

Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating degenerative disease characterized by progressive loss of motor neurons in the motor cortex, brainstem, and spinal cord. Although defined as a motor disorder, ALS can arise concurrently with frontotemporal lobal dementia (FTLD). ALS begins focally but disseminates to cause paralysis and death. About 10% of ALS cases are caused by gene mutations, and more than 40 ALS-associated genes have been identified. While important questions about the biology of this disease remain unanswered, investigations of ALS genes have delineated pathogenic roles for (a) perturbations in protein stability and degradation, (b) altered homeostasis of critical RNA- and DNA-binding proteins, (c) impaired cytoskeleton function, and (d) non-neuronal cells as modifiers of the ALS phenotype. The rapidity of progress in ALS genetics and the subsequent acquisition of insights into the molecular biology of these genes provide grounds for optimism that meaningful therapies for ALS are attainable.
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive, fatal disorder of motor neurons that begins focally and spreads, leading to weakness of limb, respiratory, and bulbar muscles. Immediately preceding death, there is a near total loss of limb and respiratory function, as well as a loss of the ability to chew, swallow, and speak. ALS is defined as an “orphan disease,” with approximately 2 per 100,000 new cases per year and a prevalence of about 5 per 100,000 total cases each year (1). In the United States (2) and Europe (3, 4), ALS is diagnosed in about 1 in 500 to 1 in 1,000 adult deaths, implying that 500,000 people in the United States will develop this disease in their lifetimes. About 10% of ALS cases are inherited, usually as dominant traits (5, 6). Both familial ALS (fALS) and sporadic ALS (sALS) can develop concurrently with frontotemporal lobar dementia (FTLD). By contrast with the dementia of Alzheimer disease (AD), in which the cardinal finding is memory loss, FTLD is characterized by behavioral changes and progressive aphasia, sometimes accompanied by movement disorders (7, 8). While AD involves prominent pathology in the hippocampus, the essential finding in FTLD is, as the name suggests, early atrophy of the frontal and temporal lobes.
Four recurring themes have emerged from the pathological analysis of autopsied cases with sALS, fALS, or ALS-FTLD with diverse genetic causes. First, the motor neuron death usually entails deposition of aggregated proteins, often ubiquitinated and predominantly cytoplasmic. Second, in ALS, the levels and functions of RNA and RNA-binding proteins are abnormal. Aggregates of protein and RNA are detected both in motor neurons and non-neuronal cells, such as astrocytes and microglia. Third, most cases entail some disturbance of neuronal cytoskeletal architecture and function. Additionally, in almost all cases, motor neuron death is influenced by non-neuronal cells, including oligodendroglia and cells involved in neuroinflamation (e.g., astroglia and microglia).
Protein toxicity: protein aggregation and prion domains
An important theme in ALS pathogenesis is that several species of protein (both WT and mutant) are dysfunctional in both fALS and sALS, as evidenced by the formation of aggregates, abnormal cleavage events, or distinctive posttranslational modifications (e.g., ubiquitination or hyperphosphorylation). These changes occur both as primary consequences of mutations in the affected proteins and as secondary phenomena induced by the underlying disease process.
Protein aggregation and inclusion bodies.
It has been apparent for decades that protein pathology is important in ALS, as suggested by an early pathological finding of deposition of threads of ubiquitinated material in motor neurons (9). This has been interpreted as denoting the presence of proteins that are conformationally unstable or modified, and so are destined for degradation. In later stages of motor neuron disease, dense aggregates of ubiquitinated protein are seen, sometimes in association with eosinophilic aggregates described as “Bunina bodies” (10). Whether these deposits are toxic or reflect a cellular response to a more primary pathology remains unclear. Indeed, the possibility that some aggregates may reflect beneficial, compensatory events has also been considered.
Cytosolic superoxide dismutase [Cu/Zn] (SOD1) was the first gene and protein whose mutations were demonstrated to cause ALS (Table 1; ref. 11). Of the more than 160 different missense and 12 truncation mutations detected in SOD1 (12), most impair conformational stability of this abundant protein, triggering its deposition in inclusion bodies within spinal motor neurons (13) both in vitro and in vivo (14, 15). There is a general correlation between the degree of mutation-induced conformational instability and the rate of clinical progression (15). Mutant SOD1 also spontaneously forms oligomers in vivo and in vitro (16, 17) that are submicroscopic and often soluble (18) and may be more toxic than larger, visible aggregates (19, 20).
Table 1. Genes whose mutations cause ALS: Part 1 of 2.
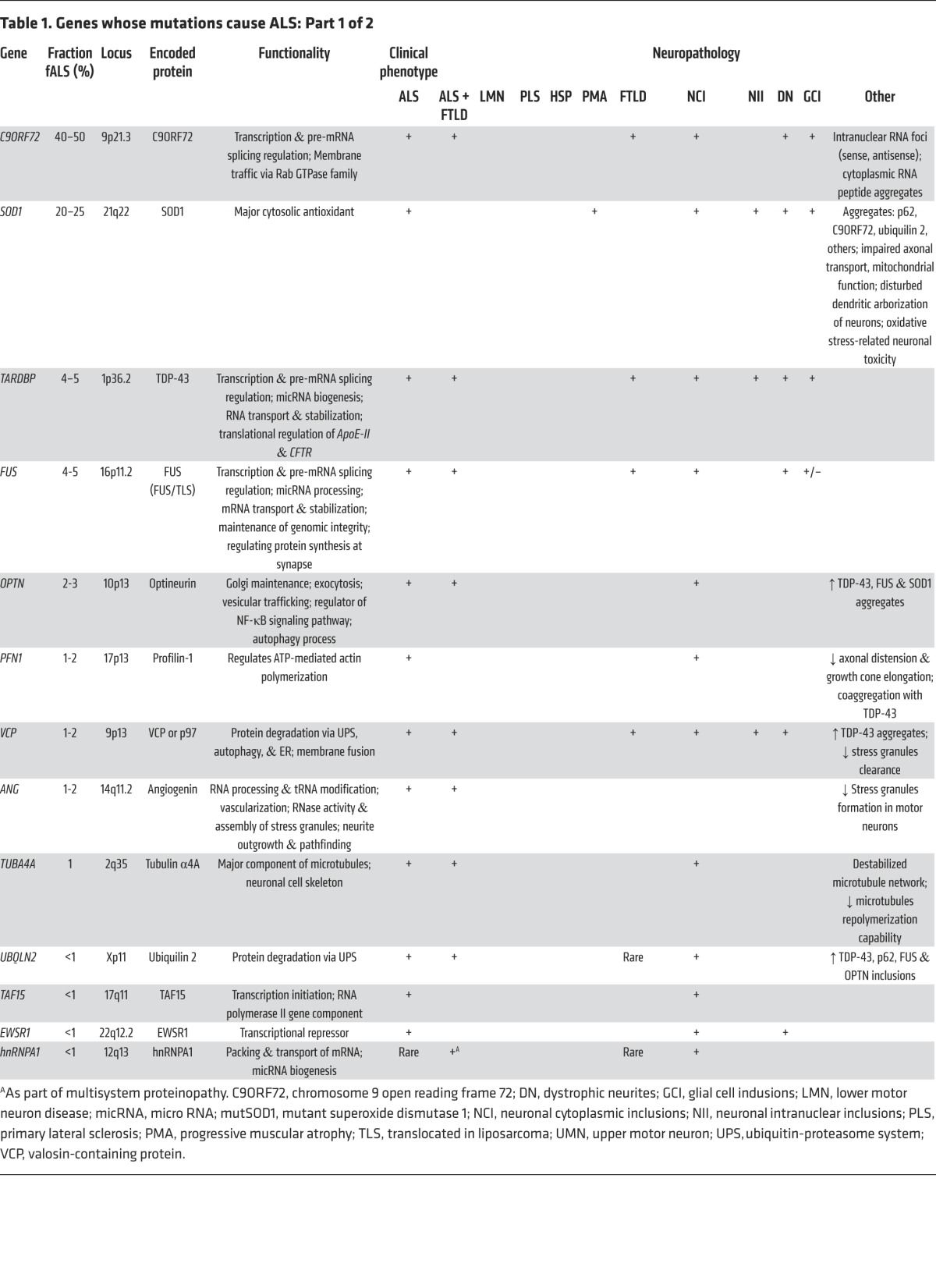
Aggregates of posttranslationally modified WT SOD1 can also be detected in the spinal cords of many sALS patients (21–23). This finding has been predicated on the use of antibodies to detect epitopes that are specific for misfolded SOD1, regardless of whether the misfolding events reflect mutations altering the structure of the SOD1 protein (in fALS) or modifications of WT SOD1 (in sALS). Importantly, the misfolded WT species can be cytotoxic, for example, by impairing axonal transport (21). A role for WT SOD1 in sALS is further suggested by the finding that sALS patients with antibodies against oxidized, WT SOD1 have a slight but statistically significant survival advantage (24).
