

Abstract
Tumor cells can engage in a process called collective invasion, in which cohesive groups of cells invade through interstitial tissue. Here, we identified an epigenetically distinct subpopulation of breast tumor cells that have an enhanced capacity to collectively invade. Analysis of spheroid invasion in an organotypic culture system revealed that these “trailblazer” cells are capable of initiating collective invasion and promote non-trailblazer cell invasion, indicating a commensal relationship among subpopulations within heterogenous tumors. Canonical mesenchymal markers were not sufficient to distinguish trailblazer cells from non-trailblazer cells, suggesting that defining the molecular underpinnings of the trailblazer phenotype could reveal collective invasion-specific mechanisms. Functional analysis determined that DOCK10, ITGA11, DAB2, PDFGRA, VASN, PPAP2B, and LPAR1 are highly expressed in trailblazer cells and required to initiate collective invasion, with DOCK10 essential for metastasis. In patients with triple-negative breast cancer, expression of these 7 genes correlated with poor outcome. Together, our results indicate that spontaneous conversion of the epigenetic state in a subpopulation of cells can promote a transition from in situ to invasive growth through induction of a cooperative form of collective invasion and suggest that therapeutic inhibition of trailblazer cell invasion may help prevent metastasis.
Keywords: Cell Biology, Oncology
Introduction
The hollow tubular architecture of epithelial tissue is fundamentally disrupted during malignant progression, culminating with the invasion of neoplastic cells into the surrounding stromal tissue (1). Once out of the epithelial niche, invasive tumor cells can intravasate into blood vessels, disseminate throughout the body, and form macrometastases that are responsible for patient deaths (2). Determining how tumor cells initiate and sustain invasive behavior may help improve patient diagnosis (3) and lead to the development of new intervention modalities (4). Given the key role of tumor cell invasion in the progression to metastasis, we sought to determine how alterations to the tumor cell-autonomous signaling circuitry promote invasive behavior.
We focused our investigation on collective invasion, which is a form of multicellular migration that is involved in tissue morphogenesis and can contribute to the spread of tumor cells through the stroma (5). Collective invasion is functionally distinct from single-cell invasion (6). During single-cell invasion, solitary tumor cells independently follow their own unique paths as they invade through the extracellular matrix (ECM), without forming any cell-cell contacts (7). By comparison, collective invasion is a process in which adherent cells migrate along a shared path through the ECM (8). The topology of collectively invading tumor cells is reminiscent of that of strands (also referred to as sprouts) and can range from a single cell to multiple cells in diameter (8, 9). Collectively invading cell strands can remain attached to a large tumor mass or may detach and continue to invade as a smaller group of cells (8). The cells in the front of a collectively invading strand, in which there is more extensive contact with the surrounding ECM, are often referred to as leader cells (6). The cells that trail behind leader cells are referred to as follower cells (6).
Leading tumor cells can initiate invasion by forming cellular protrusions, which provide traction and deliver MMP14 to cleave ECM fibers (9). The formation of these protrusions is dependent on the expression of N-WASP, which induces an ARP2/3-dependent nucleation of actin filaments (10). The induction of these protrusions may also require the activity of LIM kinases 1 and 2, which phosphorylate cofilin to increase actin filament stability (11). As leading tumor cells invade, they reorganize the ECM into paths through which follower cells can migrate (7). The follower tumor cells can further reshape the ECM and expand the width of the path (9). The ability of tumor cells to act as leader cells during collective invasion is dependent on the composition and organization of the surrounding ECM (12). For example, basal-type breast cancer cells are limited to collectively invading through paths in the ECM that have been patterned by fibroblasts (13). Similarly, KRT14-expressing breast cancer cells are only capable of leading invasion through a stromal ECM that is rich in fibrillar collagen and deficient in collagen IV (14). The relative ability of tumor cells to lead invasion can also vary among tumor cell populations originally isolated from different patients (12, 13, 15, 16). This heterogeneity indicates that there are distinct molecular states that confer an enhanced invasive ability. The nature of the traits that promote aggressive tumor cell–collective invasion are poorly understood.
To better understand the characteristics of highly invasive tumor cells, we chose to functionally annotate the capacity of 7 different breast cancer cell lines to invade using an organotypic culture model. To determine the capacity of these cell lines to collectively invade, we evaluated the invasion of multicellular breast cancer spheroids (approximately 20–100 cells) into a 1-mm-thick layer of extracellular matrix (13). Organotypic culture models, such as this, have previously revealed functional requirements for collective invasion that are conserved in vivo (9, 13). The ECM used was a mixture of collagen I and Matrigel (largely collagen IV and laminin), which are constituents of the ECM that surrounds normal mammary epithelial tissue and breast tumors (17, 18). This composition of ECM induces mammary epithelial cells to form spherical duct-like structures (19) and sustains the ductal architecture of mouse (12) and human (20) mammary epithelial tissue explants. In addition, basal-type and KRT14-expressing breast cancer cells are not capable of leading invasion into this ECM (13, 14). Therefore, our organotypic culture system was designed to reveal the traits of a distinct type of breast cancer cell that has an enhanced capacity to lead collective invasion.
In our initial investigation, we detected collective invasion within a subpopulation of spheroids in the triple-negative breast cancer (TNBC) cell lines. To better understand the molecular underpinnings of collective invasion, we sought to define the functional characteristics of this highly invasive TNBC subpopulation. We termed this subpopulation “trailblazer” cells, because of their enhanced ability to act as leader cells during collective invasion, compared with basal-type and KRT14-expressing breast cancer cells. We discovered that the trailblazer cells are an epigenetically distinct subpopulation and can be defined by a conserved pattern of gene expression. A cohort of genes that specified the trailblazer cell phenotype were necessary for the formation of cellular protrusions into the ECM. Importantly, the increased expression of genes necessary for trailblazer cell invasion correlated with poor outcome in patients with TNBC. One of these genes, the guanine nucleotide exchange factor DOCK10, was required for spontaneous metastasis. In heterogeneous populations, trailblazer cells induced the invasion of non-trailblazer cells, thus revealing a new type of commensal relationship among naturally existing tumor subpopulations. Together, these results demonstrate how the epigenetic alteration of the signaling circuitry in a subpopulation of tumor cells can promote collective invasion through cell-autonomous and non-cell-autonomous mechanisms.
Results
A distinct subpopulation of trailblazer cells has enhanced invasive ability.
To begin defining the molecular traits that confer tumor cells with invasive ability, we analyzed spheroid invasion in an organotypic culture system that reconstitutes key features of collective invasion that are conserved in vivo (9, 13). Normal mammary epithelial cells form duct-like spheroids in this system (Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/JCI77767DS1), indicating that our model was testing for unique traits of tumor cells that promote cell-autonomous invasion, potentially during the transition from ductal carcinoma in situ (DCIS) to invasive breast cancer (13). To begin defining traits that promote collective invasion, we determined the percentage of invasive trailblazer spheroids that were formed in 7 different breast cancer cell lines that represent key known features of intertumor molecular diversity. Invasive spheroids were detected in 3 of the 7 cell lines evaluated, with the percentage of invasive spheroids ranging between 8% and 75% of the total population (Figure 1, A and B). None of the cell lines contained a 100% pure population of invasive spheroids (Figure 1B). The 3 cell lines that contained invasive spheroids were derived from patients with TNBC (no detectable estrogen receptor [ER], progesterone receptor, or human epidermal growth factor receptor 2 [HER2] expression) (Figure 1A). TNBC accounts for 10% to 20% of diagnosed breast cancers and has a relatively worse outcome compared with that of ER+ breast cancer (21). Importantly, the strand-like organization of the collectively invading cells observed in organotypic culture was also detected in primary breast tumors (Figure 1C). Thus, our results indicate that there can be a distinct subpopulation within a community of tumor cells that has an enhanced capacity to lead collective invasion. We refer to this intrinsically invasive subpopulation as trailblazer cells to distinguish them from other types of leader cells, such as KRT14-expressing breast cancer cells, that are unable to invade under these conditions. The noninvasive subpopulation, which may require additional extrinsic factors to invade, is referred to as “opportunist” cells herein.
Figure 1. Breast cancer cell lines contain subpopulations of invasive trailblazer and noninvasive opportunist cells.

(A) Representative images of breast cancer spheroids in organotypic culture stained as indicated (n = 3). Masks show the outline of the spheroids. Inset regions are indicated by dashed boxes. Solid arrows identify representative invasive trailblazer spheroids. Arrows with dashed tails identify representative noninvasive opportunist spheroids. Scale bar: 50 μm. PR, progesterone receptor. (B) The percentage of invasive trailblazer spheroids in each cell line. Error bars indicate SD, n = 3. *P < 0.05, ***P < 0.001, unpaired Student’s t test, compared with T47D. (C) Representative H&E-stained primary breast tumors. Arrows indicate patterns of tumor organization that are consistent with collective invasion. Scale bar: 50 μm. (D) Representative images of SUM159 spheroids (day 5) stained as indicated (n = 3). Inset regions are indicated by dashed boxes. Solid arrows indicate where collagen I is being reorganized into parallel tracks by trailblazer cells. Arrows with dashed tails indicate where collagen I is arranged perpendicular to the edge of the noninvasive opportunist spheroid. Scale bar: 50 μm. (E) Time-lapse phase images of the SUM159 spheroids and cell displacement over 18 hours (mean ± SD, n = 3, 15 spheroids total per condition). Solid arrows indicate a leading cell. The arrow with a dotted tail indicates a following cell. Scale bar: 50 μm. **P < 0.01, unpaired Student’s t test.
