

ELMOD2, a putative Arf1–GTPase-activating protein, was found to control recruitment of adipocyte triglyceride lipase to lipid droplets (LDs). ELMOD2 was found in LDs, endoplasmic reticulum, and mitochondria, but palmitoylation was required only for LD distribution. Because palmitoylation-deficient ELMOD2 was defective in this functionality, ELMOD2 is likely to regulate the Arf1–coatomer protein complex I mechanism operating in LDs.
Abstract
Adipocyte triglyceride lipase (ATGL) is the major enzyme involved in the hydrolysis of triglycerides. The Arf1–coat protein complex I (COPI) machinery is known to be engaged in the recruitment of ATGL to lipid droplets (LDs), but the regulatory mechanism has not been clarified. In the present study, we found that ELMOD2, a putative noncanonical Arf–GTPase activating protein (GAP) localizing in LDs, plays an important role in controlling ATGL transport to LDs. We showed that knockdown of ELMOD2 by RNA interference induced an increase in the amount of ATGL existing in LDs and decreased the total cellular triglycerides. These effects of ELMOD2 knockdown were canceled by transfection of small interfering RNA-resistant cDNA of wild-type ELMOD2 but not by that of mutated ELMOD2 lacking the Arf-GAP activity. ELMOD2 was distributed in the endoplasmic reticulum and mitochondria as well as in LDs, but palmitoylation was required only for distribution to LDs. An ELMOD2 mutant deficient in palmitoylation failed to reconstitute the ATGL transport after the ELMOD2 knockdown, indicating that distribution in LDs is indispensable to the functionality of ELMOD2. These results indicate that ELMOD2 regulates ATGL transport and cellular lipid metabolism by modulating the Arf1-COPI activity in LDs.
INTRODUCTION
The lipid droplet (LD) is an organelle that exists in most cells and stores lipids mainly as triacylglycerols (TAGs) and sterol esters (Fujimoto and Parton, 2011; Walther and Farese, 2012). Recent studies have revealed that LDs are active organelles engaged in a wide range of functions, but many of these functions are related to the stored lipids, which are utilized for various cellular activities, including β-oxidation, membrane biogenesis, and synthesis of signaling molecules. Thus, to study lipid metabolism, and also to understand LD functions, it is important to clarify the molecular mechanism that regulates the lipid stores.
Hydrolysis of TAG is the first step of lipid utilization and is mediated by three enzymes: adipocyte triglyceride lipase (ATGL), hormone-sensitive lipase (HSL), and monoglyceride lipase, which work in this order to hydrolyze one TAG molecule to generate one glycerol and three free fatty acids (Zechner et al., 2012). Among the three enzymes, HSL has been studied most extensively: HSL exists as a soluble protein in the cytosol in the resting condition, but upon a β-adrenergic stimulus, HSL is phosphorylated by protein kinase A and recruited to LDs by binding to an LD-resident protein, perilipin1, which is also phosphorylated by protein kinase A. Because of this mechanism, fatty acid release after β-adrenergic stimulation is enhanced to 50–100 times that of the resting state (Londos et al., 1999).
In comparison with HSL, the molecular mechanism that recruits ATGL to LDs is less well understood. ATGL may partially exist in the cytosol (Villena et al., 2004; Zimmermann et al., 2004), but most ATGL is likely to exist in a membrane-bound form (Soni et al., 2009). Transport of ATGL to LDs has been thought to require vesicular trafficking, because knockdown of Arf1, coat protein complex I (COPI), or GBF1, an Arf–guanine nucleotide exchange factor, induced a decrease of ATGL in LDs (Beller et al., 2008; Soni et al., 2009) and/or generation of fewer and larger LDs (Beller et al., 2008; Guo et al., 2008), but the detailed mechanism has not been clarified. The simplest model of this mechanism may be that COPI-coated vesicles originating in the Golgi and/or endoplasmic reticulum–Golgi intermediate compartment (ERGIC) carry ATGL to LDs. Alternatively, the COPI (and COPII) coat might create an environment in the ERGIC (and/or ER exit site) that facilitates lateral conveyance of ATGL to adjacent LDs (Soni et al., 2009). In these proposed model hypotheses, the Arf1-COPI machinery would function in locations other than LDs. The existence of Arf1 and COPI in LDs (Bartz et al., 2007; Soni et al., 2009), however, suggested another possibility: the Arf1-COPI system functions in LDs themselves.
In the present study, we examined the function of ELMOD2 with regard to the recruitment of ATGL to LDs. ELMOD2 is a member of the ELMO domain–containing protein family protein, and despite the lack of the signature sequence of the canonical Arf–GTPase-activating proteins (GAPs), the ELMO domain of ELMOD1 has Arf-GAP activity (East et al., 2012), and recombinant ELMOD2 exhibits in vitro GAP activity to Arf1 as well as to Arl2, Arf3, and Arf6 (Bowzard et al., 2007; Ivanova et al., 2014). We found that endogenous ELMOD2 has Arf-GAP activity and that knockdown of ELMOD2 leads to an increase of ATGL in LDs and a decrease of the cellular TAG content. We also found that localization of ELMOD2 in LDs depends on palmitoylation and that both the Arf-GAP activity and palmitoylation are critical for ELMOD2 functionality. The results indicated that ELMOD2 down-regulates the recruitment of ATGL to LDs by suppressing the Arf1-COPI activity in LDs. ELMOD2 is the first protein that has been shown to be targeted to LD by palmitoylation and could be a new drug target for controlling cellular lipid metabolism.
RESULTS
Subcellular distribution of ELMOD2
We identified ELMOD2 as an LD protein via proteomic analysis of an LD preparation obtained from human hepatocellular carcinoma cell line Huh7 cells (Suzuki et al., 2012). Another proteomic study using Caco-2/TC7 cells also identified ELMOD2 as an LD-associated protein (Bouchoux et al., 2011). To study ELMOD2, we first generated a polyclonal antibody against a synthetic peptide corresponding to amino acids 191–202 of the ELMOD2 protein. In Western blotting, the antibody gave a positive band at the range consistent with the molecular mass (35 kDa) of ELMOD2 (Figure 1A). The antibody also reacted with expressed ELMOD2 tagged with green fluorescent protein (GFP) at the N terminus (GFP-ELMOD2) and ELMOD2 tagged with V5 at the C-terminus (ELMOD2-V5) at the expected region of the gel (Figure 1A). The specificity of the antibody was confirmed by a marked decrease of the reaction intensity in Western blotting when ELMOD2 was knocked down with RNA interference (RNAi; see Figure 5A later in this article).
