

1. Introduction
Post-translational modifications (PTMs) of histone proteins are a hallmark of epigenetic regulation. They provide a mechanism to modulate chromatin structure and constitute the main features of the so-called “histone code”.1 The proposed function of this code is to integrate exogenous and endogenous signals into a diverse set of histone PTM patterns to enable the epigenetic control of gene expression. The key regulators of this process are the so-called “writers” and “erasers”, which act by dynamically modifying histones, and other chromatin-associated proteins, as well as the “readers”, which interpret these PTMs, thereby facilitating the downstream activation or repression of gene expression.2
The writers are histone-modifying enzymes that can be grouped according to their amino acid substrate preference, affecting mainly lysine, arginine, and serine residues.3 These enzymes can be further classified according to the type of covalent modification that they catalyze. Histone modifications include acetylation, methylation, phosphorylation, and the more recently described modifications of citrullination, ubiquitination, SUMOylation, proline isomerization, O-GlcNAcylation, and ADP-ribosylation.1b,3 On the basis of detailed mass spectrometric analyses, there are at least 15 different types of covalent histone modifications,4 and since histone proteins are modified at multiple sites, and different stoichiometries, the total number of histone marks is >160.5 Although our understanding of how histone modifications contribute to the epigenetic control of gene transcription has grown immensely over the past ∼15 years, the precise impact of this vast number of modifications, not to mention the crosstalk between them, has yet to be fully realized.
Histone proteins are small, highly basic proteins consisting of a globular domain and flexible N-terminal and C-terminal tails that protrude from the nucleosome. The core histone proteins (histones H2A, H2B, H3, and H4) form an octameric particle consisting of two H2A–H2B dimers and an H3–H4 tetramer, around which wrap two helical turns of DNA (∼150 bp).6 This structure, which is generally termed a nucleosome, comprises the basic building block of higher order chromatin structures that are further organized through the function of linker histones such as histone H1. On the basis of nucleosome positioning studies, around 80% of the yeast genome and even 99% of the mappable genome of human granulocytes is occupied by nucleosomes, thereby highlighting the importance of nucleosome-packaged DNA for eukaryotic cells.7 Importantly, while histone PTMs are found throughout the entire protein, they are most often clustered within the N-terminal tail. Although research on histone lysine modifications has drawn considerable attention and even resulted in the approval of novel anticancer drugs,8 the modification of histone arginine residues is a recently emerging nucleosomal mark of similar importance (Figure 1).
Figure 1.
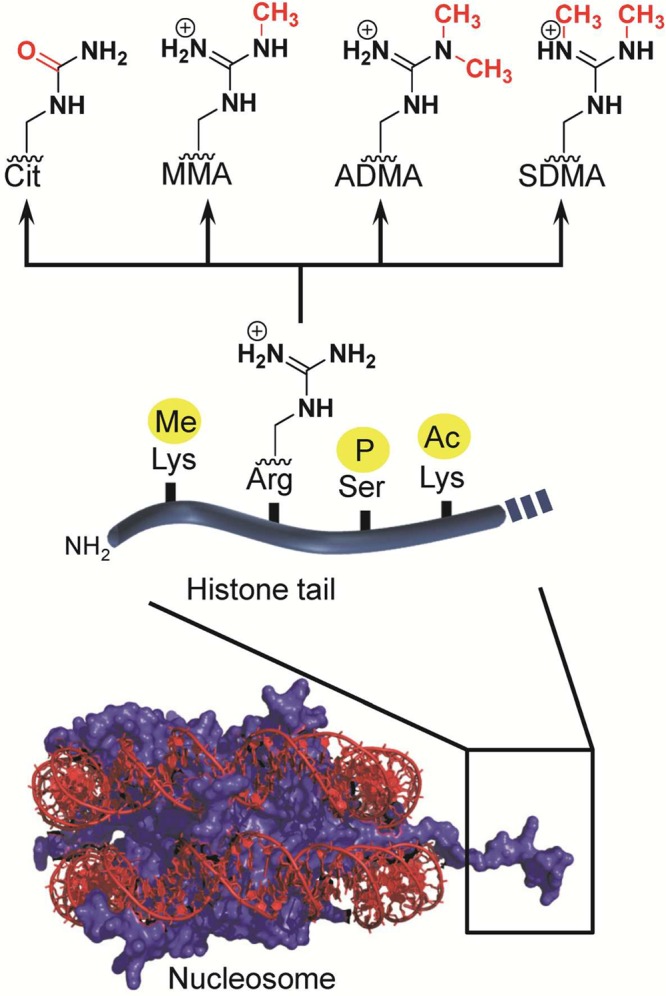
N-terminal tails of histone proteins are the preferred targets of histone-modifying enzymes. The major modifications of histone arginine residues are citrullination and methylation. Abbreviations: Cit, citrulline; MMA, monomethylarginine; ADMA, asymmetric dimethylarginine; SDMA, symmetric dimethylarginine.
Arginine residues possess a characteristic guanidinium-containing side chain that has one of the highest pKa values (pKa = 12.5; this value refers to the side chain of the free amino acid in aqueous solution at 25 °C) of all amino acids;9 thus, these residues are protonated and positively charged at physiological pH. The high pKa value also renders the guanidinium group a poor nucleophile and as such presents a considerable challenge for modifying this residue. In addition to its positive charge, the arginine guanidinium contains five potential hydrogen bond donors that can be used to interact with other polar groups (Figure 2). Notably, the strong charge favors the location of arginine residues to the outer hydrophilic surfaces of proteins. Consequently, they are readily accessible for binding to molecules with negative charges, e.g., nucleic acids. In this respect, arginines are one of the most common residues involved in the formation of protein/DNA and protein/RNA complexes.10 The frequent use of arginine for this type of interaction can be explained by opposite charge attraction, the length and flexibility of the side chain, and the ability to produce excellent hydrogen-bonding geometries with nucleobases or phosphate groups (Figure 2). For example, the orientation of the planar guanidinium nitrogen atoms perfectly matches with the oxygen atoms of phosphate (present in nucleic acids and phosphoproteins) to form a stable bidentate salt bridge that is at least 2-fold stronger than the interaction between a lysine ammonium group and a phosphoryl group.11 In addition, the possible bidentate hydrogen-bonding ability of arginine not only confers a more economical way to optimize bond energies but also increases specificity in the recognition of specific DNA sequences, as exemplified by the recognition of guanine (Figure 2).10a
Figure 2.

