

Abstract
Recently, we reported that human leukocyte antigen (HLA) class I expression is predominantly regulated by the mitogen-activated protein kinase (MAPK) pathway as one of the oncogenic regulations of HLA class I expression. In the present study, we examined mechanisms of how HLA class I and PD-L1 are regulated by MAPK inhibitors and interferon-γ (IFN-γ). Furthermore, we evaluated the expression of major signal transduction molecules by Western blot and anti-tumor CTL activity by a cytotoxic assay when HLA class I and PD-L1 were modulated by MAPK inhibitors and/or IFN-γ. As a result, we confirmed, as a more general phenomenon, that the inhibition of MAPK could upregulate HLA class I expression in a panel of human solid tumors (n = 26). Of note, we showed that MAPK inhibitors act on the upregulation of HLA class I expression through a different pathway from IFN-γ; there was an additive effect in the upregulation of HLA class I when treated with the combination of MAPK inhibitors and IFN-γ, and there was no overlapping activation of JAK2/STAT1 and Erk1/2 molecules when treated with either IFN-γ or MAPK inhibitors. Furthermore, we showed that IFN-γ–treatment impaired the tumor-specific CTL activity due to the upregulation of PD-L1 in spite of the upregulation of HLA class I, while MAPK inhibitors can augment the tumor-specific CTL activity due to the upregulated HLA class I without PD-L1 alterations. In conclusion, in addition to the original anti-proliferative activity, MAPK inhibitors may work toward the enhancement of T-cell-mediated anti-tumor immunity through the upregulation of HLA class I without the upregulation of PD-L1.
Keywords: Cytotoxic T lymphocyte, human leukocyte antigen-A, interferon-γ, mitogen-activated protein kinase, PD-L1
Reduced expression of human leukocyte antigen (HLA) class I on tumors is often associated with disease progression and a poor prognosis in patients with diverse cancers.1–3 It is generally accepted that downregulations of HLA class I expression and antigen-processing machinery (APM) components are associated with reduced sensitivity to lysis by anti-tumor-specific CTLs.2,4 Factors like genomic alteration, transcriptional regulation, protein transportation, and oncogene regulation may all be involved in the inactivation of HLA class I molecules.4,5 Recently, we showed that HLA class I expression is predominantly regulated by the ras/mitogen-activated protein kinase (MAPK) pathway in gastric and esophageal cancer as one of the oncogenic regulations of HLA class I expression, and the inhibition of the MAPK pathway with specific inhibitors leads to the upregulation of HLA class I expression on tumors.6 Since it has been shown that upregulation of the MAPK pathway occurs in the majority of tumors, owing to oncogenic activating mutations in KRAS, NRAS, and BRAF,7 manipulation of the MAPK pathway is a promising strategy for cancer treatment.
Interferon-γ is well-known to upregulate HLA class I expression as well as APM components on tumor cells on the one hand, while IFN-γ is one of the most potent inducers of inhibitory B7-family molecules such as programmed death ligand-1 (PD-L1) (also known as B7-H1) on the other hand.8–10 Although there have been a number of clinical trials of cancer therapy with IFN-γ, it showed very limited clinical effects.11,12 Thus, it is suggested that the double-edged sword effects of IFN-γ, which are the upregulation of HLA class I and PD-L1, are one of the reasons why IFN-γ has not been effective as a therapeutic agent for human cancer.10 In fact, it has been shown that PD-L1 expression on tumors inhibits the effector phase of CTL function through anergy or the apoptosis of T cells,5,13–15 and blockade of PD-L1 can augment the tumor-specific CTL response in vitro.16 Moreover, evidence has recently been accumulated from several clinical trials of immune checkpoint blockade with therapeutic mAbs, including the interaction between PD-L1 and PD1, with encouraging clinical effects such as prolonged overall survival and an improved response rate,17–20 suggesting that the immune checkpoint may play a pivotal role in anti-tumor immunity.
In the present study, we examined mechanisms of how HLA class I and PD-L1 are regulated by IFN-γ or MAPK inhibitors in a panel of human solid tumors. Furthermore, we evaluated the down-stream functional consequences of modulating HLA class I and PD-L1 by IFN-γ and/or MAPK inhibitors.
Materials and Methods
Tumor cell lines and HLA typing
Esophageal squamous cell carcinoma cell lines (ESCC), TE-1, TE-4, TE-5, and TE-14, and a lung cancer cell line, PC-9, were purchased from the RIKEN BioResource Center (Ibaraki, Japan). ESCC lines, KYSE30, KYSE50, KYSE70, and KYSE110, gastric cancer cell lines, MKN7, MKN45, MKN74, NUGC-3, and OCUM-1, and a breast cancer cell line, MRK-nu-1, were purchased from the Japanese Collection of Research Bioresources Cell Bank (Osaka, Japan). Gastric cancer cell lines, NCI-N87 and KATO III, and a pancreatic cancer cell line, PANC-1, breast cancer cell lines, BT-549, MCF7, and MDA-MB-231, a lung cancer cell line, NCI-H520, hepatocellular carcinoma cell lines, SNU-449 and SNU-475, an epidermoid carcinoma cell line, A431NS, and human B-lymphoblastoid cell lines, EB-3 and Jiyoye, were purchased from the American Type Culture Collection (Manassas, VA, USA). Another gastric cancer cell line, OE19, was purchased from Sigma-Aldrich (St. Louis, MO, USA). An HLA-A24 expressing TAP mutant human B-lymphoblastoid cell line, TISI, was purchased from the IHWG Cell and Gene Bank (Seattle, WA, USA). All cell lines were cultured in RPMI-1640 containing L-glutamine (Invitrogen, Carlsbad, CA, USA) with 5% FCS (Invitrogen) and Penicillin-Streptomycin (Sigma-Aldrich), and were regularly authenticated and matched to short tandem repeat DNA profiles of the original cell lines in each cell bank.
