

Background: Elongin is both a Pol II elongation factor and part of a ubiquitin ligase targeting stalled Pol II.
Results: Elongin ubiquitin ligase assembly is driven by signals that provoke Pol II stalling and/or activate Elongin-dependent transcription.
Conclusion: Elongin ligase assembly is a regulated process.
Significance: This study provides insight into Elongin ubiquitin ligase functions and general mechanisms of ubiquitin ligase activation.
Keywords: confocal microscopy, endoplasmic reticulum stress (ER stress), fluorescence resonance energy transfer (FRET), gene transcription, RNA polymerase II, stress response, transcription elongation factor, ubiquitin ligase
Abstract
Elongin A performs dual functions in cells as a component of RNA polymerase II (Pol II) transcription elongation factor Elongin and as the substrate recognition subunit of a Cullin-RING E3 ubiquitin ligase that has been shown to target Pol II stalled at sites of DNA damage. Here we investigate the mechanism(s) governing conversion of the Elongin complex from its elongation factor to its ubiquitin ligase form. We report the discovery that assembly of the Elongin A ubiquitin ligase is a tightly regulated process. In unstressed cells, Elongin A is predominately present as part of Pol II elongation factor Elongin. Assembly of Elongin A into the ubiquitin ligase is strongly induced by genotoxic stress; by transcriptional stresses that lead to accumulation of stalled Pol II; and by other stimuli, including endoplasmic reticulum and nutrient stress and retinoic acid signaling, that activate Elongin A-dependent transcription. Taken together, our findings shed new light on mechanisms that control the Elongin A ubiquitin ligase and suggest that it may play a role in Elongin A-dependent transcription.
Introduction
Elongin A plays multiple roles in regulation of transcription elongation by RNA polymerase II (Pol II).3 Elongin A was first identified as a subunit of the Elongin complex, a Pol II elongation factor that interacts directly with transcribing Pol II and stimulates the overall rate of RNA chain synthesis (1, 2). The Elongin complex is a heterotrimer composed of the large, transcriptionally active Elongin A protein and two smaller proteins, Elongins B and C, which form a stable subcomplex that binds to Elongin A through a short sequence motif referred to as the BC-box and stimulates Elongin A transcription activity (3, 4). Elongin C is similar in sequence to the SCF ubiquitin ligase subunit Skp1, whereas Elongin B is a ubiquitin-like protein (5, 6). Elongin A has been implicated in activation of Pol II transcription in response to a variety of signals, including retinoic acid, ecdysone, notch, EGF, and TGF-β, as well as stresses, such as heat shock, DNA damage, nutrient deprivation, and endoplasmic reticulum (ER) stress (7–12). In at least some cases, Elongin A is thought to function in the release of promoter-proximally paused Pol II (9).
In addition to its function as a Pol II elongation factor, Elongin A has been shown to function as the substrate recognition subunit of a Cullin-RING ubiquitin ligase that targets the Rpb1 subunit of transcriptionally stalled Pol II for ubiquitylation and degradation by the proteasome (13–17). The Elongin A-containing ubiquitin ligase belongs to the large BC-box (SOCS-box) family of Cullin-RING ubiquitin ligases (reviewed in Ref. 18). Members of this family include a substrate recognition subunit that binds via a BC-box to an Elongin BC heterodimer, which functions as an adaptor to link the BC-box protein to a heterodimeric submodule composed of a Cullin protein (CUL2 or CUL5) and one of two RING finger proteins, RBX1 or RBX2 (19). The RING domains of RBX1 or RBX2 interact directly with an E2 ubiquitin-conjugating enzyme to promote transfer of ubiquitin to substrate bound by the ligase (18). In the Elongin A ubiquitin ligase, CUL5 links a CUL5-RBX2 module to the Elongin ABC complex through interactions with Elongin C and a short amino sequence motif referred to as the Cullin-box located just C-terminal to the BC-box in Elongin A (Fig. 1A) (10, 19).
FIGURE 1.
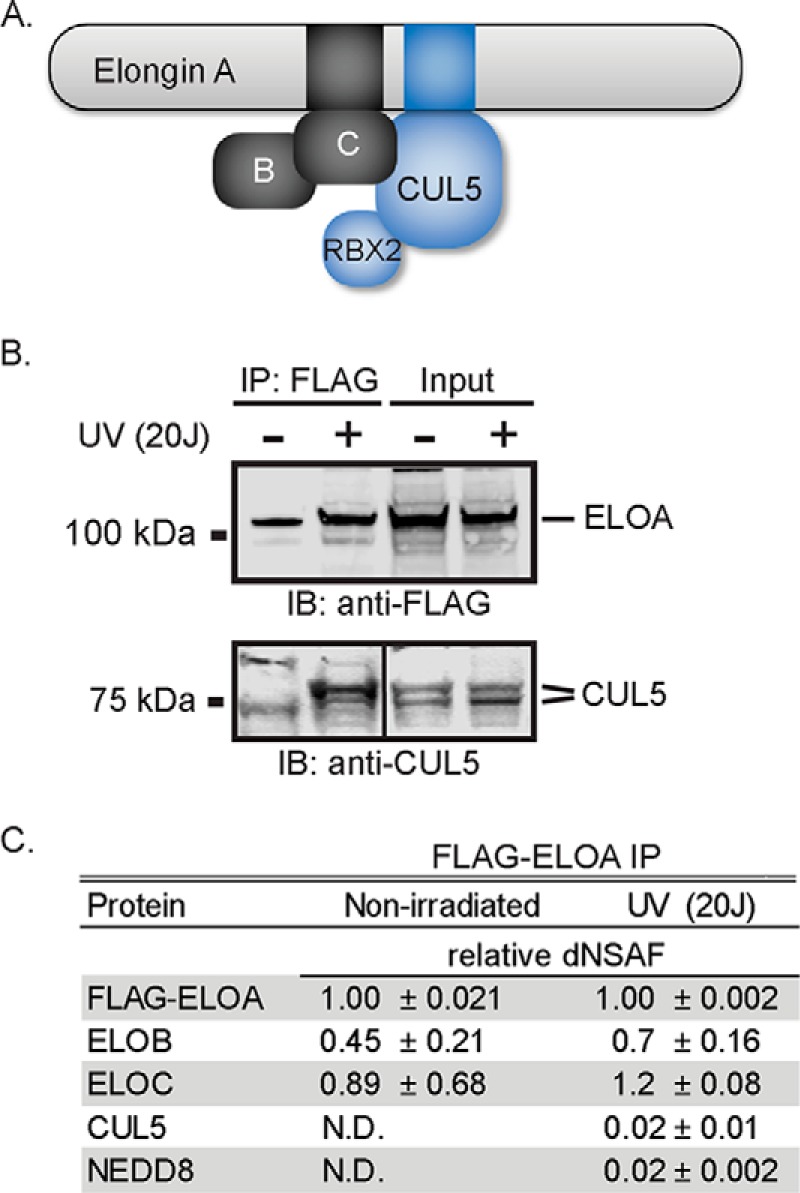
Increased co-immunoprecipitation of Elongin A and CUL5 after UV-irradiation. A, diagram showing organization of the Elongin A ubiquitin ligase. The black box in Elongin A represents the BC-box, and the blue box represents the Cullin-box. B, Western blots of anti-FLAG immunoprecipitates (IP) from Flp-In-HEK293 cells stably expressing FLAG-Elongin A. UV-irradiated cells were subjected to 20 J/cm2 of 254-nm UV irradiation and allowed to recover for 10 min. Anti-FLAG immunoprecipitations were performed with nuclear extracts from irradiated or non-irradiated cells. For the anti-FLAG immunoblot, input and immunoprecipitates corresponding to material from ∼2 × 107 cells were applied to the gel. For the anti-CUL5 immunoblot, material from ∼2 × 107 and ∼6 × 107 cells of the input and immunoprecipitate samples, respectively, were used. Lanes showing CUL5 input and immunoprecipitates are from non-adjacent lanes of the same exposure of the same immunoblot. C, UV-enhanced binding of CUL5 to Elongin A assessed by MudPIT mass spectrometry. Values represent the average and S.D. of the dNSAF for each protein from three biological replicates, normalized to Elongin A. ELOA, Elongin A; ELOB, Elongin B; ELOC, Elongin C; IB, immunoblot; N.D., not detected.
Exposure of cells to UV irradiation and other DNA-damaging agents or to drugs that induce Pol II stalling and/or arrest induces ubiquitylation and degradation of Rpb1 (20–22). Similarly, transcription-coupled ubiquitylation of Rpb1 in vitro is strongly stimulated by conditions that provoke transcriptional stalling/arrest (23–26). In both yeast and mammalian cells, DNA damage-induced Rpb1 ubiquitylation is impaired by mutation or silencing of the Elongin A gene (14, 16). In addition, mutation of the ELC1 or CUL3 genes, which encode the yeast orthologs of Elongin C and CUL5, respectively, leads to defects in DNA damage-induced Rpb1 ubiquitylation (16, 17). The HECT domain E3 ubiquitin ligase NEDD4 (referred to as Rsp5 in yeast) has also been implicated in Pol II ubiquitylation (27–29). Biochemical experiments have revealed that Pol II ubiquitylation is a two-step process initiated by monoubiquitylation of Rpb1 by NEDD4/Rsp5 (15), followed by polyubiquitylation by the Elongin A ubiquitin ligase (14, 15). Based on these observations, it has been proposed that NEDD4/Rsp5 and the Elongin A ubiquitin ligase function together as part of a “fail-safe” mechanism for ubiquitylation and removal of Pol II that is stalled at sites of DNA damage or other impediments and would otherwise block further transcription of genes (15, 30).