Bidendate interactions of the arginine guanidinium group exemplified by (i) the carboxyl group of aspartate (trypsin–peptide complex, PDB code 1OX1) (left), (ii) the phosphoryl group of phosphotyrosine (SH2–pTyr, PDB code 4F5B) (middle),227 and (iii) atoms O6 and N7 of guanine (p53–DNA, PDB code 3TS8) (right).228
Given the ability to form these types of interactions, it is clear that the post-translational modification of an arginine residue could have dramatic effects on cell signaling and, like other histone modifications, contribute to disease pathogenesis. In fact, there is increasing experimental evidence to suggest that the dysregulation of arginine-modifying enzymes plays pivotal roles in cancer, inflammatory diseases, neurodegenerative diseases, and other conditions.8b,12 In this review, we aim to summarize the current knowledge surrounding the post-translational modification of histone arginine residues, focusing on enzyme classes that catalyze the citrullination and methylation of arginine residues as well as noncanonical arginine modifications such as phosphorylation, ADP-ribosylation, and arginylation. The major focus will be given to the PADs (protein arginine deiminases) and PRMTs (protein arginine methyltransferases).
2. Histone Citrullination (Arginine Deimination)
2.1. Overview of Protein Citrullination
Protein citrullination is an emerging PTM that results from the conversion of peptidyl arginine to peptidyl citrulline. This PTM is catalyzed by the calcium-regulated PAD family of enzymes (Figure 3).13 Due to the exchange of an imine for a carbonyl group, this reaction is referred to as deimination. The PAD-catalyzed hydrolysis of a guanidinium group has a profound effect on the electrostatics and the hydrogen-bonding potential of the original side chain as citrulline is neutral and contains two hydrogen bond acceptor sites and only three potential hydrogen bond donors compared to the five present in arginine (Figure 4). Despite the large electronic effects, the overall change in mass is marginal: −0.02 Da, accounting for the extra proton in the charged guanidinium group, or +0.98 Da, as encountered during mass spectrometric analyses of the neutral guanidine form.
Figure 3.
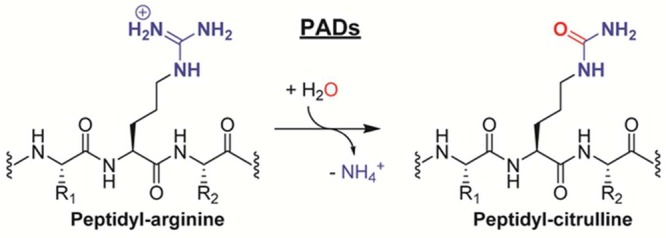
PAD enzymes catalyze the hydrolytic conversion of peptidyl arginine residues into citrulline.
Figure 4.

Electrostatic surface potential and hydrogen bond donor/acceptor sites of the side chains of arginine and citrulline. Hydrogen bond donor sites are highlighted in red, whereas hydrogen bond acceptor sites are depicted in blue. Cα denotes the α-carbon. Charge potentials were rendered by using SPARTAN (Wavefunction Inc.), with negative electrostatic charges shown in red, positive charges in blue, and neutral charges in green.
There are five human PAD isozymes, including PAD1, PAD2, PAD3, and PAD4, which are catalytically active, and PAD6, for which no activity has been detected.13,14 On the basis of the historic nomenclature, human PAD5 was thought to represent a novel PAD family member that differed from mouse PAD4.15 However, detailed sequence and expression analysis revealed that human PAD5 was the mouse PAD4 orthologue. As such, it was renamed PAD4, leaving PAD5 unused.13 The PADs share a high degree of sequence conservation (70–95% identity among each isozyme in different mammals and 50–55% identity between individual isozymes within one species) (Figure 5) and possess low pI values, typically around 5.8.13 The net negative charge is thought to be instrumental for recognizing the positive charge of a substrate arginine residue as well as for the binding of essential calcium ions (see below).
Figure 5.

Sequence alignment of human PAD family members. Catalytic residues are highlighted with red asterisks below the alignment. The sequence alignment was generated using Clustal Omega and visualized using Espript 3.0.229 The consensus sequence is abbreviated as follows: uppercase letters indicate identical residues, lowercase letters indicate consensus level >0.5, “!” represents any conserved residue of isoleucine (I) or valine (V), “$” represents any conserved residue of leucine (L) or methionine (M), “%” represents any conserved residue of phenylalanine (F) or tyrosine (Y), and “#” represents any conserved residue of asparagine (N), aspartate (D), glutamine (Q), or glutamate (E). The relative accessibility of each residue is depicted below the consensus motif: blue indicates accessible residues, cyan marks intermediately accessible residues, white stands for buried residues, and red indicates that the accessibility is not predicted.
The PADs are widely distributed in higher eukaryotes, and their expression as well as activity is associated with the regulation of gene expression and various developmental stages (see below).13 PAD1 is highly expressed in the epidermis and uterus, PAD2 is expressed in numerous tissues, PAD3 is mainly found in the skin and hair follicles, PAD4 is primarily expressed in granulocytes, macrophages, and neutrophils, and PAD6 is expressed in oocytes and embryos.15,16 Within the cell, PAD protein and/or activity are detected in various cellular compartments, including the cytoplasm, mitochondria, and nucleus.17 Although PAD4, which contains a canonical nuclear localization signal (NLS; P56PAKKKST63) was long thought to be the only nuclear PAD enzyme,17a emerging evidence indicates that other PAD isozymes can localize to the nucleus as well.17b For example, PAD2 was recently found in the nucleus of murine mammary epithelial cells, and in nuclear fractions of astrocytes as well as hippocampal neurons of scrapie-infected mice.17b,17c
2.2. Structure and Mechanism of the PADs
PADs (EC 3.5.3.15) belong to the guanidino-group-modifying branch of the pentein superfamily.18 They are calcium-regulated cysteine hydrolases that act on carbon–nitrogen bonds. Other members of this family include the amidinotransferases (ATs), which catalyze the transamidination reaction of nonpeptidyl arginine to glycine, thereby generating the creatine precursor guanidinoacetate, the arginine deiminases (ADIs), which act on nonpeptidyl arginine and the dimethylarginine dimethylaminohydrolase (DDAH) enzymes, which are highly selective for methylated nonpeptidyl arginine.18a In contrast to these enzyme groups, the PADs only act on peptidyl arginine residues and require at least an N-terminal amide bond and a C-terminal carbonyl for efficient substrate recognition.19 The PADs are highly efficient enzymes as exemplified by PAD1, which exhibits a rate enhancement over the noncatalyzed reaction (kcat/knon) under near-neutral pH conditions of ∼8.5 × 1011 (kcat = 0.45 s–1 versus knon = 5.3 × 10–13 s–1). In terms of catalytic proficiency (i.e., (kcat/KM)/knon), evaluated by comparing the second-order rate constant (kcat/KM = 4.1 × 103 M–1 s–1) with the rate of the spontaneous reaction in neutral solution in the absence of a catalyst (knon = 5.3 × 10–13 s–1), the rate enhancement is 8 × 1015 M–1 (Table 1).20
Table 1. Rate Constant for PAD1-Catalyzed (kcat at pH 7.6) and Noncatalyzed (knon at pH 7) Citrullination Reaction.