Human leukocyte antigen genotyping of all tumor cell lines was performed using the PCR-Luminex method in the HLA laboratory (Kyoto, Japan).
Cell treatment with inhibitors
Tumor cells were cultured in a 12-well plate and exposed to various concentrations of IFN-γ with or without the MAPK signal inhibitors, PD98059 (Cell Signalling Technology, Danvers, MA, USA), UO126 (Cell Signalling Technology), and PD0325901 (Cayman Chemical, Ann Arbor, MI, USA). DMSO (Sigma-Aldrich) was used as a negative control for MAPK inhibitor- and IFN-γ-treatments, since both were dissolved in DMSO. Cells were analyzed by Western blotting after 1-h incubation with each reagent and were used for flow cytometry and cytotoxicity assays after 48-h incubation.
Under doses of MAPK inhibitors used in the present study, percentages of dying cells treated with MAPK inhibitors accounted for <8%, when analyzed by Annexin-V and 7-AAD staining.
Gene silencing using small interfering RNA
Tumor cells were grown at approximately 60% confluence in a 12-well plate and transfected with 100 nmol of ON-TARGETplus SMARTpool siRNA Human MAPK3 (Erk1) (Thermo Scientific, Rockford, IL, USA) or ON-TARGETplus SMARTpool siRNA Human MAPK1 (Erk2) (Thermo Scientific) in Opti-MEN I Reduced Serum Medium (Invitrogen) including Lipofectamine 2000 (Invitrogen), according to the manufacturer's protocol. For control transfectants, 100 nmol of ON-TARGETplus siCONTROL Non-Targeting siRNA Pool (Thermo Scientific) was used for transfection in parallel. After 48 h, cells were analyzed for p44/42 MAPK and phospho-p44/42 MAPK by Western blotting and for HLA class I and HLA-A expression by flow cytometry. Control-, Erk1-, and Erk2-siRNA transfectants were always cultured in parallel for the same period of time.
Western blotting
Cell were lysed in CelLytic lysis buffer (Sigma-Aldrich) with a protease inhibitor cocktail (Sigma-Aldrich) and a phosphatase inhibitor cocktail (Sigma-Aldrich). The protein concentration of cell lysates was measured using a BCA protein assay (Thermo Scientific). Five micrograms of protein was separated on 4–12% NuPage Bis-Tris gels (Invitrogen) and transferred to a polyvinylidene fluoride (PVDF) membrane using the iBlot Dry Blotting System (Invitrogen), according to the manufacturer's protocol. Blots were blocked in PBS (−) with 0.05% Tween 20 (Sigma-Aldrich) buffer (PBST) with 2% nonfat dry milk, followed by incubation overnight in PBST with 2% nonfat dry milk and primary antibodies. The following were used as primary antibodies and purchased from Cell Signalling Technology: JAK2 mAb, phospho-JAK2 mAb, STAT1 mAb, phospho-STAT1 mAb, p44/42 MAPK (Erk1/2) mAb, phospho-p44/42 MAPK (Erk1/2) mAb, and β-actin antibody. After washing in PBST, membranes were incubated with the HRP-linked anti-rabbit IgG (Cell Signalling Technology). Blots were visualized by ECL Prime (Amersham Pharmacia, Uppsala, Sweden) according to the manufacturer's protocol and with a film processor (Konica SRX-101A; JZ Imaging & Consulting, Willoughby, OH, USA).
Flow cytometry
Cells were stained with antibodies as previously described.6 The following antibodies were used for staining: Annexin V-FITC (Becton Dickinson, San Jose, CA, USA), 7-AAD (Becton Dickinson), anti-human HLA-ABC (clone: W6/32)-FITC and -PE (eBioscience, Santa Clara, CA, USA), anti-human HLA-A2 (clone: BB7.2)-PE (Becton Dickinson), anti-human HLA-A24 (clone: 22E1)-PE (Medical & Biological Laboratories, Nagoya, Japan), and anti-human CD274 (B7-H1) (clone: MIH1)-PE (eBioscience). Isotype-matched immunoglobulin served as a negative control, and staining was detected with an LSR II flow cytometer (Becton Dickinson).
Dead and/or apoptotic cells were excluded using Annexin-V and 7-AAD. The relative mean fluorescence intensity (rMFI) was calculated according to the formula: [(MFI with specific mAb − MFI with isotype mAb)/MFI with isotype mAb]/[(MFI with specific mAb of control treatment − MFI with isotype mAb of control treatment)/MFI with isotype mAb of control treatment].
Generation of HLA-A24 restricted, cancer-testis antigen-specific CTL clone
HLA-A24-positive PBMCs were collected using Ficoll Paque (GE Health Care, Uppsala, Sweden) gradient centrifugation. Monocytes were enriched by adherence to a plastic tissue culture flask for 1 h at 37°C and cultured in AIM-V (Invitrogen) supplemented with 5% human serum Type AB (Lonza, Walkersville, MD, USA), 1000 units/mL of GM-CSF (R&D Systems, Minneapolis, MN, USA) and 1000 units/mL of IL-4 (R&D Systems). Five days later, 20 μg/mL of OK-432 (Chugai Pharmaceutical, Tokyo, Japan) was added for the maturation of dendritic cells. After 48-h incubation, mature dendritic cells were pulsed with 20 μg/mL of HLA-A24-restricted LY6K-peptide (RYCNLEGPPI),21 which is the immunodominant peptide derived from cancer-testis antigen, and 3μg/mL of β2-microglobulin (Sigma-Aldrich) for 3 h at 37°C and co-incubated with autologous CD8 T cells at a 1:10 ratio in a 24-well plate. T cells were cultured in AIM-V supplemented with 5% human serum Type AB supplemented with 20 IU/mL interleukin (IL)-2 (CHIRON, Emeryville, CA, USA). Cultured T cells were re-stimulated with LY6K-peptide-loaded, Mitomycin C (Kyowa Hakko Kirin, Tokyo, Japan)-treated autologous mature dendritic cells every 7 days. IL-2 (40–80 IU/mL) was replenished after each re-stimulation. One week after the third stimulation, the CTL lines were tested for their antigen specificity for LY6K peptide using the ELISpot assay in combination with irrelevant HLA-A24-binding peptides, TTK (SYRNEIAYL), CDCA1 (VYGIRLEHF), KOC1 (KTVNELQNL), and TOMM34 (KLRQEVKQNL). A peptide-specific CTL clone was established from an HLA-A24-restricted, LY6K-peptide-specific CTL line using a limiting dilution method. Log-phase T cells were incubated with Mitomycin C-treated Jiyoye and EB-3 as feeders in a 96-well U-bottomed plate in AIM-V supplemented with 5% human serum Type AB in the presence of 30 ng/mL of anti-human CD3 mAb (Becton Dickinson) and 125 IU/mL of IL-2.