As part of our effort to understand the function of the Elongin A ubiquitin ligase, we have been investigating mechanism(s) that control its assembly and activation. Here, we report that Elongin A and CUL5 are rapidly recruited in cells to regions of localized DNA damage. Assembly of the Elongin A ubiquitin ligase is triggered following DNA damage as well as by treatment of cells with drugs that block Pol II elongation. Ligase assembly is also triggered by induction of ER and nutrient stress and by retinoic acid signaling, all of which can activate Elongin A-dependent transcription. Taken together, these findings argue that assembly of the Elongin A ubiquitin ligase is a tightly regulated process that can be driven at least in part by the availability of its substrate, transcriptionally stalled Pol II. In addition, they raise the possibility that the Elongin A ubiquitin ligase not only contributes to degradation of stalled Pol II but might also play a more active role in Elongin A-dependent transcription.
Experimental Procedures
Materials
Monoclonal anti-FLAG® M1 antibody produced in mice (F3040) was from Sigma. Rabbit anti-Cul5 antibody (NBP1-22970) was from Novus Biologicals, and goat anti-Elongin A (R-19) C-terminal antibody was purchased from Santa Cruz Biotechnology, Inc. Anti-(6-4) photoproduct (6-4 PP) monoclonal antibody (clone 64M-2) (CAC-NM-DND-002) and anti-cyclobutane pyrimidine dimer (CPD) monoclonal antibody (clone TDM-2) (CAC-NM-DND-001) were purchased from Cosmo Bio Co., Ltd. Rabbit anti-γ-H2A.X (phospho-Ser-139) antibody (DNA double strand break marker; ab2893) was from Abcam. Donkey anti-rabbit IgG Alexa Fluor 568 (A-10042), chicken anti-goat IgG Alexa Fluor 488 (A-21467), and chicken anti-mouse IgG (H + L) (A-21463) were purchased from Life Technologies and used as secondary antibodies for indirect immunofluorescence. HaloTag® R110DirectTM ligand G3221 was purchased from Promega. Hoechst solution (33258; used at 1:1000 dilution), 5,6-dichloro-1-β-ribofuranosyl benzimidazole (DRB; D1916; used at 25 μm), l-histidinol dihydrochloride (H-6647; used at 2 mm), (S)-(+)-camptothecin (C-9911; used at 5 μm), aphidicolin (A-4487; used at 4 μm), hydroxyurea (H-8627; used at 200 μm), all-trans-retinoic acid (R-2625; used at 10 μm), methyl methanesulfonate (129925; used at 0.1 μm), hydrogen peroxide (16911; used at 300 μm), bleomycin sulfate (B-5507; used at 20 μm), triptolide (T-3652; used at 10 μm), and thapsigargin (T-9033; used at 300 nm) were all from Sigma. KU60019 (S1570; used at 10 μm) and PJ34 (S2886; used at 10 μm) were purchased from Selleckchem. α-Amanitin (used at 10 μm) was purchased from Sigma (A2263) or EMD/Millipore (129741). Etoposide (341205; used at 100 μm) was purchased from Millipore. FuGENE HD and ViaFect transfection reagents were obtained from Promega.
Cell Culture and Stable Cell Lines
HEK293 and HeLa cells were cultured in DMEM and U2OS cells in McCoy's medium (Gibco), at 37 °C in 5% CO2. All media were supplemented with 5% Glutamax, 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin (Gibco). HEK293 cells stably expressing FLAG-ELOA were generated using pcDNA5/FRT/FLAG-ELOA and the Flp-in system (Invitrogen). For some experiments, culture medium for U2OS cells contained phenol red-free McCoy's medium and charcoal-stripped One ShotTM fetal bovine serum (Gibco).
Plasmids
To generate Halo pcDNA5 plasmids encoding Halo-tagged versions of rat Elongin A (GenBankTM accession number AAA82095), DNA fragments flanked by SgfI and PmeI restriction sites were amplified from plasmids encoding wild type and mutant rat Elongin A (4) and transferred into the SgfI and PmeI sites in the plasmid Halo pcDNA5/FRT PacI PmeI (31). pFN21A-TCEB3, encoding Halo-tagged human ELOA (GenBankTM accession number NP_003189.1) was purchased from Promega (catalog no. FHC03618). A plasmid encoding mCherry-tagged CUL5 (mCherry-CUL5) was constructed by introducing a PCR-generated DNA fragment encoding human CUL5, flanked by BglII and KpnI restriction sites, into pmCherry-C1 (Clontech, 632524). Plasmids pCI-Neo-ELOB and pCI-Neo-ELOC were generated by introducing cDNAs encoding human Elongins B and C, respectively, into pCI-Neo (Promega, E1841) and were a gift from Takumi Kamura. To generate the plasmid encoding FLAG-Halo-tagged ALC1, ALC1 was PCR-amplified and introduced into a derivative of pcDNA5/FRT.
Immunopurification of FLAG-Elongin A-associated Proteins
Parental HEK293 cells or HEK293 cells stably expressing FLAG-ELOA were grown to 70–80% confluence in 4–5 15-cm plates and, where indicated, exposed to 20 J/cm2 UVC in a UVP CL-1000 cross-linker and allowed to recover for 10 min in a 37 °C, 5% CO2 incubator. Nuclear extracts were prepared as described (32), except that they were extracted with buffer containing 0.42 m NaCl instead of KCl. The resulting nuclear extracts were mixed with anti-FLAG (M2)-agarose beads (Sigma) equilibrated in 10 mm HEPES-NaOH, pH 7.9, 0.2% Triton X-100, 0.3 m NaCl, 10 mm KCl, 1.5 mm MgCl2, and protease inhibitor mixture (Sigma; catalog no. P8340) in a ratio of 100 μl of packed beads/3 ml of nuclear extract and gently rocked overnight at 4 °C. The beads were washed four times with a 50-fold excess of the same buffer. Proteins were eluted by incubation for 30 min at 4 °C with one packed bead volume of 10 mm HEPES-NaOH, pH 7.9, 0.05% Triton X-100, 0.1 m NaCl, 1.5 mm MgCl2, protease inhibitor mixture, and 0.2 mg/ml FLAG peptide (Sigma). Beads were removed by centrifugation, and the elution was repeated an additional two times. Purified proteins were subjected to immunoblotting or analyzed by multidimensional protein identification technology (MudPIT) mass spectrometry.
Mass Spectrometry
Identification of proteins associated with FLAG-Elongin A was accomplished using a modification of the MudPIT procedure (33, 34) using an LTQ ion trap mass spectrometer equipped with a nano-LC electrospray ionization source (Thermo Scientific). Tandem mass spectra (MS/MS) were interpreted using SEQUEST (35) against a database of 29,178 human proteins (downloaded from NCBI on August 16, 2011) and complemented with 177 sequences from frequent contaminants (human keratins, IgGs, proteolytic enzymes). To estimate false discovery rates, each sequence was randomized (keeping amino acid composition and length the same), and the resulting “shuffled sequences” were added to the forward database and searched at the same time. Peptide/spectrum matches were sorted and selected using DTASelect (36) with the following criteria set. The DeltCn was required to be at least 0.08, with minimum XCorr value of 1.8 for singly charged, 2.0 for doubly charged, and 3.0 for triply charged spectra, and a maximum Sp rank of 10. Using these criteria, the overall false discovery rates for samples prepared from UV-treated or non-irradiated cells were 0.11 or 0.17%, respectively. Peptide hits from multiple runs were compared using CONTRAST (36). Relative protein levels were estimated using distributed normalized spectral abundance factors (dNSAFs) calculated for each protein as described (37–40).
Microirradiation, Live Imaging, Acceptor Photobleaching (AP)-FRET, and Image Analysis
Time lapse movies, UV microirradiation, and AP-FRET measurements were performed on a PerkinElmer Life Sciences UltraVIEW VoX spinning disk microscope, which included a Yokagawa CSU-X11 spinning disk, an ORCA-R2 camera (Hamamatsu), and a PerkinElmer Life Sciences PhotoKinesis accessory. The microscope base was a Carl Zeiss Axiovert 200M equipped with a ×40, 1.3 numerical aperture plan-apochromat objective. All emission was collected through a multiband dichroic (405/488/561/640 nm). R110Direct was excited with the 488-nm laser and was imaged with a 500–550-nm emission filter. mCherry was excited with the 561-nm laser and imaged through a 415–475-nm, 580–650-nm multiband emission filter. Hoechst was excited with the 405-nm laser and imaged through a 415–475-nm, 580–650-nm multiband emission filter. Imaging of singly labeled cells demonstrated that these filter settings eliminated bleed-through. Laser power and exposure time were adjusted beforehand to maximize image quality and eliminate significant photobleaching; the absence of photobleaching was confirmed by observing unperturbed cells in the acquisition field of view.