| type | rate constant | half-life |
|---|---|---|
| knon | 5.3 × 10–13 s–1 | ∼42000 years |
| kcat | 0.45 s–1 | 1.54 s |
2.2.1. PAD Structure
As with other members of the guanidino-group-modifying pentein family, PADs contain a catalytic α/β propeller domain that is located on its C-terminal half.18b,21 PADs typically exist as homodimeric proteins in solution and, according to crystallographic studies, bind in a head-to-tail fashion (Figure 6A).21 The dissociation constant (Kd) for PAD4 dimer formation was estimated to be ∼450 nM.22 Moreover, dimerization was shown to be important for activity and cooperativity, since disruption of the dimer interface reduces the activity by 50–75%.22 The dimerization interface covers a large surface area of ∼2000 Å2 comprising multiple contacts between the N-terminal domain of one protomer and the catalytic domain of the other protomer. Each subunit comprises two domains, including the N-terminal domain, which can be further subdivided into two immunoglobulin-like (Ig) subdomains that are proposed to be important for protein–protein interactions and to facilitate substrate selection.21 The C-terminal catalytic domain harbors all the active site residues, and the dimeric PAD4 structure revealed that both active site cavities are located on the same dimer face and are separated by ∼65 Å (Figure 6B).
Figure 6.
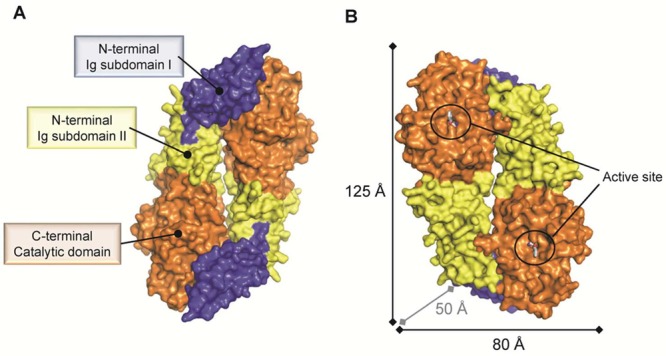
Surface representation of the dimeric PAD4 C645A mutant bound to the substrate BAA (PDB code 1WDA). (A) PAD4 exists as a head-to-tail dimeric protein that comprises three domains as indicated for one protomer. (B) The catalytic sites of both protomers are located on the same dimer face and are separated by ∼65 Å. Abbreviation: Ig subdomain, immunoglobulin-like subdomain.
2.2.2. Calcium Dependency of PAD Enzymes
The crystal structure of PAD4 further showed that each protomer contains two bound calcium ions (Ca1, Ca2) in the catalytic domain and three additional calcium ions (Ca3, Ca4, and Ca5) located in the Ig II subdomain (Figure 7). Although calcium binding is crucial for catalytic activity, it has no influence on dimer formation.21,22 Detailed comparisons of calcium-free and calcium-bound PAD4 and PAD2 indicate that calcium binding has a profound effect on the PAD structure, and is critical for forming a catalytically competent active site (Figure 8).21,23 Notably, loop regions comprising the active site cavity are largely disordered in the absence of calcium and only become visible (ordered) in the presence of calcium. Calcium ions Ca3, Ca4, and Ca5 bind to a conserved negatively charged region and are thought to stabilize the structure, whereas Ca2 and in particular Ca1 are positioned close to the bottom of the active site cleft and are thus essential for maintaining the architecture of the active site (Figure 8).21 This calcium-induced conformational change is also highlighted by the fact that C645 in PAD4 and C647 in PAD2, the nucleophilic cysteine residue, moves ≥5 Å into the active site when calcium binds to the enzyme. The organization of the active site cavity of calcium-bound, substrate-free PAD4 is almost identical to that of the substrate-bound complex, indicating that substrate binding has little effect on the formation of the active site.21 On the basis of calcium titration experiments with PAD2, which were monitored by X-ray crystallography, Ca1 and Ca6 bind first, and Ca3, Ca4, and Ca5 bind next and cause a conformational change that generates the Ca2 site. Calcium binding at Ca2 is critical for triggering the movement of the active site cysteine into a catalytically competent position. Despite Ca2 binding last, these titration experiments, coupled with mutagenesis and hydrogen/deuterium exchange experiments, indicate that Ca3, Ca4, and Ca5 act as a calcium switch to control the overall calcium dependence of the enzyme.
Figure 7.
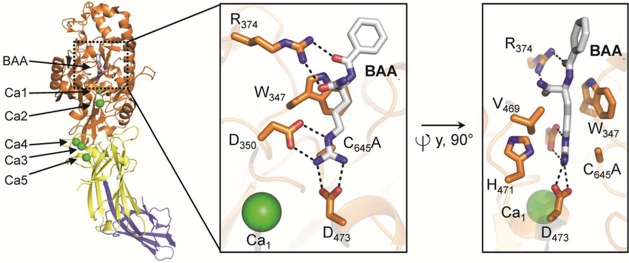
Domain organization and calcium-binding sites of the PAD4 C645A protomer bound to the substrate BAA (PDB code 1WDA). The structural elements are color coded according to Figure 6. The insets on the right depict two orientations of the PAD4 active site bound to BAA, highlighting critical residues for substrate binding and catalysis. Polar contacts of <3.5 Å are represented as dashed lines.
Figure 8.
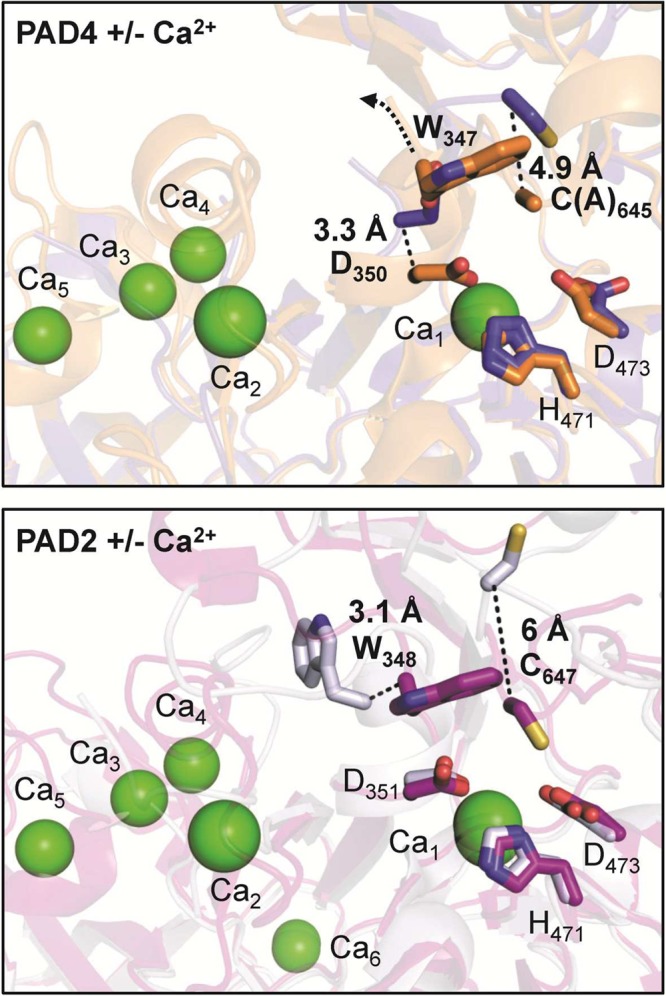
The image on top represents the structure of PAD4 without Ca2+ (blue) (PDB code 1WD8) superimposed onto the structure of PAD4 with Ca2+ (orange) (PDB code 1WD9). The image on the bottom depicts the structure of apoPAD2 without Ca2+ (gray) (PDB code 4N20) overlaid onto the structure of holoPAD2 F221/222A mutant with Ca2+ (purple) (PDB code 4N2C). The movements of active site residues (bold) are highlighted as dashed lines. Ca2+ ions are illustrated as green spheres, whereas active site residues are shown as sticks.
