

Abstract
Background
The PathoLogic program constructs Pathway/Genome databases by using a genome's annotation to predict the set of metabolic pathways present in an organism. PathoLogic determines the set of reactions composing those pathways from the enzymes annotated in the organism's genome. Most annotation efforts fail to assign function to 40–60% of sequences. In addition, large numbers of sequences may have non-specific annotations (e.g., thiolase family protein). Pathway holes occur when a genome appears to lack the enzymes needed to catalyze reactions in a pathway. If a protein has not been assigned a specific function during the annotation process, any reaction catalyzed by that protein will appear as a missing enzyme or pathway hole in a Pathway/Genome database.
Results
We have developed a method that efficiently combines homology and pathway-based evidence to identify candidates for filling pathway holes in Pathway/Genome databases. Our program not only identifies potential candidate sequences for pathway holes, but combines data from multiple, heterogeneous sources to assess the likelihood that a candidate has the required function. Our algorithm emulates the manual sequence annotation process, considering not only evidence from homology searches, but also considering evidence from genomic context (i.e., is the gene part of an operon?) and functional context (e.g., are there functionally-related genes nearby in the genome?) to determine the posterior belief that a candidate has the required function. The method can be applied across an entire metabolic pathway network and is generally applicable to any pathway database. The program uses a set of sequences encoding the required activity in other genomes to identify candidate proteins in the genome of interest, and then evaluates each candidate by using a simple Bayes classifier to determine the probability that the candidate has the desired function. We achieved 71% precision at a probability threshold of 0.9 during cross-validation using known reactions in computationally-predicted pathway databases. After applying our method to 513 pathway holes in 333 pathways from three Pathway/Genome databases, we increased the number of complete pathways by 42%. We made putative assignments to 46% of the holes, including annotation of 17 sequences of previously unknown function.
Conclusions
Our pathway hole filler can be used not only to increase the utility of Pathway/Genome databases to both experimental and computational researchers, but also to improve predictions of protein function.
Background
Genome sequencing projects generate large numbers of nucleotide sequences each year [1]. Once the sequences are obtained, functions must be assigned to these new sequences. This is typically accomplished by searching large, public databases for similar sequences. Assessment methods include hidden Markov models and identification of functional motifs [2-4]. These methods assign functional annotations based on sequence alone and do not incorporate non-sequence-based information.
Most genome annotation efforts fail to assign function to 40 – 60% of the new sequences. Even when annotated, many functions remain incomplete (only one function of a multidomain protein) or nonspecific (e.g., "thiolase family protein"). Operon- and pathway-based information can provide additional clues about protein function, and can clarify incomplete or nonspecific annotations. For instance, the annotation of CC1617 (a Caulobacter cresentus gene) as guaB (E.C.# 1.1.1.205) is supported by the fact that CC1617 is part of a predicted operon with guaA (E.C.# 6.3.5.2) and the pathway for de novo biosynthesis of purine nucleotides (I) includes both of these reactions.
The PathoLogic program [5] constructs Pathway/Genome Databases (PGDBs) by using a genome's annotation to predict the set of metabolic pathways present in an organism. PathoLogic determines the set of reactions catalyzed by an organism from the enzymes annotated in its genome. For each pathway in a set of reference pathways, if one or more reactions is present in the organism, PathoLogic adds that pathway to the set of pathways present in that organism. Figure 1 shows an example reference pathway and the reactions that PathoLogic has identified in the organism's genome. PathoLogic has assigned genes to three of the six reactions. When PathoLogic includes a pathway in a PGDB, the pathway may be missing one or more enzymes; we refer to these as "missing enzymes" or "pathway holes". We present a fully computational approach for finding missing enzymes (i.e., filling pathway holes), thus improving both the completeness and accuracy of the PGDB and the annotation of its associated genome.
Figure 1.
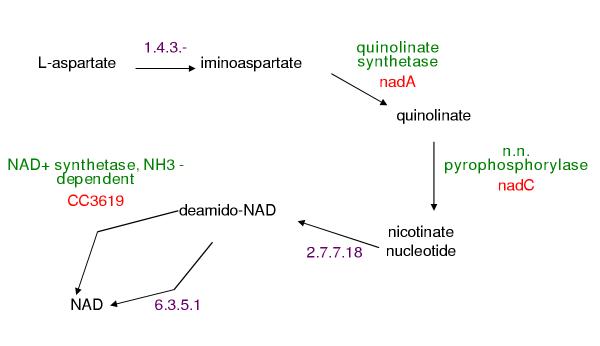
Example pathway created by PathoLogic for the Caulobacter cresentus PGDB, CauloCyc The enzymes for the quinolinate synthetase, nicotinate-nucleotide pyrophosphorylase, and NAD(+) synthetase reactions are known. The enzymes for the 1.4.3.-, nicotinate-nucleotide adenylyltransferase, and NAD(+) synthase (glutamine-hydrolyzing) reactions are missing.
Previous computational efforts to identify missing enzymes in metabolic pathways have focused solely on sequence similarity as a means to find candidate enzymes. Other methods have proposed the use of additional types of evidence, but have not provided a computational algorithm for combining these data [6-8]. Rather than merely providing a score of the similarity of a sequence from the genome of interest to sequences that catalyze the same reaction, we use a Bayes classifier to determine the probability that the candidate protein has the function required to fill the pathway hole. Our computational method focuses on identification of potential candidates to fill pathway holes and evaluation of these candidates by combining homology-, operon-, and pathway-based data. This approach will refine metabolic maps available in PGDBs and improve annotations for previously unannotated or incompletely annotated proteins.
Implementation
The pathway hole filler is implemented as part of the Pathway Tools software [5], a software environment for creating, editing, and querying PGDBs, such as EcoCyc and MetaCyc http://biocyc.org[9].
Overall algorithm
Figure 2 shows the overall algorithm used for filling a pathway hole. The steps of the algorithm applied to each reaction lacking an enzyme are
Figure 2.