The molecular basis for the toxicity of mutant SOD1 is multifactorial and elusive. Because SOD1 detoxifies superoxide anion (a by-product of aerobic metabolism), it is likely that mutant SOD1 provokes oxidative stress. This is exaggerated when SOD1 is zinc-depleted, a condition that accelerates misfolding (25). Secondly, mutant SOD1 can form toxic nitrotyrosines (26). A third factor in SOD1 pathology is aberrant cellular buffering and trafficking of copper and zinc. A fourth aspect is that unstable mutant SOD1 protein acquires a capacity to bind to substances with hydrophobic surfaces, to which WT SOD1 is less adherent (27). Whatever the molecular underpinnings of its cytotoxicity, miscreant forms of SOD1 can disrupt a wide set of cellular functions. While a complete recounting of these pathways is beyond the scope of this review, some of the adverse effects include: provocation of cellular hyperexcitability (28), disruption of mitochondrial function (29, 30), induction of the unfolded protein response (UPR) and ER stress (31), impairment of molecular motors and axonal transport (32), and early disruption of the neuromuscular synaptic structures (6, 33).
Central in the investigations of these phenomena has been the robust finding that transgenic mice that express high levels of mutant SOD1 protein can develop motor neuron disease with a temporal, clinical, and pathological pattern that recapitulates much of human ALS. The most widely investigated mouse expresses high levels of the SOD1G93A allele (34); however, several other ALS-associated SOD1 alleles have also been incorporated into ALS mouse models.
Accumulation of ubiquitinated protein is a key characteristic of early ALS pathology (33). A major breakthrough in understanding this was the 2006 discovery by Neumann and colleagues that trans-active response (TAR) DNA-binding protein (TDP-43) is the ubiquitinated target in both sALS and FTLD (35). The TDP-43 gene (TARDBP) encodes a 43-kDa protein containing two RNA recognition domains; a glycine-rich, putative prion-like domain; and both nuclear localization and nuclear export motifs (Figure 1). Under normal conditions, TDP-43 is located in the nucleus, where it is involved in DNA/RNA binding, transcription, RNA splicing, miRNA biogenesis, RNA transport, and RNA transcript stability (reviewed in ref. 36). TDP-43 has a pivotal role in nonsense-mediated decay, the process whereby abnormal mRNA transcripts (e.g., base-pair substituted or truncated) are rapidly degraded.
Figure 1. Structure and functional domains of DNA/RNA-binding proteins associated with fALS.

Several functional domains are common to the ALS-associated DNA/RNA-binding proteins, including TDP-43, hnRNPA1, FUS, and TAF15. These common domains include a nuclear localization motif, RNA-repeat binding domains, and glycine-rich domains of low structural complexity that possess prion-like activity. ALS-related mutations are annotated in their corresponding residues. Structurally ordered, disordered and prion-like domains are noted beneath each peptide. Sources of sequence data: Uniprot (domain prediction; http://www.uniprot.org), FoldIndex (ordered/disordered domain prediction; http://bip.weizmann.ac.il/fldbin/findex). Prion domain mapping data provided by refs. 85, 89, and 92. Adapted with permission from Elsevier (139).
In most cases of sALS, fALS, and FTLD, hyperphosphorylated, cleaved TDP-43 accumulates diffusely in the cytoplasm of neurons and glia, where it assembles into round and thread-like inclusions. These pathological forms of TDP-43 are first evident within spinal motor neurons but then disperse rostrally into the brain and throughout the CNS (37). TDP-43 has now been identified as a component in cytoplasmic deposits in disparate neurodegenerative diseases (36). The depletion of TDP-43 from the nucleus in these disorders has suggested that TDP-43–mediated toxicity may reflect either loss of its function in the nucleus, an acquired adverse effect of its pathological presence in the cytoplasm (gain-of-function), or both.
Following the identification of TDP-43 protein in inclusion bodies in sALS, several investigators identified ALS-associated mutations in TARDBP in cases of fALS (38), accounting for 4%–5% of dominantly inherited fALS, approximately 1% of sALS (39), and rare cases of FTLD (Table 1; ref. 40). To date, more than 35 dominantly transmitted coding missense mutations have been described in TARDBP; a single premature stop codon was also identified (12). With only three known exceptions, these mutations alter residues encoding the C-terminal glycine-rich domain, the site of a putative prion-like domain.
The proximal pathobiology of TDP-43 and SOD1 are fundamentally different. Motor neuron death by mutant SOD1 generally is a consequence of the abundance of the mutant protein: the higher the burden of mutant SOD1G93A protein in mice, the more fulminant the disease. (An exception is a truncated mutant form of SOD1 that causes ALS, although at low levels; ref. 41). To a remarkable degree, cellular function in vivo is relatively unaffected by rather dramatic increases and decreases in levels of the WT protein. SOD1 KO mice demonstrate a late-life, slow motor neuropathy (42) but do not develop fulminant ALS as seen in the transgenic SOD1G93A mice. By contrast, both reduction and elevation of levels of WT TDP-43 can be devastating, leading to frank motor neuron disease (43–46). It thus seems unlikely that mutant TDP-43 toxicity bears a simple relationship to the dose of the protein or its RNA transcript.
Shortly after the identification of TDP-43, a second RNA-binding protein, fused in sarcoma/translocated in liposarcoma (FUS/TLS or just FUS), was implicated through analysis of a fALS pedigree from Cape Verde (47, 48). Structurally comparable to TDP-43 (Figure 1), FUS is localized within the nucleus, where it interacts with DNA and RNA in a broad range of processes. It is able to translocate to the cytoplasm, where it is required for RNA processing. Almost half of the FUS mutations are within the C-terminal nuclear localization sequence, with a smaller cluster found within the glycine-rich domain. Mutant FUS, like mutant SOD1 and TDP-43 — and WT TDP-43 in sALS — is detected in diverse types of intracellular inclusions. Perhaps most notably, FUS is a major component of stress granules (below); mutations in FUS that lead to intracellular retention of this protein increase its propensity to form these cytoplasmic structures. Mutant FUS is also a major component of ubiquitin- and p62-positive cytoplasmic inclusions in both fALS and FTLD patients. These aggregates are detected in both neurons and glia of the brain and spinal cord (47, 48). By contrast with TDP-43 inclusions, the frequency of FUS-positive inclusion bodies in sALS is low (49–51). There is little experience with mouse models of FUS-dependent ALS. Fus-null mice are viable but sterile, and susceptible to chromosomal instability (52). A transgenic rat expressing mutant FUS developed active motor neuron disease (53).
In 2011, a trio of teams made the discovery that 40%–50% of fALS and 8%–10% of sALS cases, as well as cases of ALS-FTLD, are caused by mutations in the gene C9orf72 (Figure 2; refs. 54–56). This gene encodes a protein of approximately 54.3 kDa (481 amino acids) whose function remains unknown. Bioinformatic analyses suggest that this protein shares structural features with differentially expressed in normal and neoplastic (DENN) cells and GDP/GTP exchange factors (GEF), and may regulate membrane cell trafficking (57). The distinctive mutation in this gene is an expansion of an intronic hexanucleotide GGGGCC (G4C2) repeat motif. Normally present in 30 or fewer copies, in C9orf72-associated ALS, the repeat domain expands to encompass hundreds of tandem repeats (58). The expanded segment is transmitted as a dominant trait in ALS and ALS-FTLD pedigrees. Affected individuals are heterozygous for the expanded allele; homozygous cases are infrequent (59, 60).
Figure 2. Pathogenic mechanisms associated with hexanucleotide repeat-expanded C9orf72 and various DNA/RNA-binding proteins.

(A) The mutant C9orf72 gene associated with fALS contains an intronic G4C2 motif often expanded to several hundred (and even several thousand) repeats. This GC-rich domain is transcribed in both the sense and antisense directions, producing mRNA prone to forming large intranuclear foci that are believed to sequester some RNA-binding proteins. The sense or antisense transcripts undergo noncanonical RAN translation in all six possible reading frames, generating five dipeptide-repeat peptides (GA, GR, GP, PR, PA), which form inclusions that are associated with the protein p62. (B) Under normal conditions, the DNA/RNA-binding proteins mutated in fALS, most notably TDP-43 and FUS, are typically located within the nucleus, where they serve multiple functions. These proteins are also able to translocate to the cytoplasm, where they may localize to stress granules under some adverse cellular conditions. When these proteins are defective (e.g., bearing ALS-related mutations) the normal range of interactions with DNA and RNA are disrupted; this can lead to marked changes in transcription, splicing, and translation. Furthermore, the presence of the low-complexity, prion-like domains is thought to facilitate oligomer self-assembly under conditions of cellular stress, thereby promoting prion-like toxicity and intercellular spread.