Notably, the metal dependency of PAD4 was tested using various cations, including barium, calcium, magnesium, manganese, samarium, strontium, and zinc.19 None of the examined metals except calcium could efficiently activate the catalytic activity of PAD4. Conversely, some of these metals were even decent PAD inhibitors. The most potent inhibition was accomplished using samarium (present as Sm3+ ion in samarium sulfate, Sm2(SO4)3) and zinc (present as Zn2+ ion in zinc chloride, ZnCl2), which possessed IC50 values of 40 and 750 μM, respectively. Thus, calcium binding is a critical prerequisite for proper substrate binding.
The strict calcium dependency of PAD activity is further reflected by the fact that calcium activates the enzyme by more than 10000-fold. Also notable is the fact that full PAD4 activity requires a high concentration of calcium and the half-saturation constant for calcium binding (K0.5,Ca) ranges from 130 to 710 μM depending on the substrate.19,22 How the PADs are activated in cells is, however, still unknown. For instance, the concentration of calcium required for maximal PAD4 activity is 100–1000-fold higher than that observed in activated cells. Therefore, it is currently unclear how PADs are efficiently activated in vivo and/or how the calcium dependency is lowered inside cells. It has been proposed that PADs may temporarily locate to intracellular calcium channels that can provide local calcium concentrations in the millimolar range upon channel opening,24 sufficient to activate PAD activity.23 In addition, one can speculate that the PADs’ calcium dependency may be altered by specific PTMs or via the interaction of binding proteins. In this regard, it was recently shown that antibodies, isolated from patients with rheumatoid arthritis, can bind and activate PAD4 by lowering the concentration of calcium required for activity.25
2.2.3. PAD Substrate Recognition
In contrast to other members of the guanidino-group-modifying pentein family such as ADI and DDAH, where peptidyl arginine is occluded from the solvent by a loop closed over the active site, PAD4 substrate binding occurs at the molecular surface of the enzyme, which is accessible for peptide/protein interactions.26 Thus, steric effects provide at least a partial explanation for why peptidyl arginine residues are recognized by PADs but not by ADI or DDAH.21
The active site of PAD4 has a characteristic U-shaped tunnel containing two entrance doors (Figure 9). This tunnel is also found in other hydrolases of the pentein superfamily such as DDAH.27 The “front door” is the actual substrate-binding site accommodating the arginine side chain residue and derived inhibitors, while the “back door” provides solvent access through a highly polar tunnel connected to the base of the active site. This narrow solvent channel presumably allows the ammonia generated by the hydrolysis of arginine to diffuse away and provide access for a water molecule for the subsequent hydrolytic phase of the reaction to occur.28 As a result, the access of other small molecules is restricted and thereby prevents their reaction with the S-alkylthiouronium intermediate during enzyme catalysis (see below), only allowing water to enter the active site. Interestingly, in related amidinotransferases that do not utilize a hydrolytic mechanism but transfer the activated amidine via a reactive cysteine thiouronium intermediate onto their respective substrate molecule such as glycine, the amidine-donor arginine substrate occupies a position similar to that of the back door solvent channel in PAD4.18c This observation raises the possibility that the solvent channel in PAD4 and related hydrolases is a vestigial substrate-binding site that is retained in amidinotransferases from a common promiscuous ancestor.18c Although the front door in PAD4 represents the major site of inhibitor binding, it has been proposed that the back door solvent channel might represent an alternative target for inhibitor development.29
Figure 9.

Top view of the PAD4 C645A mutant bound to BAA colored according to its electrostatic surface potential, highlighting two connected cavities that form a continuous tunnel (orange rod) of ∼21 Å (PDB code 1WDA). The lower image illustrates the side view of the active site cavity (front door), occupied by BAA, and the back door tunnel, presumably involved in incoming water channeling and ammonia (product) extrusion.
The structure of PAD4 bound to benzoyl-l-arginine amide (BAA), a small-molecule mimic of peptidyl arginine, illustrates key residues that are important for substrate recognition (Figure 7). For example, aspartates D473 and D350 form two bidentate salt bridges with the substrate guanidinium group, positioning it for nucleophilic attack by C645. Notably, C645 and H471, which are involved in general acid/base catalysis, are located on opposite sides of the guanidinium carbon center. The aliphatic portion of the substrate arginine side chain is clamped between W347 and V469 via hydrophobic interactions. W347, along with R372, also appears to be a key player in generating a catalytically competent active site since mutation of either residue decreases activity to near background levels.30 In the case of W347, this effect is best interpreted by the importance of this large hydrophobic side chain forming the wall of the substrate-binding pocket. Although R372 does not directly hydrogen bond to the substrate, except for a water-mediated interaction, it directly interacts with D345 and E351, which binds Ca2 and is close to the active site residue D350. Therefore, R372 is critical for maintaining the structural organization of the active site, but plays only a minor role in substrate binding.
As mentioned above, PAD4 is highly selective for peptidyl arginine substrates over nonpeptidyl arginine. This observation can be rationalized by the engagement of the peptide backbone. Specifically, the main chain carbonyl oxygen of the preceding N-terminal residue and the arginine backbone carbonyl oxygen atom form hydrogen bond interactions with the side chain of R374 (see Figure 7), thereby conferring specificity toward peptidyl arginine as opposed to free arginine, which lacks these amide bonds.21 Consistent with this residue being important for substrate recognition, mutation of R374 to alanine results in a ∼20–50-fold reduction in enzymatic activity.31 The crystal structures of PAD4 bound to several histone substrate peptides further confirmed this observation and revealed that the great majority of contacts occur between the backbone carbonyl groups of the peptide and the side chains of residues surrounding the active site, i.e., Q346, W347, R372, and R374 (Figure 10).31
Figure 10.