FIGURE 1:

Subcellular distribution of ELMOD2. (A) Validation of the rabbit anti-ELMOD2 antibody by Western blotting. The antibody recognized endogenous ELMOD2 in Huh7 cells. The positive reaction was observed at the mobility expected from the molecular mass of 35 kDa (arrowhead). ELMOD2-V5 and GFP-ELMOD2 expressed by cDNA transfection were also recognized by the anti-ELMOD2 antibody as extra bands corresponding to the size of the tagged protein, which was confirmed using anti-V5 and anti-GFP antibodies, respectively. A nonspecific reaction is shown by an asterisk (*). (B) Endogenous ELMOD2 in Huh7 cells was enriched in the LD fraction. Subcellular fractions of Huh7 cells were obtained either by sucrose density–gradient ultracentrifugation or by differential centrifugation. With either method, LDs were concentrated in the fraction of the lowest density, which was confirmed by the enrichment of ADRP (perilipin2). Note that ELMOD2 was also detected in the microsomal and mitochondrial fractions obtained by differential ultracentrifugation. (C) Endogenous ELMOD2 in HeLa cells was also detected in the LD fraction. Fractions enriched with LDs (L), membranes (M), and the cytosol (C) were obtained by OptiPrep density–gradient centrifugation and subjected to Western blotting with anti-ELMOD2 antibody. HeLa cells harbor few LDs in the normal culture condition (−OA), and the reaction in the LD fraction was apparent only when cells were cultured with 0.4 mM OA for 12 h (+OA). The cytosol fraction was overloaded to show a paucity of soluble ELMOD2. (D) ELMOD2, either conjugated with GFP at the N-terminus (a, green) or tagged with V5/His at the C-terminus (b, red), showed concentration around LDs in Huh7 cells. LDs were stained with BODIPY558/568-C12 (a, red) or BODIPY493/503 (b, green). (E) GFP-ELMOD2 (green) showed colocalization with SERCA2 and Tom20 (red), indicating distribution in the ER and mitochondria, respectively. HeLa cells were cultured with 0.4 mM OA for 3 h to induce LD formation. (F) Preembedding immunoelectron microscopy of GFP-ELMOD2 in HeLa cells cultured with 0.4 mM OA for 3 h. The results confirmed the presence of GFP-ELMOD2 in LDs (red arrowhead), the ER (yellow arrowheads), and mitochondria (green arrowheads).
FIGURE 5:

Effect of ELMOD2 knockdown on Arf1 and ATGL. (A) GST-GGA3 pull down. Huh7 cells were transfected with either control or ELMOD2 siRNA. ELMOD2 was reduced significantly by RNAi (left panel). Arf1-HA precipitated with GST-GGA3 was increased significantly by ELMOD2 knockdown, indicating that ELMOD2 functions as an Arf1-GAP in the cell (middle panel). The relative reaction intensity of Arf1-HA pulled down with GST-GGA3 increased significantly after RNAi (right panel; mean ± SD; n = 3; Student's t test; *p < 0.05). (B) ATGL in LDs was increased by knockdown of ELMOD2. HeLa cells transfected with control or ELMOD2 siRNA were cultured with 0.4 mM OA for 12 h. The total cell and the LD fraction were examined by Western blotting. The expression level of ATGL in the cell was not influenced by ELMOD2 knockdown. The relative reaction intensity of ATGL to TIP47 in the LD fraction increased significantly after RNAi (mean ± SD; Student's t test; *p < 0.05). The experiment was repeated three times. (C) The increase of ATGL in LDs after ELMOD2 RNAi was confirmed by immunofluorescence labeling. HeLa cells transfected with control or ELMOD2 siRNA were doubly labeled for ATGL (red) and TIP47 (green). The relative labeling intensity of ATGL to TIP47 in LDs increased significantly after ELMOD2 knockdown (mean ± SD; Student's t test; *p < 0.05). The experiment was repeated three times, and in each experiment, 50 LDs chosen randomly from five micrographs were examined. (D) The TAG content was decreased by knockdown of ELMOD2. The total lipid was extracted from HeLa cells that were transfected with control or ELMOD2 siRNA and treated with 0.4 mM OA for 3 h. The amount of TAG was normalized to the protein content (mean ± SD; Student's t test; *p < 0.05). The experiment was repeated three times. (E) Both the number and size of LDs were decreased by knockdown of ELMOD2. HeLa cells were treated using the same protocol as in D and labeled with BODIPY493/503. LDs in 10 randomly taken micrographs were examined (mean ± SD; Student's t test; *p < 0.05).
The anti-ELMOD2 antibody was used to examine subcellular fractions of Huh7 cells by Western blotting. Consistent with the result of the proteomic analysis, ELMOD2 was detected in the LD fraction obtained either by sucrose density–gradient ultracentrifugation or by differential ultracentrifugation (Figure 1B). ELMOD2 was also found in the microsome and mitochondrial fractions obtained by differential ultracentrifugation (Figure 1B).
In comparison with Huh7 cells, HeLa cells harbor a far smaller number of LDs under normal culture conditions. Thus, when HeLa cells were fractionated by OptiPrep density–gradient ultracentrifugation, ELMOD2 was found largely in the membrane fraction (Figure 1C). But after cells were cultured with 0.4 mM oleic acid (OA) for 12 h to increase LDs, ELMOD2 was observed in the LD fraction as well as in the membrane fraction (Figure 1C), indicating that ELMOD2 in HeLa cells also distributes in LDs.
Because the anti-ELMOD2 antibody did not work in immunofluorescence labeling, we observed the distribution of GFP-ELMOD2 and ELMOD2-V5. Consistent with the results of Western blotting for endogenous ELMOD2, both GFP-ELMOD2 and ELMOD2-V5 showed prominent accumulation around LDs (Figure 1D). A similar distribution was also reported for ELMOD2-hemagglutinin (HA; East et al., 2012).
In addition to their dense distribution around LDs, the tagged ELMOD2 proteins were also found to distribute in a reticular pattern. To identify the location of this non-LD distribution, we observed GFP-ELMOD2 correlatively with various organelle markers in HeLa cells. The results showed that the reticular distribution of GFP-ELMOD2 frequently overlaps with that of SERCA2 (ER) and Tom20 (mitochondria) (Figure 1E) but not with that of ERGIC53 (ER–Golgi intermediate compartment), GM130 (Golgi), EEA1 (early endosome), Lamp1 (late endosome), or tubulin (microtubule) (Supplemental Figure S1).
Preembedding immunoelectron microscopy using anti-GFP antibody confirmed that GFP-ELMOD2 distributed on the LD surface (Figure 1F). Consistent with the Western blotting and immunofluorescence microscopic results, the labeling for GFP-ELMOD2 was also observed in the ER and mitochondria (Figure 1F).
ELMOD2 is released from LDs and membranes only by detergent treatment
Proteins are targeted to LDs by several different mechanisms (Walther and Farese, 2012), but the amino acid sequence of ELMOD2 does not imply any known mechanism. To study how ELMOD2 is bound to LDs and membranes, we treated subcellular fractions with a nonionic detergent (1% Triton X-100), an alkali (0.1 M Na2CO3, pH 11), or a high-salt solution (1 M NaCl) and then examined them to determine whether ELMOD2 was released to a soluble fraction.
ELMOD2 in the LD fraction was scarcely solubilized when treated with either the alkali or the high-salt solution, but it was completely solubilized by Triton X-100 (Figure 2A). This behavior was similar to that of ATGL, whereas adipose differentiation-related protein (ADRP), which is bound to LDs via several hydrophobic segments (McManaman et al., 2003; Nakamura and Fujimoto, 2003; Targett-Adams et al., 2003), was solubilized to a certain degree by the alkali or high-salt treatment.