The G4C2 expansion produces both nuclear RNA foci and cytoplasmic protein inclusions. The expansion is located within a normally noncoding intron within the C9orf72 promoter and produces a large RNA transcript containing the motif. At autopsy, C9orf72 mutant brains show widespread intranuclear RNA foci (54, 61) generated from both sense and antisense transcripts across the expanded G4C2 segment. Foci have been described in fibroblasts (62–64) and motor neurons derived from induced pluripotent cells generated from fibroblasts of C9orf72 ALS cases (62–65). Notably, the expansion shows some instability; its size varies modestly between different cell types in the same individual (65). In addition to these intranuclear RNA foci, histopathological studies have documented that these cases have at least three types of protein inclusions. Two of these are TDP-43–positive inclusions and distinctive spheroidal inclusions that are positive for a 62 kDa protein (discussed below) that is abundant in hippocampus and cerebellum (66). Several components of these p62-positive inclusions have since been identified, including RNA-binding motif 45 (RBM45) (67) and heterogeneous nuclear ribonucleoprotein A3 (hnRNPA3) (68). Unexpectedly, neurons from cases with the C9orf72 intronic expansion also possess a third type of inclusion: intracytosolic aggregates composed of dipeptide repeat proteins encoded by the intronic hexanucleotide repeat that are produced through noncanonical, repeat-associated non-ATG–mediated (RAN-mediated) translation. These atypical peptides reflect translation of amino acids from all possible reading frames of the G4C2 expanded domains (69–73). Some RAN-produced peptides, such as those containing repeated arginines, are neurotoxic in flies and mammalian neurons in vitro (74, 75)
Protein degradation.
In ALS, as in many other neurodegenerative diseases, it remains to be determined if inclusion bodies are directly cytotoxic or whether they reflect alternative pathogenic events, such as the presence of oligomeric species of the offending proteins. Many aspects of these models remain unclear. We do not know how well neurons can metabolize the visible or submicroscopic aggregates. Conceivably, the inability to metabolize abundant misfolded proteins will also impair the routine turnover of other proteins, a secondary adverse effect. Moreover, if a more generalized impairment of protein degradation ensues, this may induce ER stress and the UPR.
Mutations in other fALS genes are likely to impair protein degradation. One example is valosin-containing protein (VCP) whose mutations not only cause ALS, but also FTLD, inclusion body myositis, and Paget’s disease (76). VCP is a ubiquitin-dependent segregase that facilitates the segregation and proteosomal degradation of ubiquitinated proteins. Rare mutations in ubiquilin-2 (UBQLN2) and its homolog UBQLN1, which regulate ubiquitination of protein targets, have been associated with a rare, dominantly inherited, X-linked form of fALS (77) and the infantile-onset motor neuron disease Brown-Vialetto-Van Laere syndrome (78), respectively. UBQLN2 is a component of inclusion bodies in the brains and spinal cords of patients harboring UBQLN2 mutations that impair normal proteasome-mediated protein degradation. Polymorphisms in sequestosome-1 (SQSTM1, p62) are also associated with fALS and sALS (79–82). This ubiquitin-binding scaffold protein decorates the surface of inclusion bodies in many neurodegenerative diseases.
Prion-like domains in ALS proteins.
ALS usually begins focally and spreads in a pattern that implicates contiguous pools of motor neurons. The possibility that a pattern of dissemination of pathology and then motor neuron death in ALS might be prion-like was first suggested more than a decade ago in the context of SOD1-associated disease (83). Indeed, cell-to-cell spread and propagated misfolding of both mutant (84) and WT (22) SOD1 have been reported. The prion hypothesis in ALS has recently been underscored by the observation that TDP-43 and FUS have low-complexity, glycine-rich domains that enhance aggregate formation (Figure 1). Indeed both proteins emerge in an in silico screen for proteins that harbor domains comparable to known yeast prion peptides (85–87). Many proteins contain both RNA-binding and prion-like activities; of approximately 250 human genes predicted to contain prion-like domains (88), 12%–20% are predicted to possess RNA-binding activity (89). Overexpression of DNA/RNA-binding proteins in yeast identified two prion-like proteins that were structural homologues to fALS-associated FUS: RNase polymerase II, TATA box binding protein–associated (TBP-associated) factor 68 kDa (TAF15) (90), and Ewing sarcoma breakpoint region 1 (EWSR1) (91). Mutational analyses have defined rare coding sequence variants in both TAF15 and EWSR1 in ALS; moreover, mutant TAF15 mislocalizes from the nucleus to form cytoplasmic punctae in spinal cord neurons (90, 92).
More recently, two heterogeneous nuclear ribonucleoproteins (hnRNPA1 and hnRNPA2B1) with prion-like domains have been genetically linked with fALS and the VCP-linked multisystem phenotype described previously (93). Intriguingly, the ribonucleoprotein hnRNPA3 is reported to bind the C9orf72 fALS-associated G4C2 repeat and accumulate in cytoplasmic inclusions unique to C9orf72 patients (68). The prion domain–containing protein CREST has been identified as a sALS risk factor in a trio analysis exome screen (94). CREST is a calcium-activated transcriptional activator and essential component of the nBAF (SWI/SNF) chromatin-remodeling complex.
One caveat arises in considering the model of ALS as a prion-driven disease. In prion disease (e.g., Creutzfeldt-Jakob disease, Kuru, or Gerstmann-Sträussler-Scheinker syndrome), the templated misfolding propagates not only between cells within an individual but also between individuals within a species, and even between species (95, 96). While data support the contention that prion-like misfolding of ALS proteins can propagate from cell to cell, transmission between individuals or species has not been documented in ALS.
Role of prion-like domains in RNP granule formation.
Many prion-like domains containing DNA/RNA-binding proteins are components of ribonucleoprotein granules (RNP granules), which maintain RNA homeostasis during cellular stress (reviewed in ref. 89). Stress granules, a form of RNP granule that rapidly assembles in response to a range of stressors, are particularly relevant to ALS pathology. The interaction and self-assembly of the prion-like domains of cytosolic RNA-binding proteins facilitates the rapid assembly of stress granules, allowing the sequestration of RNA into these inclusions — in some cases within minutes of toxic insult (97). The majority of RNA/DNA-binding, prion-like domain–containing proteins associated with ALS can be incorporated into stress granules (88, 97, 98). Thus, in response to stress, mutant forms of TDP-43 (100) and FUS (97) incorporate rapidly into persistent stress granules or form small oligomeric aggregates that interact with the granules. This brings together the prion-like domains of the RNA-binding proteins and thus is potentially permissive for prion assembly and propagation. An abnormally strong interaction of the mutant prion-like domains may prevent the disassembly of these granules, resulting in the persistent sequestration of mRNAs and thereby impeding their translation. Alternatively, their incorporation into granules might facilitate the conversion of prion-like domains to amyloid states, seeding larger, fibrillary oligomers and inclusion bodies. Furthermore, the presence of ubiquitin-modifying proteins, kinases, and proteases within stress granules might modify FUS or TDP-43 within inclusions, stabilizing the protein through phosphorylation or removal of ubiquitin.
RNA biology
RNA/DNA binding function.
As noted above, numerous DNA/RNA interacting proteins have now been associated with fALS, most commonly TDP-43 and FUS, with more rare examples, including TAF15, EWSR1, ANG, SETX, ELP3, and ataxin-1 and -2, hnRNPA1 and hnRNPA2B1, and CREST (Tables 1 and 2). TDP-43 and FUS function in almost every aspect of DNA and RNA processing, including transcription, splicing, RNA transport, miRNA processing, and translation. Our understanding of the role of these proteins in ALS pathogenesis is limited; we do not yet know which, if any of the myriad functions served by these proteins, is the Achilles heel in fALS. Under normal physiological conditions, these fALS-associated DNA/RNA-binding proteins are largely nuclear; a fraction of these proteins translocate to the cytoplasm under physiological conditions. The mutant, ALS-associated variant proteins (e.g., TDP-43, FUS, TAF15, hnRNPA1, hnRNA2B1) are frequently mislocalized to the cytoplasm, often leaving the nucleus entirely depleted of the affected protein (Figure 2; refs. 35, 47). It is unclear whether the primary pathology is loss of function in the nucleus or an acquired cytotoxic effect of the mutant protein in the myriad functions normally served by these proteins. What is clear is that the function of these proteins can be substantially impaired by ALS-associated mutations. As one example, TDP-43 and hnRNPA1 play a role in the recognition of splice sites on RNA transcripts (101, 102). In mice that overexpress the human Q331K mutant, the splicing of over 1,000 different RNA transcripts is altered (103).