PAD4 (orange) bound to histone H3 peptide (gray) (PDB code 2DEW). Waters are depicted as red spheres. Adapted with permission from ref (31). Copyright (2006) National Academy of Sciences, U.S.A.
The lack of significant contacts between the enzyme and side chains of the substrate may explain why sequence-specific substrate recognition elements have been difficult to identify. While it was suggested that PAD4 recognizes five successive residues with the consensus sequence of ΦXRXX, where Φ denotes amino acids with small side chain moieties and X denotes any amino acid, it is evident from both the lack of strong sequence specificity and the available PAD4 crystal structures that there are no obvious substrate-specificity determinants apart from the arginine-binding site.31 These structures do, however, indicate that PAD4 peptide substrates adopt a β-turn-like conformation in which the peptide backbone is kinked to allow proper penetration of the arginine side chain into the deep active site cavity. Therefore, in contrast to most histone-modifying enzymes that interact extensively with their peptide ligands and recognize their substrates in a sequence-specific manner, the PADs likely modify exposed arginine residues that can adopt the type of β-turn-like structure described above.32 Thus, the enzyme has a fairly broad sequence specificity and could target multiple arginine sites in histones. However, specificity may be provided by controlling access of the enzyme to a limited subset of arginine residues in higher order chromatin structures, by crosstalk with other PTMs, or through cooperation of PAD4 with additional factors. In this respect, the Ig-like domains might also contribute to substrate selection. For example, the binding affinity between PAD4 and HDAC1 was decreased by 3.3-fold upon introduction of an autocitrullination-mimicking glutamine residue at position R123, while there was no effect on the PAD4–H3 interaction.30b
2.2.4. Proposed Catalytic Mechanism of PADs
On the basis of the available crystal structures and biochemical studies, the following catalytic mechanism has been proposed (Figure 11).21,33 Briefly, PAD4 catalyzes arginine citrullination by using a nucleophilic cysteine residue that forms a covalent reaction intermediate, which is hydrolyzed by an incoming water molecule. Substrate binding is initiated by strong electrostatic interactions between D350 and D473, the two active site aspartate residues that coordinate the guanidinium group. The carboxylate of D473 binds to both terminal ω-nitrogens of the guanidinium group, whereas D350 coordinates to one ω-nitrogen and the δ-nitrogen. Consequently, the thiolate of the active site cysteine, C645, is appropriately positioned to promote nucleophilic attack on the guanidinium carbon, which results in the formation of a covalent tetrahedral intermediate. H471, which is located on the opposite side of the guanidinium group, is thought to promote catalysis by protonating the tetrahedral intermediate, either concomitantly with nucleophilic attack by the active site cysteine or in a stepwise fashion, thereby generating a better leaving group and promoting cleavage of the scissile C–N bond. After the collapse of this reaction intermediate, ammonia is released. Thereafter, the covalent S-alkylthiouronium intermediate is subsequently hydrolyzed by the attack of an H471-activated water molecule, forming a second tetrahedral intermediate that collapses to eliminate the C645 thiolate, ultimately generating citrulline. Support for this mechanism comes from initial studies confirming that PADs are cysteine-dependent enzymes as evidenced by the strong inhibition afforded by iodoacetate or PCMB and the requirement of a reducing agent such as DTT.34 Detailed mutagenesis, pH rate profile, solvent isotope effect, and solvent viscosity effect studies also support this mechanism.33 In addition, several inhibitor-bound structures, i.e., F-amidine (N-alpha-Benzoyl-N5-(2-fluoro-1-Iminoethyl)-L-Ornithine Amide), Cl-amidine (N-alpha-Benzoyl-N5-(2-Chloro-1-Iminoethyl)-LOrnithine Amide), and TDFA (threonine-aspartate-F-amidine) (see below),30a,35 are consistent with the proposed mechanism.
Figure 11.

Proposed catalytic mechanism for PAD enzymes.
Notably, PAD4 has a pH optimum of ∼7.6 and uses a reverse protonation mechanism that is manifested by the high pKa of the active site cysteine C645 (pKa ≈ 8.3); this pKa value was measured by pH-dependent kinetic inactivation studies using the cysteine reactive compound iodoacetamide.33a In addition, H471 has to be protonated for efficient catalysis to take place. However, the pKa for H471 is ∼7.3 and is therefore below the pH optimum of the enzyme. Consequently, there is just a small pH window for optimal PAD4 activity, indicating that only a fraction of PAD4 (∼15%) exists in the proper deprotonated C645-thiolate and protonated H471-imidazolium forms at the pH optimum. Mechanistic studies on PAD1 and PAD3 have confirmed that they also proceed through a similar reverse protonation mechanism.33b Interestingly, however, PAD2 appears to use a substrate-assisted mechanism, in which the positively charged substrate guanidinium promotes catalysis by depressing the pKa of the nucleophilic cysteine.36 This conclusion was mainly based on pH-dependent inactivation studies by comparing the guanidinium group mimicking compound 2-chloroacetamidine with the neutral iodoacetamide. Consistent with PAD4, iodoacetamide inactivation revealed a pKa of 8.2 for the active site cysteine, whereas the use of 2-chloroacetamidine yielded a pKa value of 7.2 for the same cysteine. Therefore, it was proposed that PAD2, like DDAH,37 uses a substrate-assisted mechanism rather than a reverse-protonation mechanism.36
2.3. Do PADs Function as “Demethyliminases”?
There has been some controversy regarding whether the PADs act on methylated arginine residues. One study claimed that PAD4 catalyzes the conversion of monomethylarginine into citrulline.38 However, the “demethylimination” reaction occurs at rates that are several orders of magnitude (100–10000-fold) slower for peptide substrates containing either a single monomethyl or a single asymmetrically dimethylated arginine residue than the actual deimination reaction of unmodified arginine residues.19 Two further studies report that methylation of the guanidinium group even prevents and inhibits citrullination.39 Using synthetic peptides that contain methylated arginine residues, neither human PAD2, PAD3, PAD4, and PAD6 enzymes nor PADs present in mouse tissue extracts are capable of generating peptidyl citrulline or peptidyl methylcitrulline from either mono- or dimethylated peptidyl arginine.39b
Notably, structural comparison of the active site cavity of PAD4 with DDAH further revealed striking differences that might explain the individual substrate preferences (Figure 12A). DDAH is very selective for asymmetric dimethylated nonpeptidyl arginines to ensure that DDAH only hydrolyzes methylated arginine, which acts as a physiological inhibitor of nitric oxide synthase, while sparing unmodified arginine, which is the substrate for nitric oxide synthase.27b The active site residues in PAD4 and DDAH are highly conserved; however, PAD4 has a much smaller active site pocket, containing an aspartate at the bottom. This aspartate directly forms a bidentate hydrogen bond to the substrate guanidinium group, whereas DDAH possesses a lysine at the corresponding position that bends away from the substrate-binding site, thereby forming a larger active site pocket that can accommodate methylated arginine residues or even an iminopentyl group as illustrated in Figure 12A.27b,40 In the case of PAD4, a methylated guanidinium group would clash with the active site aspartate residues, thereby preventing proper substrate alignment (Figure 12B). Taken together, methylarginines are unlikely to represent physiologically relevant substrates, and it is more likely that arginine methylation antagonizes citrullination as proposed by Cuthbert et al. and Kearney et al.19,28,41 Notably, since citrulline residues are not methylated by the PRMTs, these two modifications are mutually exclusive.