FIGURE 2:
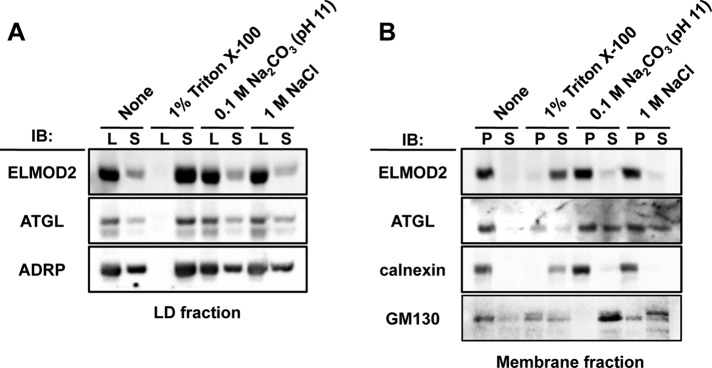
ELMOD2 and ATGL in isolated LDs and membranes. (A) LDs were isolated from Huh7 cells cultured with 0.4 mM OA for 12 h by OptiPrep density–gradient centrifugation. The LD preparation was treated with either 1% Triton X-100, 0.1 M sodium carbonate (pH 11), or 1 M NaCl for 30 min at 4ºC and ultracentrifuged to separate the LD fraction (L), which floated to the top surface, from the underlying soluble fraction (S). Both ELMOD2 and ATGL were released from LDs with Triton X-100 but not with the alkali or the high-salt treatment. In contrast, ADRP was partially solubilized with either the alkali or the high-salt treatment. (B) The membrane fraction obtained from Huh7 cells cultured with 0.4 mM OA for 12 h by OptiPrep density–gradient centrifugation was treated in the same manner as in A and then ultracentrifuged to obtain the pellet (P) and the supernatant (S). ELMOD2 was released from membranes only with the Triton X-100 treatment, whereas a significant portion of ATGL was released with the alkali or the high-salt treatment. Calnexin and GM130 were used as examples of transmembrane and peripheral membrane proteins, respectively.
ELMOD2 in the membrane fraction showed a similar behavior to ELMOD2 in LDs, in that it was solubilized only with the Triton X-100 treatment. On the other hand, ATGL in the membrane fraction showed resistance to extraction with Triton X-100, but it was solubilized at least partially with alkali or high-salt treatments (Figure 2B; Soni et al., 2009). Calnexin and GM130, a transmembrane protein in the ER and a peripheral membrane protein in the Golgi, respectively, were examined as controls to validate the rationale of the experiment; as expected, calnexin was solubilized only by Triton X-100, whereas GM130 was largely solubilized by either alkali or high salt (Figure 2B). These results suggested that ELMOD2 is bound to both LDs and membranes through hydrophobic interactions. They also implied that the anchorage of ATGL to LDs and membranes may be mediated by different mechanisms.
ELMOD2 is anchored to LDs with palmitoylation
The above results were intriguing, considering that ELMOD2 lacks hydrophobic sequences that are likely to mediate direct membrane anchorage (Bowzard et al., 2007). Actually, ELMOD2 mutants lacking either the C-terminal ELMO domain (aa 1–125) or the N-terminal non-ELMO domain region (aa 126–293) or a mutant made of the central portion alone (aa 70–200) were not distributed in LDs (Supplemental Figure S2). Although these results may have been caused by a gross change in protein conformation, they were consistent with the supposition that no particular hydrophobic portion anchors ELMOD2 to LDs.
We hypothesized that anchorage of ELMOD2 to LDs and/or membranes may occur through lipid modification. As ELMOD2 lacks typical consensus sequences for N-terminal myristoylation and C-terminal prenylation, we examined whether ELMOD2 is palmitoylated. HEK293T cells were cotransfected with cDNAs of GFP-ELMOD2 and DHHC3, a palmitoyltransferase, and incorporation of palmitate to GFP-ELMOD2 was examined by metabolic labeling with [3H]palmitate (Fukata et al., 2004). GFP-ELMOD2 was found to incorporate [3H]palmitate, although the signal intensity was less than that of a representative DHHC3 substrate, PSD-95 (Figure 3A). The result indicated that ELMOD2 is palmitoylated in cells.
FIGURE 3:

Palmitoylation of ELMOD2. (A) GFP-ELMOD2 cDNA or PSD-95 cDNA was transfected to HEK293T cells with HA-DHHC3 cDNA or HA-GST cDNA. After metabolic labeling with [3H]palmitate, proteins were separated by SDS–PAGE, which was followed by autoradiography (top panel). Western blotting (IB) for GFP, PSD-95, and HA is shown (bottom panels). When DHHC3 cDNA was cotransfected, [3H]palmitate was incorporated to GFP-ELMOD2 (arrowhead), under the conditions PSD-95 was palmitoylated (arrow). (B) cDNA of GFP alone or that of various GFP-ELMOD2 constructs was transfected to HEK293T cells with either HA-DHHC3 cDNA or HA-GST cDNA. Autoradiography and Western blotting were detected as described in A. GFP-ELMOD2 proteins incorporating [3H]palmitate are shown by an arrowhead. (C) Huh7 cells expressing either GFP-ELMOD2 (WT) or GFP-ELMOD2 (−5Cys) were lysed and examined by the ABE method for palmitoylation. Only the lysate of the GFP-ELMOD2 (WT)-expressing cell showed intense labeling at the expected molecular mass, indicating palmitoylation of GFP-ELMOD2 in cells.
Palmitoylation of ELMOD2 was further confirmed by replacing all five cysteines with alanine (GFP-ELMOD2 [−5Cys]): that is, the incorporation of [3H]palmitate was significantly reduced in GFP-ELMOD2 (−5Cys) in comparison with that in GFP-ELMOD (wild-type [WT]) (Figure 3B). Replacement of two cysteine residues at the 94th and 98th positions with alanine (GFP-ELMOD2 [–2Cys]) decreased the labeling to some extent but not to the basal level, suggesting that ELMOD2 may be palmitoylated in other cysteine residues as well (Figure 3B).
To examine whether ELMOD2 is palmitoylated without overexpression of DHHC3, we analyzed Huh7 cells expressing GFP-ELMOD2 (WT) and GFP-ELMOD2 (−5Cys), using the acyl-biotinyl exchange (ABE) method (Noritake et al., 2009). The results clearly indicated that GFP-ELMOD2 (WT) but not GFP-ELMOD2 (−5Cys) was palmitoylated in Huh7 cells (Figure 3C).
To examine whether the targeting of ELMOD2 to LDs is dependent on palmitoylation, we observed distribution of GFP-ELMOD2 with or without replacement of cysteine residues. In this experiment, to exclude the possibility that endogenous ELMOD2 makes complexes with the GFP-tagged ELMOD2 mutants, we took advantage of the molecular replacement approach. HeLa cells were first treated with ELMOD2 small interfering RNA (siRNA) and then transfected with siRNA-resistant ELMOD2 cDNAs. Replacement of one or two of the five cysteine residues did not cause an obvious change in distribution of GFP-ELMOD2 (Supplemental Figure S3). In contrast, accumulation around LDs was significantly reduced for GFP-ELMOD2 (−5Cys) (Figure 4A). It was notable, however, that GFP-ELMOD2 (−5Cys) was still distributed in the ER and mitochondria (Figure 4B). These results indicated that palmitoylation is specifically required for targeting to LDs. Consistently, treatment with 2-bromopalmitate, which inhibits protein palmitoylation, abrogated distribution of GFP-ELMOD2 in LDs, whereas it did not influence the distribution in the ER and mitochondria (Supplemental Figure S4).