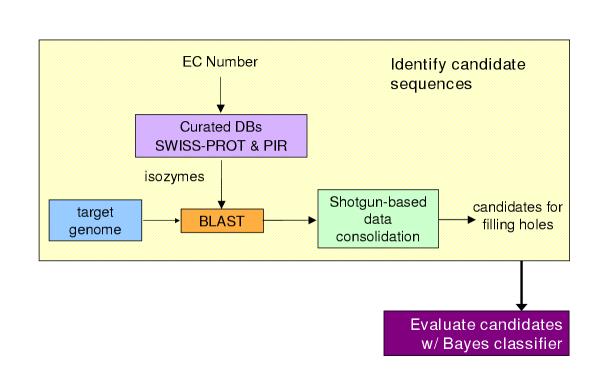
Overall algorithm for filling pathway holes
1. Sequence retrieval – Retrieve from Swiss-Prot [10] and PIR [11] sequences for enzymes that catalyze the desired reaction in other organisms. Because these sequences are not necessarily homologs, we will refer to enzymes with the same function in a variety of organisms as isozymes. For Swiss-Prot, the program retrieves Swiss-Prot IDs directly from the ENZYME database. For PIR sequences, the program retrieves IDs from the MetaCyc PGDB. Sequences are then retrieved directly from the most recent version of each database.
2. Homology search – BLAST [12,13] each query isozyme sequence against the genome of the organism of interest.
3. Data consolidation – Congruence analysis of the resulting BLAST hits to consolidate data reported for sequences that align with one or more query isozymes.
4. Candidate evaluation – Determine the probability that each candidate protein has the activity required by the missing reaction.
Steps 1 through 3 compose the "candidate identification" phase of the process and generate the evidence used in the fourth step, "candidate evaluation". Our implementation of the homology search and data consolidation steps is based on the Shotgun congruence analysis algorithm [14]. We applied our method to PGDBs developed by SRI's Bioinformatics Research Group for three organisms – CauloCyc (Caulobacter cresentus [15]), MtbRvCyc (Mycobacterium tuberculosis [16]), and VchoCyc (Vibrio cholerae [17]). All three PGDBs are available through the BioCyc Web site at http://BioCyc.org
In this report, we will describe both the candidate identification and evaluation steps of our algorithm, including the Bayesian network used for evaluation and the calculation of the conditional probability distributions used for the network. We will also present the results of a five-fold cross-validation study completed to evaluate the predictive value of various network structures. Our prediction of putative hole-fillers for three PGDBs will demonstrate the utility of the method for identifying functions for unannotated genes and for increasing the completeness of the metabolic map provided by a PGDB.
Candidate identification
To determine the function of a sequence, researchers typically use a single sequence of unknown function to search for potential homologs in a large, public database. We have, in effect, reversed this search process to fill pathway holes in a PGDB. The activity of the missing reaction is known from the inferred pathway; we search the genome for a sequence that will provide that function. Our method uses multiple isozymes from other organisms to search for similar sequences in the genome used to build the PGDB. Searching the smaller genome database greatly increases search sensitivity by reducing the probability of finding a match by chance [18,19]. Also, if a sequence with the desired function exists in the genome, its alignment with multiple isozymes will be more credible than an alignment between a single sequence queried against a large sequence database [4,13,14].
For each pathway hole, we retrieve isozyme sequences from the Swiss-Prot and PIR databases. Next, we use BLAST (version 2.0.10) to search the genome of interest with each of the query sequences. The BLAST queries use the default search parameters (e.g., filtering on, gapped) with the E-value cutoff reduced to 1.0 (from the default of 10) to reduce the number of false hits returned but maintain the ability to identify hits with remote similarity.
Our program evaluates each candidate based on the data collected from congruence analysis of the set of BLAST output files (one output file per isozyme sequence). For each candidate, the congruence analysis groups together the BLAST hits to the candidate in each output file. If a query isozyme's output file does not include a hit to the candidate, no data from that query is included. The evidence for each candidate hole-filler is a summary of this group of hits. For example, candidate Z is the first hit in output file A and the second hit in output file B, and none of the remaining output files include a hit to the candidate. The evidence for candidate Z will include data (E-values, alignment lengths, etc.) for the hit in output file A and the hit in output file B. Table 1 describes the parameters calculated for use in the Bayes classifier.
Table 1.
Node names and descriptions for Bayes classifier.
| node | description |
| has-function | true if the protein has the function required to fill the pathway hole, false if it does not |
| Shotgun-score | the number of query sequences whose BLAST output included the candidate sequence |
| best-E-value | negative log of the E-value for the best alignment of the candidate with a query sequence |
| average-rank | the average rank of the candidate sequence in the BLAST output lists (e.g., if a candidate is the best hit in each search, the average rank for the candidate is 1) |
| average-fraction-aligned | the average of each alignment length normalized by the length of the query sequence |
| pathway-directon | true if the hit is in the same directon as another gene in the same pathway; a directon is a contiguous series of genes transcribed in the same direction |
| adjacent-rxns | true if the hit is adjacent to one of the genes coding the enzyme for an adjacent reaction in the pathway |
Table 2 shows an example of a data consolidation step and Figure 3 is a graphic representation of the consolidation process. In the example in Table 2, five isozyme sequences were retrieved from Swiss-Prot and PIR. The five BLAST output files included hits to a total of seven candidate sequences from the Caulobacter genome – CC3619, CC1620, CC1541, CC3460, CC2963, CC3013, and CC0705. Because each of the five output files included hits to CC3619, its Shotgun-score is five (5). Likewise, the five values for the fraction of the query isozyme aligned to the hit are used to calculate the average-fraction-aligned for CC3619.
Table 2.
Example of consolidation of BLAST output data for glutamine-hydrolyzing NAD(+) synthase.