Preparation of IL-2-stimulated NK cells
Natural killer (NK) cells were isolated using Dynabeads Untouched Human NK cells (Invitrogen) from PBMCs. To generate IL-2-stimulated NK cells, isolated NK cells were cultured in RPMI-1640 medium containing 10% FCS and 1000 IU/mL of IL-2 (CHIRON). Every 2 days, half of the tissue culture medium was exchanged with fresh medium including IL-2.
Cytotoxic assay
The cytotoxic activity of the CTL clone and IL-2-stimulated NK cells was measured using a calcein-release assay, as previously described.6 Briefly, target cells were incubated with 5 μM of calcein-AM (Dojindo Laboratories, Kumamoto, Japan) for 30 min at 37°C and 5% CO2. Stained targets (5 × 103/well) were co-cultured at various ratios of the CTL clone or IL-2-stimulated NK cells in 200 μL of culture medium for 4 h. Assays were performed in triplicate in a 96-well U-bottomed plate. After 4-h incubation, 100 μL of the supernatant was transferred from the culture plate to a 96-well flat-bottomed plate and the fluorescence of each supernatant was measured at 485-nm excitation and 528-nm emission using Infinite 200 (Tecan Group, Männedorf, Switzerland). Spontaneous release was obtained from target cells incubated without effector cells, and maximum release was obtained from detergent-released target cells. The percentage of specific lysis was calculated according to the formula: % specific lysis = 100 × (experimental release − spontaneous release)/(maximum release − spontaneous release).
For the anti-PD-L1 blocking experiments, tumor cells were pre-incubated with aniti-MIH1 mAb (10 ng/mL; eBioscience) for 1 h. Thereafter, treated tumor cells and effectors were subjected to a cytotoxic assay.
ELISpot assay
The cancer-testis antigen specific response was determined by the IFN-γ enzyme-linked immunospot (ELISpot) assay, and the ELISpot assay was performed according to the manufacturer's protocol with a commercial kit (Mabtech, Stockholm, Sweden). Briefly, 96-well plates with a nitrocellulose membrane (Millipore, Bedford, MA, USA) were coated with anti-IFN-γ capture mAb overnight at 4°C. After blocking with AIM-V containing 5% human AB serum for 90 min, target cells (2 × 104 cells per well) were added together with the CTL clone (2 × 103 cells per well) to each well and incubated overnight at 37°C. The cells were then removed, and the plates were incubated with biotinylated secondary anti-human-IFN-γ mAb and streptavidin-alkaline phosphatase. The plates were developed with substrate (Sigma-Aldrich) at room temperature in the dark. The spots were quantified using an ELISPOT reader, KS ELISPOT Compact (ZEISS, Göttingen, Germany). The SFC (Spot Forming Cell) was calculated according to the formula: (the number of spots in a well containing the CTL clone and target cells) − (the number of spots in a well containing the CTL clone without target cells) − (the number of spots in a well containing target cells without the CTL clone).
Statistics
Student's unpaired t-test was performed to determine significance, and analyses were performed at a significance level of 5% (P < 0.05) using SPSS statistics 20.0 (SPSS Inc.).
Results
Upregulation of HLA class I molecules by inhibition of MAPK pathway in tumor cells
Information on the tumor type and HLA-A allele genotype in all tested cell lines are presented in Supplemental Table S1.
According to our previous findings in gastric and esophageal cancer, upregulation of HLA class I on tumor cells induced by MAPK inhibitors occurred in a dose-dependent manner.6 We could further confirm the upregulation of HLA class I expression by inhibition of the MAPK pathway with PD98059 (optimal dose, 50 μM) in several different types of cancer (n = 26), such as breast, pancreas, lung, and liver cancer cells (Fig.1a). For the cancer cell lines with HLA-A02 or/and HLA-A24-positive cell lines (n = 19), the upregulation of HLA-A02 as well as HLA-A24 was also confirmed (Fig.1a). Based on our previous reports6 and the present study (Fig. S1a), tumor cells were treated with optimal doses of several different MAPK inhibitions, PD98059 (50 μM), UO0126 (2 μM), and PD0325901 (1 μM). MAPK inhibitors can significantly induce the upregulation of total HLA class I (W6/32), HLA-A02, and HLA-A24 in all cell lines tested (Fig.1a,b and Supplemental Fig. S2) and the degree of increased MHC class I with MAPK inhibitors differed among cell lines.
Figure 1.