FIGURE 4:

Palmitoylation and subcellular distribution of ELMOD2. (A) Replacement of five cysteine residues with alanine significantly decreased the accumulation of ELMOD2 in LDs. Huh7 cells expressing either GFP-ELMOD2 (WT) or the mutant, GFP-ELMOD2 (−5Cys) (green) were incubated with BODIPY558/568-C12 to label LDs (red; arrowheads) and treated with 0.1% saponin for 5 min before fixation to extract cytosolic proteins. (B) GFP-ELMOD2 (−5Cys) as well as GFP-ELMOD2 (WT) (green) showed colocalization with SERCA2 and Tom20 (red), indicating distribution in the ER and mitochondria, respectively. (C) Huh7 cells expressing either GFP-ELMOD2 (WT) or GFP-ELMOD2 (−5Cys) were fractionated by ultracentrifugation and analyzed by Western blotting. GFP-ELMOD2 (−5Cys) lacking palmitoylation was scarcely found in the LD fraction but was observed in the mitochondria and microsomal fractions. Consistent with the decrease in the LD fraction, the proportion of GFP-ELMOD2 (−5Cys) in the cytosol was increased in comparison with that of GFP-ELMOD2 (WT). The expression level of GFP-ELMOD2 (−5Cys) was lower than that of GFP-ELMOD2 (WT), as seen previously for other palmitoylated proteins (Tanimura et al., 2006). ADRP, Tom20, calnexin, and tubulin were used as markers of LDs, mitochondria, microsome, and cytosol, respectively.
Distribution of GFP-ELMOD2 (−5Cys) was also examined by Western blotting of subcellular fractions. In comparison with GFP-ELMOD2 (WT), a much smaller proportion of GFP-ELMOD2 (−5Cys) was partitioned to the LD fraction (Figure 4C). In contrast, the proportion of GFP-ELMOD2 (−5Cys) in the membrane and cytosolic fractions was equivalent and larger than that of GFP-ELMOD2 (WT), respectively (Figure 4C). These results confirmed the microscopic observations, in that ELMOD2 in LDs was decreased significantly by the lack of palmitoylation, whereas ELMOD2 in the ER and mitochondria was not.
Knockdown of ELMOD2 increases ATGL in LDs and reduces TAG
Previous studies showed that down-regulation of the Arf1-COPI machinery decreases ATGL in LDs and/or promotes LD enlargement (Beller et al., 2008; Guo et al., 2008; Soni et al., 2009). Considering that ELMOD2 was shown to have Arf1-GAP activity in vitro (Bowzard et al., 2007; Ivanova et al., 2014), we hypothesized that ELMOD2 is related to the regulation of Arf1-COPI machinery in LDs. We thus examined whether endogenous ELMOD2 has Arf1-GAP activity by using a pull down with GST-GGA3, which binds to the GTP form but not to the GDP form of Arf1 (Santy and Casanova, 2001). The amount of Arf1-HA that was precipitated with GST-GGA3 was significantly greater in cells treated with ELMOD2 siRNA than in cells treated with control siRNA (Figure 5A), indicating that endogenous ELMOD2 in HeLa cells may function as GAP to Arf1.
If ELMOD2 exerts Arf1-GAP activity, a decrease of ELMOD2 should activate Arf1 and increase ATGL in LDs, thereby leading to a decrease of TAG. To test this hypothesis, we examined the effect of ELMOD2 knockdown on the amount of ATGL in LDs and TAG in the total cell in HeLa cells. Through the knockdown of ELMOD2, the protein expression level of ATGL was not changed, but the relative proportion of ATGL in the LD fraction was increased (Figure 5B). The increase of ATGL in LDs after ELMOD2 depletion was also observed by immunofluorescence labeling. In this experiment, LD-resident TIP47 (perilipin3) was taken as a reference, and the labeling intensity of ATGL relative to that of TIP47 was measured. The relative labeling intensity of ATGL to TIP47 increased significantly in cells transfected with ELMOD2 siRNA but not in cells transfected with control siRNA (Figure 5C).
Consistent with the increase of ATGL in LDs, knockdown of ELMOD2 caused a significant decrease of the cellular TAG content (Figure 5D). Both the number and size of LDs were also decreased significantly by the treatment (Figure 5E). These results suggested that ATGL recruited to LDs as a result of ELMOD2 knockdown was functional and that a physiological function of ELMOD2 in the resting condition is to regulate the amount of ATGL in LDs and to maintain TAG storage.
For verification of the specificity of the above experiment, siRNA-resistant ELMOD2 cDNA was transfected to determine whether the effect of ELMOD2 knockdown on the ATGL distribution can be canceled. We discovered that the increase of ATGL in LDs caused by ELMOD2 RNAi was suppressed significantly in cells expressing GFP-ELMOD2, thus excluding the possibility of off-target effects of siRNA (Figure 6A).
FIGURE 6:

ELMOD2 functionality and ATGL transport. (A) The effect of ELMOD2 expression on the amount of ATGL in LDs. After knockdown of endogenous ELMOD2 with RNAi, GFP cDNA or siRNA-resistant cDNA of either GFP-ELMOD2 (WT) or GFP-ELMOD2 (R167K) was transfected to HeLa cells. The cells were treated with 0.4 mM OA for 3 h and doubly labeled for ATGL (green) and TIP47 (red). In comparison with cells expressing GFP alone, the expression of GFP-ELMOD2 (WT) significantly decreased the relative labeling intensity of ATGL to TIP47 in LDs, whereas the expression of GFP-ELMOD2 (R167K) did not (mean ± SD; Student's t test; *p < 0.05). The results indicated that the Arf-GAP activity is indispensable for the effect of ELMOD2 on ATGL. The experiment was repeated three times, and 50 LDs chosen randomly from five micrographs were examined. (B) The same experiment as in A was performed to examine the effect of siRNA-resistant GFP-ELMOD2 (−5Cys). The expression of GFP-ELMOD2 (−5Cys) did not affect the relative labeling intensity of ATGL to TIP47, indicating that palmitoylation is critical for the functionality of ELMOD2 (mean ± SD; Student's t test; *p < 0.05). The experiment was repeated three times, and 100 LDs chosen randomly from 10 micrographs were examined.
The experiment was extended to test whether the GAP activity of ELMOD2 is necessary for functionality. Expression of a mutant ELMOD2 that lacks the GAP activity (R167K; East et al., 2012; Ivanova et al., 2014) failed to decrease the amount of ATGL in LDs (Figure 6A), indicating the Arf-GAP activity is essential. Likewise, expression of the palmitoylation-deficient GFP-ELMOD2 (−5Cys) was also unable to decrease the amount of ATGL in LDs (Figure 6B). These results showed that both the Arf-GAP activity and distribution to LDs by palmitoylation are critical for ELMOD2 to recruit ATGL to LDs.