Prior to microirradiation and/or AP-FRET, HeLa or U2OS cells were plated at 50–60% confluence in MatTek glass bottom dishes (35 mm, No. 2 14-mm diameter glass) and were transfected using FuGENE HD (HeLa cells) or ViaFect (U2OS cells) with plasmids encoding Halo-ELOA (100 ng), mCherry-CUL5 (400 ng), ELOB (100 ng), and ELOC (100 ng). To label Halo-tagged proteins with rhodamine 110 in living cells, medium was changed after 24 h, HaloTag® R110DirectTM ligand was added to a final concentration of 100 nm, and cells were allowed to incubate overnight without washing as directed in the manufacturer's protocol. Except where indicated in the figure legends, cells were stained for 30 min with Hoechst dye to mark nuclei and/or sensitize cells to UV irradiation 48 h post-transfection.
Microirradiation was performed as follows. Cells were subjected to laser UV microirradiation in a 200 × 3-pixel (34 × 0.51-μm) stripe or a diffraction-limited spot centered in the nucleus. The microirradiation was performed with 100% 405-nm laser power, and cells were exposed to 500–700 microwatts for ∼3 s (40 iterations) and 1.5 s (250 iterations) for the stripe and spot, respectively. Before and after microirradiation, one image/s was collected. Under these conditions, normal cell and nuclear morphology was preserved over the time scale of the experiment. Quantitative analysis of protein recruitment following microirradiation was performed with custom analysis plugins in ImageJ (National Institutes of Health, Bethesda, MD); plugins are available for download at the Stowers Institute Web site. Camera offset and uniform background were subtracted by manually selecting a background region and subtracting its average from each channel and frame. In some cases, StackReg (P. Thévanez, Biomedical Imaging Group, Swiss Federal Institute of Technology Lausanne) was used to correct for translational drift. The fluorescence intensity of the microirradiated stripe as a function of time (I(t)) was measured as the average intensity of a manually selected region corresponding to the visible bleached region immediately after microirradiation. Total nuclear fluorescence intensity (T(t)) was measured in the same way, selecting the nuclear boundary. Normalized recruitment (Rt) values were calculated using the equation,
where I(0) and T(0) are the average fluorescence intensities of the microirradiated and total nuclear region, respectively, averaged over the preirradiation time period.
For measurements of AP-FRET, a sequence of at least three images of each region of interest was collected before and after photobleaching of the mCherry photoacceptor with 15 iterations of 100% 561-nm laser power. Under these conditions, we observed no photobleaching of the rhodamine 110 donor fluorophore in control experiments performed with cells expressing rhodamine 110-labeled, Halo-tagged protein without a mCherry photoacceptor. FRET efficiencies (E) were calculated as follows,
where the brackets represent a temporal average, and Ibefore and Iafter refer to the donor fluorescence intensity before and after acceptor photobleaching.
Indirect Immunofluorescence
Untransfected HeLa cells were plated on either 4-well chamber slides (BD Falcon) or MatTek Glass bottom dishes, 35 mm, No. 2 (14-mm diameter glass) gridded dishes and grown to ∼60% confluence. Cells were stained or not with Hoechst dye and subjected to laser UV microirradiation as described above. For the experiments in Table 1, a preselected diffraction limited spot in the nucleus was irradiated as described above. Cells were then fixed in 2% paraformaldehyde for 20 min, washed in ice-cold PBS, permeabilized using 0.5% Triton in phosphate-buffered saline, and incubated with a blocking buffer containing 5% BSA, 100 μm MgCl2, and 0.1% Triton for 1 h at room temperature. Dishes or slides were incubated overnight with primary antibodies as indicated. Cells were washed in blocking buffer without BSA and then incubated for 1 h in the dark with appropriate secondary antibodies at a final concentration of 0.005 mg/ml. After washing with phosphate-buffered saline, ProLong® gold antifade reagent with DAPI (Life Technologies, P36935) was added. The same microirradiated cells were located for imaging by a combination of the relative grid positioning and morphology. To compare levels of 6-4 PPs, CPDs, and γ-H2A.X in Hoechst-sensitized and unsensitized U2OS cells, cells were either treated or not with Hoechst dye for 30 min and exposed or not to 20 J/cm2 UVC in a UVP CL-1000 cross-linker and allowed to recover for 10 min in a 37 °C, 5% CO2 incubator. After cells were fixed and permeabilized as described above, 2 m HCl was added for 30 min to denature cellular DNA. Cells were washed, blocked as described above, and incubated with anti-6-4 PPs, anti-CPDs, or γ-H2A.X overnight. After incubation with appropriate secondary antibodies and washing with phosphate-buffered saline, fixed and stained cells were treated with ProLong® gold antifade reagent with DAPI. Anti-CUL5 and γ-H2A.X staining was visualized with donkey anti-rabbit Alexa Fluor 568, anti-Elongin A was visualized with chicken anti-goat Alexa Fluor 488, and anti-6-4 PPs and CPD were visualized with chicken anti-mouse Alexa Fluor 647. Regions of cells growing in a monolayer were selected based on postfixation DAPI staining. Individual nuclei were outlined manually, and quantitative analysis of fluorescence intensity was performed using custom analysis plugins in ImageJ (National Institutes of Health). Camera offset and uniform background were subtracted by manually selecting a background region and subtracting its average from each channel and frame.
TABLE 1.
Co-localization of Elongin A and CUL5 immunostaining
Spatial image Pearson cross-correlation (Pearson (x, y)) between green (anti-Elongin A immunostaining) and red (anti-CUL5 immunostaining) fluorescence was determined for individual cells (n = 10).
| Pearson correlation coefficient (x, y) | |
|---|---|
| Non-irradiated cells | 0.31 ± 0.14 |
| Whole cell irradiation (20 J) | 0.79 ± 0.14 |
| Microirradiated cells | |
| Crossing irradiation site | 0.93 ± 0.05 |
| Not crossing irradiation site | 0.87 ± 0.02 |
Results
Isolation of the Elongin A Ubiquitin Ligase from UV-irradiated Cells
In initial experiments to determine whether assembly of the Elongin A ubiquitin ligase is a regulated process, we observed that Elongin A copurified with substantially more of the ubiquitin ligase subunit CUL5 from extracts of UV-irradiated cultured cells than from extracts of untreated cells. Elongin A and associated proteins were purified by anti-FLAG-agarose immunoaffinity chromatography from HEK293 Flp-In cells stably expressing N-terminally FLAG-tagged human Elongin A. As shown in the Western blots of Fig. 1B, little or no CUL5 copurified with FLAG-Elongin A from nuclear extracts prepared from untreated cells; however, CUL5 was readily detected in FLAG immunoprecipitates from cells harvested 10 min after exposure to UV irradiation. Elongin A-associated proteins were also analyzed by MudPIT mass spectrometry (33, 34). Consistent with Western blotting results, CUL5 was detected by mass spectrometry only in FLAG immunoprecipitates from UV-irradiated cells (Fig. 1C). In MudPIT data sets, the relative amount of a particular protein in a sample can be estimated from a normalized spectral abundance factor, or dNSAF (40). Comparison of relative dNSAF values in samples prepared from control and UV-irradiated cells suggests that Elongins B and C are constitutively bound to Elongin A, in roughly stoichiometric amounts. In contrast, CUL5 was detected only in samples prepared from UV-irradiated cells, with a normalized spectral abundance factor value ∼2% of the dNSAFs of Elongins A, B, and C. Thus, even after UV irradiation, only a small fraction of FLAG-Elongin A was stably associated with CUL5. We also note the appearance in FLAG-Elongin A immunoprecipitates of the ubiquitin-like NEDD8 protein, which is conjugated to Cullin proteins in a process that is important for the assembly and/or activity of Cullin-RING ubiquitin ligases (reviewed in Ref. 41). Taken together, these findings suggest that UV irradiation of cells either induces assembly of the Elongin A ubiquitin ligase or, alternatively, increases the amount of ligase complex that can be solubilized during preparation of nuclear extracts.
Elongin A and CUL5 Are Recruited to Sites of Laser Microirradiation
Many proteins with roles in DNA damage repair and/or the cellular response to DNA damage are rapidly recruited to regions of DNA damage induced by UV irradiation of cells sensitized by treatment with intercalating dyes. UV irradiation of sensitized cells results in the formation of double and single strand DNA breaks as well as other forms of DNA damage (42, 43). Because the Elongin A ubiquitin ligase participates in Pol II ubiquitylation following DNA damage (11, 14–17), we asked whether we could detect recruitment of Elongin A and CUL5 to sites of DNA damage. In these experiments, the nuclei of HeLa cells that had been pretreated with the DNA-intercalating Hoechst dye were subjected to UV microirradiation with a 405-nm laser to induce localized DNA damage (Fig. 2). Five minutes after irradiation, cells were fixed and analyzed by indirect immunofluorescence using antibodies against endogenous Elongin A, CUL5, and γ-H2A.X, which marks sites of nuclear DNA damage (44). Elongin A and γ-H2A.X were both enriched at sites of microirradiation (Fig. 2, B–D and F), as was CUL5 (Fig. 2G). Notably, there was a significant increase in the spatial correlation, assessed by Pearson correlation, of anti-Elongin A and anti-CUL5 immunostaining throughout the entire nuclei of sensitized cells exposed to localized UV microirradiation or to whole cell UV irradiation (Table 1).
FIGURE 2.