Figure 12.

Comparison of PAD4 and DDAH active site pockets. (A) Active site of PAD4 with bound BAA (left side) (PDB code 1WDA), bovine DDAH with bound citrulline abbreviated as Cit (middle panel) (PDB code 2C6Z), and human DDAH with bound N5-(1-iminopentyl)-l-ornithine abbreviated as LN6 inhibitor (right side) (PDB code 3P8P). All protein structures are colored according to their electrostatic surface potential. (B) Bidendate recognition of the substrate arginine guanidinium group (gray) by the carboxyl groups of PAD4 (orange, PDB code 1WDA) D473 and D350, left panel. Methylation of the arginine guanidinium group would disfavor and preclude tight interactions with the aspartate residues, right panel.
2.4. Is Protein Citrullination a Reversible Modification?
Although many histone PTMs are reversibly regulated by the action of writers and erasers, there is currently no known citrulline eraser. However, the level of H3 citrullination is dynamically controlled, indicating that citrulline deposition is transient during gene expression.41 For example, PAD4 was shown to be recruited to the pS2 promoter and to citrullinate histone H3 when MCF-7 cells were stimulated with estrogen. Notably, this promoter region elicits a strong signal for H3 citrullination 40 min poststimulation; however, after an additional 10 min, the amount of H3 citrullination drops rapidly to its original levels, observed before estrogen stimulation, suggesting that a “decitrullinase” may exist.41 While the dynamic nature of citrulline marks could be caused by histone tail clipping, epitope occlusion, or nucleosome displacement,42 the existence of a decitrullinase remains a formal possibility.
Precedence for such a reaction comes from the urea cycle where nonpeptidyl citrulline is converted to free arginine by the combined actions of argininosuccinate synthetase and argininosuccinate lyase (Figure 13).43 Argininosuccinate synthetase catalyzes the conversion of l-citrulline into argininosuccinate (Figure 13A). The first step involves the activation of the urea oxygen of citrulline via covalent modification by an adenosine monophosphate (AMP) moiety.43b Subsequently, the α-amine of aspartate acts as the nucleophile and attacks the carbon center of the urea, thereby displacing the AMP moiety and generating argininosuccinate. Thus, argininosuccinate synthetase converts the neutral urea of citrulline into a guanidinium connected to succinate. To remove the succinate moiety, the second enzyme, argininosuccinate lyase, catalyzes C–N bond cleavage with the subsequent release of fumarate and arginine (Figure 13B). The catalytic mechanism proceeds through the deprotonation of the β-carbon of succinate, thereby forming a highly reactive carbanion intermediate.43a Redistribution of the negative charge into the carboxylate group generates the aci-carboxylate intermediate, which provides the driving force for the cleavage of the fumarate group. Protonation of the guanidinium by a general-acid catalyst further facilitates the reaction.
Figure 13.

(A) Active site of argininosuccinate synthetase with bound citrulline, aspartate, and ATP (PDB 1J1Z) and proposed mechanism. (B) Active site of the argininosuccinate lyase homologue δ crystalline T161D mutant with bound argininosuccinate highlighted in gray (PDB 1TJW) and proposed mechanism. Abbreviations: Cit, citrulline; Asp, aspartate; ATP, adenosine triphosphate; PPi, pyrophosphate; AMP, adenosine monophosphate, AS, argininosuccinate; H–B, general acid.
Whether a similar set of enzymes might act on peptidyl citrulline is unclear, but this mechanism provides the principle requirements for promoting a decitrullination reaction, i.e., covalent modification of the urea oxygen atom coupled to incorporation of ammonia, donated by an aspartate-, glutamate-, or glutamine-dependent enzyme. Thus, one could imagine a variety of alternative strategies to achieve the same outcome, including activation by phosphorylation to generate a better leaving group and cleavage of the phospho intermediate by ammonia.
2.5. Inhibitors and Chemical Probes of PADs
2.5.1. Reversible Inhibitors of PADs
In recent years, numerous PAD inhibitors have been developed (Figures 14–16). Initial studies focused on reversible inhibitors (Figure 14). For example, Taxol (the generic name is paclitaxel) inhibited PAD4 in the low millimolar range.44 The authors suggested that paclitaxel acts as a noncompetitive inhibitor of PAD4, since the KM was not affected while the Vmax was reduced. Given the absence of citrullination when testing methylated arginine residues as PAD4 substrates (see above), Hidaka and colleagues also determined whether these arginine derivatives can inhibit PAD4 activity.39a They observed that both benzoyl-Nω-monomethylarginine (Bz-MMA) and benzoyl-Nω,Nω-dimethylarginine (Bz-ADMA) inhibit the enzymatic activity of PAD4. However, these compounds are relatively modest inhibitors. The IC50 value of Bz-ADMA was estimated to be ∼400 μM, whereas the IC50 value for the weaker inhibitor Bz-MMA was not determined.
Figure 14.

Reversible PAD inhibitors. The presence of a guanidine group is highlighted in blue.
Figure 16.

(A) Covalent, irreversible inhibitors of the PADs. The presence of an amidine group is highlighted in blue. The potency toward the individual PAD isozymes is represented below the compounds. (B) Potential mechanisms of PAD inactivation by chloroacetamidine-based inhibitors.