Immunofluorescence analysis and time-lapse imaging showed that the leader trailblazer cells formed long cellular protrusions (LCPs) into the ECM before invading away from the main mass of cells (Figure 1, D and E, and Supplemental Video 1), similar to previous reports (9, 10). Additional trailblazer cells could then migrate into the space within the ECM created by the first invading cell, indicating that invading breast cancer cells could engage in a leader-follower relationship, similar to that observed during angiogenesis (Figure 1E and Supplemental Video 1). By comparison, the noninvasive opportunist spheroids had a cellular organization similar to that of noninvasive DCIS (Figure 1D). Cells within the opportunist TNBC spheroids were capable of migrating within the space generated by the proliferative expansion of the cellular mass but did not extend LCPs into the surrounding ECM (Figure 1, D and E, and Supplemental Video 2). This intraspheroid movement indicated that cell motility itself did not confer the capacity to invade, similar to previous results (13). The collagen fibers in the ECM were oriented perpendicular to the surface of the noninvasive spheroids, potentially preventing the motile cells from migrating into the stroma (Figure 1D). This perpendicular organization of collagen fibers around the spheroids is similar to the arrangement of collagen fibers around noninvasive regions of primary tumors (14, 22). These findings indicate that the trailblazer cells are a distinct invasive subpopulation within a larger group of motile breast cancer cells.
Trailblazer and opportunist cells are epigenetically distinct subpopulations.
We next sought to define how trailblazer ability was specified within a subpopulation of cells. Phenotypic heterogeneity within a tumor cell population can be heritable, if induced by genetic and epigenetic variability, or stochastic, if in response to fluctuations within cell signaling pathways (23, 24). To distinguish between heritable and stochastic heterogeneity, we manually isolated the trailblazer and opportunist spheroids from HCC1143 and SUM159 breast cancer cells and 4T1 mouse mammary carcinoma cells. The spheroids were then enzymatically dissociated and recultured as a monolayer for 30 population doublings (Figure 2A). Both the trailblazer and opportunist phenotypes were enriched in the respective daughter cell lines after multigenerational subculturing, demonstrating that the ability to collectively invade was a heritable phenotype (Figure 2, B and C). The SUM159 trailblazer cells were also more invasive than the SUM159 opportunist cells in a second type of invasion model in which cells invade vertically from a monolayer into the ECM (Supplemental Figure 1B). The rate of trailblazer invasion was influenced by the composition of ECM in spheroid culture and in our vertical invasion assay (Supplemental Figure 1C and Supplemental Figure 2). However, SUM159 trailblazer cells invaded into ECM more efficiently than SUM159 opportunist cells in all conditions (Supplemental Figure 1C and Supplemental Figure 2).
Figure 2. The trailblazer and opportunist phenotypes are heritable.

(A) Model depicting the methodology for generating the trailblazer and opportunist subpopulations (also see the Methods). (B) Quantification of invasion (mean + SD, n = 3). **P < 0.01, ***P < 0.001, unpaired Student’s t test. (C) Representative images of 4T1, SUM159, and HCC1143 parental, daughter trailblazer, and daughter opportunist subpopulation spheroids stained with phalloidin (n = 3). Masks show the outline of the spheroids. Arrows identify invasive spheroids. Scale bar: 50 μm. (D) Representative time-lapse imaging of SUM159 trailblazer (SUM159-T) and normal mammary gland explants (n = 3). Imaging began 24 hours after plating explants in organotypic culture. Arrow shows an example area of collective invasion. Scale bar: 50 μm.
To determine whether the trailblazer phenotype could be sustained in vivo, we analyzed the invasiveness of explants derived from SUM159 trailblazer cell primary tumors (Figure 2D). As was observed with cells in culture, the SUM159 trailblazer explants invaded extensively into the ECM (Figure 2D). By comparison, normal mammary epithelial tissue, which contains KRT14-expressing cells, was not invasive when isolated and cultured under the same conditions (Figure 2D). Thus, the trailblazer cells could retain their unique invasive characteristics in vivo.
The genetic fingerprints of the HCC1143 trailblazer and HCC1143 opportunist subpopulations showed high identity (Supplemental Figure 3A), demonstrating that the HCC1143 trailblazer subpopulation was not a contaminating cell line. To determine whether smaller-scale genetic changes were contributing to the trailblazer and opportunist phenotypes or whether there was an epigenetic basis for the distinct traits, we examined the invasive characteristics of low passage daughter cell lines derived from opportunist cell clones. Low passage clonal populations have nearly identical genotypes. Therefore, the presence of heterogeneity within a clonal population would demonstrate that that the trailblazer phenotype could be induced by nongenetic factors (25). Indeed, a subpopulation of trailblazer-type cells was detected in low passage clonal SUM159 opportunist populations (Supplemental Figure 3B). Thus, these combined results demonstrate that the trailblazer phenotype could be induced through nongenetic factors and could be maintained during cell division and, therefore, was under epigenetic control (26). Together, these results indicate that the trailblazer cells are an epigenetically distinct subpopulation that can spontaneously arise from a pure opportunist cell population.
Trailblazer cells are a distinct subpopulation within a larger cohort of cells that adopted mesenchymal traits.
To determine whether the trailblazer and opportunist populations were components of an epigenetic state defined by cell surface markers, we analyzed the expression the integrin CD49f and the transmembrane glycoprotein epithelial cell adhesion molecule (EpCAM). The expression level of these proteins can be used to differentiate among phenotypically distinct subpopulations of breast cancer cells (27–29). A consistent feature of our analysis was that both the HCC1143 trailblazer and SUM159 trailblazer subpopulations had relatively low levels of EpCAM expression (Figure 3A). Low EpCAM surface levels were also detected on SUM159 opportunist cells; however, this subpopulation differed from SUM159 trailblazer cells in its distribution of CD49f (Figure 3A). Similar results were obtained when evaluating the expression of EpCAM and the glycoprotein CD24 (Supplemental Figure 3C). These results indicate that trailblazer cells are a subpopulation within the EpCAMlo cohort of cells.
Figure 3. The expression of canonical mesenchymal traits is not sufficient to induce the trailblazer phenotype.

(A) Representative FACS analysis of EpCAM and CD49f expression in the indicated cells (n = 3). (B) Representative FACS analysis of EpCAM and CD49f expression in parental SUM149 and SUM229 cells (n = 3). (C) HCC1143, SUM149, and SUM229 cells were sorted into daughter subpopulations based on the level of EpCAM expression. Sorted EpCAMhi and EpCAMlo cells grown in organotypic culture and stained with phalloidin and the percentage of invasive spheroids for the EpCAMhi and EpCAMlo subpopulations derived from the HCC1143, SUM149, and SUM229 cells (mean + SD, n = 3) are shown. Scale bar: 50 μm. (D) Heat map showing the mRNA expression of EMT-related genes, including epithelial markers (CDH1, CLDN3, CLDN7, and EPCAM) and mesenchymal markers (FOXC1, FOXC2, SNAI1, SNAI2, TWIST1, VIM, ZEB1, and ZEB2). Expression is the mean of 2 biological replicates. (E) Heat map showing the mRNA expression of basal cytokeratins (KRT14 and KRT5), the basal transcription factor p63, and epithelial cytokeratins (KRT8 and KRT18). Expression is the mean of 2 biological replicates. T, trailblazer; O, opportunist; P, parental.
These FACS results, showing that EpCAMhi subpopulations were opportunist cells, suggested to us that the level of EpCAM expression could be used to enrich for trailblazer and opportunist cells from populations consisting of a mixture of EpCAMhi and EpCAMlo cells. To test this possibility, we first confirmed previous observations that the SUM149 and SUM229 cells lines contained both EpCAMhi and EpCAMlo subpopulations (ref. 28 and Figure 3B). Flow cytometry was then used to sort the HCC1143, SUM149, and SUM229 parental cell lines into EpCAMhi and EpCAMlo subpopulations. Each subpopulation was then grown in organotypic culture. The EpCAMlo subpopulations were enriched with trailblazer cells relative to the EpCAMhi subpopulations (Figure 3C), consistent with our analysis of the SUM159 trailblazer and HCC1143 trailblazer cells. In addition, the EpCAMlo subpopulations maintained this enrichment of trailblazer cells for least 10 population doublings in monolayer culture. Thus, standard flow cytometry could be used to isolate trailblazer-enriched cell populations.