| Sequences producing significant alignments | E-value | rank in output | fraction aligned | |
| Hits found by Query Isozyme Q9CBZ6 | ||||
| CC3619 NAD(+) synthetase, putative | 0.0 | 1 | 0.99 | |
| CC2963 hydrolase, carbon-nitrogen family | 0.010 | 2 | 0.06 | |
| CC3013 TonB-dependent receptor | 0.34 | 3 | 0.07 | |
| Hits found by Query Isozyme Q58747 | ||||
| CC3619 NAD(+) synthetase, putative | 3e-07 | 1 | 0.70 | |
| CC1620 GMP synthase | 0.029 | 2 | 0.14 | |
| CC0705 ORF | 0.15 | 3 | 0.34 | |
| Hits found by Query Isozyme F86762 | ||||
| CC3460 ORF | 0.024 | 1 | 0.43 | |
| CC3619 NAD(+) synthetase, putative | 0.092 | 2 | 0.48 | |
| CC1541 2-isopropylmalate synthase | 0.80 | 3 | 0.16 | |
| Hits found by Query Isozyme P18843 | ||||
| CC3619 NAD(+) synthetase, putative | 2e-04 | 1 | 0.49 | |
| CC1541 2-isopropylmalate synthase | 0.36 | 2 | 0.43 | |
| Hits found by Query Isozyme P47623 | ||||
| CC3619 NAD(+) synthetase, putative | 6e-05 | 1 | 0.55 | |
| CC1620 GMP synthase | 0.24 | 2 | 0.09 | |
| Results of data consolidation | ||||
| consolidated data for hits from target genome | shotgun-score | best-E-value | average-rank | average-fraction-aligned |
| CC3619 | 5 | 0.0 | 1.2 | 0.64 |
| CC1620 | 2 | 0.029 | 2 | 0.12 |
| CC1541 | 2 | 0.36 | 2 | 0.30 |
| CC3460 | 1 | 0.024 | 1 | 0.43 |
| CC2963 | 1 | 0.010 | 2 | 0.06 |
| CC3013 | 1 | 0.34 | 3 | 0.07 |
| CC0705 | 1 | 0.15 | 3 | 0.34 |
Figure 3.
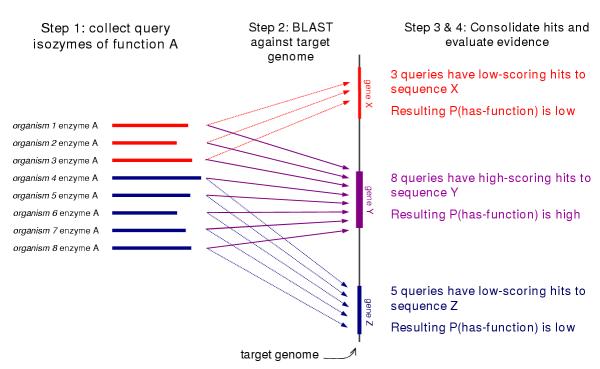
A graphical representation of the data consolidation process
Candidate evaluation
Bayesian network structure
Each of the candidate hits is evaluated by calculating the probability that the sequence encodes the desired function based on operon-, homology- and pathway-based data. We use a Bayesian network to calculate this probability.
Based on background knowledge of protein function prediction, we initially specified a Bayesian network [20] to capture the factors involved in predicting protein function from sequence alignment data. Part of our initial network, N0, is shown in Figure 4. The network includes nodes for E-values, alignment length, and the rank of the candidate protein in the isozyme's BLAST output. Each node is conditionally dependent on the has-function node, as indicated by the arcs connecting the node to the has-function node. Additional arcs indicate other conditional dependencies; for example, because alignment length affects the E-value of an alignment and the E-value affects the rank of the candidate in the BLAST output, N0 includes arcs connecting these nodes. If a hit has a very low E-value, it is more likely to be one of the first hits listed in the BLAST output (low rank). If the alignment between an isozyme and the candidate protein is longer, it may be more likely to have a lower E-value.
Figure 4.
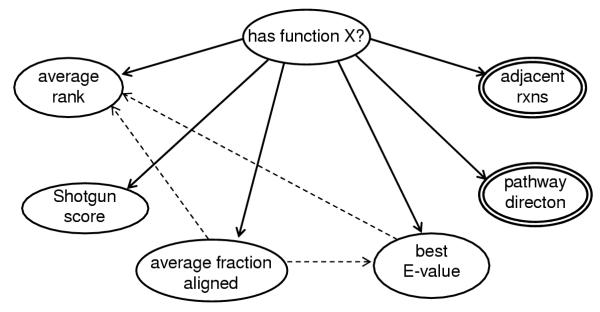
Network N0 includes all single-outline nodes plus both the solid and dashed arcs The simple Bayes classifier used by our program (N1) includes all nodes but excludes the dashed arcs. We simplified the network, excluding the dashed arcs to reduce the amount of data required to accurately construct the conditional probability distributions needed for the network.
While the N0 network may more accurately capture the relationship between these variables, large amounts of data would be required to determine the probability distribution for each node in the network. Thus, we instead chose to assess the adequacy of a drastically simpler network. Figure 4 shows both N0 and the static naïve Bayes classifier we evaluated, N1. We eliminated all conditional dependencies between the child nodes (shown as dashed lines in Figure 4). We also added pathway-based data to the network (shown as double-lined nodes in Figure 4). Each node in our model is described in Table 1. Since the naïve classifier has only one parent node (has-function), the probability distributions can be obtained from a much smaller dataset.
Calculating the probability that a candidate catalyzes the missing reaction
In evaluating the candidate sequences identified by our searches, we are primarily concerned with the network's parent node, has-function. The probability that this state is true, P(has-function), or P, is the probability that a candidate protein has the function needed for the missing reaction. The probability that the protein does not have the needed function is 1-P. Each of the other nodes in the network will have probability distributions conditioned on the state of the has-function node. For example, suppose our network structure included only the parent node, has-function, and two child nodes, average-rank and pathway-directon, as defined in Table 1. To calculate the probability that a candidate has the desired function given the evidence for that candidate, we need the following data: the evidence for the particular candidate (i.e., the candidate's values for average-rank and pathway-directon), the probability of finding that evidence if the candidate has the desired function or if it does not, and the prior probability that any candidate has the desired function. For our example network and candidate we need
I – The average-rank of the candidate and the value of pathway-directon. Let's assume that the values of average-rank and pathway-directon for our example candidate are 1.5 and "true", respectively.
II – The probability that a protein has a particular value for each child node given that it has or does not have the desired function. For our example candidate, P(average-rank = 1.5 | has-function) = 0.40 and P(average-rank = 1.5 | ¬has-function) = 0.03. Also, P(pathway-directon = true | has-function) = 0.24 and P(pathway-directon = true | ¬has-function) = 0.04.
III – The prior probability distributions are calculated from a data set where 4.1% of the candidates are true hits and 95.9% are false hits; thus, P(has-function) = 0.041 and P(¬has-function) = 0.959.
(I) is the evidence collected from the BLAST searches for each candidate protein. The values for (II) are taken from the conditional probability distributions. Forty percent of the candidates have an average-rank between 1.0 and 2.0 when the candidate is a true hit. Likewise, pathway-directon = "true" for 24% of the candidates that are true hits (conversely, pathway-directon is "false" for 76% of the candidates when the candidate is a true hit). Since our network assumes conditional independence among the child nodes, we use Bayes' Rule to determine P(has-function | average-rank = 1.5 and pathway-directon = "true"):

Calculation of conditional distributions
To perform this calculation for each candidate, we must determine a conditional probability distribution for each node in the network (conditioned on the state of the has-function node). We derived the data used to calculate these distributions from the known reactions in each PGDB.