Toxicity of RNA foci.
Both sense and antisense RNA transcripts are present in intranuclear foci in cases of C9orf72-mediated ALS. The possibility that these foci are directly unfavorable is suggested by studies in myotonic dystrophy type 1, wherein RNA foci transcribed from CTG expansions sequester the transcriptional splice factor muscleblind-like 1 (MBNL-1), depleting the pool of the protein in the nucleus (104). The possibility that this model pertains to C9orf72 fALS patients has led to efforts to identify protein-binding partners of the sense and antisense foci generated from the expanded G4C2 repeat domain of C9orf72. Candidates identified to date include ADARB2 (62), hnRNP-H, and SF2 (59, 61), though the consequence of this binding is presently unclear. The C9orf72 expansions might compromise cellular viability via mechanisms other than sequestering transcription factors. It is conceivable that the expanded G4C2 domain and/or the foci reduce expression of the normal C9orf72 protein and that this loss of function of the C9orf72 protein impairs motor neuron viability. Another possibility is that the pathological DNA expansions and RNA foci alter transcript splicing. Still another conjecture is that the G4C2 expansions elaborate functional short RNA fragments that act as siRNA or miRNA. Clearly, further studies will be required to characterize the C9orf72 expansion, its RNA and peptide products, and their respective potentials for cytotoxicity of both neuronal and non-neuronal cells.
Cytoskeletal function
Cytoskeletal structure.
A defining feature of motor neurons is the length of their axons, rendering them highly dependent on intracellular transport mechanisms to maintain normal structure and function. Critical in sustaining this extraordinary architecture is the cytoskeleton and associated molecular scaffolds and motors. Early in the genetic studies of ALS, several rare mutations were detected in genes encoding neurofilament heavy chain (105–107) and peripherin (108–110); both components were detected in cytoplasmic inclusions (Table 2; ref. 111). Additional cytoskeletal components have been identified as rare causes or epistatic modifiers of disease onset in ALS. Exome sequencing of fALS DNA identified four mutations in the gene encoding profilin-1 (Table 1), an essential regulatory component in the conversion of monomeric actin (G-actin) to its filamentous form (F-actin) (Figure 3; ref. 112). Components of the cytoskeletal system also act as modifiers of the ALS phenotype. EphA4, an ephrin receptor tyrosine kinase that regulates developmental axon outgrowth, was identified in a zebrafish screen of genes that rescue motor axon abnormalities induced by expression of SOD1 (113). In a Dutch cohort, expression of EphA4 was inversely correlated with both onset and survival in ALS (Table 3). EphA4 normally repels axons, serving as a molecular brake on outgrowth. It is plausible that reduced EphA4 activity through reduced expression or loss-of-function mutations enhances axonal outgrowth in response to injury. It is also possible that this, in turn, accounts for the enhanced survival in ALS mice hemizygous for this gene. Identification of other disease-modifying factors like EphA4 will increase our understanding of molecular events that contribute to ALS and provide targets for therapeutic intervention.
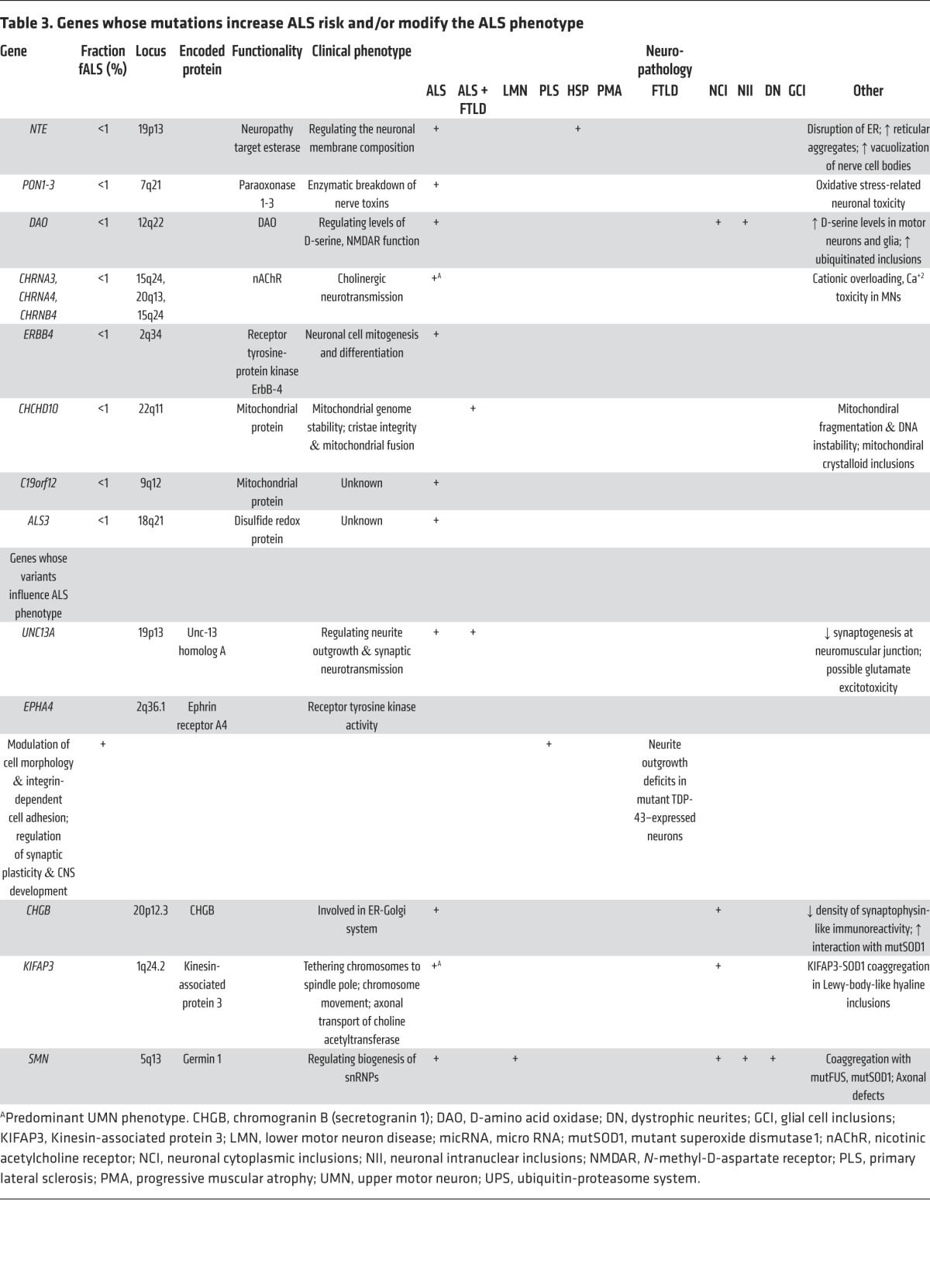
Table 2. Genes whose mutations cause ALS: Part 2 of 2.