DISCUSSION
ELMOD2 and the Arf1-COPI machinery in LDs
In the present study, ELMOD2, a putative Arf-GAP protein, was found to be present in LDs. In conjunction with the presence of Arf-GEF (GBF1) in LDs (Soni et al., 2009), the results showed that LDs harbor both positive and negative regulators of Arf1, indicating that the Arf1-COPI machinery may be regulated locally in LDs.
Recent studies showed that Arf1 and COPI induce budding of small-sized LDs (nanodroplets) from artificial LDs (Thiam et al., 2013) and isolated natural LDs (Wilfling et al., 2014). The budding from LDs should entail the reduction of phospholipid packing and the increase of the surface tension in the LD surface; this increased surface tension is thought to induce membrane bridges between LDs and the ER membrane through which membrane-bound ER proteins like ATGL may laterally diffuse to LDs (Wilfling et al., 2014). The present results were consistent with this hypothesis: down-regulation of ELMOD2 probably increased the nanodroplet budding by activating the Arf1-COPI machinery in LDs, thus increasing the ATGL recruitment (Figure 7).
FIGURE 7:
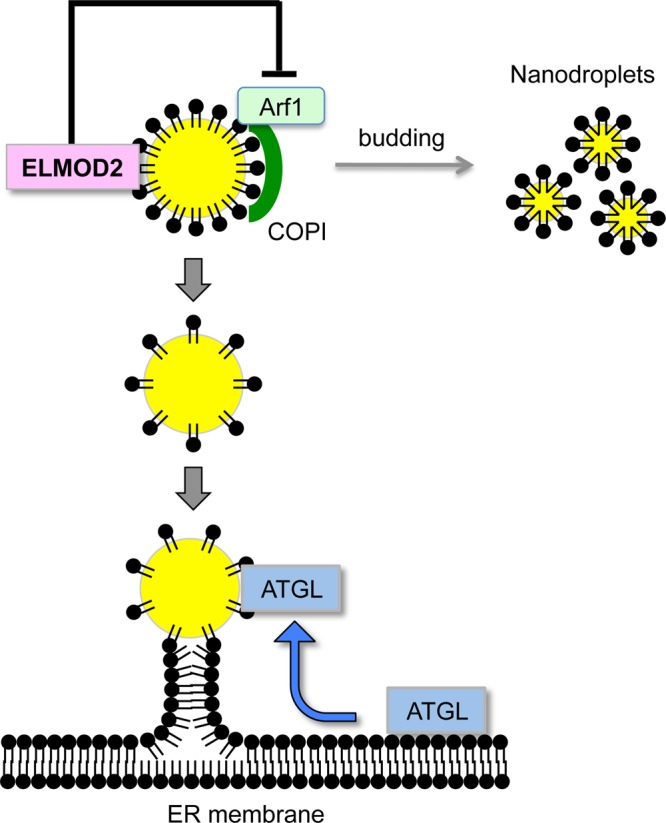
Schematic diagram that illustrates the ELMOD2 function on ATGL transport. The active Arf1-COPI complex in LDs is thought to facilitate budding of nanodroplets, thereby increasing the surface tension of the LD surface. This likely induces the formation of ER–LD membrane bridges through which ATGL is recruited to LDs. The Arf-GAP activity of ELMOD2 likely down-regulates these processes by inactivating Arf1 in LDs.
In relation to the transport of ATGL from the ER to LDs, it was notable that ATGL in LDs and ATGL in membranes exhibited different behaviors to the alkali and high-salt treatments (Figure 2). This difference may be correlated with two mechanisms of ATGL anchorage, that is, one by hydrophobic binding via a C-terminal domain (Lu et al., 2010; Murugesan et al., 2013) and the other by a protein–protein interaction with an LD protein G0/G1 switch gene-2 (G0S2; Lu et al., 2010; Cornaciu et al., 2011). The interaction with G0S2 that occurs only in LDs is likely to change the conformation of ATGL so that the C-terminal domain binds more tightly to LDs than to membranes. This may help transport ATGL from membranes to LDs unidirectionally and more efficiently than other proteins.
ELMOD2 is anchored to LDs by palmitoylation
The present study showed that palmitoylation is necessary for ELMOD2 to localize in LDs. Palmitoylation is known to play active roles in the sorting and trafficking of many proteins (Linder and Deschenes, 2007; Fukata and Fukata, 2010), but, to our knowledge, this is the first example of protein distribution in LDs that hinges on palmitoylation. Although the mechanism by which palmitoylation dictates LD localization of ELMOD2 is not clear, palmitoylation may induce partitioning of ELMOD2 into a raft-like microdomain in the ER membrane (Levental et al., 2010), which may then lead to distribution to LDs as observed for caveolins (Fujimoto et al., 2001). Alternatively, palmitoylation may cause a change in the molecular orientation of ELMOD2 on the membrane (Hayashi et al., 2005), which may induce a new set of molecular interactions to favor LD distribution.
Detailed mechanisms aside, it is important that palmitoylation and depalmitoylation are reversible and regulatable processes that may occur quickly. In this respect, palmitoylation differs from hydrophobic peptide sequences that are likely to conduct targeting of many LD proteins constitutively. Thus it is possible that palmitoylation/depalmitoylation cycles regulate distribution of ELMO2 dynamically according to the cellular condition. Most of ∼23 S-palmitoyl transferases in mammals are thought to exist in the ER and/or the Golgi (Ohno et al., 2006). Considering the critical role of palmitoylation in dictating the distribution of ELMOD2, it would be important to identify an enzyme(s) engaged in palmitoylation of ELMOD2 and to examine whether and how the enzymatic activity is regulated with regard to lipid metabolism.
It is not likely, however, that palmitoylation/depalmitoylation is the only mechanism that regulates the function of ELMOD2. In fact, it was reported that the GAP activity of recombinant ELMOD2 was inhibited by binding to sigma-1 receptor (Ivanova et al., 2014). Sigma-1 receptor, a transmembrane protein in the mitochondria-associated membrane (Hayashi and Su, 2007), may not bind directly to ELMOD2 in LDs, but other binding partners, including lipids, might modulate the LD-resident ELMOD2 function.
ELMOD2 and human idiopathic pulmonary fibrosis
ELMOD2 is a candidate gene for susceptibility to idiopathic pulmonary fibrosis (Hodgson et al., 2006), and herpesvirus saimiri infection was shown to be linked with the etiology of the disease (Folcik et al., 2014), but the manner in which ELMOD2 is related to the disease is not known (East et al., 2012). The present results indicated that down-regulation of ELMOD2 causes a decrease of LDs. This finding led us to note that association with LDs is functionally important for viperin, an antiviral protein that is induced by interferon and inhibits a broad range of viruses (Seo et al., 2011). We speculate that reduction of ELMOD2 may compromise viperin's antiviral activity by reducing LDs. Although this does not explain why the disease occurs specifically in the lung, we hope that further studies on ELMOD2 may help clarify the pathogenesis of this fatal human disease.