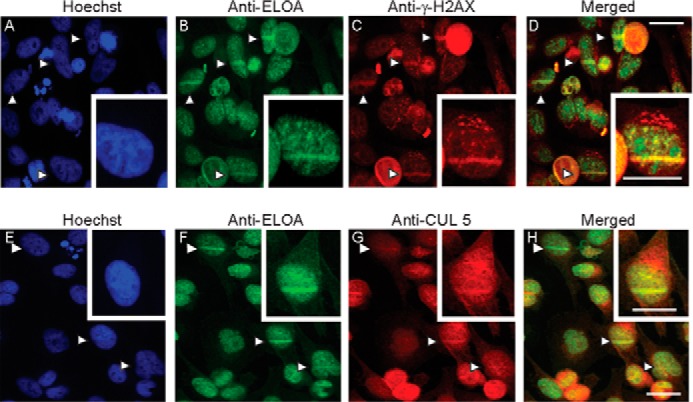
Enrichment of Elongin A and CUL5 at sites of localized DNA damage induced by laser microirradiation. HeLa cells were stained with Hoechst dye, and some cells in each field were microirradiated with a 405-nm UV laser. Cells were fixed and analyzed by indirect immunofluorescence using anti-Elongin A (B and F) and anti-γ-H2A.X (C) or anti-CUL5 (G). Merged images are shown in D and H, and Hoechst staining is shown in A and E. Arrows, microirradiated regions. Scale bars, 26 μm.
We also assessed the recruitment of Elongin A and CUL5 to sites of DNA damage in living cells using mCherry-tagged CUL5 (mCherry-CUL5) and Elongin A fused to an N-terminal Halo tag (Halo-Elongin A) covalently labeled with a cell-permeable rhodamine 110 derivative (HaloTag R110Direct) (45). These exogenously expressed proteins behaved similarly to their endogenous counterparts; Halo-Elongin A, like endogenous Elongin A, exhibited a largely nuclear localization, whereas mCherry-CUL5 was distributed throughout both the nucleus and cytoplasm (Fig. 3A). In addition, both Halo-Elongin A and mCherry-CUL5 were rapidly enriched at laser-targeted regions after microirradiation of nuclei in Hoechst-sensitized cells (Fig. 3B). Of note, enrichment of Halo-Elongin A or mCherry-CUL5 was not detected in unsensitized cells subjected to UV laser microirradiation (Fig. 3C).
FIGURE 3.

Rapid recruitment of Elongin A and CUL5 after microirradiation of living cells. A, localization of Halo-Elongin A and mCherry-CUL5 in U2OS cells. B, Hoechst-sensitized U2OS cells expressing Halo-Elongin A and mCherry-CUL5 were microirradiated and imaged 1 s before (−1 s) or at the times indicated after initiation of microirradiation. C, unsensitized U2OS cells expressing Halo-Elongin A and mCherry-CUL5 were microirradiated along a stripe running between the white arrows and imaged as in B.
Genotoxic Stress Induces Assembly of Elongin A and CUL5 in Living Cells
To monitor assembly of the Elongin A ubiquitin ligase in living cells, we used FRET to measure interaction of Halo-Elongin A with mCherry-CUL5. During FRET, energy is transferred by a donor fluorophore to an acceptor fluorophore. As a result of the energy transfer, the emission of the donor is quenched, and that of the acceptor is enhanced in a strongly distance-dependent fashion; for typical fluorescent proteins, FRET occurs only at distances less than 100 Å (46). The emission spectrum of the donor fluorophore rhodamine 110 (R110) on Halo-Elongin A overlaps the excitation spectrum of the mCherry acceptor on CUL5, allowing energy transfer when donor and acceptor are in close proximity. In these experiments, we used a variant of FRET called AP-FRET (47). In AP-FRET, FRET efficiency is determined by comparing the donor fluorescence emission before acceptor photobleaching (when energy can be transferred to acceptor molecules) with donor emission after acceptor photobleaching (when the acceptor can no longer absorb energy emitted from the donor). As shown in the representative images in Fig. 4, A and B, and in the data shown in Fig. 4C, an increase in fluorescence from the R110-labeled Halo tag was readily detected after acceptor photobleaching of UV-irradiated, Hoechst-sensitized HeLa or U2OS cells expressing both Halo-Elongin A and mCherry-CUL5. This AP-FRET signal reflects an interaction between Elongin A and CUL5, because we found in control experiments that it was not observed after photobleaching of cells expressing free Halo tag and mCherry, Halo-Elongin A and free mCherry, or free Halo tag and mCherry-CUL5 (Fig. 4, A–C). As shown in Fig. 4C, the AP-FRET signal was ∼4–7-fold smaller in non-irradiated sensitized cells than in sensitized cells subjected to UV irradiation. Taken together, these results argue that assembly of the Elongin A ubiquitin ligase is strongly induced following UV irradiation of Hoechst-sensitized cells. The biochemical experiments shown in Fig. 1 indicate that UV-induced DNA damage alone is sufficient to induce some Elongin A-CUL5 interaction. Nevertheless, we detected little to no AP-FRET signal in cells that had not been presensitized with Hoechst, although γ-H2A.X and UV-induced lesions, including CPDs and 6-4 PPs, still accumulated in unsensitized cells, albeit to a somewhat lesser amount than in cells treated with Hoechst (Fig. 4, D and E). Thus, it appears that formation of UV-induced lesions, such as CPDs, 6-4 PPs, and other photoproducts, does not provide a sufficiently potent signal to drive enough ligase assembly to be detected in single cell, AP-FRET assays.
FIGURE 4.

AP-FRET reveals genotoxic stress-induced interaction of Elongin A with CUL5. A, representative images of donor (R110-labeled Halo-Elongin A or Halo tag) and acceptor (mCherry-CUL5 or mCherry), pre- and post-acceptor bleaching. B, heat maps showing relative pixel intensities of R110-Halo signals, before and after acceptor bleaching. C, FRET efficiency in HeLa and U2OS cells expressing the indicated proteins. Hoechst-sensitized cells were subjected or not to 20 J/cm2 UV irradiation and allowed to recover for 10 min. Values represent average ± S.E. (error bars) (n ≥ 18). D, FRET efficiency in U2OS cells subjected to UV microirradiation (microirrad) or treatment with the indicated genotoxic agent for 60 min. + Hoechst, Hoechst-sensitized cells; − Hoechst, unsensitized cells; Bleo, bleomycin; Eto, etoposide; Campto, camptothecin; Aphid, aphidicolin; HU, hydroxyurea; MMS, methylmethane sulfate. Values represent average ± S.E. (n ≥ 10). E, Hoechst-sensitized or unsensitized cells were subjected or not to 20 J/cm2 of UV irradiation, allowed to recover for 10 min, fixed, and immunostained with the indicated antibodies. γ-H2A.X, 6-4 PP, and CPD immunostaining was quantitated in individual nuclei. Box and whisker plots show the resulting pixel intensities (n = 50); boxes show the median (central horizontal line), average (black dot), and interquartile ranges, whereas the whiskers show the full range of data.
We also measured Elongin A-CUL5 AP-FRET in cells treated with a variety of agents that induce genotoxic stress. These agents included hydrogen peroxide; the topoisomerase 1 inhibitor camptothecin; the topoisomerase 2 inhibitor etoposide; the DNA-alkylating agent methyl methanesulfonate; bleomycin, which catalyzes formation of single-stranded and double-stranded DNA lesions; and aphidicolin and hydroxyurea, which interfere with DNA replication and can cause formation of DNA lesions including single and double strand breaks. As shown in Fig. 4D, after all of these treatments, the Elongin A-CUL5 AP-FRET signal was increased compared with that seen in cells treated with vehicle alone, indicating that multiple genotoxic stresses can drive assembly of the Elongin A ubiquitin ligase.
To begin to address the mechanism by which genotoxic stress leads to assembly of the Elongin A ubiquitin ligase, we asked whether it is restricted to DNA damage regions. To do so, we compared AP-FRET at sites of microirradiation and elsewhere in the nucleus (Fig. 5A). We observed that the AP-FRET signal measured at damage regions reached a maximum within 2 min after laser microirradiation and persisted for at least 15 min. When measured at positions in the nucleus distant from sites of microirradiation, AP-FRET signal was increased to a similar extent and with similar kinetics, indicating that the Elongin A ubiquitin ligase is not restricted to sites of microirradiation-induced DNA damage and raising the possibility that signaling pathways activated by DNA damage might contribute to ligase assembly.
FIGURE 5.