Employing a PAD4-targeted activity-based protein profiling (ABPP) inhibitory screen (described below), Thompson and colleagues screened a small library of therapeutics used for the treatment of rheumatoid arthritis (RA).45 Given that streptomycin contains two guanidinium groups, the authors also considered the possibility that streptomycin might act as an alternative substrate; however, streptomycin was not deiminated by PAD4. In general, the potency of the tested compounds is relatively weak, ranging from low millimolar to mid-micromolar. Notably, streptomycin is a competitive inhibitor of PAD4, with a Ki value of ∼0.56 mM, and the tetracycline derivative minocycline was shown to be a mixed-type inhibitor with a Ki value of ∼0.63 mM. A similar compound, chlortetracycline, which only differs from minocycline by the addition of a hydroxyl and methyl group at position 6 and a chloro group replacing the dimethylamine moiety at position 7, is a significantly more potent inhibitor (Ki value of ∼0.11 mM). Kinetic studies further revealed that chlortetracycline is also a mixed inhibitor, similar to minocycline. Although the detailed mechanism of inhibition and the specific binding site of the tetracycline derivatives are currently unknown, the tetracycline scaffold might be exploited in the future design of reversible PAD4 inhibitors. Although it is tempting to speculate that the efficacy of the most effective PAD4 inhibitors, identified in this screen, minocycline and other tetracycline derivatives, is due in part to their ability to inhibit cellular PAD4, we note that it is not clear whether the high concentrations of compound needed to inhibit PAD4 in vitro could be achieved systemically. Additionally, these compounds are known to inhibit a wide range of other enzymes, including collagenase, poly-ADP-ribose polymerase-1 (PARP-1), Arachidonate 5-lipoxygenase, and several cysteine proteinases, that may also contribute to their efficacy as RA therapeutics and their ability to impair neutrophil chemotaxis and act as anti-inflammatory agents.
In addition to these compounds, the guanidine derivative 6 (Figure 14) was recently shown to inhibit PAD4 activity (8% inhibition at 1 μM and 36% inhibition at 10 μM).46 The authors claim that 6 most likely acts through a noncovalent mechanism to block the activity of PAD4; however, the detailed mode of action and inhibition studies were not performed by the authors. Ferretti and colleagues also recently described a novel PAD inhibitor (7) comprising a 3,5-dihydroimidazol-4-one ring that replaces the acyclic guanidine moiety present in arginine residues.47 This new small-molecule PAD3 inhibitor was reported to show inhibition at 100 nM using cell extracts containing recombinant PAD3. The high potency of this inhibitor is rather surprising since the pKa of acylguanidines is typically 4–5 orders of magnitude lower than that of the corresponding guanidines.48 Additionally, it is noteworthy that this strong level of inhibition has not been replicated in our hands.
More recently, an ABPP-based inhibitor screening strategy to identify inhibitors that target the calcium-free form of PAD2 identified ruthenium red (8) as a PAD2 inhibitor.49 This compound preferentially binds the apoenzyme with a Ki of 17 μM for PAD2 and was shown to be competitive with calcium, presumably binding at the calcium 3, 4, and 5 sites. Ruthenium red is also a potent inhibitor for the other PAD isozymes with apparent Ki values of 30 μM for PAD1, 25 μM for PAD3, and 10 μM for PAD4. The ability to identify inhibitors targeting the apo form of the PADs holds great promise for developing highly potent non-active-site-directed reversible inhibitors targeting the PADs.
Lewis and colleagues recently described a highly potent and reversible inhibitor that shows remarkable selectivity for PAD4.50 In this study, the authors screened a DNA-encoded small-molecule library for PAD4 inhibitors in the absence and presence of calcium. Optimization of the primary hits yielded GSK199 (9) and GSK484 (10) (Figure 15A). Notably, inhibition is calcium dependent for both compounds. In the absence of calcium, GSK199 and GSK484 inhibit PAD4 with IC50 values of 200 and 50 nM, respectively, while in the presence of calcium their potencies were reduced by ≥5-fold. Interestingly, detailed kinetic analysis demonstrated a mixed mode of inhibition for these compounds and showed that they possess more than 35-fold selectivity for PAD4 compared to the other PADs. The crystal structure of PAD4 bound to GSK199 revealed that the inhibitor directly interacts with the active site residues D473 and H471 (Figure 15B). Comparison of inhibitor- and substrate-bound PAD4 shows that an active site α-helical region, present in the substrate-bound form, adopts a new conformation and is reordered to form a β-hairpin in the inhibitor-bound structure (Figure 15C). In addition, detailed inspection of the orientation of GSK199 highlights a partial overlap between the aminopiperidine group of 9 with the substrate guanidinium group. However, in contrast to the substrate side chain that occupies the front door channel, the benzimidazole and pyrrolopyridine moieties of 9 protrude out into the back door solvent exchange channel (Figure 15C). Notably, these compounds bind a form of PAD4 that lacks calcium at the Ca2 site, mimicking our calcium titration data with PAD2, and again highlighting the importance of Ca2 for generating a catalytically competent conformation. Overall, these inhibitors represent a great example of a successful combination of high-throughput screening efforts with detailed biochemical and structural characterizations to yield novel compounds with potential therapeutic applications.
Figure 15.

(A) Reversible, mixed-type PAD4 inhibitors. Kis is the dissociation constant for the enzyme–inhibitor complex. (B) Crystal structure of PAD4 bound to inhibitor 9, GSK199 (PDB code 4X8G). GSK199 (gray) directly interacts with active site residues H471 and D473, and is further stabilized by binding to F634 and N588. Hydrogen bonds of <3.5 Å are represented as dashed black lines. (C) The image on the left side depicts the structure of PAD4 (orange) bound to inhibitor GSK199 (gray, PDB code 4X8G) superimposed onto the structure of PAD4 (green) bound to BAA substrate (green stick model, PDB code 1WDA). Residues 633–640 (red, denoted by α) of PAD4 bound to BAA adopt an α-helical conformation, while residues 633–645 (yellow, denoted by β) of PAD4 bound to GSK199 form an antiparallel β-sheet. The image on the right side compares the binding sites of BAA (green) and GSK199 (gray) mapped onto the structure of PAD4 (PDB code 1WDA), colored according to its electrostatic surface potential.