Although some reactions in a PGDB are missing, other reactions in the pathways have proteins assigned to them. We call these "known" reactions. To generate data for the conditional distributions for our classifier, we ran the "candidate identification" step of our program on the set of known reactions in a PGDB, identifying a list of candidate hole-fillers for each reaction. Since each known reaction has been assigned one or more enzyme sequence by PathoLogic, we can identify true and false hits in the list of candidates identified for each reaction. For each reaction, all enzymes previously assigned by PathoLogic are considered true hits; all other hits are designated false hits. Given the set of true hits and false hits, we calculated conditional distributions for the network. As an example, the distributions for the average-fraction-aligned are shown in Figure 5.
Figure 5.
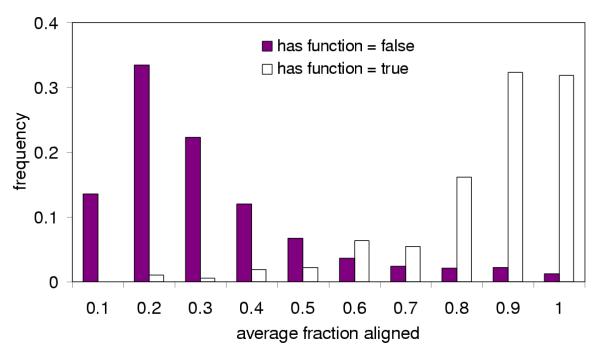
Example of conditional probability distribution calculated from the candidates identified for the known reactions in CauloCyc This figure shows the probability distribution for the average-fraction-aligned node. The set of candidates for all known reactions in the PGDB was divided into two subsets – true hits (those candidates that are assigned to a particular reaction in the PGDB) and false hits (those candidates that are not assigned to a particular reaction in the PGDB). We partition the values into nonoverlapping bins and determine the frequency of candidates within each bin for the two sets of hits. These frequencies make up the conditional probability distributions used for our Bayesian network. For example, if the avg fraction of query aligned for a candidate is 0.84, the P(average-fraction-aligned = 0.84 | has-function) = 0.16 and P(average-fraction-aligned = 0.84 | ¬has-function) = 0.02.
Cross-validation and statistical evaluation
To evaluate our model structure, we divided the set of known reactions in a PGDB and their candidate proteins randomly into five separate sets. Given the network and the associated distributions, we then calculated P for each candidate sequence from the data collected for the sequence. Using these true hits, false hits, and their associated P's, we evaluated various network structures. For each set, the probability distributions were derived from the other four.
Since classifying each protein (has-function or ¬has-function) depends on the chosen probability threshold, a single assessment of specificity and sensitivity will not completely describe the predictive power of the model. Hence, we compared models by plotting the number of true positives as a function of the number of false positives. We used McNemar's test [21] to determine statistically significant differences between the models at various false positive rates.
Results
While our program is generally applicable to any organism's PGDB, we evaluated its ability to match proteins to a missing enzyme using three PGDBs: CauloCyc (Caulobacter cresentus), MtbRvCyc (Mycobacterium tuberculosis strain H37Rv), and VchoCyc (Vibrio cholerae). The number of pathways, missing reactions, known reactions, and a summary of the data collected during the validation process for each PGDB are shown in Table 3.
Table 3.
PGDB and cross-validation statistics.
| Pathway/Genome Database | |||
| MtbRv | Caulo | Vcho | |
| pathways in PGDB | 146 | 139 | 172 |
| incomplete pathways | 118 | 99 | 116 |
| known reactions | 327 | 354 | 417 |
| missing reactions | 313 | 255 | 276 |
| cross-validation | |||
| # true hits | 491 | 469 | 506 |
| # false hits | 10093 | 9624 | 9151 |
| positive predictive value at P > 0.9 | 0.66 | 0.67 | 0.79 |
| true-positives at P > 0.9 | 325 | 371 | 449 |
Cross-validation and model exploration
To explore the adequacy of the model proposed for the Bayes classifier, we performed fivefold cross-validation on the known reactions from the three PGDBs.
The candidate identification step of our program generates an average of 27 candidate proteins per pathway hole. Although, we identify one true hit for most reactions, some reactions (e.g., 101 for VchoCyc) have multiple true hits (when multiple genes are assigned to a reaction either as part of a multimeric complex or as separate polypeptides that catalyze the same reaction), while other reactions (e.g., 44 for VchoCyc) have no true hits identified (i.e., either no isozyme sequences were available or all candidate proteins for that reaction are false hits).
Using data from the CauloCyc PGDB, we explored various network configurations to determine their effect on the predictive value of the classifier. The following three models were included in our investigation:
model 1 – classification of each hit by the Bayes classifier shown in Figure 4, N1
model 2 – classification of each hit by the Bayes classifier excluding the best-E-value node
model 3 – classification of each hit by E-value of the best alignment alone (i.e., choosing an E-value cutoff to classify hits as positive or negative)
Results for the three models are shown in Figure 6. Although model 3 performs better at low numbers of false positives (10 false positives, p = 0.04), at higher numbers (>31 false positives, p < 1e-5), model 1 outperforms both model 2 and model 3. The inset in Figure 6 displays results for all three PGDBs (CauloCyc, MtbRvCyc, and VchoCyc) predicted using model 1.
Figure 6.
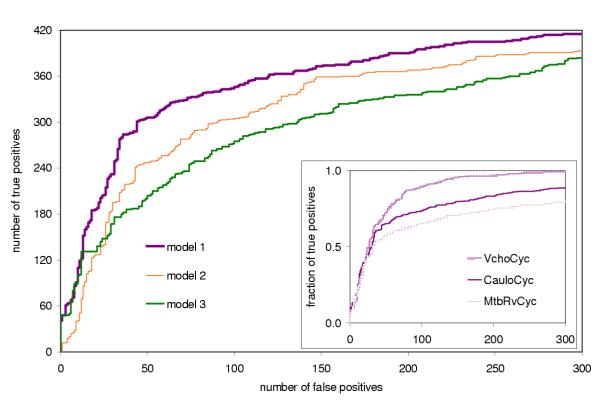
True positives versus false positives for classification using E-value cutoff alone, and using the Bayes classifier model without E-valuesThe inset shows the fraction of true positives versus number of false positives as determined by model 1 for all three PGDBs evaluated.