EpCAMlo breast cancer cells can have mesenchymal traits, including reduced expression of E-cadherin and increased expression of vimentin (27). Collective invasion has previously been investigated in cells with such mesenchymal features (9). However, the contribution of epithelial-to-mesenchymal transition (EMT) characteristics to collective invasion is poorly understood. To determine whether trailblazer cells had acquired mesenchymal characteristics, we evaluated the expression of transcription factors and cell-cell adhesion proteins that have previously been used to distinguish between cells with epithelial or mesenchymal traits (30). The trailblazer cells did have mesenchymal features; however, the SUM159 opportunist cells also had the same mesenchymal molecular traits, consistent with the SUM159 opportunist cells expressing a low level of the epithelial marker protein EpCAM. These results indicate that, while the expression of these EMT-related genes may be necessary for trailblazer function, they are not sufficient to confer the trailblazer phenotype (Figure 3D). In addition, we also did not detect an increase in the expression of basal cell cytokeratins (KRT5 and KRT14) in the trailblazer cells (Figure 3E). Taken together, these results suggest that the trailblazer cells are a distinct subpopulation among cells that have adopted a canonical mesenchymal molecular phenotype, such as can occur during the epithelial-to-mesenchymal transition.
Trailblazer cells express higher levels of genes required for collective invasion.
To identify molecular traits that confer trailblazer cells with invasive ability, we analyzed the gene expression profiles of the combined trailblazer and opportunist subpopulations derived from SUM159 and HCC1143 cell lines. We hypothesized that the epigenetic conversion to the trailblazer state involves an increase in the expression of genes that are necessary for collective invasion. To begin testing this possibility, we used significance analysis of microarrays to identify genes that were 4-fold overexpressed in the trailblazer subpopulations relative to the opportunist subpopulations, with a FDR <5% (Supplemental Figure 4). This analysis produced a list of 49 probe sets for 44 genes (Supplemental Table 1 and Supplemental Figure 4) that were highly expressed in trailblazer cells. To prioritize for functional testing, we focused on 26 genes that contained matching probes in a publicly available patient cohort of mRNA expression with associated outcome (see Methods for details) (Figure 4A). This prioritization strategy would allow us to later determine how the expression of genes that contributed to trailblazer invasion correlated with patient characteristics.
Figure 4. Trailblazer cells express higher levels of genes required for collective invasion.

(A) Heat map showing the expression of the 26 genes that are specifically elevated in SUM159 and HCC1143 trailblazer cells. Expression is the mean of 2 biological replicates. (B) The relative invasion of SUM159 trailblazer cells ≥50 μm into the ECM. The number of invasive cells is normalized to the total cell number in the field of view for each condition. Relative invasion is the normalized invasive value of the test condition divided by the normalized invasion value of the control cells from the experimental replicate (median ± range, n = 3). The dashed red box indicates genes that were further investigated. Images are representative x-z views of SUM159 trailblazer invasion into ECM after transfection with the indicated siRNAs. For images of additional siRNA-transfected cells, see Supplemental Figure 5. Scale bar: 50 μm. (C) The relative invasion of SUM229 trailblazer cells ≥40 μm into the ECM normalized to the total cell number and compared with control cells (mean ± SD, n = 3). (D) Heat map showing that genes required for trailblazer cell invasion are highly expressed in 578T cells. Expression is the mean of 2 biological replicates. (E) The relative invasion of 578T cells ≥50 μm into the ECM normalized to the total cell number and compared with control cells (mean ± SD, n = 3).
To evaluate a requirement for invasion, we determined the invasive ability of SUM159 trailblazer cells transfected with siRNAs targeting the 26 candidate genes (Figure 4B and Supplemental Figures 4 and 5). A median relative invasion value of 0.5 was used to prioritize further investigation of candidates. This thresholding yielded 7 genes (DOCK10, DAB2, ITGA11, PDGFRA, LPAR1, VASN, and PPAP2B) for further testing (Figure 4B and Supplemental Figure 4). Two individual nonoverlapping siRNAs targeting each gene recapitulated the results observed with siRNA pools (Supplemental Figure 6A), and target depletion was >70% (Supplemental Figure 6B). In addition, the elevated expression of DOCK10, DAB2, LPAR1, ITGA11, PDGFRA, VASN, and PPAP2B in SUM159 trailblazer cells was confirmed by quantitative PCR and immunoblot (Supplemental Figure 6, C and D). The increased expression of these 7 genes in multiple trailblazer populations (SUM159 and HCC1143) suggested that these genes may be required for the invasion of additional trailblazer populations. Indeed, depletion of each gene resulted in a >50% reduction in SUM229 trailblazer cell invasion (Figure 4C and Supplemental Figure 7A). We then further tested the requirement of the 7 genes in 578T breast cancer cells for invasion. 578T cells contained a large percentage of trailblazer cells. Importantly, each of the 7 genes was expressed at a high level in 578T cells relative to SUM159 opportunist cells (Figure 4D). Depletion of 6 of the 7 genes reduced invasion by >50% (Figure 4E and Supplemental Figure 7B), consistent with the results observed in the SUM159 trailblazer and SUM229 trailblazer populations. However, depletion of LPAR1 resulted in a modest increase in invasion, which suggests that the requirement of LPAR1 is dependent on additional factors (Figure 4E and Supplemental Figure 7B). Nevertheless, our analysis indicates that all 7 genes were necessary for invasion in at least two populations, with 6 of the genes having a broader functional role across trailblazer populations. Together, these results suggest that there was an increase in the expression of genes that can contribute to collective invasion during the conversion to the trailblazer phenotype.
Increased expression of genes required for trailblazer invasion correlates with poor breast cancer patient outcome.
We next evaluated how the expression of the 7 genes that were required for invasion (DOCK10, DAB2, ITGA11, LPAR1, PPAP2B, VASN, and PDGFRA) correlated with patient outcome. We termed this set of 7 genes our “trailblazer signature.” The expression of the 7 trailblazer signature genes in the 2 trailblazer subpopulations, 2 opportunist subpopulations, and 7 noninvasive cell lines was used to develop a prediction model for high trailblazer gene expression using a Random Forest approach (31). The prediction model was then applied to the expression profiles of 161 primary tumor samples (27), and patients were classified into “Trailblazer-high” and “Trailblazer-low” groups. Interestingly, both recurrence-free and overall survival times were shorter for Trailblazer-high patients compared with those for Trailblazer-low patients (Figure 5A). Importantly, multivariate analysis showed that the 7-gene trailblazer expression signature was associated with survival when adjusting for ER, HER2, and TNBC status (Supplemental Figure 8A). Analysis of clinically relevant molecular subtypes in the two groups showed that Trailblazer-high patients were more likely to be diagnosed with TNBC, consistent with our discovery of trailblazer cells in TNBC cell lines (Table 1). By comparison, Trailblazer-low patients were more frequently found to have ER+/HER2– disease (Table 1). Notably, patients from all 4 molecular subtypes were classified as Trailblazer-high (Table 1). This suggests that more extensive testing of additional ER+ and HER2+ cells lines may reveal the presence of ER+ or HER2+ trailblazer cells. It is also possible that ER+ and HER2+ trailblazer cells were lost during the establishment of the cell lines that we evaluated.
Figure 5. The elevated expression of genes required for trailblazer cell invasion correlates with poor patient outcome.

(A) Patients were classified as Trailblazer high and Trailblazer low using the 7 genes that were required for invasion in the SUM159 trailblazer and SUM229 trailblazer cells (DOCK10, DAB2, LPAR1, PPAP2B, ITGA11, VASN, and PDGFRA). Kaplan-Meier curves were drawn for both the Trailblazer-high and Trailblazer-low groups. Survival differences were compared using the log-rank (Mantel-Cox) test. (B) Kaplan-Meier survival curves for Trailblazer-high and Trailblazer-low TNBC patients. Survival differences were compared using the log-rank (Mantel-Cox) test.
Table 1. Clinical characteristics of the Trailblazer-high and Trailblazer-low patient groups.