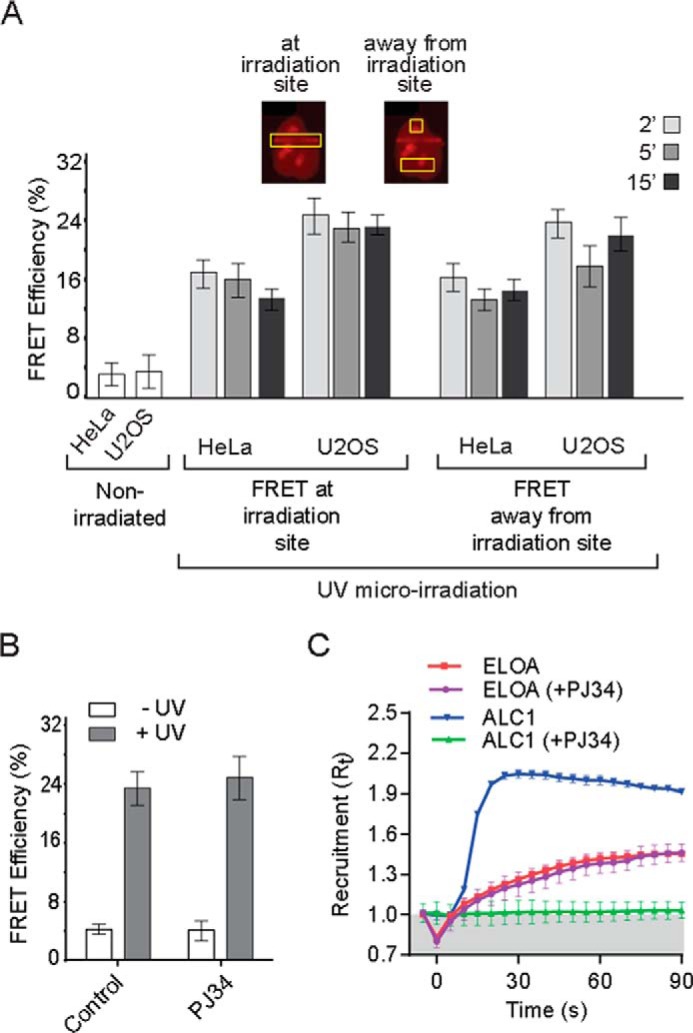
A, the Elongin A-CUL5 interaction is not restricted to DNA damage regions. FRET efficiencies were measured at or away from regions of laser microirradiation (n ≥ 18). Cells were allowed to recover for 2, 5, or 15 min after microirradiation. Inset, representative images of Halo-Elongin A in microirradiated cells. Yellow boxes, regions of FRET measurements at or away from sites of laser microirradiation. B, assembly of the Elongin A ubiquitin ligase is independent of PARP activity. AP-FRET was measured 5 min after microirradiation of Hoechst-sensitized U2OS cells that had been treated or not with PARP inhibitor PJ34. Values represent average ± S.E. (error bars) (n = 15). C, Elongin A recruitment to microirradiated regions is not inhibited by PJ34. Kinetics of Elongin A or ALC1 recruitment (n = 15) to microirradiated regions of Hoechst-sensitized U2OS cells, with or without PJ34. Cells were imaged every second, and intensity values were binned over 5-s intervals. Microirradiation was initiated at time = 0 s.
One of the earliest steps in DNA damage signaling involves recruitment and activation of poly(ADP-ribose) polymerases (PARPs) at DNA lesions, and the resulting localized synthesis of poly(ADP-ribose) contributes to rapid recruitment of enzymes with roles in multiple DNA repair pathways. To determine whether PARP activity is needed for recruitment and/or assembly of the Elongin A ubiquitin ligase, we treated cells with the PARP inhibitor PJ34. Under conditions where PJ34 completely inhibited recruitment of the PARP-activated chromatin remodeling enzyme ALC1 to regions of microirradiation-induced DNA damage, PJ34 had no effect on either recruitment or assembly of the Elongin A ubiquitin ligase (Fig. 5, B and C).
Microirradiation-induced Elongin A Recruitment and Assembly of the Elongin A Ubiquitin Ligase Are Independent Processes
To explore the relationship between recruitment of Elongin A to regions of DNA damage and microirradiation-enhanced assembly of the Elongin A ubiquitin ligase, we took advantage of Elongin A mutants that we have shown previously are defective in their abilities to bind Elongins B and C and/or CUL5 and to stimulate Pol II elongation and/or support ubiquitylation of Rpb1 (4, 14) (Fig. 6A). Elongin A mutant Δ546–565, which lacks the BC-box, fails to bind to Elongins B and C or to CUL5 and is inactive in transcription and Rpb1 ubiquitylation in vitro (4, 14). A second mutant, Elongin A Δ566–585, binds to Elongins B and C but is unable to stimulate Pol II elongation or to support ubiquitylation in vitro. A third mutant, Elongin A Δ401–440, binds normally to Elongins B and C and to CUL5, and it supports stimulation of Pol II elongation and Rpb1 ubiquitylation in vitro. Comparison of fluorescence intensity indicated that there were no significant differences between the average levels of expression of the Halo-tagged wild type and mutant Elongin A proteins, although the expression level of each protein varied in individual cells. Importantly, in single cell measurements, we observed no correlation between levels of wild type or mutant Halo-Elongin A expression and AP-FRET signals (Fig. 6, B and C).
FIGURE 6.

Elongin A mutations differentially affect assembly with CUL5 and recruitment to sites of microirradiation. A, schematic representation of wild type Elongin A and mutants analyzed in this study and their abilities to bind Elongins B and C and to stimulate Pol II elongation or support Rpb1 ubiquitylation in vitro. B, FRET efficiencies in U2OS cells (non-irradiated or 5 min after microirradiation) expressing wild type or mutant Halo-Elongin A and mCherry-CUL5. Values represent average ± S.E. (error bars) (n ≥ 12); *, p ≤ 0.005. C, scatter plot showing Halo-Elongin A relative pixel intensity, measured before acceptor photobleaching, as a function of FRET efficiency in individual cells. D, kinetics of recruitment (n ≥ 12) to microirradiated regions of wild type and mutant Halo-Elongin A. Cells were imaged every second, and intensity values were binned over 5-s intervals. Microirradiation was initiated at time = 0 s.
As expected, deletion of the BC-box led to a complete loss of microirradiation-induced binding of Elongin A to CUL5 in living cells as detected by AP-FRET (Fig. 6B). In contrast, deletion of the BC-box had no significant effect on Elongin A enrichment at regions of laser-induced DNA damage (Fig. 6D), arguing that recruitment of Elongin A to regions of DNA damage does not require prior assembly of the ubiquitin ligase. Deletion of Elongin A residues 566–585 or 401–440 did not prevent UV-induced binding of Elongin A to CUL5, although the average AP-FRET signal was reduced by 50–60% relative to that observed in cells expressing full-length Halo-Elongin A (Fig. 6B). On the other hand, Elongin A Δ566–585 was only very weakly enriched at regions of laser-induced DNA damage, and there was no detectable enrichment of Elongin A Δ401–440 (Fig. 6D). Thus, assembly of the Elongin A ubiquitin ligase does not depend on stable recruitment of Elongin A to microirradiated regions, supporting the idea that it can occur at a distance from sites of DNA damage.
Drugs That Interfere with Transcript Elongation, but Not Initiation, by Pol II Induce Assembly of the Elongin A Ubiquitin Ligase
Because of prior evidence that Pol II stalling can induce Rpb1 ubiquitylation (23–26, 48), we wished to test the possibility that Pol II stalling can drive assembly of the Elongin A ubiquitin ligase independent of DNA damage. To do so, we took advantage of two drugs known to interfere with Pol II elongation by different mechanisms. The first, α-amanitin, is a general Pol II inhibitor that binds near the Pol II active site and reduces the rate of nucleotide addition from several thousand to just a few nucleotides/min (49–51). The other, DRB, is a cyclin-dependent kinase inhibitor that inhibits the P-TEFb kinase Cdk9 (52, 53). P-TEFb activity is required for efficient elongation through promoter-proximal regions of genes but appears to be dispensable for elongation throughout the remainder of the gene body (54–57). As shown in Fig. 7A, Elongin A-CUL5 AP-FRET signal could be detected as early as 5 min after the addition of either α-amanitin or DRB to cells and reached levels almost as high as that attained after UV irradiation of sensitized cells, suggesting that Pol II stalling or arrest either near the promoter or throughout the transcribed region of genes can provoke assembly of the Elongin A ubiquitin ligase. We also used the Pol II transcription inhibitor triptolide, which has little or no effect on Pol II elongation but prevents transcript initiation by inhibiting the ATPase activity of the XPB subunit of the general transcription factor TFIIH (57, 58). Unlike with α-amanitin or DRB, we observed no increase in Elongin A-CUL5 AP-FRET signal after treating cells for 60 min with triptolide, suggesting that inhibition of transcript initiation is not sufficient to drive ligase assembly.
FIGURE 7.
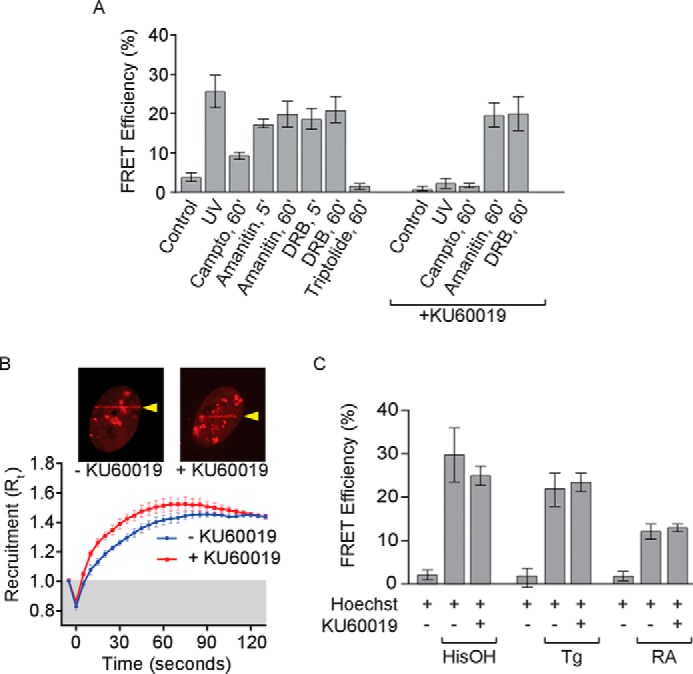
Multiple stresses induce assembly of the Elongin A ubiquitin ligase. A, FRET efficiencies in U2OS cells expressing Halo-Elongin A and mCherry-CUL5. AP-FRET was measured in control cells or 5 min after microirradiation or after treatment for the indicated times with camptothecin, α-amanitin, DRB, or triptolide, with or without KU60019 added 1 h prior to the addition of drugs or microirradiation. Data for cells treated with camptothecin without KU60019 are from the same cells as in Fig. 4D and are included for comparison. Values represent average ± S.E. (error bars) (n ≥ 18). B, kinetics of recruitment of Halo-Elongin A after microirradiation of U2OS cells (n ≥ 10) treated or not with KU60019; data were analyzed as in Fig. 5C. The insets show representative images taken 60 s after microirradiation. Arrows, regions of microirradiation. C, AP-FRET was measured in U2OS cells after 12, 4, or 1 h of treatment with histidinol (HisOH), thapsigargin (Tg), or retinoic acid (RA), respectively, with or without KU60019. In each case, Hoechst dye was added to cultures 1 h before the addition of compound or vehicle. Values represent average ± S.E. (n = 10).