Candidate identification for missing reactions
We used model 1 to identify enzymes for the missing reactions in MtbRvCyc, CauloCyc, and VchoCyc.
Table 4 summarizes the results of filling pathway holes in the three databases. About 53% of the pathway holes in the three databases were filled using a cutoff of P > 0.9. A few additional pathway holes were filled with P(has-function) lower than our cutoff. All of these had nonspecific or unknown functions and in most cases P(has-function) was greater than 0.7. Specific changes made to the three PGDBs can be found in Additional files 1, 2, 3 and 4. Figure 7 shows the percent of pathway holes filled as a function of the probability threshold chosen for the three PGDBs. For about half of the pathway holes filled, the separation between the most likely candidate and the second most likely candidate was greater than 0.9, demonstrating that the highest scoring candidate is often the only reasonable contender for the missing enzyme.
Table 4.
Summary of hole-filling results.
| Pathway/Genome Database | |||
| MtbRv | Caulo | Vcho | |
| missing reactions w/ isozyme seqs | 195 | 162 | 156 |
| putative enzymes found at P > 0.9 | 84 | 71 | 82 |
| fillers with borderline P (0.5 to 0.9) | 37 | 17 | 11 |
| holes with no BLAST hits | 8 | 7 | 7 |
| holes filled with ORFs at P > 0.8 | 9 | 2 | 6 |
| newly completed pathways at P > 0.9 | 18 | 17 | 17 |
| newly completed pathways at P > 0.8 | 20 | 14 | 17 |
Figure 7.
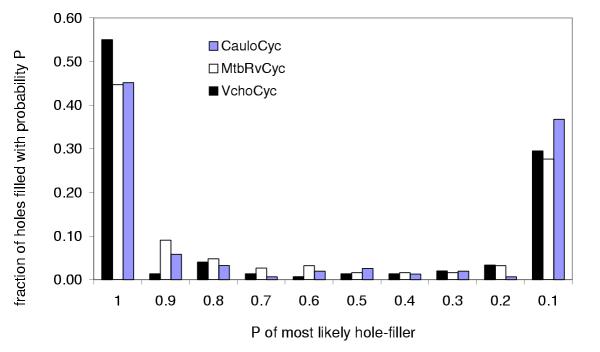
Fraction of pathway holes filled as a function of probability threshold
As a result of applying our program to the three PGDBs, we linked 17 missing reactions with sequences of previously unknown function. Our program identified putative enzymes for a total of 266 missing reactions across the three databases. These 266 instances fall into four categories explaining the absence missing enzyme.
1. The function of the protein was correctly annotated; however, due to inconsistent and nonstandardized annotations, the PathoLogic name-matcher did not recognize or understand the annotation, leading to a "missing enzyme" when the enzyme had, in fact, been identified in the genome already.
When creating a PGDB, PathoLogic uses an enzyme name-matcher and an EC-number-based-matcher to link enzymes in the genome annotation to reactions in the reference pathway database (i.e., MetaCyc). For each reaction, the name-matcher tries to match one of the protein products or its synonyms in the annotation to the activity of the reaction. If no gene's annotation matches any of the names/synonyms for the reference reaction, no enzyme will be linked to that reaction in the PGDB resulting in a missing enzyme in all pathways including that reaction.
2. The annotated activity is nonspecific (e.g., "thiolase family protein" or "probable phosphoribosyltransferase"). Our program provides a more exact specification of the enzyme's function than the original annotation.
3. The annotated activity is incomplete (e.g., a multifunctional protein annotated with only one function). Our program can identify additional functions that were missed by the original annotation process.
4. The functional annotation of the protein is inconsistent with the activity required for the reaction. These instances may represent either false positives classified by our program or, perhaps, incorrectly annotated proteins.
Example pathway: pyridine nucleotide biosynthesis
PathoLogic's prediction of the pyridine nucleotide biosynthesis pathway in CauloCyc is shown in Figure 1. Enzymes have been assigned for the quinolinate synthetase, nicotinate-nucleotide pyrophosphorylase, and NH3-dependent NAD+ synthetase reactions. The other three reactions in the pathway (L-aspartate oxidase, nicotinate-nucleotide adenylyltransferase, and glutamine-dependent NAD+ synthetase) remain as missing reactions. Our hole-filling program has identified enzymes in the Caulobacter genome for each of these missing reactions as shown in Figure 8.
Figure 8.
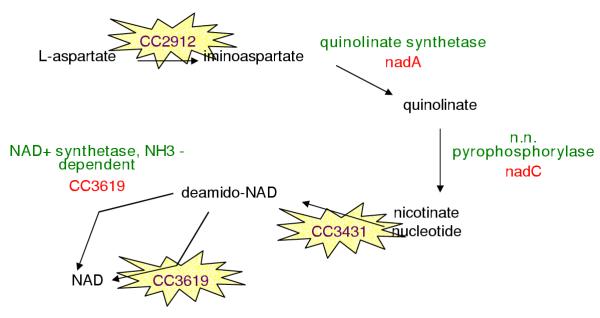
Pyridine biosynthesis pathway with putative enzyme assignments identified by our program
The first missing enzyme, nadB (CC2913), was actually annotated as L-aspartate oxidase in the Caulobacter genome. Because the reaction (and the E.C. number of the reaction) catalyzed by nadB changes depending on whether it acts as a monomer (1.4.3.-) or in complex with nadA (1.4.3.16) [22,23], CauloCyc includes two different reactions for L-aspartate oxidase. During the creation of CauloCyc, PathoLogic matched CC2913 to the monomeric reaction, but did not match the enzyme to the reaction catalyzed by the complex (complexes must be created by the user), leaving a hole in the pathway.
Our program assigns putative nicotinate-nucleotide adenylyltransferase activity to CC3431 for the second missing reaction in the pathway. CC3431 lacked any functional description in the original annotation. When queried against GenBank, the highest scoring hit to CC3431 is a nicotinate-nucleotide adenylyltransferase with an E-value of 3e-38. Against the nicotinate (nicotinamide) nucleotide adenylyltransferase TIGRFAM [24], CC3431 scores just above the trusted cutoff score. The sequence has been assigned to the NadD COG (COG1057) [25].