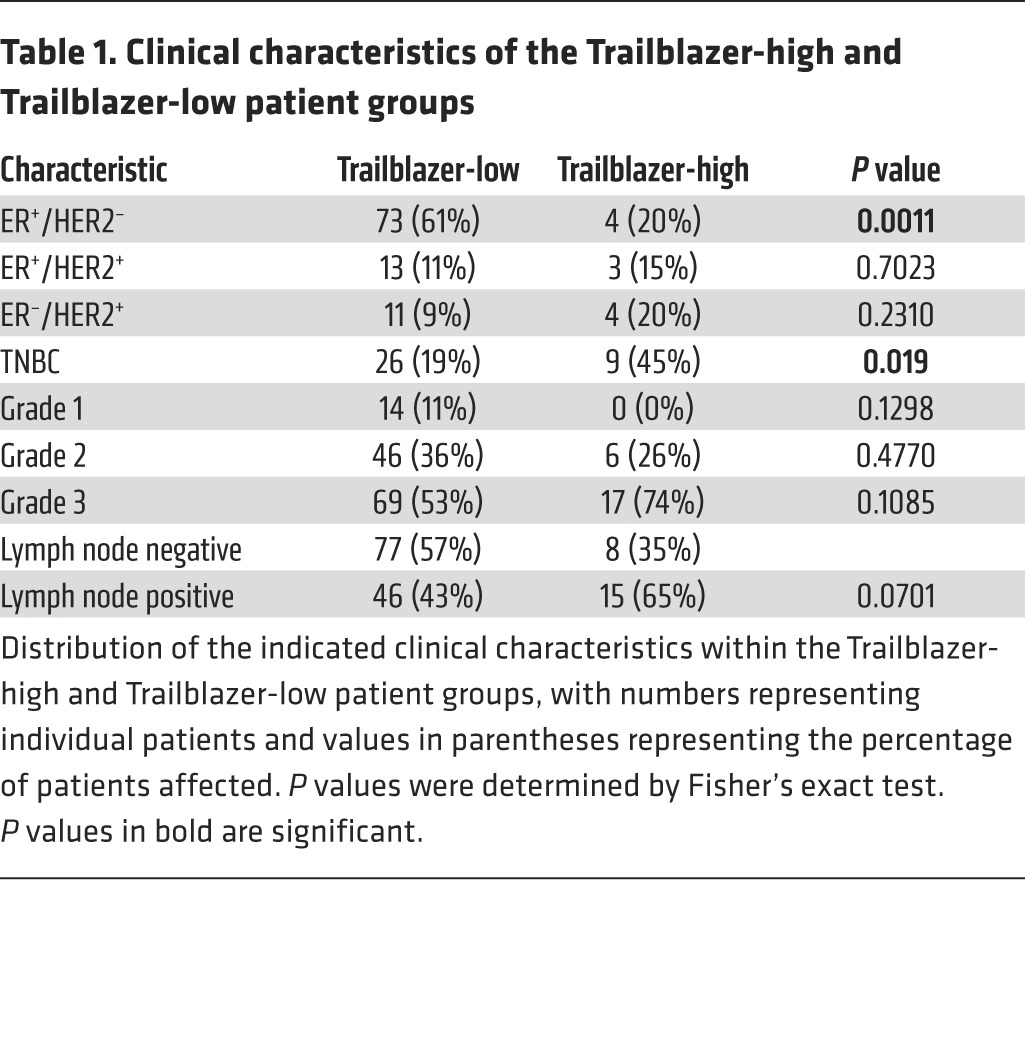
There are no clearly defined metrics for determining the relative outcome within the TNBC patient population (32). We therefore determined whether the trailblazer expression signature could refine the prognosis of patients with TNBC. The Trailblazer-high TNBC patient group had a shorter time to relapse and shorter overall survival time (Figure 5B). A similar trend was observed when analyzing a second TNBC patient group (Supplemental Figure 8B). The Trailblazer-high and Trailblazer-low groups in the HER2+ and ER+ subtypes did not show differences in relapse or survival periods (Supplemental Figure 8C). Thus, the ability of trailblazer gene expression to refine outcome within the molecular subtypes was specific to TNBC. Together, these results suggest that the 7-gene trailblazer expression signature has the potential to serve as a prognostic indicator for patients with TNBC.
Trailblazer signature genes control parallel signaling pathways that are integrated together to promote LCP formation and invasion.
We next focused on determining how trailblazer signature genes could control collective invasion. The formation of cellular protrusions during collective invasion can be dependent on N-WASP (10). Consistent with these prior findings, collective invasion and LCP formation in SUM159 trailblazer cells also required N-WASP expression (Supplemental Figure 9, A and C). N-WASP is activated by a conformational change triggered by direct binding to active GTP-bound CDC42 (33), which can be triggered by DOCK10 in melanoma cells (34). This suggested to us that DOCK10 may promote a CDC42- and N-WASP–dependent formation of LCPs in trailblazer cells. Indeed, DOCK10 and CDC42 were required for LCP formation in SUM159 trailblazer cells (Figure 6A and Supplemental Figure 9C), and CDC42 was required for the collective invasion of both SUM159 trailblazer cells (Figure 6B) and 578T cells (Supplemental Figure 9B). By comparison, depletion of RAC1, which is a small G protein that controls actin polymerization through WAVE proteins, did not reduce invasion (Figure 6B and Supplemental Figure 9C).
Figure 6. Multiple trailblazer genes are necessary for LCP formation.

(A) SUM159 trailblazer cells transfected with the indicated siRNAs were plated onto a layer of ECM for 24 hours and stained. Scale bar: 50 μm. Graph shows the relative length of LCPs (mean + SD of ≥20 cells, n = 3). (B) Invasion of SUM159 trailblazer cells transfected with the indicated siRNAs. Scale bar: 50 μm. Graph shows relative invasion (mean + SD, n = 3). (C) Collective invasion of SUM159 opportunist CDC42Q61L cells, as shown in B. Graph shows relative invasion compared with control SUM159 trailblazer cells (mean + SD, n = 3). (D) Relative invasion of SUM159 trailblazer and SUM159 trailblazer CDC42Q61L cells transfected with the indicated siRNAs (mean + SD, n = 3). (E) SUM159 trailblazer cells transfected with the indicated siRNAs were plated onto a layer of ECM for 24 hours and stained. Scale bar: 50 μm. Graph shows the relative length of LCPs (mean ± SD of ≥20 cells, n = 3). Horizontal bars indicate the medians, boxes indicate 25th to 75th percentiles, and whiskers indicate minimum and maximum. (F) Representative images of the movement of SUM159 trailblazer cells transfected with the indicated siRNAs over a 14-hour time period in monolayer culture. The color indicates the time period within the 14 hours of imaging. Scale bar: 50 μm. Graph shows the displacement of cells over time (mean ± SD, n = 3). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, unpaired Student’s t test.
To determine whether the activation of CDC42 was sufficient to promote invasion, we expressed a constitutively active mutant of CDC42 (CDC42Q61L) in SUM159 opportunist cells (Supplemental Figure 9D). CDC42Q61L was not sufficient to induce SUM159 opportunist cell invasion (Figure 6C), indicating that additional signaling events were required. To test this possibility, we investigated whether the expression of CDC42Q61L influenced the invasion of SUM159 trailblazer cells transfected with siRNAs targeting N-WASP, DOCK10, and the trailblazer signature genes DAB2, ITGA11, and PDGFRA. CDC42Q61L expression in SUM159 trailblazer cells decreased their dependency on DOCK10, but not N-WASP, for invasion (Figure 6D and Supplemental Figure 9D). These results further indicate that DOCK10 functions by promoting the CDC42-dependent activation of N-WASP. Interestingly, SUM159 trailblazer cells expressing CDC42Q61L required DAB2 and PDGRA for invasion (Figure 6D), suggesting that these genes regulate signaling pathways that act in conjunction with DOCK10 and CDC42. In addition, DAB2 and ITGA11 were both required for LCP formation in SUM159 trailblazer (Figure 6E) and 578T cells (Supplemental Figure 10A), indicating that, like DOCK10, DAB2 and ITGA11 contributed to invasion through promoting LCPs. By comparison, PDGRA was not required for LCP formation (Figure 6E), suggesting that trailblazer signature genes may also control LCP-independent functions that are required for collective invasion.
To determine whether the control of LCP formation by DOCK10, DAB2, and ITGA11 reflected a general function of these genes in the control of motility, we measured the motility of SUM159 trailblazer cells within a cell monolayer. Depletion of DOCK10 and DAB2 did not reduce cell motility, and a reduction in ITGA11 expression only produced a modest suppression of movement (Figure 6F). In addition, depletion of CDC42 in SUM159 trailblazer spheroids reduced collective invasion and LCP formation but not intraspheroid movement (Supplemental Figure 10B). This finding is consistent with our previous observations that the noninvasive SUM159 opportunist cells are also highly motile within spheroids (Figure 1E). These results indicate that the DOCK10/CDC42/N-WASP pathway, DAB2, and ITGA11 are specifically required for formation of LCPs and collective invasion but are not required for general cell movement.
Taken together, these results suggest that DOCK10 promotes collective invasion through the activation of CDC42 and N-WASP, which are necessary for LCP formation. Importantly, the function of this pathway in promoting LCP formation and collective invasion is dependent on parallel signaling networks that are specifically active in trailblazer cells.
The trailblazer signature gene DOCK10 is necessary for spontaneous metastasis.
Our analysis of patient tumor gene expression and functional results in organotypic culture indicated that trailblazer signature genes could promote metastasis. To test this possibility, we determined whether the expression of DOCK10 was necessary for SUM159 trailblazer cells to metastasize to the lungs in an orthotopic xenograft model. Six weeks after injection into the mammary fat pad, control tumors and tumors expressing a DOCK10 shRNA were of similar size (Figure 7A and Supplemental Figure 10C). However, the SUM159 trailblazer cells expressing an shRNA targeting DOCK10 did not spread to the lung, demonstrating that DOCK10 expression was necessary for metastatic spread from the primary tumor (Figure 7B). Thus, our results indicate that trailblazer gene expression can contribute to metastasis by promoting the dissemination of tumor cells from primary tumors and may be necessary for extravasation in distant tissues.