Human leukocyte antigen (HLA) class I expression of a panel of solid tumor cell lines is upregulated by mitogen-activated protein kinase (MAPK) inhibitors. (a) Twenty-six tumor cell lines were treated with PD98059 (MAPK signal inhibitor, 50 μM) and the expressions of HLA class I, HLA-A02, and HLA-A24 were assessed by flow cytometry. Dead and/or apoptotic cells were excluded on the basis of Annexin V and 7-AAD staining. The relative mean fluorescence intensity (rMFI) was calculated according to the formula: ([MFI with specific mAb − MFI with isotype mAb]/MFI with isotype mAb)/([MFI with specific mAb of control treatment − MFI with isotype mAb of control treatment]/MFI with isotype mAb of control treatment). **P < 0.01 between the PD98059- and dimethylsulfoxide (DMSO)-treated cells.(b) Tumor cells were treated with various MAPK inhibitors, PD98059 (50 μM), UO126 (2 μM), and PD0325901 (1 μM), or DMSO as a negative control. **P < 0.01, *P < 0.05 between the MAPK inhibitor-treated and DMSO-control cells.
To further demonstrate the involvement of MAPK signaling in the regulation of HLA class I expression, four cancer cell lines were treated with either Erk1- or Erk2-siRNA to silence Erk1/2 expression. Transfection of siRNA-Erk1 or -Erk2 reduced the basal levels of total Erk1/2 and phospho-Erk1/2 in comparison to those in the control-siRNA (Fig.2a). As a result, the upregulation of HLA-A24, HLA-A02, and total HLA class I was more marked with Erk2-silencing, while the Erk1-silencing led to a reduced HLA-A upregulation (Fig.2b). Thus, it was further confirmed that the MAPK pathway, especially the Erk-2 molecule, plays an important role in the upregulation of HLA class I.
Figure 2.

Erk1/2-silencing and human leukocyte antigen (HLA)-A expression. (a) The levels of total Erk1/2 (Erk) and phospho-Erk1/2 (p-Erk) are shown by Western blot, when tumor cells were treated with Erk1-siRNA (siErk1), Erk2-siRNA (siErk2), or control-siRNA (siCTR). Western blot data show one out of three independent experiments with comparable results. (b) The expressions of total HLA class I (W6/32), HLA-A02, and HLA-A24 were analyzed by flow cytometry and are expressed as the relative mean fluorescence intensity (rMFI) in comparison to cells treated with control-siRNA (siCTR). **P < 0.01, *P < 0.05, the Erk-siRNA-treated tumor cells versus control-siRNA-treated cells.
Inhibition of MAPK pathway can additively act on the upregulation of HLA class I expression with IFN-γ
Since IFN-γ is well-known to upregulate HLA class I expression on tumor cells,22 we treated a panel of tumor cells (n = 26) with a combination of IFN-γ and MAPK inhibitors. For IFN-γ treatments, we treated tumor cells at different doses and exposure intervals and measured the expression of HLA class I by flow cytometry. As shown in Supplemental Figure S1(b), IFN-γ could markedly upregulate the expression of HLA class I after 48 h and 0.5 ng/mL. Also, the combination of IFN-γ and PD98059 led to the most marked additive effects after 48 h and 0.5 ng/mL of IFN-γ (Suppl. Fig. S1c). The same pattern was also noted with TE4 and TE1 cells (data not shown). Tumor cells were, therefore, exposed to IFN-γ for 48 h and 0.5 ng/mL for the subsequent experiments.
When tumor cells were treated with MAPK inhibitors and/or IFN-γ, the degree of upregulation of HLA class I induced by IFN-γ was much greater, ranging from a 1.5- to 14.0-fold increase (mean = 3.9-fold increase) (Fig.3a), in comparison with those by PD98059, ranging from a 1.2- to 1.9-fold increase (mean = 1.3-fold increase) (Fig.1a). Of note, the combination of PD98059 with IFN-γ had additive effects on the upregulation of HLA class I expression in 19 out of 26 tumor cells tested (Suppl. Fig. S3a). The same trends were seen in the expressions of HLA-A02 and HLA-A24 (Fig.3a, Suppl. Fig. S3b,c). When treated with a combination of IFN-γ with different MAPK inhibitors, UO126 and PD0325901 (Fig.3b), the same additive effects were confirmed in the upregulation of HLA-class I, HLA-A02, and HLA-A24.
Figure 3.

Additive effects on the upregulation of human leukocyte antigen (HLA) class I when treated with a combination of interferon (IFN)-γ and mitogen-activated protein kinase (MAPK) inhibitors. (a) summarized data from 26 tumor cell lines in terms of upregulation of HLA class I, when treated with a combination of IFN-γ (0.5 ng/mL) and PD98059 (50 μM). rMFI, relative mean fluorescence intensity. ##P < 0.01, IFN-γ−treated cells versus IFN-γ + PD98059-treated cells. **P < 0.01, *P < 0.05 between the treated and control cells. (b) Tumor cells were treated with a combination of IFN-γ and various MAPK inhibitors, PD98059 (50 μM), UO126 (2 μM), or PD0325901 (1 μM)). **P < 0.01, *P < 0.05.
Based on the effect of HLA class I induced by IFN-γ and/or PD98059 (Suppl. Fig. S3a), we classified the 26 cell lines tested into three categories: additive effects of PD98059 with IFN-γ (n = 19), upregulation by IFN-γ but not by PD98059 (n = 4, TE-4, SNU-449, MKN45, and OCUM1), and no upregulation by either PD98059 or IFN-γ (n = 3, KYSE70, KYSE110, and MKN74) (Fig.4). There was no cell line showing the upregulation of HLA class I by PD98059 but not by IFN-γ.
Figure 4.