The third missing reaction, glutamine-dependent NAD+ synthetase, is catalyzed by the same enzyme catalyzing the NH3-dependent reaction. CC3619 includes an N-terminal domain that enables the reaction to proceed with either substrate [26]. Each of the two domains of CC3619 has been assigned to COGs (COG0388, predicted amidohydrolase and COG0171, NAD synthase). Additional files 5, 6 and 7 contain alignments [27] of the set of sequences used to query the Caulobacter genome and the putative enzyme assigned to each pathway hole in the example pathway.
Discussion
We have shown that our hole-filling program can improve the quality of PGDBs and the quality of genome annotations by clarifying nonspecific and incomplete annotations and providing annotations for sequences of previously unknown function. For the three PGDBs used in this work, an average of 27% of the pathways were complete before running our program. After filling pathway holes, an average of 38% of the pathways are complete, and an average of 28% of the previously missing reactions have putative enzymes assigned with probabilities above 0.9.
In the course of this work, we have identified additional uses for the pathway hole-filler in improving the quality of a PGDB. For example, in cases where several enzymes have been assigned to a reaction by PathoLogic, our predictor may determine which enzyme is most likely to be the correct one. When the set of enzymes all have the same general functional annotation (e.g., aldehyde dehydrogenase), we can provide a more specific function for the enzyme assigned by our predictor when the activity of the missing reaction is more specific.
Related work
Osterman and Overbeek have provided a review of comparative genomics techniques that can be applied to the identification of missing enzymes in metabolic pathways [7]. The authors provide detailed descriptions and examples of cases where homology alone was insufficient for finding a missing gene to fill a pathway hole. Their approach encompasses three phases: identifying holes in pathways, identifying and ranking candidate genes, and experimental verification. Identification and ranking of candidates includes the use of comparative genomics techniques to gather potential candidates and the evidence needed to support or refute the assignment of the missing activity to that gene. The review proposes the approach to be used by experimentalists in identifying missing genes, but does not propose a computational method. Our method employs a similar approach for gathering candidates and evidence, but then combines these elements in a way that can be efficiently applied to large-scale predictions.
Reed et al. [28] have completed a "network gap analysis" for E. coli similar to our search for missing enzymes to fill pathway holes. As a result of this analysis, they made putative functional assignments to 55 ORFs in the E. coli K-12 genome, using an approach similar to the candidate identification step of our algorithm. For a particular hole of interest, a set of orthologous sequences was used to find sequences with that function in the E. coli genome. Their approach differs from ours in that they used MEME [29] and ClustalW [27] to generate profiles for the set of orthologous sequences. These profiles were then used with MAST [30] and HMMER [31] to query the genome rather than searching the genome with each sequence individually. Search results were inspected manually to confirm the putative functional assignments, and the three top-scoring hits were reported for putative reannotations. The study provided no specific evaluation criteria for recommended reannotations (e.g., E-value cutoff) besides the appearance of the sequence within the top three hits for a search.
Our algorithm provides several improvements over this approach. Our Bayesian network can be used programmatically to rigorously combine disparate sources of evidence including sequence-based and non-sequence-based sources. MAST and HMMER consider only sequence-based information, and Reed et al. do not provide an automated means of combining the evidence from their searches. Following the identification of candidate sequences, our program calculates a probability from homology-, operon-, and pathway-based data. The probability P(has-function) gives a clear indication of how likely it is that each candidate provides a particular function; thus, for each reaction, the set of potential candidates can be directly compared to each other based on all of the evidence included in our network. Comparing E-values from MAST and HMMER excludes valuable contextual information. Instead of gauging the likelihood that a sequence has the desired function, E-values reflect the likelihood that each match might be found by chance in the database, which is not the question most searches intend to ask. Also, our method does not restrict the search to only the elements common to all sequences used to build the profile (i.e., the query sequences are not assumed to be evolutionarily related), nor does it require multiple sequences. The program evaluates candidates found by a single query sequence or multiple query sequences in the same way. The Shotgun-score node appropriately adjusts the P(has-function) based on the number of query sequences identifying a candidate.
To compare the two methods, we used our program to identify enzymes for a subset of the reactions evaluated by Reed et al. and currently included in EcoCyc [32]. Table 5 compares predictions for five reactions for which a gene has been experimentally identified. In one case, E.C.# 1.3.99.3, neither method correctly classifies the enzyme known to possess acyl CoA dehydrogenase activity (b0221). For the four remaining reactions, both methods agree on the enzymes that have been experimentally confirmed (i.e., those shown in bold). Discrepancies between the two methods appear in classifying additional candidates. The profile method makes several putative assignments to reactions 1.3.99.3, 1.6.6.9, 2.7.1.23, and 2.8.1.2 where our program has computed a probability of less than 0.2 for all of those assignments (shown in italics in Table 5), suggesting that they are false hits.
Table 5.
Probabilities calculated for putative functional assignments made by Reed et al. Bnumbers (unique IDs for E. coli genes) shown in italics are below the P cutoff (P > 0.8). Those in bold correspond to the gene assigned to the reaction in EcoCyc when one is assigned.
| E.C. # | Bnumber | P(has-function) | MAST E-val | HMMER E-val |
| 1.3.99.3 | b0221 | 0.05 | -- | 1e-4 |
| b0039 | 0.80 | 1e-7 | 10e-13 | |
| b1695 | 0.93 | 2e-9 | 4e-16 | |
| b4187 | 0.003 | 6e-4 | 3e-4 | |
| 1.6.6.9 | b0997 | 1.0 | 1e-145 | 0 |
| b1872 | 1.0 | 6e-147 | 0 | |
| b3551 | 0.93 | 5e-95 | 0 | |
| b1587 | 0.17 | 5e-16 | 2e-28 | |
| b1588 | 0.05 | 6e-16 | 3e-26 | |
| 2.7.1.23 | b2615 | 1.0 | 3e-63 | 1e-144 |
| b3916 | 2e-5 | 8e-4 | 1.0 | |
| 2.7.2.1 | b3115 | 1.0 | 5e-69 | 2e-170 |
| b2296 | 1.0 | 2e-95 | 1e-261 | |
| 2.8.1.2 | b2521 | 0.91 | 1e-133 | 2e-208 |
| b1757 | 0.05 | 2e-4 | 4e-12 |
The results for E.C.#'s 1.2.7.1, 3.2.1.68, and 2.8.1.2 demonstrate the power of our method to discriminate between likely true and false hits. Results from our program and from the profile-based method for these three reactions are shown in Table 6. The MAST and HMMER E-values for the three candidates (b1378, b3431, and b1757) are comparable; however, the values for P(has-function) calculated by our program differ substantially between b3431 and the other two. With a threshold of P > 0.9, our program assigns isoamylase activity to b3431, but classifies b1378 as a false hit for pyruvate synthase, and b1757 as a false hit for 3-mercaptopyruvate sulfurtransferase. While the profile-based E-values agree, the features used by our network (specifically average-rank and pathway-directon) do not, resulting in distinct classifications for the candidates.