Figure 7. The trailblazer signature gene DOCK10 is required for metastasis.

(A) Representative bioluminescence imaging of control and DOCK10 shRNA–expressing SUM159 trailblazer-Luc-GFP primary tumors. Graph shows the relative photon flux of the primary tumors (mean ± SD, control = 7 mice, DOCK10 shRNA = 8 mice). NS, unpaired Student’s t test. (B) Representative bioluminescence imaging (BLI) and fluorescent imaging of the lungs from mice bearing control and DOCK10 shRNA–expressing SUM159 trailblazer-Luc-GFP primary tumors. Images show bioluminescence imaging of lungs immediately after mice were sacrificed and GFP expression of SUM159 trailblazer cells in the lungs immediately after mice were sacrificed (control = 7 mice, DOCK10 shRNA = 8 mice; scale bar: 100 μm) as well as lungs immunostained with anti-GFP antibody and counterstained with phalloidin. (control = 7 mice, DOCK10 shRNA = 8 mice; scale bar: 50 μm). Graphs show the relative photon flux and number of micrometastases in the lungs normalized to the photon flux of the corresponding primary tumor (mean ± SD, control = 7 mice, DOCK10 shRNA = 8 mice). ***P < 0.001, Mann Whitney U test.
Trailblazer cells induce opportunist cell invasion through a commensal relationship.
Our data suggested how a large population of trailblazer cells could directly contribute to disease progression by collectively invading through the ECM and then disseminating to the lungs. We next asked how the trailblazer cells could act within a heterogenous population containing a majority of opportunist cells. To do this, we generated multicellular clusters of cells in suspension for 24 hours before growth in organotypic culture. Using this approach, we were able to generate multicellular heterogeneous spheroids that consisted of 25% SUM159 trailblazer cells and 75% SUM159 opportunist cells. Invasion into the ECM was detected in the heterogenous spheroids (Figure 8A), indicating that the SUM159 trailblazer cells could be the minority population and still invade into the ECM. Interestingly, the invasive projections contained both SUM159 trailblazer and SUM159 opportunist cells (Figure 8A), indicating that the two subpopulations could engage in a cooperative relationship that promoted SUM159 opportunist invasion. SUM159 trailblazer cells also induced SUM159 opportunist cells to invade from a cell monolayer into a layer of ECM (Figure 8B), demonstrating that the cooperative induction of invasion was a general behavior and not a phenotype specific to clustered spheroids. Time-lapse imaging showed that the invasive projections were initiated by one or more SUM159 trailblazer cells forming LCPs before invading into the ECM (Figure 8C and Supplemental Videos 3–6). The SUM159 opportunist cells were then able to invade by following along the path created by the SUM159 trailblazer cells (Figure 8C and Supplemental Videos 3–6). Once out of the spheroids, the SUM159 opportunist cells did not generate LCPs, indicating that escape from the spheroid did not promote trailblazer-type behavior (Figure 8C and Supplemental Videos 3–6). When grown together in close proximity, the SUM159 trailblazer cells did not induce SUM159 opportunist cell invasion, suggesting that paracrine signaling was not sufficient to induce invasion (Supplemental Figure 11A). The SUM159 opportunist cells were unable to invade into the surrounding layer of ECM when DOCK10, CDC42, or N-WASP expression was reduced in the SUM159 trailblazer cells (Figure 8D), indicating that the SUM159 trailblazer cells facilitated SUM159 opportunist cell invasion by forming paths into the ECM. SUM159 trailblazer cells transfected with CDC42 siRNAs were unable to invade on their own; however, these cells were capable of opportunistically invading when clustered with control SUM159 trailblazer cells (Supplemental Figure 11B). These results indicate that trailblazer traits, such as DOCK10-dependent regulation of CDC42, were not required for opportunistic invasion. 4T1 trailblazer cells induced the invasion of 4T1 opportunist cells in spheroid culture (Supplemental Figure 11C), indicating that the commensal relationship observed was a general characteristic shared between trailblazer and opportunist subpopulations. 4T1 trailblazer cells also induced the opportunistic invasion of HC-11 mouse mammary epithelial cells (Supplemental Figure 11D), demonstrating that opportunistic invasion did not require a specific set of genetic perturbations or for opportunist cells to be descendants of trailblazer cells. Taken together, these results show that trailblazer and opportunist subpopulations can engage in a commensal relationship in which the invasion of trailblazer cells promotes the invasion of nearby opportunist cells.
Figure 8. Trailblazer cells induce the collective invasion of opportunist cells through a commensal relationship.

(A) Representative images of SUM159 spheroids composed of 100% trailblazer, 100% opportunist, or 25% trailblazer/75% opportunist cells in organotypic culture. Spheroids were formed in hanging drops for 24 hours before plating in organotypic culture. Arrows identify invasive projections. Scale bar: 50 μm. Graph shows the percentage of invasive spheroids (mean ± range of 30 spheroids per condition, n = 2). (B) Representative x-z views of the invasion into ECM of 100% trailblazer, 100% opportunist, or 25% trailblazer/75% opportunist SUM159 cells. Fluorescent nuclei are shown. Scale bar: 50 μm. Graph shows the number of SUM159 opportunist cells that invaded ≥50 μm into the ECM (mean ± SD, n = 3). Unpaired Student’s t test. (C) Representative time-lapse images of spheroids composed of 100% trailblazer, 100% opportunist, or 25% trailblazer/75% opportunist cells. Solid arrows indicate cells leading invasion. Arrows with dashed tails indicate cells following into an existing projection (100 spheroids per condition total, n = 3). Scale bar: 25 μm. (D) SUM159 trailblazer cells were transfected with the indicated siRNAs for 24 hours before clustering with untransfected SUM159 opportunist cells in spheroids at 1:4 ratio. Spheroid clusters grown for 48 hours in organotypic culture and representative images are shown. Arrow identifies invasive projection. Scale bar: 50 μm. Graphs show the percentage of invasive spheroids (mean + SD, 50 spheroids per condition, n = 3). *P < 0.05, **P < 0.01, ****P < 0.0001, unpaired Student’s t test.
Trailblazer cells induce opportunistic collective invasion in vivo.
To determine whether trailblazer cells could induce opportunistic invasion in vivo, we examined the interaction between the SUM159 trailblazer cells and MCFDCIS cells when coinjected into the mammary fat pads of immune compromised mice. MCFDCIS cells are a unique bipotential cell type that has basal characteristics and expresses KRT14 (35, 36). When grown as xenografts, MCFDCIS cells formed noninvasive DCIS lesions that were surrounded by myoepithelial cells and a contiguous layer of collagen I and collagen IV (Figure 9A and Supplemental Figure 12), thus allowing the investigation of factors that can promote the transition from in situ to invasive growth (13, 35, 36). The SUM159 trailblazer cells promoted a disruption in the organization of collagen IV and induced the MCFDCIS cells to collectively invade through channels surrounded by collagen I (Figure 9A and Supplemental Figure 12). The SUM159 trailblazer cells were within the same channels as the collectively invading MCFDCIS cells, possibly in the front edge of the invasive projection, as indicated by the strands of tumor cells connected to a larger lesion of DCIS cells (Figure 9A and Supplemental Figure 12). Thus, the pathology of the tumors is consistent with the induction of opportunist cell invasion through of paths generated by a trailblazer cell population, analogous to the commensal relationship observed during collective invasion in organotypic culture (Figure 8, A and D).
Figure 9. Cooperation between tumor cell subpopulations during collective invasion in vivo.

(A) Immunostaining of primary tumors composed of MCFDCIS cells alone or a mixture of MCFDCIS and SUM159 trailblazer cells (1:1 ratio). Representative images are shown. Arrows indicate representative areas of MCFDCIS invasion (MCFDCIS, n = 10 mice; MCFDCIS+SUM159 trailblazer, n = 10 mice). (B) Representative images of patient samples immunostained with anti-CD44 antibody. The dashed line shows the boundary of the DCIS tumor. Solid arrows indicate CD44+ tumor cells. The dashed arrow indicates CD44+ stromal cells. Scale bar: 50 μm.
To determine whether heterogeneous populations of tumor cells can collectively invade together in tumors, we immunostained patient tumor samples with an anti-CD44 antibody. The cell surface glycoprotein CD44 is heterogeneously expressed in breast tumors and can be used to distinguish epigenetically distinct subpopulations of tumor cells with unique phenotypes (37). CD44+ tumor cells were frequently intermixed with CD44– cells within strands of invading cells, indicating that epigenetically distinct tumor cells can invade together within primary tumors (Figure 9B). A mixture of CD44+ and CD44– cells was also observed in DCIS tumors, which suggests the potential for epigenetically distinct subpopulations of tumor cells to collectively invade together during the transition from in situ to invasive growth (Figure 9B). Together, our results suggest that trailblazer cells can induce the collective invasion of opportunist cells in vivo.