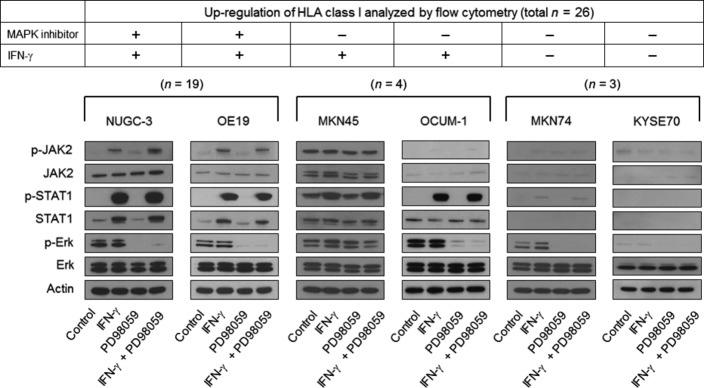
Western blot analysis of major signaling molecules when treated with a combination of interferon (IFN)-γ and mitogen-activated protein kinase (MAPK) inhibitors. Tumor cell lines were treated with IFN-γ (0.5 ng/mL) and/or PD98059 (50 μM), and the levels of total Erk1/2 (Erk) and phospho-Erk1/2 (p-Erk) as well as total STAT1 and phospho-STAT1 (p-STAT1) are shown by Western blot. Data from Western blot show one out of three independent experiments with comparable results.
MAPK inhibitors and IFN-γ independently act on distinct signal pathways
In order to further investigate the additive effects of MAPK inhibitors with IFN-γ on the upregulation of HLA class I, we evaluated major signal transduction molecules, JAK2 and STAT1 for IFN-γ treatment,22 and Erk as an MAPK inhibitor,6 by Western blot.
In 18 out of 19 cell lines with the additive effect of MAPK inhibitors with IFN-γ, both phospho-STAT1/JAK2 activation and inhibition of phospho-Erk were seen independently, as noted in the representative Western blot for NUGC3 and OE19 (Fig.4); when cells were treated with IFN-γ, there was no alteration of p-Erk expression and, in turn, there was no activation of phospho-STAT1 or phospho-JAK2 when treated with MAPK inhibitors. Of interest, only MKN7 out of the 19 cell lines with the additive effect of MAPK inhibitors with IFN-γ showed that the MAPK inhibitor, PD98059 induced the upregulation of phospho-STAT1 (Suppl. Fig. S4a,b). The upregulation of phospho-STAT1 was also confirmed in the Erk-siRNA treatment to silence the MAPK pathway (Suppl. Fig. S4c). The observation only seen in MKN7 suggests that PD98059 treatment may partially act through the JAK-STAT pathway in MKN7. Taken together, it is most likely that IFN-γ and MAPK inhibitors can independently act on the upregulation of HLA class I, and the mechanisms underlying the upregulation of HLA class I may be different in most cases tested.
Of note, among the 26 tumor cell lines tested, there were three IFN-γ-resistant cell lines in terms of the upregulation of HLA class I, KYSE70, KYSE110, and MKN74 (Suppl. Fig. S3). The lack of IFN-γ–sensitivity is due to defects in the IFN-γ signal cascade, in which neither phospho-STAT1 nor phospho-JAK2 was detected, as well as a constitutive lack of STAT1 and JAK2 (Fig.4).
Furthermore, among seven PD98089-resistant cell lines, there were two patterns of MAPK-signaling molecules (Fig.4): one showed no inhibition of p-Erk in spite of treatment with MAPK inhibitors (MKN45), and the other showed clear inhibition of p-Erk with MAPK inhibitors (MKN74, OCUM-1 and KYSE70).
MAPK inhibitors, but not IFN-γ treatment, can enhance tumor-specific CTL activity
To investigate the functional consequence of upregulated HLA class I on immune recognition, we examined antigen-specific CTL responses of tumor targets treated with IFN-γ and/or PD98059. HLA-A24-restricted CTL clones against LY6K (lymphocyte antigen-6 complex locus K)-peptide were generated by repeated stimulation with mature DCs pulsed with LY6K-peptide.21 As shown in Supplemental Figure S5(a), ELISPOT assays revealed that the CTL clone specifically reacted with TISI targets pulsed with the LY6K-peptide in comparison to the TISI targets pulsed with several HLA-A24-binding, irrelevant peptides (TTK, CDCA1, IMP3, and TOMM34).
HLA-A24-positive, LY6K-positive tumor cells, MKN7, KYSE30, and TE1 (Suppl. Fig. S5b), were pre-treated with IFN-γ and/or PD98059 and subjected to ELISPOT and cytotoxic assays. Of importance, an ELISPOT assay showed marked upregulation of CTL reactivity when treated with PD98059, while CTL reactivity was completely lost when treated with IFN-γ (Fig.5a). Similarly in a cytotoxic assay, PD98059-pre-treated MKN7 showed much higher susceptibility to the CTL clone compared to the control target, while IFN-γ-pre-treated MKN7 showed lower susceptibility to the CTL clone compared to the control target (Fig.5b). These results indicate there was a markedly contrasting phenomenon in terms of CTL reactivity between IFN-γ and PD98059 treatment, in spite of the upregulation of HLA class I in both treatments.
Figure 5.
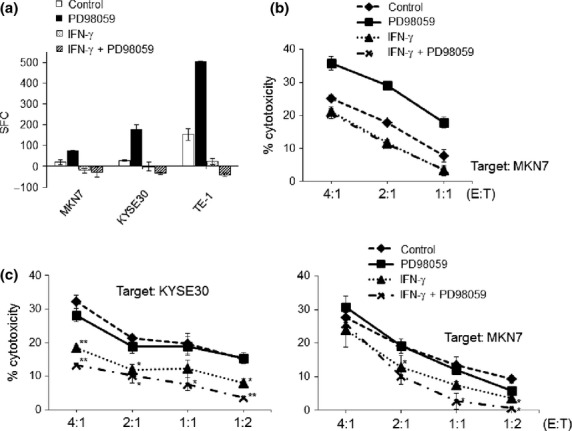
CTL reactivity and NK sensitivity when treated with a combination of interferon (IFN)-γ and mitogen-activated protein kinase (MAPK) inhibitors.(a,b) MKN7, KYSE30, and TE1 were pre-treated with IFN-γ (0.5 ng/mL) and/or PD98059 (50 μM), and subjected to an ELISPOT assay (a) and cytotoxic assay (b) against the HLA-A24-positive, LY6K-specific CTL clones. SFC, spot forming cells. E/T, effector: target. (c) KYSE30 and MKN7 were pre-treated with IFN-γ (0.5 ng/mL) and/or PD98059 (50 μM), and subjected to a cytotoxic assay against IL-2-activated NK cells. **P < 0.01, *P < 0.05 between the treated and control cells.