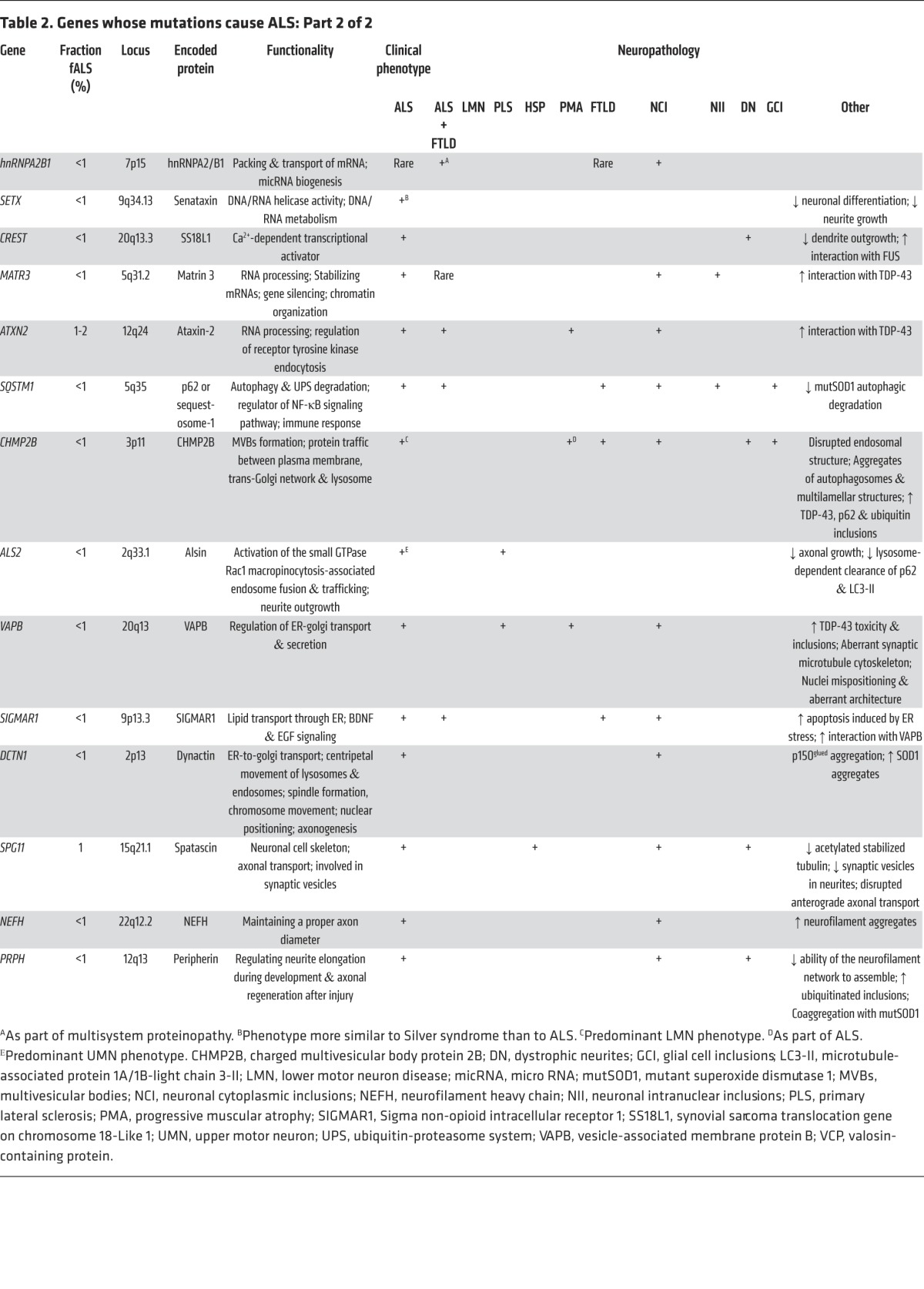
Figure 3. Disruption of neuroaxonal structure and axonal vesicle trafficking in ALS.
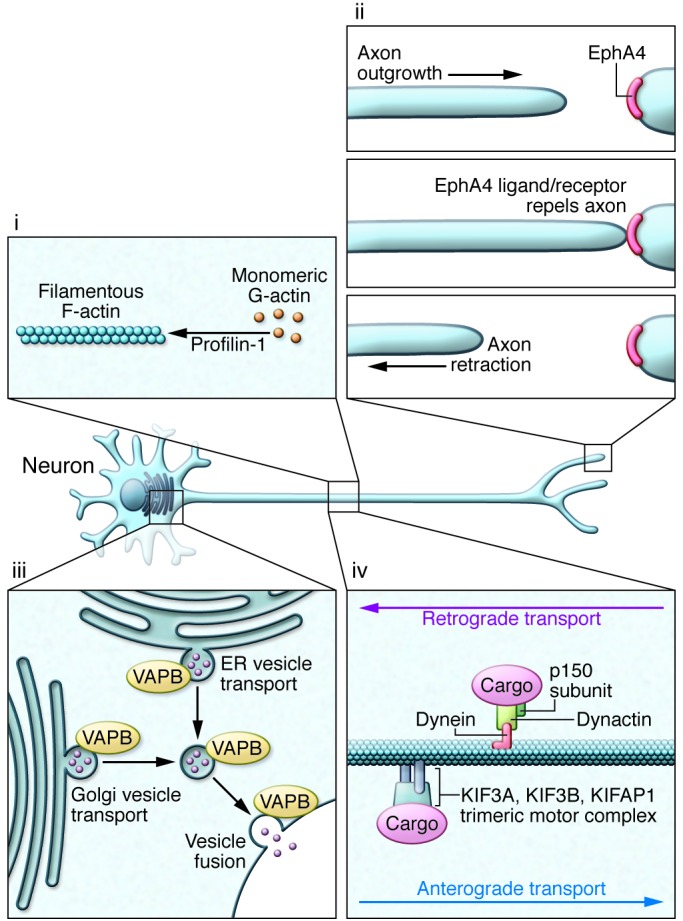
(i) Mutations in profilin-1 disrupt multiple functions of this protein, including its role in facilitating polymerization of monomeric G-actin to its filamentous F-actin form. (ii) Reduced expression of EphA4 correlates with increased survival in sALS, hypothetically through reducing this protein’s normal activity in signaling axon repulsion and arresting axonal growth. Reduction in levels of EphA4 or its activity is believed to enhance the capacity of a distal motor terminal to extend, remodel, and potentially reinnervate the target muscle. (iii) Disruption in endocytosis and the transport of vesicles from the golgi apparatus and ER are a likely consequence of ALS-associated mutations in VAPB. (iv) Mutations have been detected in the p150 subunit of dynactin, which is required for tethering cargos to the dynein retrograde transport complex. Some studies have suggested that altered expression of KIFAP1 enhances ALS risk by impairing function of the anterograde transport motor complex.
Table 3. Genes whose mutations increase ALS risk and/or modify the ALS phenotype.
Axonal transport.
Like the cytoskeleton, efficient axonal transport is critical for axonal function. ALS-related mutations have now been identified in components of the axonal transport system. Dynactin is required for tethering cargos of vesicles and organelles to the retrograde transport motor dynein. Introduction of mutations in DCTN1 led to dysfunction of dynactin in rodents, with defective vesicular transportation, degeneration of motor neurons, and premature death (Table 2; refs. 114, 115). A second component of axonal transport, kinesin-associated protein 3 (KIFAP3), was identified as a modifier of survival in sALS (Table 3). A polymorphism associated with reduced expression of KIFAP3 was found to correlate with an extension of lifespan by over a year in sALS cases (116), although this association is not seen in all populations (117). Forming a trimeric motor complex with KIF3A and KIF3B, KIFAP3 functions in anterograde transport and chromosome cytokinesis. The role of KIFAP3 in disease is unclear; however, increases in its expression have been noted early in pathology in SOD1 transgenic mice (118). Finally, fALS-associated mutations have been identified in vesicle-associated membrane protein/synaptobrevin-associated membrane protein B (VAPB), a ubiquitously expressed protein that is particularly important during golgi and ER vesicle transport and secretion (Table 2; ref. 119). Secreted VAPB is a ligand for Eph receptors; its secretion is blocked by introduction of ALS-associated mutations (120). In drosophila, expression of mutant VAPB induced deficits in synaptic morphology and neurodegeneration (121).
Non–cell-autonomous influences on motor neuron death
An important insight from the transgenic SOD1 ALS mice is that cells other than the motor neuron can modify the rate of progression of the disease. This was first convincingly demonstrated by a report that survival of mice expressing mutant SOD1G93A in motor neurons was dramatically improved when the motor neurons were surrounded by WT cells (122). Survival in SOD1G93A transgenic ALS mice is remarkably prolonged by eliminating expression of the SOD1 gene from astrocytes (123), microglia (124), or oligodendroglia (125); reciprocally, it is worsened when mutant SOD1 is largely eliminated from Schwann cells in these transgenic mice (126). The motor neuron death process provokes a neuroinflammatory reaction that recruits and activates astrocytes and microglia. The impact of the microglia, which can be neuroprotective as well as toxic (127, 128), is determined by many factors including the phenotype of incoming reactive T cells (129, 130). That ALS-derived astrocytes can be toxic to motor neurons has been robustly demonstrated in vitro using human astrocytes and motor neurons derived from induced pluripotent stem cells (131–133). The toxic influences derived from cultured ALS astrocytes may involve signaling via prostaglandin receptors (134).
Conclusion
Like other neurodegenerative disorders, ALS poses challenges in therapy development. Not least is the inadequacy of our understanding of the basic biology of this disease. Perhaps most importantly, it is unclear what causes sALS. Are there fundamentally distinct mechanisms in motor neuron death yet to be discovered, possibly involving neurotoxic environmental factors? Do genetic factors define susceptibility to such toxins? Why is onset in fALS delayed for many decades when the mutant genes are expressed from the earliest embryonic stages of development? Does this reflect an age-dependent accumulation of injury in post-mitotic motor neurons, an age-dependent loss of capacity to compensate for the toxic genes, a requirement for a concomitant adverse environmental stimulus, or some combination of all of these factors? Will it be possible to define complex, multigenic or epistatic gene-by-gene interactions in ALS? What are the roles of epigenetic factors or disturbances of the microbiome in fALS and sALS?