Double strand breaks and other DNA lesions are thought to cause Pol II to become stalled or arrested during transcript elongation, both because they can prevent Pol II from transcribing past the lesion (30, 59, 60) and because they induce checkpoint kinase-dependent transcriptional silencing on regions of chromatin proximal to the lesion (61, 62). To begin to explore the possible relationship between mechanism(s) responsible for assembly of the Elongin A ubiquitin ligase after UV-induced DNA damage and Pol II stalling, we tested the effects on the Elongin A-CUL5 interaction of the small molecule inhibitor KU60019, which preferentially inhibits ATM kinase (63, 64). Treatment with KU60019 had no effect on AP-FRET signal after the addition of α-amanitin or DRB (Fig. 7A). In addition, it did not prevent enrichment of Elongin A at regions of localized DNA damage induced by UV microirradiation (Fig. 7B). In contrast, treatment with KU60019 completely blocked the increase in AP-FRET signal after microirradiation or the addition of the topoisomerase inhibitor camptothecin. Taken together, these findings suggest that DNA damage or Pol II stalling/arrest drive assembly of the Elongin A ubiquitin ligase by different mechanisms or, alternatively, that KU60019 blocks a step upstream of Pol II stalling in the pathway leading to ligase assembly. In addition, they provide further evidence that the recruitment of Elongin A to DNA damage regions does not require prior assembly with CUL5 to form the ubiquitin ligase complex.
Retinoic Acid and ER- and Nutrient Stress-induced Assembly of Elongin A with CUL5
Results of previous studies have implicated Elongin A in activating transcription in response to retinoic acid signaling and to multiple stresses, including DNA damage, ER stress, and amino acid limitation (10–12). As we have shown, Elongin A and CUL5 are rapidly recruited to sites of laser microirradiation-induced DNA damage, and treatment of cells with various genotoxic agents drives assembly of Elongin A with the ubiquitin ligase subunit CUL5. We therefore asked whether assembly of Elongin A with CUL5 might also be provoked by treatment of cells with thapsigargin to induce ER stress, with histidinol to mimic amino acid limitation, or with retinoic acid. Strikingly, treatment of cells with either thapsigargin, histidinol, or retinoic acid led to increases in Elongin A-CUL5 AP-FRET signal comparable to those observed after DNA damage or treatment with drugs that cause Pol II stalling. Unlike the UV-induced Elongin A-CUL5 interaction but similar to that induced by α-amanitin or DRB, the interaction between Elongin A and CUL5 induced by retinoic acid, thapsigargin, or histidinol was not blocked by the ATM inhibitor KU60019 (Fig. 7C).
Discussion
Previous studies revealed that the Elongin A protein exists in cells in at least two functionally distinct protein complexes. In the Elongin ABC complex, Elongin A functions as the transcriptionally active subunit of a Pol II transcription elongation factor. In the Elongin A ubiquitin ligase complex, Elongin A serves as the substrate recognition subunit of a CUL5/RBX2-containing Cullin-RING ubiquitin ligase that targets the Rpb1 subunit of Pol II stalled at sites of DNA damage and perhaps other impediments. In this report, we have used AP-FRET to demonstrate that assembly of the Elongin A ubiquitin ligase is a tightly regulated process that can be induced in living cells by a variety of cellular stresses, including genotoxic and transcriptional stresses, thapsigargin-induced ER stress, histidinol-induced amino acid starvation, and retinoic acid signaling. To our knowledge, these studies provide the first evidence for regulated assembly of any of the large family of BC-box-Cullin-RING ligases and thus provide evidence for a new mechanism by which members of this family can be activated in response to diverse signaling pathways.
What signal or signals drive assembly of the Elongin A ubiquitin ligase? Our finding that the binding of Elongin A to CUL5 can be provoked not only by genotoxic stress but also by treating cells with either DRB or α-amanitin is consistent with the idea that the interaction of the Elongin ABC complex with its substrate, stalled Pol II, can induce its binding to CUL5 to form the ubiquitin ligase. There is precedent for the idea that substrate binding can promote formation of a ubiquitin ligase. For example, binding of the F-box protein and substrate recognition subunit Fbxl3 to its target, Cry1, has been shown to promote binding of Fbxl3 to Skp1 and CUL1 to form a functional SCF (Skp1-CUL1-F-box protein) ubiquitin ligase (65).
The observation that Elongin A and CUL5 are co-recruited to regions of localized DNA damage induced by laser microirradiation raises the possibility that the ligase assembles at or near DNA lesions. Notably, however, we observed that the Elongin A-CUL5 AP-FRET signal appears with similar kinetics and to a similar extent at sites of microirradiation and at distant regions within the same nuclei, indicating that the ligase is not restricted to regions of DNA damage. In addition, we identify Elongin A mutants that exhibit little to no enrichment at microirradiated regions but are still able to assemble with CUL5. Although we cannot rule out the possibility that ligase assembly is initiated during interactions with damage sites too transient to be detected in our assays, these results taken together are most consistent with the idea that DNA damage-induced assembly of the Elongin A ubiquitin ligase is controlled in part by diffusible signals within the cell or nucleus and might involve transcription-independent DNA damage signaling. Indeed, we find that assembly of the Elongin A ubiquitin ligase induced by DNA-damaging agents, but not by drugs that interfere with Pol II elongation, is blocked by treating cells with the protein kinase inhibitor KU60019, which is reported preferentially to inhibit the DNA damage checkpoint kinase ATM at the concentrations used in our experiments (63, 64). We cannot conclude from inhibitor experiments alone that the block to DNA damage-induced assembly of the Elongin A ubiquitin ligase is specifically due to ATM-dependent checkpoint signaling. Nevertheless, our results do suggest that DNA damage promotes ligase assembly via a KU60019-sensitive mechanism independent of Pol II stalling or, alternatively, that the KU60019-sensitive step in the pathway leading to ligase assembly is upstream of Pol II stalling. The latter possibility would be consistent with prior evidence that DNA damage can interfere with Pol II transcript elongation via a checkpoint kinase-dependent mechanism(s) (61, 62).
Finally, our observation that retinoic acid signaling as well as stresses including thapsigargin-induced ER stress and histidinol-induced amino acid starvation also induce assembly of the Elongin A ubiquitin ligase was somewhat surprising, because to our knowledge these stimuli have not been shown to induce Pol II stalling or arrest. Taken together with evidence that activation of some genes in response to retinoic acid signaling or to ER stress and amino acid starvation is defective in cells depleted of Elongin A (10–12), our finding that ligase assembly is induced under these conditions raises the intriguing possibility that Rpb1 ubiquitylation by the Elongin A ubiquitin ligase could participate in the transcription activation process, either via an active mechanism that directly increases transcriptional efficiency or through the removal of aberrantly stalled or arrested Pol II that would otherwise block activation of the gene. Indeed, transcriptional activation of any given gene probably involves an increase in the number of elongating Pol II molecules and a concomitant increase in the probability that a Pol II molecule might become stalled or arrested, thereby blocking further transcription. In addition, actively transcribed regions are subject to increased rates of DNA damage that might reasonably be anticipated to increase the likelihood of Pol II stalling (reviewed in Ref. 66). Further, DNA breaks and other lesions are introduced in both the promoters and transcribed regions of some genes during transcriptional activation and have been proposed to play an integral role in the activation process (66–72). Thus, one could imagine that signaling pathways that lead to substantial increases in transcription might also increase assembly and recruitment of the Elongin A ubiquitin ligase as a means of assuring that it is immediately available when needed to remove stalled or arrested Pol II.
Although our data raise the possibility that the Elongin A ubiquitin ligase might contribute to gene activation, it is important to emphasize that ligase-independent functions of Elongin are also likely to be important in this process, because transcription defects in Elongin A(−/−) or Elongin A knockdown cells can be rescued in whole or in part by Elongin A mutants that can stimulate Pol II elongation in vitro but do not support efficient Rpb1 ubiquitylation (10, 11). Future studies investigating in more detail the biochemical mechanism(s) underlying regulation of assembly of the Elongin A ubiquitin ligase, the signals responsible for its induction, and the potential role of the ligase in Pol II transcription activation will be needed to resolve these issues.
Acknowledgments
We thank Tari Parmley, Maria Katt, Alexis Murray, and members of the tissue culture core for help with cells and tissue culture; Kym Delventhal, Kyle Weaver, MaryEllen Kirkman, and Dominic Heinecke for assistance with mutagenesis; Anoja Perera and Michael Peterson for assistance with DNA sequencing; Shigeo Sato, Chieri Tomomori-Sato, and Aaron J. Gottschalk for the FLAG-Halo-ALC1 plasmid; and Sara Jackson for help with figure preparation.