NK sensitivity of tumor cells treated with IFN-γ or MAPK inhibitors
Since the interaction between the killer immunoglobulin-like receptor family on NK cells and HLA class I molecules results in inhibitory signals,23,24 we evaluated the sensitivity of tumor cells to IL-2-activated NK cells when treated with IFN-γ or MAPK inhibitors. As shown in Figure5(c), NK sensitivities of IFN-γ-treated KYSE30 and MKN7 were downregulated in comparison to control targets, while PD98059-treated targets remained at almost similar levels compared to the control targets.
Upregulation of PD-L1 expression induced by IFN-γ but not MAPK inhibitors
In order to further analyze the contrasting phenomenon of CTL reactivity between IFN-γ and PD98059 treatment, we measured the expression of PD-L1 on tumor cells by flow cytometry. As shown in Figure6(a) and Supplemental Figure S2(c), PD-L1 expressions were consistently upregulated in all tested cells when treated with IFN-γ. In contrast, there was no significant alteration or only a slight increase of PD-L1 expression when treated with MAPK inhibitors, PD98059, UO126, and PD0325901 (Fig.6a). There was no additive or synergic effect on the upregulation of PD-L1 between IFN-γ and MAPK inhibitors.
Figure 6.
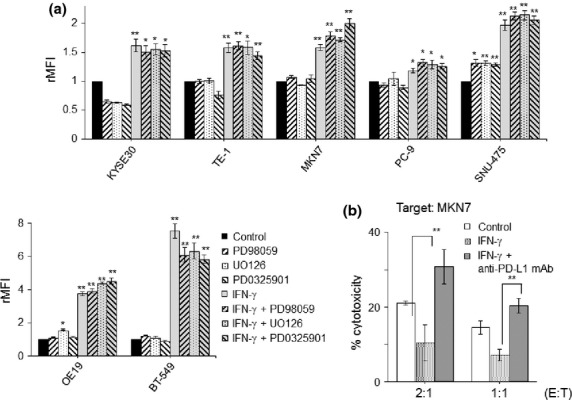
Expression of PD-L1 on tumor cells when treated with interferon (IFN)-γ and/or mitogen-activated protein kinase (MAPK)inhibitors. (a) Tumor cells were treated with a combination of IFN-γ (0.5 ng/mL) with various MAPK inhibitors, PD98059 (50 μM), UO126 (2 μM), and PD0325901 (1 μM). The expression of PD-L1 was analyzed by flow cytometry. Dead and/or apoptotic cells were excluded on the basis of Annexin V and 7-AAD staining. rMFI, relative mean fluorescence intensity. (b) MKN7 was pre-treated with IFN-γ (0.5 ng/mL) and/or anti-PD-L1 mAb (anti-MIH1 mAb, 10 ng/mL), and subjected to a cytotoxic assay against the HLA-A24-restricted, LY6K-specific CTL clones. **P < 0.01, *P < 0.05 between the treated and control cells.
In order to further evaluate the involvement of PD-L1 in the anti-tumor CTL activity when treated with IFN-γ as shown in Figure5(b), cytotoxic assays were performed in the presence of anti-PD-L1 mAb. As a result, the impaired anti-tumor CTL activities induced by IFN-γ were completely restored and even reached to higher levels than non-treatment control targets, when pre-treated with anti-PD-L1 mAb (Fig.6b). Taken together, the upregulated PD-L1 by IFN-γ impaired the anti-tumor CTL activity and blocking with anti-PD-L1 mAb could restore the anti-tumor activity of CTLs.
Discussion
We recently showed that inhibition of the MAPK pathway with specific inhibitors could upregulate HLA class I expression on gastric and esophageal cancers.6 In the present study, we further expanded this observation to a more general phenomenon, showing that the inhibition of MAPK can upregulate HLA class I expression in a panel of human solid tumors. Of note, we showed for the first time that MAPK inhibitors act on the upregulation of HLA class I expression through a distinct pathway different from IFN-γ, in which MAPK inhibitors did not stimulate the JAK/STAT pathway. The observation is based on two facts: there was an additive effect in the upregulation of HLA class I when treated with the combination of MAPK inhibitors with IFN-γ, and there was no overlapping activation in major signal transduction molecules in most cases when treated with either IFN-γ or MAPK inhibitors.
As an important result in the present study, tumor-antigen specific CTL activities were markedly impaired when tumor cells were treated with IFN-γ, as analyzed by both cytotoxic and ELISPOT assays. Although it is suggested that IFN-γ treatment may have double-edged sword effects for anti-tumor immunity,10 involving the upregulation of HLA class I and APM component and the upregulation of PD-L1, there is still limited information on how IFN-γ acts on the immune checkpoint, especially in a human study. It has been shown that co-culture of PD-L1-expressing tumor cells with tumor-specific T cells resulted in the increased apoptosis of T cells.14,15 In the present study, IFN-γ-treatment resulted in a significant impairment of tumor-specific CTL activity, in spite of the upregulation of HLA class I. We clearly showed that the impairment of CTL activity induced by IFN-γ was due to the upregulation of PD-L1, as shown in the blocking experiments with anti-PD-L1 mAbs. This is also supported by the comparative experiments between IFN-γ and MAPK inhibitors, in which MAPK inhibitors induced the upregulation of HLA class I, but did not alter PD-L1 expression, leading to the enhancement of tumor-specific CTL activity.