MATERIALS AND METHODS
Cell culture
Huh7 and HeLa cells were obtained from the Japanese Collection of Research Bioresources Cell Bank and cultured in DMEM and the original Eagle's minimum essential medium, respectively, supplemented with 10% fetal calf serum and antibiotics at 37°C in a humidified atmosphere containing 5% CO2. OA (Sigma-Aldrich, St. Louis, MO) in complex with fatty acid-free bovine serum albumin (Wako, Osaka, Japan) at a molar ration of 6:1 was added to the culture medium at a final concentration of 400 μM.
Antibodies
Rabbit anti-ELMOD2 antibody was raised against a peptide (ILSRSNHPKLGY) corresponding to amino acids 191–202 of the human ELMOD2 protein and affinity purified with the antigen peptide. Rabbit anti-TIP47 was obtained as previously described (Ohsaki et al., 2006). Rabbit anti-ERGIC53 and rabbit anti-GM130 antibodies were kindly provided by Nobuhiro Nakamura (Kyoto Sangyo University). Mouse anti-ADRP, guinea pig anti-TIP47 (Progen, Heidelberg, Germany), rabbit anti-ATGL (Cell Signaling Technology, Danvers, MA), rabbit anti-actin, mouse anti-α-tubulin (Sigma-Aldrich), mouse anti-calnexin (BD Biosciences, San Jose, CA), mouse anti-EEA1 (BD Biosciences), rabbit anti-GFP (Frontier Institute, Sapporo, Japan), mouse anti-HA (Nacalai Tesque, Kyoto, Japan), mouse anti-SERCA2 (Affinity Bioreagents, Rockford, IL), rabbit anti-Tom20 (Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-PSD-95 (Thermo Scientific, Waltham, MA), mouse anti-V5 (Invitrogen, Waltham, MA), and fluoronanogold-conjugated goat anti-rabbit immunoglobulin G (IgG; Nanoprobes, Yaphank, NY) antibodies were obtained from the respective suppliers.
Transfection
A plasmid to transduce HA-tagged Arf1 was kindly provided by Kazuhisa Nakayama (Kyoto University). Plasmids encoding GFP-ELMOD2 and ELMOD2-V5 cDNAs were prepared via a conventional method. Control and ELMOD2 siRNA were obtained from Dharmacon. Plasmids and siRNA were transfected using Lipofectamine 2000, Lipofectamine RNAiMAX (Invitrogen), or X-TREMEGENE HP (Roche, Basel, Switzerland) according to the manufacturers' instructions.
Subcellular fractionation
Subcellular fractions of Huh7 cells cultured with OA for 12 h were obtained by two different methods. In the method using a sucrose density–gradient ultracentrifugation (Figure 1B), the cells were disrupted by nitrogen cavitation, and the postnuclear supernatant adjusted to 0.27 M sucrose was overlaid with 0.135 M sucrose in a disruption buffer and the buffer alone and centrifuged for 1 h at 154,000 × g in an SW41 rotor (Beckman, Indianapolis, IN). Eight fractions were obtained from the top of the sucrose gradient. In the method using differential centrifugation (Figures 1B and 4C), the cells were disrupted in homogenization buffer (30 mM Tris-HCl, pH 7.4, 225 mM mannitol, 75 mM sucrose, 0.1 mM ethyleneglycoltetraacetic acid, and protease inhibitor) by being passed through a 26-G needle repeatedly. The mitochondrial fraction was obtained as a pellet by centrifuging the postnuclear supernatant for 10 min at 8000 × g. The supernatant of the centrifugation was overlaid with the homogenization buffer, omitting sucrose, and centrifuged at 166,000 × g for 5 h in a TLS-55 rotor (Beckman). The LD fraction (floating on the top surface), the cytosol fraction (collected using a Centritube Slicer [Beckman]), and the microsome fraction (pellet) were obtained.
For analysis of the behavior of ELMOD2 in response to various treatments (Figure 2), LDs and membranes were collected from the top-floating layer and the pellet of the sucrose density–gradient centrifugation. They were treated for 1 h at 4°C with buffer alone, 1% Triton X-100 in buffer, 1 M NaCl in buffer, or 0.1 M Na2CO3 (pH 11). After incubation, LDs were separated from solubilized proteins using the same sucrose-density centrifugation described above, whereas membranes were separated from solubilized proteins by centrifugation at 166,000 × g for 1 h.
For subcellular fractionation of HeLa cells (Figure 1C), the cells were disrupted in homogenization buffer by being passed through a 26-G needle. The postnuclear supernatant, adjusted to 30% OptiPrep (Axis Shield, Dundee, Scotland), was overlaid with 20% OptiPrep and then 10% OptiPrep in a homogenization buffer and the buffer alone and subsequently centrifuged at 166,000 × g or 5 h at 4ºC in a TLS-55 rotor (Beckman). Fractions enriched with LDs (floating on the top surface), membranes (collected at the 10%/20% OptiPrep interface), and the cytosol (the 30% OptiPrep layer) were collected using a Centritube Slicer.
Pull-down analysis
Huh7 cells transfected with control or ELMOD2 siRNA were transduced with Arf1-HA cDNA. The cells were lysed in a pull-down buffer (25 mM Tris-HCl, pH 7.2, 150 mM NaCl, 5 mM MgCl2, 1% NP-40, 5% glycerol, and protease inhibitor), and the cleared lysate was reacted with GST-GGA3 coupled with glutathione-Sepharose 4B (GE Healthcare, Little Chalfont, UK) for 1 h at 4°C. After washing, the bound proteins were released by boiling in an SDS sample buffer.
Western blotting
Each sample was boiled with SDS sample buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 10% glycerol). SDS–PAGE and Western transfer were performed using a Mini-PROTEAN and a Trans-BLOT apparatus (Bio-Rad, Hercules, CA), respectively. The Western blot signal was detected by chemiluminescence and captured either by Hyperfilm (GE Healthcare) or by a Light-Capture II imager (ATTO, Tokyo, Japan). The band intensity was measured using CS Analyzer 3 software (ATTO).
Metabolic labeling with palmitate
The labeling with [3H]palmitic acid was performed as described previously (Fukata et al., 2013). Briefly, HEK293T cells were transfected with HA-DHHC3 cDNA together with cDNA of either PSD-95-GFP or GFP-ELMOD2. The cells were labeled with 0.25 mCi/ml [3H]palmitic acid for 4 h, and the cell lysate was resolved by SDS–PAGE followed by fluorography and Western blotting.