Discussion
Tumor cells can collectively invade as cohesive strands of cells. Our discovery of a highly invasive trailblazer subpopulation in multiple breast cancer cell lines suggests that the induction of collective invasion was influenced by the epigenetic control of a lynchpin invasive signaling network. Indeed, analysis of the gene expression profiles of multiple trailblazer populations identified a cohort of genes, which were highly expressed in trailblazer cells, and key functional regulators of collective invasion. Importantly, this gene expression signature was active in patients with a greater risk of recurrent tumor growth and death from breast cancer, suggesting that trailblazer cell–driven collective invasion may affect the nature of disease progression. Finally, the ability of trailblazer cells to induce the collective invasion of non-trailblazer cells suggests that invasion, metastasis, and patient outcome may be influenced by commensal relationships among epigenetically distinct tumor cell subpopulations (Figure 10).
Figure 10. Model of the commensal relationship between trailblazer and opportunist subpopulations.

Heterogenous tumors may contain subpopulations of trailblazer (green) and opportunist (red) cells. Over time, the trailblazer subpopulation can invade, creating paths through which the opportunist cells migrate away from the primary tumor. Once into the vasculature, both populations may seed organs (such as the lung) and form metastatic lesions.
Our discovery of phenotypic heterogeneity with respect to collective invasion is consistent with mounting observations of functional diversity for a suite of traits, including proliferation rate and metastatic colonization (38–40). In particular, there has been extensive investigation into the regulation and function of mesenchymal subpopulations of breast cancer cells, which can be resistant to chemotherapy and initiate the growth of tumors (41–44). Our results now indicate that these mesenchymal subpopulations can contain trailblazer cells. Importantly, not all cells with canonical mesenchymal characteristics are trailblazer cells, indicating that there is functional heterogeneity within the mesenchymal subpopulation. Whether traits previously attributed to mesenchymal-type cells, such as resistance to chemotherapy, are conferred to trailblazer or non-trailblazer mesenchymal subpopulations remains to be determined.
The trailblazer phenotype was induced by an epigenetic conversion in cell state, which allowed the phenotype to be heritable, long-lived, and dynamic. This heritable epigenetic control of invasive behavior is functionally distinct from the transient induction of single-cell invasive heterogeneity that occurs in response to stochastic fluctuations in signaling activity and paracrine signals from macrophages in the tumor microenvironment (24, 45). The ability of trailblazer cells to autonomously invade has the potential to make tumors refractory to intervention strategies designed to target the microenvironment. Our results suggest that drugs that perturb the epigenetic regulation of gene expression, which are currently being investigated in preclinical and clinical settings (46), may prove useful in preventing tumor cells from becoming trailblazer cells.
DOCK10, DAB2, ITGA11, PDGFRA, VASN, PPAP2B, and LPAR1 expression was increased at least 4-fold in trailblazer cells relative to that in opportunist cells and required for collective invasion.
Further investigation revealed that DOCK10, DAB2, and ITGA11 promoted invasion through the regulation of LCPs, which provide traction and contribute to the reorganization of the ECM during collective invasion (9, 10). DOCK10 controlled LCP activation by activating CDC42, which can subsequently promote actin polymerization through triggering a conformational change in ARP2/3 regulator N-WASP. Importantly, the DOCK10/CDC42/N-WASP pathway required the expression of additional trailblazer signature genes to promote collective invasion. This suggests that trailblazer genes coordinate the regulation of multiple parallel signaling pathways to induce collective invasion.
The function of PDGFRA, VASN, PPAP2B, and VASN in promoting invasion remains to be determined. Interestingly, PDGFRA was required for invasion in 3 distinct cell populations but was not required for LCP formation. PDGFRA is required for ECM degradation in cells that have undergone EMT (47), which suggests that PDGFRA and other trailblazer genes regulate ECM proteolysis during collective invasion. Additional genes beyond the 7 validated in this study may also be required for invasion. For instance, siRNAs targeting 8 genes, which had 4-fold increased expression in trailblazer cells, suppressed invasion by at least 40%. Upon further validation of siRNA specificity, and a demonstration of function in additional trailblazer populations, one or more of these 8 genes may prove to also be a bona fide regulator of invasion. In addition, negative results in siRNA experiments may be a consequence of incomplete knockdown of gene expression or the kinetics of loss of expression (48). Also, siRNAs targeting 3 genes highly expressed in trailblazer cells enhanced invasion in at least 2 experiments. This suggests that genes highly expressed in the trailblazer cells may be components of negative feedback loops that dampen the extent of invasion.
Instead of competing against each other to achieve clonal dominance, the trailblazer and opportunist subpopulations engaged in a cooperative relationship known as commensalism (49), which is characteristic of tumor-microenvironment interactions (13, 35, 50) and has recently been shown to exist between tumor cells (51, 52). The ability of the trailblazer subpopulation to induce opportunist cells to invade demonstrates that there are distinct functional requirements for collective invasion depending on the position of a cell within the invading cell collective. This observation is consistent with previous results in other models, indicating that proteins can have distinct functional requirements within an invasive projection (10, 11). Our results now show that there can be cooperation among naturally existing subpopulations with different heritable traits during collective invasion. Further, trailing cells do not require canonical mesenchymal traits or the accumulation of genetic abnormalities, as normal mammary epithelial cells were capable of opportunistic invasion. Functional heterogeneity is a key characteristic of developmental programs, such as border cell migration and angiogenesis, in which “leader” cells are transiently defined by paracrine signals from the stroma (53, 54). Thus, the trailblazer and opportunist subpopulations were employing the basic characteristics of a preexisting biological program; but rather than having their “leader” and “follower” roles transiently defined, the traits were epigenetically hardwired.
The expression of genes associated with the trailblazer phenotype correlated with poor patient outcome, which suggests that the mechanism of collective invasion can contribute to the characteristics of disease progression. A possible explanation for the worse survival of Trailblazer-high patients is that the epigenetic induction of the trailblazer phenotype promotes an earlier transition to invasive growth compared with tumors that require contributions from the microenvironment. A more rapid progression to invasive disease could result in the extensive dissemination of tumor cells to distant organs prior to diagnosis.
Our in vivo analysis showed that, while the SUM159 trailblazer cells were capable of disseminating to the lung and forming micrometastases, they were inefficient at colonization and metastatic growth within the time frame of our experiments. This finding is consistent with those in previous reports showing that the sustained retention of mesenchymal traits can suppress macrometastatic growth and that cells may need to revert to a more epithelial phenotype before colonization (52, 55, 56). It is possible that trailblazer cells also can lie dormant before converting to a state that gives rise to colonization. However, our functional data suggest an intriguing alternative model, in which the invasion of the trailblazer subpopulation of tumor cells from the ductal epithelium creates new avenues for the secondary invasion of opportunist cells away from the primary tumor (Figure 10). Once into the stroma, both the trailblazer and opportunist cells could then intravasate into the vasculature and disseminate throughout the body. Consistent with this model, our results, as well as those of others (37), show that epigenetic heterogeneity precedes invasion and that epigenetically distinct tumor cells can comingle in invasive regions of breast tumors and at metastatic sites (57). Because gene expression profiles measure the population averaged molecular characteristics of heterogenous primary tumors, not the traits of individual cells (58), some Trailblazer-high patient tumors may contain both trailblazer and opportunist cells. Similarly, a small subpopulation of trailblazer cells could contribute to invasion and metastasis in a subset of the Trailblazer-low patients. The potential for cooperation between the trailblazer and opportunist subpopulations is consistent with results showing that genetically distinct subpopulations of mouse small-cell lung cancer cells and clones derived from a human prostate tumor can cooperate to promote metastasis (51, 52). The mechanistic basis for cooperation between the subpopulations was not determined in these models, so it is possible that these subpopulations use a form of collaboration similar to that observed between the trailblazer and opportunist cells. Taken together, these results support the investigation of the potential interactions among tumor cell subpopulations during tumorigenesis.
In summary, our identification and functional investigation of a highly invasive trailblazer subpopulation has provided new insight into the molecular traits that confer tumor cells with the ability to collectively invade. Importantly, we further discovered that collective invasion can be the product of a commensal relationship between trailblazer cells and other non-trailblazer subpopulations. This cooperation during collective invasion has the potential to promote the dissemination of tumor cells with a diverse range of molecular features, which could increase the risk of metastasis and promote resistance to treatment. Thus, the therapeutic targeting of trailblazer cell subpopulations may have direct and collateral benefits that improve patient outcome.
Methods
Further information can be found in the Supplemental Methods.
Cell culture and reagents.