Another important challenge in developing ALS therapeutics is the inherent difficulty in accessing the CNS. The brain and spinal cord are protected by the blood-brain barrier, which limits permeation of many small molecules, as well as macromolecular therapies. Two other major challenges in ALS therapy are diagnosing the disease rapidly enough to allow early intervention and the lack of sensitive ALS biomarkers.
These challenges notwithstanding, it is encouraging that there has been substantial progress in delineating genetic factors that modulate ALS risk and phenotype. Each of the more than 40 ALS genes now reported (Tables 1–3) implicates primary events in ALS pathogenesis; the primary events converge on multiple downstream processes (e.g., mitochondrial failure, excitotoxicity, ER stress, or altered axonal transport). Each ALS gene discloses potential therapeutic targets. Most ALS genes are dominantly transmitted and thus are likely to be cytotoxic because of acquired properties that impair motor neuron viability. Of these, some such as SOD1 and C9orf72 show dose dependence of pathology: the more mutant the gene product (whether RNA or protein), the more severe the phenotype. This implies that strategies to silence the offending mutant genes may be beneficial. Fortunately, there are multiple new approaches to gene silencing, such as small, intrathecally delivered antisense oligonucleotides, and both shRNA and synthetic miRNAs delivered to the CNS via adeno-associated viral vectors. Indeed, a pilot trial of silencing of mutant SOD1 with antisense oligonucleotides has been reported (135). Another approach that has yielded positive pilot data is the use of antibodies to reduce the levels of mutant proteins in the CNS (136). For genes such as TARDBP, for which there is not a clear dose dependence of the mutant gene product to phenotype, other strategies will be needed. It is of interest that forced expression of the regulator of nonsense transcripts 1 (hUPF1) can ameliorate the toxicity of mutant TDP-43 in yeast (137) and in a rat model (138). To the extent that the pathophysiology of fALS and sALS overlap, therapeutic discoveries in the domain of ALS genetics may apply to sALS. These developments and, more generally, the expanding insights into pathophysiology allow optimism that meaningful therapies will be forthcoming, at least for inherited forms of ALS.
Acknowledgments
R.H. Brown Jr. receives support from the NINDS, the ALS Therapy Alliance, the ALS Association, the Angel Fund, the Al-Athel Foundation, the Pierre L. de Bourgknecht ALS Research Foundation, Project ALS, and P2ALS. RHB also receives support from the National Institute of Neurological Disorders and Stroke (NINDS) awards 1RC2NS070342-01, 1RC1NS068391-01, R01NS050557-05, and R01NS065847-01A1. O.M. Peters is supported by the ALS Therapy Alliance, the Angel Fund, Project ALS, and the Michael J. Fox Foundation.
Footnotes
Conflict of interest: Robert H. Brown Jr. serves as an ad hoc consultant for Biogen Inc. and Voyager Therapeutics.
Reference information:J Clin Invest. 2015;125(5):1767–1779. doi:10.1172/JCI71601.
References
- 1.O’Toole O, et al. Epidemiology clinical features of amyotrophic lateral sclerosis in Ireland between 1995 and 2004. J Neurol Neurosurg Psychiatry. 2008;79(1):30–32. doi: 10.1136/jnnp.2007.117788. [DOI] [PubMed] [Google Scholar]
- 2.Armon C. Sports and trauma in amyotrophic lateral sclerosis revisited. J Neurol Sci. 2007;262(1–2):45–53. doi: 10.1016/j.jns.2007.06.021. [DOI] [PubMed] [Google Scholar]
- 3.Alonso A, Logroscino G, Jick SS, Hernán MA. Incidence and lifetime risk of motor neuron disease in the United Kingdom: a population-based study. Eur J Neurol. 2009;16(6):745–751. doi: 10.1111/j.1468-1331.2009.02586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnston CA, et al. Amyotrophic lateral sclerosis in an urban setting: a population based study of inner city London. J Neurol. 2006;253(12):1642–1643. doi: 10.1007/s00415-006-0195-y. [DOI] [PubMed] [Google Scholar]
- 5.Leblond CS, Kaneb HM, Dion PA, Rouleau GA. Dissection of genetic factors associated with amyotrophic lateral sclerosis. Exp Neurol. 2014;262(pt B):91–101. doi: 10.1016/j.expneurol.2014.04.013. [DOI] [PubMed] [Google Scholar]
- 6.Sreedharan J, Brown RH., Jr Amyotrophic lateral sclerosis: Problems and prospects. Ann Neurol. 2013;74(3):309–316. doi: 10.1002/ana.24012. [DOI] [PubMed] [Google Scholar]
- 7.Ng AS, Rademakers R, Miller BL. Frontotemporal dementia: a bridge between dementia and neuromuscular disease. Ann N Y Acad Sci. doi: 10.1111/nyas.12638. [published online ahead of print December 30, 2014]. doi: 10.1111/nyas.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol. 2014;10(11):661–670. doi: 10.1038/nrneurol.2014.184. [DOI] [PubMed] [Google Scholar]
- 9.Leigh PN, Anderton BH, Dodson A, Gallo JM, Swash M, Power DM. Ubiquitin deposits in anterior horn cells in motor neurone disease. Neurosci Lett. 1988;93(2–3):197–203. doi: 10.1016/0304-3940(88)90081-x. [DOI] [PubMed] [Google Scholar]
- 10.Okamoto K, Mizuno Y, Fujita Y. Bunina bodies in amyotrophic lateral sclerosis. Neuropathology. 2008;28(2):109–115. doi: 10.1111/j.1440-1789.2007.00873.x. [DOI] [PubMed] [Google Scholar]
- 11.Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 12.Radunović A, Leigh PN. ALSODatabase: database of SOD1 (and other) gene mutations in ALS on the Internet. European FALS Group and ALSOD Consortium. Amyotroph Lateral Scler Other Motor Neuron Disord. 1999;1(1):45–49. doi: 10.1080/146608299300080040. [DOI] [PubMed] [Google Scholar]
- 13.Shibata N, et al. Immunohistochemical demonstration of Cu/Zn superoxide dismutase in the spinal cord of patients with familial amyotrophic lateral sclerosis. Acta Histochem Cytochem. 1993;26:619–622. doi: 10.1267/ahc.26.619. [DOI] [Google Scholar]
- 14.Borchelt DR, et al. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc Natl Acad Sci U S A. 1994;91(17):8292–8296. doi: 10.1073/pnas.91.17.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prudencio M, Hart PJ, Borchelt DR, Andersen PM. Variation in aggregation propensities among ALS-associated variants of SOD1: correlation to human disease. Hum Mol Genet. 2009;18(17):3217–3226. doi: 10.1093/hmg/ddp260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung J, Yang H, de Beus MD, Ryu CY, Cho K, Colón W. Cu/Zn superoxide dismutase can form pore-like structures. Biochem Biophys Res Commun. 2003;312(4):873–876. doi: 10.1016/j.bbrc.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 17.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 18.Brown HH, Borchelt DR. Analysis of mutant SOD1 electrophoretic mobility by Blue Native gel electrophoresis; evidence for soluble multimeric assemblies. PLoS One. 2014;9(8):e104583. doi: 10.1371/journal.pone.0104583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ray SS, Nowak RJ, Strokovich K, Brown RH, Jr, Walz T, Lansbury PT., Jr An intersubunit disulfide bond prevents in vitro aggregation of a superoxide dismutase-1 mutant linked to familial amytrophic lateral sclerosis. Biochemistry. 2004;43(17):4899–4905. doi: 10.1021/bi030246r. [DOI] [PubMed] [Google Scholar]
- 20.Ray SS, Lansbury PT., Jr A possible therapeutic target for Lou Gehrig’s disease. Proc Natl Acad Sci U S A. 2004;101(16):5701–5702. doi: 10.1073/pnas.0401934101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bosco DA, et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13(11):1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grad LI, et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc Natl Acad Sci U S A. 2014;111(9):3620–3625. doi: 10.1073/pnas.1312245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forsberg K, et al. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS One. 2010;5(7):e11552. doi: 10.1371/journal.pone.0011552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Blitterswijk M, et al. Anti-superoxide dismutase antibodies are associated with survival in patients with sporadic amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2011;12(6):430–438. doi: 10.3109/17482968.2011.585163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Estévez AG, et al. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999;286(5449):2498–2500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- 26.Beckman JS, Carson M, Smith CD, Koppenol WH. ALS, SOD and peroxynitrite. Nature. 1993;364(6438):584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- 27.Tiwari A, Xu Z, Hayward LJ. Aberrantly increased hydrophobicity shared by mutants of Cu,Zn-superoxide dismutase in familial amyotrophic lateral sclerosis. J Biol Chem. 2005;280(33):29771–29779. doi: 10.1074/jbc.M504039200. [DOI] [PubMed] [Google Scholar]
- 28.van Zundert B, Izaurieta P, Fritz E, Alvarez FJ. Early pathogenesis in the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Cell Biochem. 2012;113(11):3301–3312. doi: 10.1002/jcb.24234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vande Velde C, et al. Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PLoS One. 2011;6(7):e22031. doi: 10.1371/journal.pone.0022031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vucic S, Rothstein JD, Kiernan MC. Advances in treating amyotrophic lateral sclerosis: insights from pathophysiological studies. Trends Neurosci. 2014;37(8):433–442. doi: 10.1016/j.tins.2014.05.006. [DOI] [PubMed] [Google Scholar]