This work was supported, in whole or in part, by National Institutes of Health Grant GM041628 (to R. C. C. and J. W. C.). This work was also supported by a grant from the Helen Nelson Medical Research Fund at the Greater Kansas City Community Foundation (to the Stowers Institute) and by KAKENHI Grants 24590279 (to T. Y.) and 24590357 (to T. A.).
- Pol II
- polymerase II
- ER
- endoplasmic reticulum
- 6-4 PP
- (6-4) photoproduct
- CPD
- cyclobutane pyrimidine dimer
- DRB
- 5,6-dichloro-1-β-ribofuranosyl benzimidazole
- MudPIT
- multidimensional protein identification technology
- AP
- acceptor photobleaching
- dNSAF
- distributed normalized spectral abundance factor
- R110
- rhodamine 110.
References
- 1. Bradsher J. N., Jackson K. W., Conaway R. C., Conaway J. W. (1993) RNA polymerase II transcription factor SIII: I. Identification, purification, and properties. J. Biol. Chem. 268, 25587–25593 [PubMed] [Google Scholar]
- 2. Bradsher J. N., Tan S., McLaury H.-J., Conaway J. W., Conaway R. C. (1993) RNA polymerase II transcription factor SIII: II. Functional properties and role in RNA chain elongation. J. Biol. Chem. 268, 25594–25603 [PubMed] [Google Scholar]
- 3. Aso T., Lane W. S., Conaway J. W., Conaway R. C. (1995) Elongin (SIII): a multisubunit regulator of elongation by RNA polymerase II. Science 269, 1439–1443 [DOI] [PubMed] [Google Scholar]
- 4. Aso T., Haque D., Barstead R. J., Conaway R. C., Conaway J. W. (1996) The inducible elongin A elongation activation domain: structure, function, and interaction with the elongin BC complex. EMBO J. 15, 5557–5566 [PMC free article] [PubMed] [Google Scholar]
- 5. Garrett K. P., Tan S., Bradsher J. N., Lane W. S., Conaway J. W., Conaway R. C. (1994) Molecular cloning of an essential subunit of RNA polymerase II elongation factor SIII. Proc. Natl. Acad. Sci. U.S.A. 91, 5237–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garrett K. P., Aso T., Bradsher J. N., Foundling S. I., Lane W. S., Conaway R. C., Conaway J. W. (1995) Positive regulation of general transcription SIII by a tailed ubiquitin homolog. Proc. Natl. Acad. Sci. U.S.A. 92, 7172–7176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gerber M., Eissenberg J. C., Kong S., Tenney K., Conaway J. W., Conaway R. C., Shilatifard A. (2004) In vivo requirement of the RNA polymerase elongation factor elongin A for proper gene expression and development. Mol. Cell. Biol. 24, 9911–9919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gerber M., Tenney K., Conaway J. W., Conaway R. C., Eissenberg J. C., Shilatifard A. (2005) Regulation of heat shock gene expression by RNA polymerase II elongation factor, Elongin A. J. Biol. Chem. 280, 4017–4020 [DOI] [PubMed] [Google Scholar]
- 9. Chopra V. S., Hong J. W., Levine M. (2009) Regulation of Hox gene activity by transcriptional elongation in Drosophila. Curr. Biol. 19, 688–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yasukawa T., Bhatt S., Takeuchi T., Kawauchi J., Takahashi H., Tsutsui A., Muraoka T., Inoue M., Tsuda M., Kitajima S., Conaway R. C., Conaway J. W., Trainor P. A., Aso T. (2012) Transcriptional elongation factor Elongin A regulates retinoic acid-induced gene expression during neuronal differentiation. Cell Rep. 2, 1129–1136 [DOI] [PubMed] [Google Scholar]
- 11. Kawauchi J., Inoue M., Fukuda M., Uchida Y., Yasukawa T., Conaway R. C., Conaway J. W., Aso T., Kitajima S. (2013) Transcriptional properties of mammalian elongin A and its role in stress response. J. Biol. Chem. 288, 24302–24315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fan A. X., Papadopoulos G. L., Hossain M. A., Lin I. J., Hu J., Tang T. M., Kilberg M. S., Renne R., Strouboulis J., Bungert J. (2014) Genomic and proteomic analysis of transcription factor TFII-I reveals insight into the response to cellular stress. Nucleic Acids Res. 42, 7625–7641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kamura T., Burian D., Yan Q., Schmidt S. L., Lane W. S., Querido E., Branton P. E., Shilatifard A., Conaway R. C., Conaway J. W. (2001) MUF1, a novel Elongin BC-interacting leucine-rich repeat protein that can assemble with Cul5 and Rbx1 to reconstitute a ubiquitin ligase. J. Biol. Chem. 276, 29748–29753 [DOI] [PubMed] [Google Scholar]
- 14. Yasukawa T., Kamura T., Kitajima S., Conaway R. C., Conaway J. W., Aso T. (2008) Mammalian Elongin A complex mediates DNA-damage-induced ubiquitylation and degradation of Rpb1. EMBO J. 27, 3256–3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Harreman M., Taschner M., Sigurdsson S., Anindya R., Reid J., Somesh B., Kong S. E., Banks C. A., Conaway R. C., Conaway J. W., Svejstrup J. Q. (2009) Distinct ubiquitin ligases act sequentially for RNA polymerase II poly-ubiquitylation. Proc. Natl. Acad. Sci. U.S.A. 106, 20705–20710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ribar B., Prakash L., Prakash S. (2007) ELA1 and CUL3 are required along with ELC1 for RNA polymerase II polyubiquitylation and degradation in DNA-damaged yeast cells. Mol. Cell. Biol. 27, 3211–3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ribar B., Prakash L., Prakash S. (2006) Requirement of ELC1 for RNA polymerase II polyubiquitylation and degradation in response to DNA damage in Saccharomyces cerevisiae. Mol. Cell. Biol. 26, 3999–4005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Petroski M. D., Deshaies R. J. (2005) Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6, 9–20 [DOI] [PubMed] [Google Scholar]
- 19. Kamura T., Maenaka K., Kotoshiba S., Matsumoto M., Kohda D., Conaway R. C., Conaway J. W., Nakayama K. I. (2004) VHL-box and SOCS-box domains determine binding specificity for Cul2-Rbx1 and Cul5-Rbx2 modules of ubiquitin ligases. Genes Dev. 18, 3055–3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bregman D. B., Halaban R., van Gool A. J., Henning K. A., Friedberg E. C., Warren S. L. (1996) UV-induced ubiquitination of RNA polymerase II: a novel modification deficient in Cockayne syndrome cells. Proc. Natl. Acad. Sci. U.S.A. 93, 11586–11590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nguyen V. T., Giannoni F., Dubois M. F., Seo S. J., Vigneron M., Kédinger C., Bensaude O. (1996) In vivo degradation of RNA polymerase II largest subunit triggered by α-amanitin. Nucleic Acids Res. 24, 2924–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ratner J. N., Balasubramanian B., Corden J., Warren S. L., Bregman D. B. (1998) Ultraviolet radiation-induced ubiquitination and proteasomal degradation of the large subunit of RNA polymerase II: implications for transcription-coupled DNA repair. J. Biol. Chem. 273, 5184–5189 [DOI] [PubMed] [Google Scholar]
- 23. Lee K. B., Wang D., Lippard S. J., Sharp P. A. (2002) Transcription-coupled and DNA damage-dependent ubiquitination of RNA polymerase II in vitro. Proc. Natl. Acad. Sci. U.S.A. 99, 4239–4244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang L. Y., Jiang H., Rangel K. M. (2003) RNA polymerase II stalled on a DNA template during transcription elongation is ubiquitinated and the ubiquitination facilitates displacement of the elongation complex. Int. J. Oncol. 22, 683–689 [PubMed] [Google Scholar]
- 25. Jung Y., Lippard S. J. (2006) RNA polymerase II blockage by cisplatin-damaged DNA. Stability and polyubiquitylation of stalled polymerase. J. Biol. Chem. 281, 1361–1370 [DOI] [PubMed] [Google Scholar]
- 26. Somesh B. P., Reid J., Liu W. F., Søgaard T. M., Erdjument-Bromage H., Tempst P., Svejstrup J. Q. (2005) Multiple mechanisms confining RNA polymerase II ubiquitylation to polymerases undergoing transcriptional arrest. Cell 121, 913–923 [DOI] [PubMed] [Google Scholar]
- 27. Huibregtse J. M., Yang J. C., Beaudenon S. L. (1997) The large subunit of RNA polymerase II is a substrate of the Rsp5 ubiquitin-protein ligase. Proc. Natl. Acad. Sci. U.S.A. 94, 3656–3661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Beaudenon S. L., Huacani M. R., Wang G., McDonnell D. P., Huibregtse J. M. (1999) Rsp5 ubiquitin-ligase mediates DNA damage-induced degradation of the large subunit of RNA polymerase II. Mol. Cell. Biol. 19, 6972–6979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anindya R., Aygün O., Svejstrup J. Q. (2007) Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol. Cell 28, 386–397 [DOI] [PubMed] [Google Scholar]
- 30. Wilson M. D., Harreman M., Svejstrup J. Q. (2013) Ubiquitylation and degradation of elongating RNA polymerase II: the last resort. Biochim. Biophys. Acta 1829, 151–157 [DOI] [PubMed] [Google Scholar]