T47D, HCC1143, HCC1428, HCC1569, and HCC1954 cells were a gift from Michael Peyton, Adi Gazdar, and John Minna (UTSW). 4T1 cells were a gift from Fred Miller (Wayne State University, Detroit, Michigan, USA). HC-11 cells were a gift from Jeff Rosen (Baylor College of Medicine, Houston, Texas, USA). SUM149, SUM159, and SUM229 cells were a gift from Angelique Whitehurst (UTSW). Human-derived cell lines were validated by Powerplex genotyping. Cell lines stably expressing pCLNRX-H2B:GFP and PGK-H2B:mCherry were generated as described previously (13). SUM159 trailblazer cells expressing GFP and luciferase were generated by infection with pLVTHM-luciferase (gift from Tomoyuki Mashimo, UTSW) and pBOB-GFP lentivirus (13). Growth factor–reduced Matrigel (BD Biosciences) and bovine collagen I (BD Biosciences) were used for organotypic culture experiments. Antibodies recognizing collagen I (ab292, Abcam), RAC 1/2/3 (L129, Cell Signaling), CDC42 (11A11, Cell Signaling), N-WASP (30D10, Cell Signaling), ERK1/2 (L34512, Cell Signaling), GAPDH (Calbiochem), GFP (ab13970, Abcam), mCherry (1C51, Abcam), collagen I (Abcam), β-actin (ab8227, Abcam), collagen IV (AB756P, Chemicon), human CD44 (clone 156-3C11, Thermo Scientific), ITGA11 (SC390091, Santa Cruz), PDGFRA (sc338, Santa Cruz; D1E1E, Cell Signaling), DOCK10 (A301306A, Bethyl), and tubulin (B512, Sigma-Aldrich) were used. Hoechst 33342, phalloidin, and secondary antibodies labeled with Alexa Fluor 488 nm, 546 nm, 647 nm, or 680 nm (Invitrogen) and IR Dye 800CW (Li-Cor Biosciences) were used.
Transfection of siRNAs.
Cells were transfected with 50 to 100 nM of siRNA using RNAiMax transfection reagent (Invitrogen) for 24 to 48 hours. The siRNAs were from Dharmacon and Sigma-Aldrich. Cells in all conditions designated as “control” were transfected with a pool of siRNAs that does not target human genes. The details of the sequences for each siRNA pool are shown in Supplemental Table 2.
Organotypic culture.
Single cells were plated in 8-well chamber slides (immunofluorescence staining, BD Biosciences; live-cell imaging, Nunc) onto a base layer of Matrigel (5 mg/ml) and collagen I (1.5–2.1 mg/ml) and supplemented with a 2% Matrigel/growth medium mixture as described previously (13, 59). All cultures were grown for 6 to 8 days, except where indicated in the figure legends. For spheroid cluster experiments, a 30-μl drop of a 500,000 cell/ml growth medium suspension was placed on a tissue culture dish lid and inverted onto the bottom dish containing sterile PBS to prevent sample desiccation. Drops were then incubated at 37°C for 18 to 24 hours and pipetted into a microfuge tube and pelleted. Spheroid clusters were resuspended in 50 μl Matrigel/collagen mix and plated on 30 μl of a base layer of Matrigel/collagen I. Details of quantification of the assays are in the Supplemental Methods.
Vertical invasion of tumor cells into ECM.
10,000 cells (consisting of H2B:GFP- and H2B:mCherry-labeled cells mixed at a 1:4 ratio) were reverse transfected in duplicate in a 96-well plate with 50 nM OnTargetplus siRNAs using RNAiMax transfection reagent (Invitrogen). ECM was added 48 hours after transfection, and cells were fixed and imaged 48 hours (SUM159 trailblazer, 578T) or 72 hours (SUM229 trailblazer) after the addition of ECM.
Flow cytometry and antibodies.
Antibodies to the following human antigens were used for flow cytometry analyses: CD49f-PE (GoH3, BD Biosciences), EpCAM-FITC (VU1D9, Stem Cell Technologies), and CD24–Alexa Fluor 647 (561644, BD Biosciences). Analyses were conducted following standard flow cytometry procedures.
Quantitative real-time PCR.
Total RNA was isolated with a GenElute Mammalian Total RNA Miniprep Kit (Sigma-Aldrich) and converted to cDNA using the iScript cDNA Synthesis Kit (Bio-Rad). Applied Biosystems TaqMan Gene Expression Assays were performed with 20 ng cDNA, which was amplified with Applied Biosystems 2× TaqMan using an Applied Biosystems 7500 Real-Time PCR System. GAPDH and specific transcript levels for each transfection condition were measured in triplicate. The ΔΔCT method was applied to quantify relative gene expression (60).
Patient sample analysis.
A breast cancer data set (GEO accession GSE18229, with microarray platform GEO accession GPL1390) (27) containing 161 primary tumor samples with microarray data (Agilent Human 1A Oligo Custom Microarray) and clinical outcomes was used for the primary analysis. A second breast cancer data set (GEO accession GSE20624, with microarray platform GEO accession GPL1390) was used to analyze an additional set of 45 patients with TNBC. Quantile-quantile normalization was used to normalize cell line data and primary tumor data. The expression of 7 genes (DOCK10, DAB2, ITGA11, PDGFRA, VASN, PPAP2B, and LPAR1) in the trailblazer HCC1143 and SUM159 subpopulations and 9 noninvasive cell lines was used to develop a prediction model using the Random Forest approach (31). The model was then used to classify each primary tumor as Trailblazer high or Trailblazer low. Kaplan Meier survival curves were drawn for both the Trailblazer-high and Trailblazer-low groups, and the survival differences between the groups were compared using the Mantel-Cox log-rank test. Multivariate analysis of patient outcome was performed as described previously (61).
Xenograft experiments.
Age-matched female NOD/SCID mice were used for all in vivo experiments. When possible, littermates were housed together. NOD/SCID mice were obtained from The Jackson Laboratory and bred and maintained under specific pathogen–free conditions in a barrier facility (UTSW).
Statistics.
The mRNA expression data (Human HT-12 v4 Expression BeadChip, Illumina Inc.) for cell lines were processed with a model-based background correction approach (62), quantile-quantile normalization, and log2 transformation. Heat maps showing the relative expression of genes analyzed with Illumina BeadChips were generated with GenePattern software using the Heat Map Image module (63). Unsupervised hierarchical clustering was implemented with complete linkage and Euclidean distance. Significance analysis of microarray analysis (64) was used to identify differentially expressed genes between the trailblazer and opportunist HCC1143 and SUM159 subpopulations with a FDR of less than 5%. Median values of replicate probe sets for the same genes were used to summarize expression values for each gene. The mRNA expression data are available at GEO (accession GSE58643). Survival differences were compared using the Mantel-Cox log-rank test (Graphpad, Prism). Organotypic culture and Western blot data were analyzed by 2-tailed Student’s t test (Graphpad, Prism). Extent of metastasis was determined by Mann Whitney U test. The distribution of the clinical characteristics in the Trailblazer-high and Trailblazer-low patient groups was analyzed by Fisher’s exact test (Graphpad, Prism). P values of less than 0.05 were considered significant.
Study approval.
All experiments were approved by the UTSW Institutional Animal Care and Use Committee and were performed in compliance with UTSW institutional guidelines and with relevant laws.
Supplementary Material
Acknowledgments
This work was supported by NIH grants 1RO1CA155241 (to G.W. Pearson), 5RO1CA152301 (to Y. Xie), and 5T32CA124334 (to A.M. Prechtl and M.A. Esparza); Cancer Prevention Research Institute of Texas grants RP101251 (to Y. Xie) and RP10496 (to T.T. Dang); the Mary Kay Foundation (to G.W. Pearson); and UTSW institutional support funds (to G.W. Pearson). We thank Abhijit Bugde and Kate Phelps in the UTSW Live Cell Imaging Facility for microscopy support.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(5):1927–1943. doi:10.1172/JCI77767.
Han Sun’s present address is: Department of Quantitative Health Sciences, Cleveland Clinic, Cleveland, Ohio, USA.