- 31.Hetz C, et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23(19):2294–2306. doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morfini GA, et al. Inhibition of fast axonal transport by pathogenic SOD1 involves activation of p38 MAP kinase. PLoS One. 2013;8(6):e65235. doi: 10.1371/journal.pone.0065235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanning KC, Kaplan A, Henderson CE. Motor neuron diversity in development and disease. Annu Rev Neurosci. 2010;33:409–440. doi: 10.1146/annurev.neuro.051508.135722. [DOI] [PubMed] [Google Scholar]
- 34.Gurney ME, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264(5166):1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 35.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 36.Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19(R1):R46–R64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brettschneider J, et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. 2013;74(1):20–38. doi: 10.1002/ana.23937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sreedharan J, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319(5870):1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daoud H, et al. Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J Med Genet. 2009;46(2):112–114. doi: 10.1136/jmg.2008.062463. [DOI] [PubMed] [Google Scholar]
- 40.Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol. 2013;9(11):617–628. doi: 10.1038/nrneurol.2013.203. [DOI] [PubMed] [Google Scholar]
- 41.Jonsson PA, et al. Minute quantities of misfolded mutant superoxide dismutase-1 cause amyotrophic lateral sclerosis. Brain. 2004;127(pt 1):73–88. doi: 10.1093/brain/awh005. [DOI] [PubMed] [Google Scholar]
- 42.Reaume AG, et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13(1):43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 43.D’Alton S, Altshuler M, Cannon A, Dickson DW, Petrucelli L, Lewis J. Divergent phenotypes in mutant TDP-43 transgenic mice highlight potential confounds in TDP-43 transgenic modeling. PLoS One. 2014;9(1):e86513. doi: 10.1371/journal.pone.0086513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gendron TF, Petrucelli L. Rodent models of TDP-43 proteinopathy: investigating the mechanisms of TDP-43-mediated neurodegeneration. J Mol Neurosci. 2011;45(3):486–499. doi: 10.1007/s12031-011-9610-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu YF, et al. Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci. 2010;30(32):10851–10859. doi: 10.1523/JNEUROSCI.1630-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu LS, Cheng WC, Shen CK. Targeted depletion of TDP-43 expression in the spinal cord motor neurons leads to the development of amyotrophic lateral sclerosis-like phenotypes in mice. J Biol Chem. 2012;287(33):27335–27344. doi: 10.1074/jbc.M112.359000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kwiatkowski TJ, Jr, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323(5918):1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 48.Vance C, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323(5918):1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IR. Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathologica. 2009;118(5):605–616. doi: 10.1007/s00401-009-0581-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deng HX, et al. FUS-immunoreactive inclusions are a common feature in sporadic and non-SOD1 familial amyotrophic lateral sclerosis. Ann Neurol. 2010;67(6):739–748. doi: 10.1002/ana.22051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain. 2009;132(pt 11):2922–2931. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuroda M, et al. Male sterility and enhanced radiation sensitivity in TLS(–/–) mice. EMBO J. 2000;19(3):453–462. doi: 10.1093/emboj/19.3.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang C, et al. FUS transgenic rats develop the phenotypes of amyotrophic lateral sclerosis and frontotemporal lobar degeneration. PLoS Genet. 2011;7(3):e1002011. doi: 10.1371/journal.pgen.1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dejesus-Hernandez M, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Renton AE, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. doi: 10.1016/S1474-4422(11)70261-7. Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol. 2012 ;11(1): 54–65. [DOI] [PubMed] [Google Scholar]
- 57.Marat AL, Dokainish H, McPherson PS. DENN domain proteins: regulators of Rab GTPases. J Biol Chem. 2011;286(16):13791–13800. doi: 10.1074/jbc.R110.217067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Blitterswijk M, et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol. 2013;12(10):978–988. doi: 10.1016/S1474-4422(13)70210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cooper-Knock J, et al. C9ORF72 transcription in a frontotemporal dementia case with two expanded alleles. Neurology. 2013;81(19):1719–1721. doi: 10.1212/01.wnl.0000435295.41974.2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fratta P, et al. Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol. 2013;126(3):401–409. doi: 10.1007/s00401-013-1147-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee YB, et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5(5):1178–1186. doi: 10.1016/j.celrep.2013.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Donnelly CJ, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80(2):415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lagier-Tourenne C, et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc Natl Acad Sci U S A. 2013;110(47):E4530–E4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Almeida S, et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126(3):385–399. doi: 10.1007/s00401-013-1149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sareen D, et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 2013;5(208):208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Al-Sarraj S, et al. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122(6):691–702. doi: 10.1007/s00401-011-0911-2. [DOI] [PubMed] [Google Scholar]
- 67.Collins M, et al. The RNA-binding motif 45 (RBM45) protein accumulates in inclusion bodies in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) patients. Acta Neuropathol. 2012;124(5):717–732. doi: 10.1007/s00401-012-1045-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mori K, et al. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol. 2013;125(3):413–423. doi: 10.1007/s00401-013-1088-7. [DOI] [PubMed] [Google Scholar]
- 69.Ash PE, et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77(4):639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zu T, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci U S A. 2011;108(1):260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gendron TF, et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013;126(6):829–844. doi: 10.1007/s00401-013-1192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mori K, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013;126(6):881–893. doi: 10.1007/s00401-013-1189-3. [DOI] [PubMed] [Google Scholar]
- 73.Mori K, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 74.Mizielinska S, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345(6201):1192–1194. doi: 10.1126/science.1256800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wen X, et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014;84(6):1213–1225. doi: 10.1016/j.neuron.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Watts GD, et al. Novel VCP mutations in inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia. Clin Genet. 2007;72(5):420–426. doi: 10.1111/j.1399-0004.2007.00887.x. [DOI] [PubMed] [Google Scholar]
- 77.Deng HX, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477(7363):211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gonzalez-Perez P, et al. Association of UBQLN1 mutation with Brown-Vialetto-Van Laere syndrome but not typical ALS. Neurobiol Dis. 2012;48(3):391–398. doi: 10.1016/j.nbd.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fecto F, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68(11):1440–1446. doi: 10.1001/archneurol.2011.250. [DOI] [PubMed] [Google Scholar]
- 80.Teyssou E, et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol. 2013;125(4):511–522. doi: 10.1007/s00401-013-1090-0. [DOI] [PubMed] [Google Scholar]
- 81.Shimizu H, et al. Sporadic ALS with compound heterozygous mutations in the SQSTM1 gene. Acta Neuropathol. 2013;126(3):453–459. doi: 10.1007/s00401-013-1150-5. [DOI] [PubMed] [Google Scholar]
- 82.Chen Y, et al. SQSTM1 mutations in Han Chinese populations with sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2014;35(3):726.e7–e9. doi: 10.1016/j.neurobiolaging.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 83.Bredesen DE, Ellerby LM, Hart PJ, Wiedau-Pazos M, Valentine JS. Do post-translational modifications of CuZnSOD lead to sporadic amyotrophic lateral sclerosis? Ann Neurol. 1997;42(2):135–137. doi: 10.1002/ana.410420202. [DOI] [PubMed] [Google Scholar]
- 84.Münch C, Bertolotti A. Self-propagation and transmission of misfolded mutant SOD1: prion or prion-like phenomenon? Cell Cycle. 2011;10(11):1711. doi: 10.4161/cc.10.11.15560. [DOI] [PubMed] [Google Scholar]
