rad1 (the 9-1-1 complex) in DNA repair. In: Berhardt Leon V., editor. Advances in Medicine and Biology. Nova Publishers; Hauppauge NY: 2011. pp. 41–74. [Google Scholar]
- 33.Chang DY, Shi G, Durand-Dubief M, Ekwall K, Lu AL. The role of MutY homolog (Myh1) in controlling the histone deacetylase Hst4 in the fission yeast Schizosaccharomyces pombe. J Mol Biol. 2011;405:653–665. doi: 10.1016/j.jmb.2010.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luncsford PJ, Chang DY, Shi G, Bernstein J, Madabushi A, Patterson DN, Lu AL, Toth EA. A structural hinge in eukaryotic MutY homologues mediates catalytic activity and Rad9-Rad1-Hus1 checkpoint complex interactions. J Mol Biol. 2010;403:351–370. doi: 10.1016/j.jmb.2010.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hahm SH, Park JH, Ko SI, Lee YR, Chung IS, Chung JH, Kang LW, Han YS. Knock-down of human MutY homolog (hMYH) decreases phosphorylation of checkpoint kinase 1 (Chk1) induced by hydroxyurea and UV treatment. BMB Rep. 2011;44:352–357. doi: 10.5483/BMBRep.2011.44.5.352. [DOI] [PubMed] [Google Scholar]
- 36.Turco E, Ventura I, Minoprio A, Russo MT, Torreri P, Degan P, Molatore S, Ranzani GN, Bignami M, Mazzei F. Understanding the role of the Q338H MUTYH variant in oxidative damage repair. Nucleic Acids Res. 2013;41:4093–4103. doi: 10.1093/nar/gkt130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Towbin HT, Staehlin T, Gordon J. Eletrophoretic transfer of proteins from polyacrylamide gel to nitrocellulose sheets procedure. Proc Natl Acad Sci U S A. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hirano S, Tominaga Y, Ichinoe A, Ushijima Y, Tsuchimoto D, Honda-Ohnishi Y, Ohtsubo T, Sakumi K, Nakabeppu Y. Mutator phenotype of MUTYH-null mouse embryonic stem cells. J Biol Chem. 2003;278:38121–38124. doi: 10.1074/jbc.C300316200. [DOI] [PubMed] [Google Scholar]
- 39.Unk I, Haracska L, Johnson RE, Prakash S, Prakash L. Apurinic endonuclease activity of yeast Apn2 protein. J Biol Chem. 2000;275:22427–22434. doi: 10.1074/jbc.M002845200. [DOI] [PubMed] [Google Scholar]
- 40.Repetto G, del Peso A, Zurita JL. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat Protoc. 2008;3:1125–1131. doi: 10.1038/nprot.2008.75. [DOI] [PubMed] [Google Scholar]
- 41.Gu Y, Parker A, Wilson TM, Bai H, Chang DY, Lu AL. Human MutY homolog (hMYH), a DNA glycosylase involved in base excision repair, physically and functionally interacts with mismatch repair proteins hMSH2/hMSH6. J Biol Chem. 2002;277:11135–11142. doi: 10.1074/jbc.M108618200. [DOI] [PubMed] [Google Scholar]
- 42.Parker A, Gu Y, Mahoney W, Lee SH, Singh KK, Lu AL. Human homolog of the MutY protein (hMYH) physically interacts with protein involved in long-patch DNA base excision repair. J Biol Chem. 2001;276:5547–5555. doi: 10.1074/jbc.M008463200. [DOI] [PubMed] [Google Scholar]
- 43.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 44.Modrich P. Mechanisms in eukaryotic mismatch repair. J Biol Chem. 2006;281:30305–30309. doi: 10.1074/jbc.R600022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, III, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 46.Colussi C, Parlanti E, Degan P, Aquilina G, Barnes D, MacPherson P, Karran P, Crescenzi M, Dogliotti E, Bignami M. The Mammalian mismatch repair pathway removes DNA 8-oxodGMP incorporated from the oxidized dNTP pool. Curr Biol. 2002;12:912–918. doi: 10.1016/s0960-9822(02)00863-1. [DOI] [PubMed] [Google Scholar]
- 47.Chang DK, Ricciardiello L, Goel A, Chang CL, Boland CR. Steady-state regulation of the human DNA mismatch repair system. J Biol Chem. 2000;275:18424–18431. doi: 10.1074/jbc.M001140200. [DOI] [PubMed] [Google Scholar]
- 48.Ushijima Y, Tominaga Y, Miura T, Tsuchimoto D, Sakumi K, Nakabeppu Y. A functional analysis of the DNA glycosylase activity of mouse MUTYH protein excising 2-hydroxyadenine opposite guanine in DNA. Nucleic Acids Res. 2005;33:672–682. doi: 10.1093/nar/gki214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shinmura K, Goto M, Suzuki M, Tao H, Yamada H, Igarashi H, Matsuura S, Maeda M, Konno H, Matsuda T, Sugimura H. Reduced expression of MUTYH with suppressive activity against mutations caused by 8-hydroxyguanine is a novel predictor of a poor prognosis in human gastric cancer. J Pathol. 2011;225:414–423. doi: 10.1002/path.2953. [DOI] [PubMed] [Google Scholar]
- 50.Molatore S, Russo MT, D'Agostino VG, Barone F, Matsumoto Y, Albertini AM, Minoprio A, Degan P, Mazzei F, Bignami M, Ranzani GN. MUTYH mutations associated with familial adenomatous polyposis: functional characterization by a mammalian cell-based assay. Hum Mutat. 2010;31:159–166. doi: 10.1002/humu.21158. [DOI] [PubMed] [Google Scholar]
- 51.Li X, Wright PM, Lu AL. The C-terminal domain of MutY glycosylase determines the 7,8-dihydro-8-oxo-guanine specificity and is crucial for mutation avoidance. J Biol Chem. 2000;275:8448–8455. doi: 10.1074/jbc.275.12.8448. [DOI] [PubMed] [Google Scholar]
- 52.Tominaga Y, Ushijima Y, Tsuchimoto D, Mishima M, Shirakawa M, Hirano S, Sakumi K, Nakabeppu Y. MUTYH prevents OGG1 or APEX1 from inappropriately processing its substrate or reaction product with its C-terminal domain. Nucleic Acids Res. 2004;32:3198–3211. doi: 10.1093/nar/gkh642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chang DY, Gu Y, Lu AL. Fission yeast (Schizosaccharomyces pombe) cells defective in the MutY-homologous glycosylase activity have a mutator phenotype and are sensitive to hydrogen peroxide. Mol Genet Genomics. 2001;266:336–342. doi: 10.1007/s004380100567. [DOI] [PubMed] [Google Scholar]
- 54.Jansson K, Warringer J, Farewell A, Park HO, Hoe KL, Kim DU, Hayles J, Sunnerhagen P. The tumor suppressor homolog in fission yeast, myh1(+), displays a strong interaction with the checkpoint gene rad1(+) Mutat Res. 2008;644:48–55. doi: 10.1016/j.mrfmmm.2008.07.001. [DOI] [PubMed] [Google Scholar]