ABE method
The ABE method (Noritake et al., 2009) was modified as follows. Transfected Huh7 cells were solubilized with buffer containing 4% SDS, diluted with the Triton X-100–containing buffer (final concentration: 1% SDS and 0.15% Triton X-100), and reduced with 10 mM Tris-(2-carboxyethyl) phosphine (TCEP) for 30 min. Free cysteine residues were then blocked with 40 mM N-ethylmaleimide (NEM). Excess TCEP and NEM were removed with a chloroform/methanol precipitation (CM ppt). Protein pellets were resuspended with buffer containing 4% SDS and incubated either in buffer H (1 M hydroxylamine, pH 7.0, 1 mM biotin-HPDP [N-[6-(biotinamido)hexyl]-3'-(2'-pyridyldithio)propionamide]) to cleave thioester bonds and to biotinylate newly exposed cysteines or in buffer T (1 M Tris-HCl, pH 7.0, 1 mM biotin-HPDP) as a negative control for 1 h. Excess hydroxylamine and biotin-HPDP were removed with a CM ppt. Protein pellets were resuspended with buffer containing 2% SDS and diluted with the Triton X-100–containing buffer. The resultant samples were incubated with 30 μl of NeutrAvidin-agarose (Pierce, Rockford, IL) for 1 h at 4°C. After the beads were washed, bound proteins were suspended in SDS–PAGE sample buffer and boiled at 100°C for 5 min. Samples were subjected to SDS–PAGE and Western blotting with GFP antibody.
Immunofluorescence microscopy
Cells were fixed with 3% formaldehyde with or without 0.015% glutaraldehyde in 0.1 M phosphate buffer for 15 min and permeabilized with either 0.01% digitonin in phosphate-buffered saline (PBS) for 30 min or with 0.1% Triton X-100 in PBS for 5 min before blocking and incubation with antibodies. LDs were stained with either BODIPY493/503 or BODIPY 558/568-C12 (Invitrogen). BODIPY493/503 was applied to fixed cells, while BODIPY 558/568-C12 was added to the culture medium before fixation.
Images were captured using an Axiovert 200M fluorescence microscope (Carl Zeiss, Jena, Germany) employing an Apochromat 63× lens with a 1.40 numerical aperture. Some images were obtained using the Apotome processing system. The color, brightness, and contrast of the presented images were adjusted using Adobe Photoshop 7.0. Labeling intensity in LDs was measured using MetaMorph (Molecular Devices, Sunnyvale, CA).
For measurement of the relative labeling intensity of two proteins in LDs, the experiment was repeated three times, and in each experiment, the relative fluorescence intensity was measured in more than 50–100 LDs that were chosen randomly from 10 micrographs.
Immunoelectron microscopy
HeLa cells transfected with GFP-ELMOD2 cDNA were fixed with 3% formaldehyde and 0.05% glutaraldehyde in 0.1 M HEPES for 30 min, permeabilized with 0.2% saponin for 30 min, and labeled with rabbit anti-GFP antibody followed by Fluoronanogold-conjugated goat anti-rabbit IgG antibody. The specimen was treated with GoldEnhance (Nanoprobes) to precipitate metallic gold and then postosmicated, dehydrated, and embedded for electron microscopy. Cells transfected with GFP cDNA were treated in the same manner as a control. Ultrathin sections were observed in a JEOL JEM1011 electron microscope.
Supplementary Material
Acknowledgments
We express our sincere thanks to Nobuhiro Nakamura and Kazuhisa Nakayama for their donation of antibodies and to Atsushi Sekiya for technical guidance on the ABE method. This study was supported by Grants-in-Aid for Scientific Research of the Ministry of Education, Culture, Sports, Science and Technology of the Government of Japan and by the Naito Foundation Subsidy for Promotion of Specific Research Projects.
Abbreviations used:
- ABE
acyl-biotinyl exchange
- ADRP
adipose differentiation-related protein
- Arf1
ADP-ribosylation factor 1
- ATGL
adipocyte triglyceride lipase
- CM ppt
chloroform/methanol precipitation
- COPI
coatomer protein complex I
- ER
endoplasmic reticulum
- ERGIC
endoplasmic reticulum–Golgi intermediate compartment
- G0S2
G0/G1 switch gene-2
- GAP
GTPase-activating protein
- GFP
green fluorescent protein
- HA
hemagglutinin
- HSL
hormone-sensitive lipase
- IgG
immunoglobulin G
- LD
lipid droplet
- NEM
N-ethylmaleimide
- OA
oleic acid
- PBS
phosphate-buffered saline
- RNAi
RNA interference
- siRNA
small interfering RNA
- TAG
triacylglycerol
- TCEP
Tris-(2-carboxyethyl) phosphine
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-11-1504) on April 22, 2015.
REFERENCES
- Bartz R, Zehmer JK, Zhu M, Chen Y, Serrero G, Zhao Y, Liu P. Dynamic activity of lipid droplets: protein phosphorylation and GTP-mediated protein translocation. J Proteome Res. 2007;6:3256–3265. doi: 10.1021/pr070158j. [DOI] [PubMed] [Google Scholar]
- Beller M, Sztalryd C, Southall N, Bell M, Jackle H, Auld DS, Oliver B. COPI complex is a regulator of lipid homeostasis. PLoS Biol. 2008;6:e292. doi: 10.1371/journal.pbio.0060292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchoux J, Beilstein F, Pauquai T, Guerrera IC, Chateau D, Ly N, Alqub M, Klein C, Chambaz J, Rousset M, et al. The proteome of cytosolic lipid droplets isolated from differentiated Caco-2/TC7 enterocytes reveals cell-specific characteristics. Biol Cell. 2011;103:499–517. doi: 10.1042/BC20110024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowzard JB, Cheng D, Peng J, Kahn RA. ELMOD2 is an Arl2 GTPase-activating protein that also acts on Arfs. J Biol Chem. 2007;282:17568–17580. doi: 10.1074/jbc.M701347200. [DOI] [PubMed] [Google Scholar]
- Cornaciu I, Boeszoermenyi A, Lindermuth H, Nagy HM, Cerk IK, Ebner C, Salzburger B, Gruber A, Schweiger M, Zechner R, et al. The minimal domain of adipose triglyceride lipase (ATGL) ranges until leucine 254 and can be activated and inhibited by CGI-58 and G0S2, respectively. PLoS One. 2011;6:e26349. doi: 10.1371/journal.pone.0026349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East MP, Bowzard JB, Dacks JB, Kahn RA. ELMO domains, evolutionary and functional characterization of a novel GTPase-activating protein (GAP) domain for Arf protein family GTPases. J Biol Chem. 2012;287:39538–39553. doi: 10.1074/jbc.M112.417477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folcik VA, Garofalo M, Coleman J, Donegan JJ, Rabbani E, Suster S, Nuovo A, Magro CM, Di Leva G, Nuovo GJ. Idiopathic pulmonary fibrosis is strongly associated with productive infection by herpesvirus saimiri. Mod Pathol. 2014;27:851–862. doi: 10.1038/modpathol.2013.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto T, Kogo H, Ishiguro K, Tauchi K, Nomura R. Caveolin-2 is targeted to lipid droplets, a new “membrane domain” in the cell. J Cell Biol. 2001;152:1079–1085. doi: 10.1083/jcb.152.5.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto T, Parton RG. Not just fat: the structure and function of the lipid droplet. Cold Spring Harb Perspect Biol. 2011;3:a004838. doi: 10.1101/cshperspect.a004838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata M, Fukata Y, Adesnik H, Nicoll RA, Bredt DS. Identification of PSD-95 palmitoylating enzymes. Neuron. 2004;44:987–996. doi: 10.1016/j.neuron.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Dimitrov A, Boncompain G, Vielemeyer O, Perez F, Fukata M. Local palmitoylation cycles define activity-regulated postsynaptic subdomains. J Cell Biol. 2013;202:145–161. doi: 10.1083/jcb.201302071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y, Fukata M. Protein palmitoylation in neuronal development and synaptic plasticity. Nat Rev Neurosci. 2010;11:161–175. doi: 10.1038/nrn2788. [DOI] [PubMed] [Google Scholar]