References
- 1.Burstein HJ, Polyak K, Wong JS, Lester SC, Kaelin CM. Ductal carcinoma in situ of the breast. N Engl J Med. 2004;350(14):1430–1441. doi: 10.1056/NEJMra031301. [DOI] [PubMed] [Google Scholar]
- 2.Vanharanta S, Massague J. Origins of metastatic traits. Cancer Cell. 2013;24(4):410–421. doi: 10.1016/j.ccr.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polyak K. Molecular markers for the diagnosis and management of ductal carcinoma in situ. J Natl Cancer Inst Monogr. 2010;2010(41):210–213. doi: 10.1093/jncimonographs/lgq019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen L, Yang S, Jakoncic J, Zhang JJ. Migrastatin analogues target fascin to block tumour metastasis. Nature. 2010;464(7291):1062–1066. doi: 10.1038/nature08978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rorth P. Collective cell migration. Annu Rev Cell Dev Biol. 2009;25:407–429. doi: 10.1146/annurev.cellbio.042308.113231. [DOI] [PubMed] [Google Scholar]
- 6.Rorth P. Fellow travellers: emergent properties of collective cell migration. EMBO Rep. 2012;13(11):984–991. doi: 10.1038/embor.2012.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147(5):992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 8.Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10(7):445–457. doi: 10.1038/nrm2720. [DOI] [PubMed] [Google Scholar]
- 9.Wolf K, et al. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol. 2007;9(8):893–904. doi: 10.1038/ncb1616. [DOI] [PubMed] [Google Scholar]
- 10.Yu X, et al. N-WASP coordinates the delivery and F-actin-mediated capture of MT1-MMP at invasive pseudopods. J Cell Biol. 2012;199(3):527–544. doi: 10.1083/jcb.201203025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scott RW, et al. LIM kinases are required for invasive path generation by tumor and tumor-associated stromal cells. J Cell Biol. 2010;191(1):169–185. doi: 10.1083/jcb.201002041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen-Ngoc KV, et al. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc Natl Acad Sci U S A. 2012;109(39):E2595–E2604. doi: 10.1073/pnas.1212834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dang TT, Prechtl AM, Pearson GW. Breast cancer subtype-specific interactions with the microenvironment dictate mechanisms of invasion. Cancer Res. 2011;71(21):6857–6866. doi: 10.1158/0008-5472.CAN-11-1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. 2013;155(7):1639–1651. doi: 10.1016/j.cell.2013.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedl P, et al. Migration of coordinated cell clusters in mesenchymal and epithelial cancer explants in vitro. Cancer Res. 1995;55(20):4557–4560. [PubMed] [Google Scholar]
- 16.Kenny PA, et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol Oncol. 2007;1(1):84–96. doi: 10.1016/j.molonc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bissell MJ, Radisky DC, Rizki A, Weaver VM, Petersen OW. The organizing principle: microenvironmental influences in the normal and malignant breast. Differentiation. 2002;70(9):537–546. doi: 10.1046/j.1432-0436.2002.700907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ioachim E, et al. Immunohistochemical expression of extracellular matrix components tenascin, fibronectin, collagen type IV and laminin in breast cancer: their prognostic value and role in tumour invasion and progression. Eur J Cancer. 2002;38(18):2362–2370. doi: 10.1016/S0959-8049(02)00210-1. [DOI] [PubMed] [Google Scholar]
- 19.Seton-Rogers SE, et al. Cooperation of the ErbB2 receptor and transforming growth factor beta in induction of migration and invasion in mammary epithelial cells. Proc Natl Acad Sci U S A. 2004;101(5):1257–1262. doi: 10.1073/pnas.0308090100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pasic L, et al. Sustained activation of the HER1-ERK1/2-RSK signaling pathway controls myoepithelial cell fate in human mammary tissue. Genes Dev. 2011;25(15):1641–1653. doi: 10.1101/gad.2025611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dent R, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15):4429–4434. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 22.Conklin MW, et al. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178(3):1221–1232. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Almendro V, Marusyk A, Polyak K. Cellular heterogeneity and molecular evolution in cancer. Annu Rev Pathol. 2013;8:277–302. doi: 10.1146/annurev-pathol-020712-163923. [DOI] [PubMed] [Google Scholar]
- 24.Sanz-Moreno V, et al. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135(3):510–523. doi: 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 25.Singh DK, Ku CJ, Wichaidit C, Steininger RJ, 3rd, Wu LF, Altschuler SJ. Patterns of basal signaling heterogeneity can distinguish cellular populations with different drug sensitivities. Mol Syst Biol. 2010;6:369. doi: 10.1038/msb.2010.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447(7143):433–440. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 27.Prat A, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12(5):R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keller PJ, et al. Mapping the cellular and molecular heterogeneity of normal and malignant breast tissues and cultured cell lines. Breast Cancer Res. 2010;12(5):R87. doi: 10.1186/bcr2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim E, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15(8):907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 30.Taube JH, et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc Natl Acad Sci U S A. 2010;107(35):15449–15454. doi: 10.1073/pnas.1004900107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Breiman L. Random Forests. Machine Learning. 2001;45(1):5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- 32.Lehmann BD, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Padrick SB, Rosen MK. Physical mechanisms of signal integration by WASP family proteins. Annu Rev Biochem. 2010;79:707–735. doi: 10.1146/annurev.biochem.77.060407.135452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gadea G, Sanz-Moreno V, Self A, Godi A, Marshall CJ. DOCK10-mediated Cdc42 activation is necessary for amoeboid invasion of melanoma cells. Curr Biol. 2008;18(19):1456–1465. doi: 10.1016/j.cub.2008.08.053. [DOI] [PubMed] [Google Scholar]
- 35.Hu M, et al. Regulation of in situ to invasive breast carcinoma transition. Cancer Cell. 2008;13(5):394–406. doi: 10.1016/j.ccr.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller FR, Santner SJ, Tait L, Dawson PJ. MCF10DCIS.com xenograft model of human comedo ductal carcinoma in situ. J Natl Cancer Inst. 2000;92(14):1185–1186. doi: 10.1093/jnci/92.14.1185. [DOI] [PubMed] [Google Scholar]
- 37.Park SY, Gonen M, Kim HJ, Michor F, Polyak K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J Clin Invest. 2010;120(2):636–644. doi: 10.1172/JCI40724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roesch A, et al. A temporarily distinct subpopulation of slow-cycling melanoma cells is required for continuous tumor growth. Cell. 2010;141(4):583–594. doi: 10.1016/j.cell.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fidler IJ, Kripke ML. Metastasis results from preexisting variant cells within a malignant tumor. Science. 1977;197(4306):893–895. doi: 10.1126/science.887927. [DOI] [PubMed] [Google Scholar]
- 40.Minn AJ, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436(7050):518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Creighton CJ, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci U S A. 2009;106(33):13820–13825. doi: 10.1073/pnas.0905718106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mani SA, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9(4):265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 45.Wang W, et al. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 2004;64(23):8585–8594. doi: 10.1158/0008-5472.CAN-04-1136. [DOI] [PubMed] [Google Scholar]
- 46.Azad N, Zahnow CA, Rudin CM, Baylin SB. The future of epigenetic therapy in solid tumours — lessons from the past. Nat Rev Clin Oncol. 2013;10(5):256–266. doi: 10.1038/nrclinonc.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eckert MA, et al. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell. 2011;19(3):372–386. doi: 10.1016/j.ccr.2011.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jacob LS, et al. Genome-wide RNAi screen reveals disease-associated genes that are common to Hedgehog and Wnt signaling. Sci Signal. 2011;4(157):ra4. doi: 10.1126/scisignal.2001225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Axelrod R, Axelrod DE, Pienta KJ. Evolution of cooperation among tumor cells. Proc Natl Acad Sci U S A. 2006;103(36):13474–13479. doi: 10.1073/pnas.0606053103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DeNardo DG, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16(2):91–102. doi: 10.1016/j.ccr.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Calbo J, et al. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell. 2011;19(2):244–256. doi: 10.1016/j.ccr.2010.12.021. [DOI] [PubMed] [Google Scholar]
- 52.Celia-Terrassa T, et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest. 2012;122(5):1849–1868. doi: 10.1172/JCI59218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bianco A, et al. Two distinct modes of guidance signalling during collective migration of border cells. Nature. 2007;448(7151):362–365. doi: 10.1038/nature05965. [DOI] [PubMed] [Google Scholar]
- 54.Jakobsson L, et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol. 2010;12(10):943–953. doi: 10.1038/ncb2103. [DOI] [PubMed] [Google Scholar]
- 55.Ocana OH, et al. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell. 2012;22(6):709–724. doi: 10.1016/j.ccr.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 56.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22(6):725–736. doi: 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Almendro V, et al. Genetic and phenotypic diversity in breast tumor metastases. Cancer Res. 2014;74(5):1338–1348. doi: 10.1158/0008-5472.CAN-13-2357-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Polyak K. Heterogeneity in breast cancer. J Clin Invest. 2011;121(10):3786–3788. doi: 10.1172/JCI60534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xian W, Schwertfeger KL, Vargo-Gogola T, Rosen JM. Pleiotropic effects of FGFR1 on cell proliferation, survival, and migration in a 3D mammary epithelial cell model. J Cell Biol. 2005;171(4):663–673. doi: 10.1083/jcb.200505098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. High-throughput real-time quantitative reverse transcription PCR. Curr Protoc Mol Biol Chapter. 2006;15:Unit 15.18. doi: 10.1002/0471142727.mb1508s73. [DOI] [PubMed] [Google Scholar]
- 61.Xie Y, et al. Robust gene expression signature from formalin-fixed paraffin-embedded samples predicts prognosis of non-small-cell lung cancer patients. Clin Cancer Res. 2011;17(17):5705–5714. doi: 10.1158/1078-0432.CCR-11-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xie Y, Wang X, Story M. Statistical methods of background correction for Illumina BeadArray data. Bioinformatics. 2009;25(6):751–757. doi: 10.1093/bioinformatics/btp040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38(5):500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 64.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98(9):5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.