- 31. Banks C. A., Lee Z. T., Boanca G., Lakshminarasimhan M., Groppe B. D., Wen Z., Hattem G. L., Seidel C. W., Florens L., Washburn M. P. (2014) Controlling for gene expression changes in transcription factor protein networks. Mol. Cell Proteomics 13, 1510–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dignam J. D., Lebovitz R. M., Roeder R. G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian cell nuclei. Nucleic Acids Res. 11, 1475–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Florens L., Washburn M. P. (2006) Proteomic analysis by multidimensional protein identification technology. Methods Mol. Biol. 328, 159–175 [DOI] [PubMed] [Google Scholar]
- 34. Washburn M. P., Wolters D., Yates J. R., 3rd (2001) Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 [DOI] [PubMed] [Google Scholar]
- 35. Eng J. K., McCormack A. L., Yates J. R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass. Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 36. Tabb D. L., McDonald W. H., Yates J. R., 3rd (2002) DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1, 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Florens L., Carozza M. J., Swanson S. K., Fournier M., Coleman M. K., Workman J. L., Washburn M. P. (2006) Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods 40, 303–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paoletti A. C., Parmely T. J., Tomomori-Sato C., Sato S., Zhu D., Conaway R. C., Conaway J. W., Florens L., Washburn M. P. (2006) Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proc. Natl. Acad. Sci. U.S.A. 103, 18928–18933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zybailov B., Mosley A. L., Sardiu M. E., Coleman M. K., Florens L., Washburn M. P. (2006) Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J. Proteome Res. 5, 2339–2347 [DOI] [PubMed] [Google Scholar]
- 40. Zhang Y., Wen Z., Washburn M. P., Florens L. (2010) Refinements to label free proteome quantitation: how to deal with peptides shared by multiple proteins. Anal. Chem. 82, 2272–2281 [DOI] [PubMed] [Google Scholar]
- 41. Deshaies R. J., Emberley E. D., Saha A. (2010) Control of cullin-ring ubiquitin ligase activity by nedd8. Subcell. Biochem. 54, 41–56 [DOI] [PubMed] [Google Scholar]
- 42. Dinant C., de Jager M., Essers J., van Cappellen W. A., Kanaar R., Houtsmuller A. B., Vermeulen W. (2007) Activation of multiple DNA repair pathways by sub-nuclear damage induction methods. J. Cell Sci. 120, 2731–2740 [DOI] [PubMed] [Google Scholar]
- 43. Lukas C., Bartek J., Lukas J. (2005) Imaging of protein movement induced by chromosomal breakage: tiny “local” lesions pose great “global” challenges. Chromosoma 114, 146–154 [DOI] [PubMed] [Google Scholar]
- 44. Paull T. T., Rogakou E. P., Yamazaki V., Kirchgessner C. U., Gellert M., Bonner W. M. (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 10, 886–895 [DOI] [PubMed] [Google Scholar]
- 45. Los G. V., Encell L. P., McDougall M. G., Hartzell D. D., Karassina N., Zimprich C., Wood M. G., Learish R., Ohana R. F., Urh M., Simpson D., Mendez J., Zimmerman K., Otto P., Vidugiris G., Zhu J., Darzins A., Klaubert D. H., Bulleit R. F., Wood K. V. (2008) HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem. Biol. 3, 373–382 [DOI] [PubMed] [Google Scholar]
- 46. Sekar R. B., Periasamy A. (2003) Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J. Cell Biol. 160, 629–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Van Munster E. B., Kremers G. J., Adjobo-Hermans M. J., Gadella T. W., Jr. (2005) Fluorescence resonance energy transfer (FRET) measurement by gradual acceptor photobleaching. J. Microsc. 218, 253–262 [DOI] [PubMed] [Google Scholar]
- 48. Sigurdsson S., Dirac-Svejstrup A. B., Svejstrup J. Q. (2010) Evidence that transcript cleavage is essential for RNA polymerase II transcription and cell viability. Mol. Cell 38, 202–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bushnell D. A., Cramer P., Kornberg R. D. (2002) Structural basis of transcription: α-amanitin-RNA polymerase II cocrystal at 2.8 Å resolution. Proc. Natl. Acad. Sci. U.S.A. 99, 1218–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chafin D. R., Guo H., Price D. H. (1995) Action of α-amanitin during pyrophosphorolysis and elongation by RNA polymerase II. J. Biol. Chem. 270, 19114–19119 [DOI] [PubMed] [Google Scholar]
- 51. Rudd M. D., Luse D. S. (1996) Amanitin greatly reduces the rate of transcription by RNA polymerase II ternary complexes but fails to inhibit some transcript cleavage modes. J. Biol. Chem. 271, 21549–21558 [DOI] [PubMed] [Google Scholar]
- 52. Zhu Y., Pe'ery T., Peng J., Ramanathan Y., Marshall N., Marshall T., Amendt B., Mathews M. B., Price D. H. (1997) Transcription elongation factor P-TEFb is required for HIV-1 Tat transactivation in vitro. Genes Dev. 11, 2622–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mancebo H. S. Y., Lee G., Flygare J., Tomassini J., Luu P., Zhu Y., Peng J., Blau C., Hazuda D., Price D., Flores O. (1997) P-TEFb kinase is required for HIV Tat transcriptional activation in vivo and in vitro. Genes Dev. 11, 2633–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fraser N. W., Sehgal P. B., Darnell J. E. (1978) DRB-induced premature termination of late adenovirus transcription. Nature 272, 590–593 [DOI] [PubMed] [Google Scholar]
- 55. Zhou Q., Li T., Price D. H. (2012) RNA polymerase II elongation control. Annu. Rev. Biochem. 81, 119–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rahl P. B., Lin C. Y., Seila A. C., Flynn R. A., McCuine S., Burge C. B., Sharp P. A., Young R. A. (2010) c-Myc regulates transcriptional pause release. Cell 141, 432–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Jonkers I., Kwak H., Lis J. T. (2014) Genome-wide dynamics of Pol II elongation and its interplay with promoter proximal pausing, chromatin, and exons. Elife 3, e02407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Titov D. V., Gilman B., He Q. L., Bhat S., Low W. K., Dang Y., Smeaton M., Demain A. L., Miller P. S., Kugel J. F., Goodrich J. A., Liu J. O. (2011) XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat. Chem. Biol. 7, 182–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Svejstrup J. Q. (2002) Mechanisms of transcription-coupled DNA repair. Nat. Rev. Mol. Cell Biol. 3, 21–29 [DOI] [PubMed] [Google Scholar]
- 60. Hanawalt P. C., Spivak G. (2008) Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 9, 958–970 [DOI] [PubMed] [Google Scholar]
- 61. Shanbhag N. M., Rafalska-Metcalf I. U., Balane-Bolivar C., Janicki S. M., Greenberg R. A. (2010) ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 141, 970–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pankotai T., Bonhomme C., Chen D., Soutoglou E. (2012) DNAPKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat. Struct. Mol. Biol. 19, 276–282 [DOI] [PubMed] [Google Scholar]
- 63. Golding S. E., Rosenberg E., Valerie N., Hussaini I., Frigerio M., Cockcroft X. F., Chong W. Y., Hummersone M., Rigoreau L., Menear K. A., O'Connor M. J., Povirk L. F., van Meter T., Valerie K. (2009) Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 8, 2894–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Curtin N. J. (2012) DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 12, 801–817 [DOI] [PubMed] [Google Scholar]
- 65. Yumimoto K., Muneoka T., Tsuboi T., Nakayama K. I. (2013) Substrate binding promotes formation of the Skp1-Cul1-Fbxl3 (SCF(Fbxl3)) protein complex. J. Biol. Chem. 288, 32766–32776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Fong Y. W., Cattoglio C., Tjian R. (2013) The intertwined roles of transcription and repair proteins. Mol. Cell 52, 291–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ju B. G., Lunyak V. V., Perissi V., Garcia-Bassets I., Rose D. W., Glass C. K., Rosenfeld M. G. (2006) A topoisomerase IIβ-mediated dsDNA break required for regulated transcription. Science 312, 1798–1802 [DOI] [PubMed] [Google Scholar]
- 68. Ju B. G., Rosenfeld M. G. (2006) A breaking strategy for topoisomerase IIβ/PARP-1-dependent regulated transcription. Cell Cycle 5, 2557–2560 [DOI] [PubMed] [Google Scholar]
- 69. Lin C., Yang L., Tanasa B., Hutt K., Ju B. G., Ohgi K., Zhang J., Rose D. W., Fu X. D., Glass C. K., Rosenfeld M. G. (2009) Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 139, 1069–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Perillo B., Ombra M. N., Bertoni A., Cuozzo C., Sacchetti S., Sasso A., Chiariotti L., Malorni A., Abbondanza C., Avvedimento E. V. (2008) DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 319, 202–206 [DOI] [PubMed] [Google Scholar]
- 71. Le May N., Mota-Fernandes D., Vélez-Cruz R., Iltis I., Biard D., Egly J. M. (2010) NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol. Cell 38, 54–66 [DOI] [PubMed] [Google Scholar]
- 72. Le May N., Fradin D., Iltis I., Bougnères P., Egly J. M. (2012) XPG and XPF endonucleases trigger chromatin looping and DNA demethylation for accurate expression of activated genes. Mol. Cell 47, 622–632 [DOI] [PubMed] [Google Scholar]