Table 6.
Comparison of selected examples from Reed et al. method.
| E.C.# | Bnumber | MAST E-value | HMMER E-value | P(has function) | best-E-value | average-rank | avg-frac-aligned | pathway-directon | adjacent-rxns |
| 1.2.7.1 | b1378 | 6e-8 | 3e-16 | 0.18 | 1e-36 | 2.5 | 0.75 | F | F |
| 2.8.1.2 | b1757 | 2e-4 | 4e-12 | 0.05 | 7e-16 | 2.0 | 0.92 | F | F |
| 3.2.1.68 | b3431 | 1e-11 | 4e-9 | 0.97 | 2e-69 | 1.0 | 0.74 | T | F |
Limitations of the predictor
The distributions used to determine P(has-function | evidence) for each candidate protein are calculated from the data collected for the known reactions in the PGDB. Proteins linked to reactions by PathoLogic may have stronger similarity to the set of query isozymes than the proteins that should be assigned to missing reactions. Hence, the probability distributions calculated from known reactions may not accurately reflect the distributions for missing reactions.
The classifier structure in Figure 4 assumes conditional independence among the evidence nodes. Obviously, the average-rank of a hit and the percent of the query sequence aligned are likely to correlate with the best-E-value of the hit. Like several other Bayesian models applied to biological problems, the violation of the conditional independence assumption does not preclude our model from adequately classifying potential enzymes. Future work will include learning the structure of the model, thereby incorporating the appropriate dependencies among the evidence nodes.
Our predictor is generally applicable to any organism's PGDB and is useful not only for developing a more complete picture of the organism's metabolism, but also for identifying the function of ORFs and clarifying incomplete or nonspecific annotations. We currently incorporate evidence from homology, operon, and metabolic pathway relationships into our candidate evaluation step, but those candidates are identified based solely on homology to known proteins in SWISS-PROT or PIR. With this limitation, our program will not identify enzymes for any reaction catalyzed by an enzyme with an extremely divergent sequence or an enzyme whose activity is the result of convergent evolution. Also, reactions catalyzed by enzymes lacking sequenced isozymes in other organisms will not be identified by our program. Incorporating non-homology-based data will help to address these limitations. Fortunately, given the simple structure of the classifier, additional evidence nodes can be easily integrated. Expression data and phylogenetic profiles [33] might enhance the accuracy of the predictions and allow identification of candidate proteins with little to no homology to known sequences.
Conclusions
Our pathway hole filler provides an effective, computational method for combining evidence from homology data, operon-based data, and pathway context to identify missing reactions in a Pathway/Genome database. By identifying missing enzymes in a genome, we can not only increase the completeness of the PGDB, but we can also improve existing genome annotations by identifying functions for previously unannotated proteins and clarifying non-specific annotations. With the completion of additional pathways in PGDBs and improvements in functional annotation provided by our approach, experimental and computational researchers will benefit from PGDBs that include more accurate and relevant information.
Availability and requirements
The pathway hole filler is available as part of the Pathway Tools software which is freely available freely to academics. Contact (ptools-support@ai.sri.com) for information on obtaining the software.
Authors' contributions
MG designed, implemented, and evaluated the program. All authors have read and approved the final manuscript.
Supplementary Material
Predicted pathway hole fillers in Pathway/Genome Databases A summary of the actual changes made to the three PGDBs presented in the manuscript.
Filled pathway holes in Cb. crescentus A list of the specific putative enzymes assigned in CauloCyc.
Filled pathway holes in M. tuberculosis H37Rv A list of the specific putative enzymes assigned in MtbRvCyc.
Filled pathway holes in V. cholerae A list of the specific putative enzymes assigned in MtbRvCyc.
ClustalW alignment of query sequences and putative Caulobacter L-aspartate oxidase.
ClustalW alignment of query sequences and putative Caulobacter nicotinate-nucleotide adenylyltransferase.
ClustalW alignment of query sequences and putative Caulobacter NAD+ synthetase (glutamine-hydrolyzing).
Acknowledgments
Acknowledgements
This work was supported in part by grant No. DE-FG03-01ER63219 from the Department of Energy, DARPA contract N66001-01-C-8011, and NLM National Institutes of Health Grant NLM 07033. This financial support does not constitute an endorsement of the views expressed herein.
Contributor Information
Michelle L Green, Email: green@ai.sri.com.
Peter D Karp, Email: pkarp@ai.sri.com.