- 85.Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009;137(1):146–158. doi: 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cushman M, Johnson BS, King OD, Gitler AD, Shorter J. Prion-like disorders: blurring the divide between transmissibility and infectivity. J Cell Sci. 2010;123(pt 8):1191–1201. doi: 10.1242/jcs.051672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gitler AD, Shorter J. RNA-binding proteins with prion-like domains in ALS and FTLD-U. Prion. 2011;5(3):179–187. doi: 10.4161/pri.5.3.17230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012;1462:61–80. doi: 10.1016/j.brainres.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol. 2013;201(3):361–372. doi: 10.1083/jcb.201302044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Couthouis J, et al. A yeast functional screen predicts new candidate ALS disease genes. Proc Natl Acad Sci U S A. 2011;108(52):20881–20890. doi: 10.1073/pnas.1109434108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Couthouis J, et al. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21(13):2899–2911. doi: 10.1093/hmg/dds116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ticozzi N, et al. Mutational analysis reveals the FUS homolog TAF15 as a candidate gene for familial amyotrophic lateral sclerosis. Am J Med Genet B Neuropsychiatr Genet. 2011;156B(3):285–290. doi: 10.1002/ajmg.b.31158. [DOI] [PubMed] [Google Scholar]
- 93.Kim HJ, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495(7442):467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chesi A, et al. Exome sequencing to identify de novo mutations in sporadic ALS trios. Nat Neurosci. 2013;16(7):851–855. doi: 10.1038/nn.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Prusiner SB. Biology and genetics of prions causing neurodegeneration. Annu Rev Genet. 2013;47:601–623. doi: 10.1146/annurev-genet-110711-155524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Polymenidou M, Cleveland DW. The seeds of neurodegeneration: prion-like spreading in ALS. Cell. 2011;147(3):498–508. doi: 10.1016/j.cell.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bosco DA, et al. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010;19(21):4160–4175. doi: 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ayala YM, Misteli T, Baralle FE. TDP-43 regulates retinoblastoma protein phosphorylation through the repression of cyclin-dependent kinase 6 expression. Proc Natl Acad Sci U S A. 2008;105(10):3785–3789. doi: 10.1073/pnas.0800546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dormann D, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29(16):2841–2857. doi: 10.1038/emboj.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Colombrita C, et al. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem. 2009;111(4):1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x. [DOI] [PubMed] [Google Scholar]
- 101.Buratti E, Dörk T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001;20(7):1774–1784. doi: 10.1093/emboj/20.7.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005;280(45):37572–37584. doi: 10.1074/jbc.M505557200. [DOI] [PubMed] [Google Scholar]
- 103.Arnold ES, et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci U S A. 2013;110(8):E736–E745. doi: 10.1073/pnas.1222809110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mankodi A, Teng-Umnuay P, Krym M, Henderson D, Swanson M, Thornton CA. Ribonuclear inclusions in skeletal muscle in myotonic dystrophy types 1 and 2. Ann Neurol. 2003;54(6):760–768. doi: 10.1002/ana.10763. [DOI] [PubMed] [Google Scholar]
- 105.Figlewicz DA, et al. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum Mol Genet. 1994;3(10):1757–1761. doi: 10.1093/hmg/3.10.1757. [DOI] [PubMed] [Google Scholar]
- 106.Tomkins J, et al. Novel insertion in the KSP region of the neurofilament heavy gene in amyotrophic lateral sclerosis (ALS). Neuroreport. 1998;9(17):3967–3970. doi: 10.1097/00001756-199812010-00036. [DOI] [PubMed] [Google Scholar]
- 107.Al-Chalabi A, et al. Deletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosis. Hum Mol Genet. 1999;8(2):157–164. doi: 10.1093/hmg/8.2.157. [DOI] [PubMed] [Google Scholar]
- 108.Gros-Louis F, et al. A frameshift deletion in peripherin gene associated with amyotrophic lateral sclerosis. J Biol Chem. 2004;279(44):45951–45956. doi: 10.1074/jbc.M408139200. [DOI] [PubMed] [Google Scholar]
- 109.Leung CL, et al. A pathogenic peripherin gene mutation in a patient with amyotrophic lateral sclerosis. Brain Pathol. 2004;14(3):290–296. doi: 10.1111/j.1750-3639.2004.tb00066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Corrado L, et al. A novel peripherin gene (PRPH) mutation identified in one sporadic amyotrophic lateral sclerosis patient. Neurobiol Aging. 2011;32(3):552.e–e6. doi: 10.1016/j.neurobiolaging.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 111.Corbo M, Hays AP. Peripherin and neurofilament protein coexist in spinal spheroids of motor neuron disease. J Neuropathol Exp Neurol. 1992;51(5):531–537. doi: 10.1097/00005072-199209000-00008. [DOI] [PubMed] [Google Scholar]
- 112.Wu CH, et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012;488(7412):499–503. doi: 10.1038/nature11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Van Hoecke A, et al. EPHA4 is a disease modifier of amyotrophic lateral sclerosis in animal models and in humans. Nat Med. 2012;18(9):1418–1422. doi: 10.1038/nm.2901. [DOI] [PubMed] [Google Scholar]
- 114.Puls I, et al. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33(4):455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- 115.Laird FM, et al. Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J Neurosci. 2008;28(9):1997–2005. doi: 10.1523/JNEUROSCI.4231-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Landers JE, et al. Reduced expression of the Kinesin-Associated Protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2009;106(22):9004–9009. doi: 10.1073/pnas.0812937106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.van Doormaal PT, et al. Analysis of the KIFAP3 gene in amyotrophic lateral sclerosis: a multicenter survival study. Neurobiol Aging. 2014;35(10):2420.e13–e14. doi: 10.1016/j.neurobiolaging.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dupuis L, et al. Differential screening of mutated SOD1 transgenic mice reveals early up-regulation of a fast axonal transport component in spinal cord motor neurons. Neurobiol Dis. 2000;7(4):274–285. doi: 10.1006/nbdi.2000.0292. [DOI] [PubMed] [Google Scholar]
- 119.Nishimura AL, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75(5):822–831. doi: 10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tsuda H, et al. The amyotrophic lateral sclerosis 8 protein VAPB is cleaved, secreted, and acts as a ligand for Eph receptors. Cell. 2008;133(6):963–977. doi: 10.1016/j.cell.2008.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Forrest S, et al. Increased levels of phosphoinositides cause neurodegeneration in a Drosophila model of amyotrophic lateral sclerosis. Hum Mol Genet. 2013;22(13):2689–2704. doi: 10.1093/hmg/ddt118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Clement AM, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302(5642):113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- 123.Yamanaka K, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11(3):251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Boillée S, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312(5778):1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 125.Kang SH, et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16(5):571–579. doi: 10.1038/nn.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lobsiger CS, et al. Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc Natl Acad Sci U S A. 2009;106(11):4465–4470. doi: 10.1073/pnas.0813339106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xiao Q, et al. Mutant SOD1(G93A) microglia are more neurotoxic relative to wild-type microglia. J Neurochem. 2007;102(6):2008–2019. doi: 10.1111/j.1471-4159.2007.04677.x. [DOI] [PubMed] [Google Scholar]
- 128.Beers DR, et al. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103(43):16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Henkel JS, et al. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med. 2013;5(1):64–79. doi: 10.1002/emmm.201201544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhao W, Beers DR, Liao B, Henkel JS, Appel SH. Regulatory T lymphocytes from ALS mice suppress microglia and effector T lymphocytes through different cytokine-mediated mechanisms. Neurobiol Dis. 2012;48(3):418–428. doi: 10.1016/j.nbd.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10(5):608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Haidet-Phillips AM, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29(9):824–828. doi: 10.1038/nbt.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nagai M, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10(5):615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.de Boer AS, Koszka K, Kiskinis E, Suzuki N, Davis-Dusenbery BN, Eggan K. Genetic validation of a therapeutic target in a mouse model of ALS. Sci Transl Med. 2014;6(248):248ra104. doi: 10.1126/scitranslmed.3009351. [DOI] [PubMed] [Google Scholar]
- 135.Miller TM, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12(5):435–442. doi: 10.1016/S1474-4422(13)70061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Urushitani M, Ezzi SA, Julien JP. Therapeutic effects of immunization with mutant superoxide dismutase in mice models of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2007;104(7):2495–2500. doi: 10.1073/pnas.0606201104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ju S, et al. A yeast model of FUS/TLS-dependent cytotoxicity. PLoS Biol. 2011;9(4):e1001052. doi: 10.1371/journal.pbio.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jackson KL, et al. Preservation of forelimb function by UPF1 gene therapy in a rat model of TDP-43-induced motor paralysis. Gene Ther. 2015;22(1):20–28. doi: 10.1038/gt.2014.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Peters O, Brown RH Jr. Amyotrophic lateral sclerosis. In: Zigmond MJ, Coyle JT, Rowland LP, eds. Neurology of Brain Disorders. Waltham, Massachusetts, USA: Academic Press; 2014:262–280. [Google Scholar]