PD-L1 is an inhibitory B7 family member broadly distributed in various tissues and cell types, and is often expressed after exposure to inflammatory cytokines, especially IFN-γ.22,25 PD-L1 interacts with PD-1 and can inhibit T-cell activation and CTL-mediated lysis.14,15 Marked expression of PD-L1 has been reported in various types of human cancer.10,26 It has been reported that the inhibition of PD1-PD-L1 interaction with therapeutic mAbs induced significant and durable responses in several types of refractory tumor in clinical trials.18–20 Therefore, PD-L1 expression appears to be as one of the key mechanisms for tumors to avoid the host's immune response. Herein, we have further proved that PD-L1 overexpression by IFN-γ overcomes the upregulation of HLA class I, indicating that PD-L1 expression is a key limiting factor for anti-tumor CTL activity.
It has been shown that in lung cancer and hepatocellular carcinoma cell lines, the JAK/STAT pathway is involved in IFN-γ-mediated PD-L1 upregulation.27 However, IFN-γ can also stimulate the MAPK pathway in addition to the classical JAK/STAT pathway.28 Furthermore, the MAPK pathway seems to be a major contributor to the IFN-γ-induced overexpression of PD-L1 in malignant plasma cells29 and lymphoma.30 In contrast, it has been reported the induction of PD-L1 in fibroblasts through the PI3K pathway.31 More recently, a melanoma study showed that the effects of MEK and PI3K inhibitors on expression of PD-L1 were variable from reduction to induction.32 We showed in the present study that the upregulation of PD-L1 induced by IFN-γ was not related to the MAPK pathway, but to the JAK/STAT pathway, in a panel of human solid tumors. Therefore, it seems likely that IFN-γ signaling to control PD-L1 regulation is mediated through different pathways depending on the cell types or between malignant and benign cells.
Although it is well-known that IFN-γ can upregulate the expression of HLA class I as well as APM components, some tumor cells have lost their susceptibilities to modulation by IFN-γ,33 in which a lack of phosphorylation of JAK2 and STAT1 upon treatment with IFN-γ was seen in melanoma cells and RCCs.34 In the present study, we reported that the unresponsiveness to IFN-γ treatment, which resulted in the failed upregulation of HLA class I, was only seen in three out of the 26 human cancers tested, and that the unresponsiveness depends on a lack of constitutive and phosphorylation of JAK2 and STAT1.
Although the molecular mechanisms underlying MAPK-dependent HLA class I regulation have yet to be elucidated completely, it is possible that treatment with an MAPK inhibitor has a novel therapeutic implication for anti-tumor immunity, in addition to the original anti-proliferative activity. Our findings suggest that MAPK inhibitors may enhance T-cell mediated anti-tumor immunity through the upregulation of HLA class I without any upregulation of PD-L1. Taken together, manipulation of the MAPK signaling pathway may positively act on anti-tumor T-cell immunity, in addition to direct anti-proliferative activity on tumor cells.
Acknowledgments
This work was supported by a Clinician Scientist Award (CSA) and Clinician Scientist-Individual Research Grant (CS-IGR) from the National Medical Research Council of Singapore.
Disclosure Statement
The authors have no conflicting financial interest regarding the present study.
Supporting Information
Fig. S1. Optimal conditions of MAPK inhibitors and IFN-γ.
Fig. S2. Representative histograms of KYSE30 treated with MAPK inhibitors and/or IFN-γ.
Fig. S3. Additive effects on the upregulation of HLA class I when treated with a combination of MAPK inhibitor and IFN-γ.
Fig. S4. MAPK inhibitor can stimulate phospho-STAT1 in MKN7 cells.
Fig. S5. Specificity of tumor-antigen specific CTL clone.
Table S1. HLA-A allele expression on cancer cell lines.
References
- Watson NF, Ramage JM, Madjd Z, et al. Immunosurveillance is active in colorectal cancer as downregulation but not complete loss of MHC class I expression correlates with a poor prognosis. Int J Cancer. 2006;118:6–10. doi: 10.1002/ijc.21303. [DOI] [PubMed] [Google Scholar]
- Ferrone S, Marincola FM. Loss of HLA class I antigens by melanoma cells: molecular mechanisms, functional significance and clinical relevance. Immunol Today. 1995;16:487–94. doi: 10.1016/0167-5699(95)80033-6. [DOI] [PubMed] [Google Scholar]
- Mizukami Y, Kono K, Maruyama T, et al. Downregulation of HLA Class I molecules in the tumour is associated with a poor prognosis in patients with oesophageal squamous cell carcinoma. Br J Cancer. 2008;99:1462–7. doi: 10.1038/sj.bjc.6604715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seliger B. Molecular mechanisms of MHC class I abnormalities and APM components in human tumors. Cancer Immunol Immunother. 2008;57:1719–26. doi: 10.1007/s00262-008-0515-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie Y, Yang G, Song Y, et al. DNA hypermethylation is a mechanism for loss of expression of the HLA class I genes in human esophageal squamous cell carcinomas. Carcinogenesis. 2001;22:1615–23. doi: 10.1093/carcin/22.10.1615. [DOI] [PubMed] [Google Scholar]
- Mimura K, Shiraishi K, Mueller A, et al. The MAPK pathway is a predominant regulator of HLA-A expression in esophageal and gastric cancer. J Immunol. 2013;191:6261–72. doi: 10.4049/jimmunol.1301597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzivassiliou G, Haling JR, Chen H, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013;501:232–6. doi: 10.1038/nature12441. [DOI] [PubMed] [Google Scholar]