References
- Benson Dennis A., Karsch-Mizrachi Ilene, Lipman David J., Ostell James, Wheeler David L. GenBank. Nucl Acids Res. 2003;31:23–27. doi: 10.1093/nar/gkg057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughey R, Krogh A. Hidden Markov models for sequence analysis: extension and analysis of the basic method. Comput Appl Biosci. 1996;12:95–107. doi: 10.1093/bioinformatics/12.2.95. [DOI] [PubMed] [Google Scholar]
- Karplus K, Barrett C, Hughey R. Hidden Markov models for detecting remote protein homologies. Bioinformatics. 1998;14:846–856. doi: 10.1093/bioinformatics/14.10.846. [DOI] [PubMed] [Google Scholar]
- Krogh A, Brown M, Mian IS, Sjolander K, Haussler D. Hidden Markov models in computational biology. Applications to protein modeling. J Mol Biol. 1994;235:1501–1531. doi: 10.1006/jmbi.1994.1104. [DOI] [PubMed] [Google Scholar]
- Karp PD, Paley S, Romero P. The Pathway Tools software. Bioinformatics. 2002;18:S225–32. doi: 10.1093/bioinformatics/18.suppl_1.s225. [DOI] [PubMed] [Google Scholar]
- Claudel-Renard Clotilde, Chevalet Claude, Faraut Thomas, Kahn Daniel. Enzyme-specific profiles for genome annotation: PRIAM. Nucl Acids Res. 2003;31:6633–6639. doi: 10.1093/nar/gkg847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterman A, Overbeek R. Missing genes in metabolic pathways: a comparative genomics approach. Curr Opin Chem Biol. 2003;7:238–251. doi: 10.1016/S1367-5931(03)00027-9. [DOI] [PubMed] [Google Scholar]
- Cordwell SJ. Microbial genomes and "missing" enzymes: redefining biochemical pathways. Archives Microbiology. 1999;172:269–279. doi: 10.1007/s002030050780. [DOI] [PubMed] [Google Scholar]
- The BioCyc Knowledge Library http://BioCyc.org
- Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O'Donovan C, Phan I, Pilbout S, Schneider M. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Research. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CH, Yeh LS, Huang H, Arminski L, Castro-Alvear J, Chen Y, Hu Z, Kourtesis P, Ledley RS, Suzek BE, Vinayaka CR, Zhang J, Barker WC. The Protein Information Resource. Nucleic Acids Research. 2003;31:345–347. doi: 10.1093/nar/gkg040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish Warren, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990;215:403–410. doi: 10.1006/jmbi.1990.9999. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegg SC, Babbitt PC. Shotgun: getting more from sequence similarity searches. Bioinformatics. 1999;15:729–740. doi: 10.1093/bioinformatics/15.9.729. [DOI] [PubMed] [Google Scholar]
- Nierman WC, Feldblyum TV, Laub MT, Paulsen IT, Nelson KE, Eisen JA, Heidelberg JF, Alley MR, Ohta N, Maddock JR, Potocka I, Nelson WC, Newton A, Stephens C, Phadke ND, Ely B, DeBoy RT, Dodson RJ, Durkin AS, Gwinn ML, Haft DH, Kolonay JF, Smit J, Craven MB, Khouri H, Shetty J, Berry K, Utterback T, Tran K, Wolf A, Vamathevan J, Ermolaeva M, White O, Salzberg SL, Venter JC, Shapiro L, Fraser CM, Eisen J. Complete genome sequence of Caulobacter crescentus. Proc Natl Acad Sci U S A. 2001;98:4136–4141. doi: 10.1073/pnas.061029298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry C. E., 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Barrell BG, al. et. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Heidelberg JF, Eisen JA, Nelson WC, Clayton RA, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Umayam L, Gill SR, Nelson KE, Read TD, Tettelin H, Richardson D, Ermolaeva MD, Vamathevan J, Bass S, Qin H, Dragoi I, Sellers P, McDonald L, Utterback T, Fleishmann RD, Nierman WC, White O. DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature. 2000;406:477–483. doi: 10.1038/35020000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner SE, Chothia C, Hubbard TJ. Assessing sequence comparison methods with reliable structurally identified distant evolutionary relationships. Proc Natl Acad Sci U S A. 1998;95:6073–6078. doi: 10.1073/pnas.95.11.6073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang R, Vingron M. Statistics of large-scale sequence searching. Bioinformatics. 1998;14:279–284. doi: 10.1093/bioinformatics/14.3.279. [DOI] [PubMed] [Google Scholar]
- Pearl Judea. Probabilistic Reasoning in Intelligent Systems: Networks of Plausible Inference. San Mateo, CA, Morgan Kaufmann; 1988. [Google Scholar]
- Pagano Marcello, Gauvreau Kimberlee. Principles of Biostatistics. 1st. Belmont, CA, Wadsworth Publishing Company; 1993. [Google Scholar]
- Flachmann R, Kunz N, Seifert J, Gutlich M, Wientjes FJ, Laufer A, Gassen HG. Molecular biology of pyridine nucleotide biosynthesis in Escherichia coli. Cloning and characterization of quinolinate synthesis genes nadA and nadB. Eur J Biochem. 1988;175:221–228. doi: 10.1111/j.1432-1033.1988.tb14187.x. [DOI] [PubMed] [Google Scholar]
- Nasu S, Wicks FD, Gholson RK. L-Aspartate oxidase, a newly discovered enzyme of Escherichia coli, is the B protein of quinolinate synthetase. J Biol Chem. 1982;257:626–632. [PubMed] [Google Scholar]
- Haft Daniel H., Selengut Jeremy D., White Owen. The TIGRFAMs database of protein families. Nucl Acids Res. 2003;31:371–373. doi: 10.1093/nar/gkg128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantoni R, Branzoni M, Labo M, Rizzi M, Riccardi G. The MTCY428.08 gene of Mycobacterium tuberculosis codes for NAD+ synthetase. J Bacteriol. 1998;180:3218–3221. doi: 10.1128/jb.180.12.3218-3221.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JL, Vo TD, Schilling CH, Palsson BO. An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR) Genome Biology. 2003;4:R54.. doi: 10.1186/gb-2003-4-9-r54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Bailey TL, Gribskov M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14:48–54. doi: 10.1093/bioinformatics/14.1.48. [DOI] [PubMed] [Google Scholar]
- Eddy SR. Hidden Markov models. Curr Opin Struct Biol. 1996;6:361–365. doi: 10.1016/S0959-440X(96)80056-X. [DOI] [PubMed] [Google Scholar]
- Karp PD, Riley M, Saier M, Paulsen IT, Collado-Vides J, Paley SM, Pellegrini-Toole A, Bonavides C, Gama-Castro S. The EcoCyc Database. Nucleic Acids Research. 2002;30:56–58. doi: 10.1093/nar/30.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci U S A. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Predicted pathway hole fillers in Pathway/Genome Databases A summary of the actual changes made to the three PGDBs presented in the manuscript.
Filled pathway holes in Cb. crescentus A list of the specific putative enzymes assigned in CauloCyc.
Filled pathway holes in M. tuberculosis H37Rv A list of the specific putative enzymes assigned in MtbRvCyc.
Filled pathway holes in V. cholerae A list of the specific putative enzymes assigned in MtbRvCyc.
ClustalW alignment of query sequences and putative Caulobacter L-aspartate oxidase.
ClustalW alignment of query sequences and putative Caulobacter nicotinate-nucleotide adenylyltransferase.
ClustalW alignment of query sequences and putative Caulobacter NAD+ synthetase (glutamine-hydrolyzing).