- Guo Y, Walther TC, Rao M, Stuurman N, Goshima G, Terayama K, Wong JS, Vale RD, Walter P, Farese RV. Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature. 2008;453:657–661. doi: 10.1038/nature06928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Rumbaugh G, Huganir RL. Differential regulation of AMPA receptor subunit trafficking by palmitoylation of two distinct sites. Neuron. 2005;47:709–723. doi: 10.1016/j.neuron.2005.06.035. [DOI] [PubMed] [Google Scholar]
- Hayashi T, Su TP. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- Hodgson U, Pulkkinen V, Dixon M, Peyrard-Janvid M, Rehn M, Lahermo P, Ollikainen V, Salmenkivi K, Kinnula V, Kere J, et al. ELMOD2 is a candidate gene for familial idiopathic pulmonary fibrosis. Am J Hum Genet. 2006;79:149–154. doi: 10.1086/504639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova AA, East MP, Yi SL, Kahn RA. Characterization of recombinant ELMOD (cell engulfment and motility domain) proteins as GTPase-activating proteins (GAPs) for ARF family GTPases. J Biol Chem. 2014;289:11111–11121. doi: 10.1074/jbc.M114.548529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental I, Grzybek M, Simons K. Greasing their way: lipid modifications determine protein association with membrane rafts. Biochemistry. 2010;49:6305–6316. doi: 10.1021/bi100882y. [DOI] [PubMed] [Google Scholar]
- Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- Londos C, Brasaemle DL, Schultz CJ, Adler-Wailes DC, Levin DM, Kimmel AR, Rondinone CM. On the control of lipolysis in adipocytes. Ann NY Acad Sci. 1999;892:155–168. doi: 10.1111/j.1749-6632.1999.tb07794.x. [DOI] [PubMed] [Google Scholar]
- Lu X, Yang X, Liu J. Differential control of ATGL-mediated lipid droplet degradation by CGI-58 and G0S2. Cell Cycle. 2010;9:2719–2725. doi: 10.4161/cc.9.14.12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManaman JL, Zabaronick W, Schaack J, Orlicky DJ. Lipid droplet targeting domains of adipophilin. J Lipid Res. 2003;44:668–673. doi: 10.1194/jlr.C200021-JLR200. [DOI] [PubMed] [Google Scholar]
- Murugesan S, Goldberg EB, Dou E, Brown WJ. Identification of diverse lipid droplet targeting motifs in the PNPLA family of triglyceride lipases. PLoS One. 2013;8:e64950. doi: 10.1371/journal.pone.0064950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura N, Fujimoto T. Adipose differentiation-related protein has two independent domains for targeting to lipid droplets. Biochem Biophys Res Commun. 2003;306:333–338. doi: 10.1016/s0006-291x(03)00979-3. [DOI] [PubMed] [Google Scholar]
- Noritake J, Fukata Y, Iwanaga T, Hosomi N, Tsutsumi R, Matsuda N, Tani H, Iwanari H, Mochizuki Y, Kodama T, et al. Mobile DHHC palmitoylating enzyme mediates activity-sensitive synaptic targeting of PSD-95. J Cell Biol. 2009;186:147–160. doi: 10.1083/jcb.200903101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno Y, Kihara A, Sano T, Igarashi Y. Intracellular localization and tissue-specific distribution of human and yeast DHHC cysteine-rich domain-containing proteins. Biochim Biophys Acta. 2006;1761:474–483. doi: 10.1016/j.bbalip.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Ohsaki Y, Maeda T, Maeda M, Tauchi-Sato K, Fujimoto T. Recruitment of TIP47 to lipid droplets is controlled by the putative hydrophobic cleft. Biochem Biophys Res Commun. 2006;347:279–287. doi: 10.1016/j.bbrc.2006.06.074. [DOI] [PubMed] [Google Scholar]
- Santy LC, Casanova JE. Activation of ARF6 by ARNO stimulates epithelial cell migration through downstream activation of both Rac1 and phospholipase D. J Cell Biol. 2001;154:599–610. doi: 10.1083/jcb.200104019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo JY, Yaneva R, Cresswell P. Viperin: a multifunctional, interferon-inducible protein that regulates virus replication. Cell Host Microbe. 2011;10:534–539. doi: 10.1016/j.chom.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni KG, Mardones GA, Sougrat R, Smirnova E, Jackson CL, Bonifacino JS. Coatomer-dependent protein delivery to lipid droplets. J Cell Sci. 2009;122:1834–1841. doi: 10.1242/jcs.045849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Otsuka T, Ohsaki Y, Cheng J, Taniguchi T, Hashimoto H, Taniguchi H, Fujimoto T. Derlin-1 and UBXD8 are engaged in dislocation and degradation of lipidated ApoB-100 at lipid droplets. Mol Biol Cell. 2012;23:800–810. doi: 10.1091/mbc.E11-11-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura N, Saitoh S, Kawano S, Kosugi A, Miyake K. Palmitoylation of LAT contributes to its subcellular localization and stability. Biochem Biophys Res Commun. 2006;341:1177–1183. doi: 10.1016/j.bbrc.2006.01.076. [DOI] [PubMed] [Google Scholar]
- Targett-Adams P, Chambers D, Gledhill S, Hope RG, Coy JF, Girod A, McLauchlan J. Live cell analysis and targeting of the lipid droplet-binding adipocyte differentiation-related protein. J Biol Chem. 2003;278:15998–16007. doi: 10.1074/jbc.M211289200. [DOI] [PubMed] [Google Scholar]
- Thiam AR, Antonny B, Wang J, Delacotte J, Wilfling F, Walther TC, Beck R, Rothman JE, Pincet F. COPI buds 60-nm lipid droplets from reconstituted water-phospholipid-triacylglyceride interfaces, suggesting a tension clamp function. Proc Natl Acad Sci USA. 2013;110:13244–13249. doi: 10.1073/pnas.1307685110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villena JA, Roy S, Sarkadi-Nagy E, Kim KH, Sul HS. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J Biol Chem. 2004;279:47066–47075. doi: 10.1074/jbc.M403855200. [DOI] [PubMed] [Google Scholar]
- Walther TC, Farese RV., Jr Lipid droplets and cellular lipid metabolism. Annu Rev Biochem. 2012;81:687–714. doi: 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfling F, Thiam AR, Olarte MJ, Wang J, Beck R, Gould TJ, Allgeyer ES, Pincet F, Bewersdorf J, Farese RV, Jr, Walther TC. Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. Elife. 2014;3:e01607. doi: 10.7554/eLife.01607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zechner R, Zimmermann R, Eichmann TO, Kohlwein SD, Haemmerle G, Lass A, Madeo F. FAT SIGNALS–lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012;15:279–291. doi: 10.1016/j.cmet.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science. 2004;306:1383–1386. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.