- Wei S, Shreiner AB, Takeshita N, Chen L, Zou W, Chang AE. Tumor-induced immune suppression of in vivo effector T-cell priming is mediated by the B7-H1/PD-1 axis and transforming growth factor beta. Cancer Res. 2008;68:5432–8. doi: 10.1158/0008-5472.CAN-07-6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, Curiel T, Coukos G, Liu R, Zou W. Inhibitory B7 family members in human ovarian carcinoma. Adv Exp Med Biol. 2008;622:261–71. doi: 10.1007/978-0-387-68969-2_21. [DOI] [PubMed] [Google Scholar]
- Zou W, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8:467–77. doi: 10.1038/nri2326. [DOI] [PubMed] [Google Scholar]
- Kirkwood JM, Ernstoff MS, Trautman T, et al. In vivo biological response to recombinant interferon-gamma during a phase I dose-response trial in patients with metastatic melanoma. J Clin Oncol. 1990;8:1070–82. doi: 10.1200/JCO.1990.8.6.1070. [DOI] [PubMed] [Google Scholar]
- Alberts DS, Marth C, Alvarez RD, et al. Randomized phase 3 trial of interferon gamma-1b plus standard carboplatin/paclitaxel versus carboplatin/paclitaxel alone for first-line treatment of advanced ovarian and primary peritoneal carcinomas: results from a prospectively designed analysis of progression-free survival. Gynecol Oncol. 2008;109:174–81. doi: 10.1016/j.ygyno.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Blank C, Brown I, Peterson AC, et al. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8 + T cells. Cancer Res. 2004;64:1140–5. doi: 10.1158/0008-5472.can-03-3259. [DOI] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank C, Kuball J, Voelkl S, et al. Blockade of PD-L1 (B7-H1) augments human tumor-specific T cell responses in vitro. Int J Cancer. 2006;119:317–27. doi: 10.1002/ijc.21775. [DOI] [PubMed] [Google Scholar]
- Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachy E, Coiffier B. Anti-PD1 antibody: a new approach to treatment of lymphomas. Lancet Oncol. 2014;15:7–8. doi: 10.1016/S1470-2045(13)70587-4. [DOI] [PubMed] [Google Scholar]
- Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipson EJ, Sharfman WH, Drake CG, et al. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clin Cancer Res. 2013;19:462–8. doi: 10.1158/1078-0432.CCR-12-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Tsunoda T, Daigo Y, Nakamura Y, Tahara H. Identification of human leukocyte antigen-A24-restricted epitope peptides derived from gene products upregulated in lung and esophageal cancers as novel targets for immunotherapy. Cancer Sci. 2007;98:1803–8. doi: 10.1111/j.1349-7006.2007.00603.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–86. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- Pievani A, Borleri G, Pende D, et al. Dual-functional capability of CD3 + CD56 + CIK cells, a T-cell subset that acquires NK function and retains TCR-mediated specific cytotoxicity. Blood. 2011;118:3301–10. doi: 10.1182/blood-2011-02-336321. [DOI] [PubMed] [Google Scholar]
- Vivier E, Raulet DH, Moretta A, et al. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–9. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–48. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- Song M, Chen D, Lu B, et al. PTEN loss increases PD-L1 protein expression and affects the correlation between PD-L1 expression and clinical parameters in colorectal cancer. PLoS ONE. 2013;8:e65821. doi: 10.1371/journal.pone.0065821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Jang BC, Lee SW, et al. Interferon regulatory factor-1 is prerequisite to the constitutive expression and IFN-gamma-induced upregulation of B7-H1 (CD274) FEBS Lett. 2006;580:755–62. doi: 10.1016/j.febslet.2005.12.093. [DOI] [PubMed] [Google Scholar]
- Sun D, Ding A. MyD88-mediated stabilization of interferon-gamma-induced cytokine and chemokine mRNA. Nat Immunol. 2006;7:375–81. doi: 10.1038/ni1308. [DOI] [PubMed] [Google Scholar]
- Liu J, Hamrouni A, Wolowiec D, et al. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood. 2007;110:296–304. doi: 10.1182/blood-2006-10-051482. [DOI] [PubMed] [Google Scholar]
- Yamamoto R, Nishikori M, Tashima M, et al. B7-H1 expression is regulated by MEK/ERK signaling pathway in anaplastic large cell lymphoma and Hodgkin lymphoma. Cancer Sci. 2009;100:2093–100. doi: 10.1111/j.1349-7006.2009.01302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Seo SH, Kim BS, et al. IFN-gamma regulates the expression of B7-H1 in dermal fibroblast cells. J Dermatol Sci. 2005;40:95–103. doi: 10.1016/j.jdermsci.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Atefi M, Avramis E, Lassen A, et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res. 2014;20:3446–57. doi: 10.1158/1078-0432.CCR-13-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Respa A, Bukur J, Ferrone S, et al. Association of IFN-gamma signal transduction defects with impaired HLA class I antigen processing in melanoma cell lines. Clin Cancer Res. 2011;17:2668–78. doi: 10.1158/1078-0432.CCR-10-2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dovhey SE, Ghosh NS, Wright KL. Loss of interferon-gamma inducibility of TAP1 and LMP2 in a renal cell carcinoma cell line. Cancer Res. 2000;60:5789–96. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Optimal conditions of MAPK inhibitors and IFN-γ.
Fig. S2. Representative histograms of KYSE30 treated with MAPK inhibitors and/or IFN-γ.
Fig. S3. Additive effects on the upregulation of HLA class I when treated with a combination of MAPK inhibitor and IFN-γ.
Fig. S4. MAPK inhibitor can stimulate phospho-STAT1 in MKN7 cells.
Fig. S5. Specificity of tumor-antigen specific CTL clone.
Table S1. HLA-A allele expression on cancer cell lines.