2.5.2. Irreversible, Covalent Inhibitors of PADs
Over the past several years, major progress has been made in generating irreversible inhibitors targeting the PADs (Figure 16A). Initial studies suggested that 2-chloroacetamidine, having a guanidinium-like amidinium group, represented a suitable candidate for PAD inhibition.51 In fact, 2-chloroacetamidine is a modest PAD4 inactivator, which blocks enzyme activity in a time-dependent manner, characteristic of a covalent inhibitor. Inspired by BAA, one of the best small-molecule PAD4 substrates, the Thompson group installed reactive electrophilic fluoroacetamidine or chloroacetamidine warheads onto the BAA scaffold.35a,52 The generated compounds, denoted as F-amidine or Cl-amidine, respectively, were the first highly potent PAD4 inhibitors. In the context of BAA, this haloacetamidine-based warhead is targeted to the active site of PAD4, where it reacts with C645 to form a stable thioether adduct. Indeed, in vitro studies revealed that both Cl-amidine and F-amidine act as mechanism-based inhibitors that irreversibly inactivate PAD4 and other PAD isozymes in a calcium-dependent manner via the specific modification of C645, the active site cysteine.35a,52
The alkylation of C645 proceeds through one of two potential mechanisms.53 In the first mechanism, C645 directly displaces the halide through an SN2 mechanism. Alternatively, inactivation could proceed via a multistep mechanism that involves nucleophilic attack of the cysteine thiolate on the amidinium carbon, forming a tetrahedral intermediate that mimics the initial tetrahedral intermediate formed during substrate hydrolysis. The protonation of the tetrahedral intermediate by H471, acting as a general acid, is thought to stabilize the rather unstable hemi-iminal tetrahedral intermediate such that it is long enough lived to undergo an intramolecular halide displacement reaction, which generates a three-membered sulfonium ring. Although the proposed dicationic intermediate depicted in Figure 16B is unprecedented in the literature, dianionic intermediates have been described.43a Regardless of the specific mechanism, formation of the three-membered sulfonium ring ultimately induces the collapse of the tetrahedral intermediate, leading to a 1,2-shift that generates a thioether linkage, whose existence has been verified crystallographically (Figure 17).35a Although, in principle, both mechanisms are plausible, the bell-shaped pH inactivation rate profiles observed for both F-amidine and Cl-amidine strongly support the second inactivation mechanism, especially the importance of H471 as a general-acid catalyst. Notably, the pH-dependent rate of inactivation correlates with the pKa values obtained for H471 and C645, indicating that these two residues likely possess a critical role not only for substrate turnover but also for enzyme inactivation by haloacetamidine-containing compounds.53 The second mechanism also accounts for the otherwise poor leaving group potential of the fluoride. Furthermore, crystal structures of PAD4 bound to several inhibitors (PDB codes 2DW5, 3B1T, 3B1U, and 4DKT) show that the histidine (H471) nitrogen atom Nδ1 directly points toward the Nω atom of the amidine inhibitor, as exemplified by the PAD4·F-amidine complex, where one can observe a 2.9 Å distance between Nδ1 from H471 and Nω from the amidine group of the inhibitor. More recently, however, a computational study of the PAD4 inactivation mechanism suggested that proton donation to the departing halide may, alternatively, account for the loss of reactivity at higher pH values.54 Discriminating between these two potential mechanisms will undoubtedly be the subject of future research.
Figure 17.
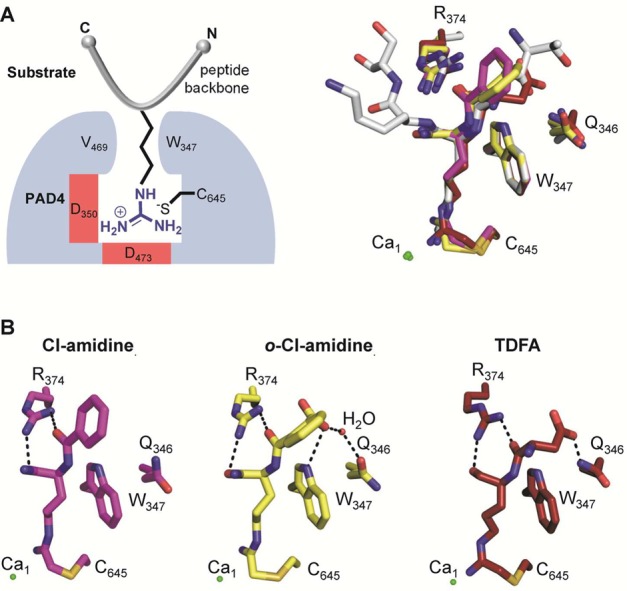
Crystal structures of PAD4 with inhibitors bound. (A) Schematic overview of PAD4 (blue) bound to a peptidyl substrate (gray) (left panel). Structural alignment of inhibitors bound to the active site of PAD4. The histone H3 peptide (sequence TARKS) bound to PAD4 (gray) (PDB code 2DEW) is included to compare the substrate-binding site. The Cl-amidine (magenta) (PDB code 2DW5), o-Cl-amidine (yellow) (PDB code 3B1T), and TDFA (red) (PDB code 4DKT) structures are aligned accordingly, including the depiction of critical PAD4-interacting residues. (B) Close-up view of PAD4 with bound inhibitors, highlighting critical inhibitor backbone interactions with PAD4 residues shown as dashed lines. Interactions between the amidine group as well as the α-amine of the inhibitor and PAD4 are omitted for clarity.
Cl-amidine and 2-chloroacetamidine possess similar maximal rates of inactivation (i.e., kinact). Cl-amidine, however, is a far more potent inhibitor due to increased binding energy. Thus, selective enzyme inactivation is driven in part by the affinity of the enzyme for the inhibitor. Further exploration of the Cl-amidine scaffold resulted in the identification of more potent PAD inactivators, such as o-carboxyl-Cl-amidine (14).30a Structural analysis of this compound bound to PAD4 revealed that the o-carboxylate forms a direct hydrogen bond with the indole NH of W347 and a water-mediated hydrogen bond with the side chain of Q346, which might explain the enhanced potency of 14 compared to Cl-amidine (Figure 17B).30a Additional selectivity studies revealed that 14 preferentially inactivates PAD1. The selectivity for PAD1 inhibition is 8-, 10-, and 3-fold higher than that obtained for PAD2, PAD3, and PAD4, respectively.30a
Using a solid-phase peptide library approach, the Thompson group also identified a novel PAD4-selective inhibitor that consists of a tripeptide comprising threonine, aspartate, and the warhead-containing F-amidine residue (TDFA).35b TDFA is highly selective for PAD4 (up to 65-fold) with excellent in vivo potency.35b The crystal structure of TDFA bound to PAD4 further revealed that the carboxylate group from the TDFA aspartate residue directly interacts with the amide nitrogen of the glutamine residue Q346 of PAD4 (Figure 17 B). Interestingly, the TDFA carboxylate adopts a position similar to that of the o-carboxylate group in o-Cl-amidine as well as the carbonyl oxygen of T7 in the H3 substrate (Figures 10 and 17), but does not directly hydrogen bond with the side chain of W347.35b The negative charge of the carboxylate might further enhance inhibitor binding through long-range electrostatic interactions with residues R374 and R639.35b
Despite the availability of several PAD inhibitors, Cl-amidine is still the most widely used compound and serves as a benchmark to estimate the potency of novel inhibitors. Although Cl-amidine was shown to reduce protein citrullination in cell and animal studies, and ameliorate disease severity in several animal models (see below), there are still several obstacles remaining to be solved before its potential clinical use, including a short in vivo half-life, poor bioavailability, and, because Cl-amidine is an irreversible inhibitor, the potential for off-target effects.55 Therefore, efforts have been undertaken to generate more stable Cl-amidine derivatives that resist proteolysis in vivo. To this end, the d-amino acid derivative of Cl-amidine (d-Cl-amidine) has been synthesized.55d-Cl-amidine is slightly less potent in vitro and preferentially inactivates PAD1. The inhibition of PAD4 by d-Cl-amidine is consistent with the observation that PADs are also active on d-arginine derivatives at a rate that is ∼5-fold weaker than that for the l-isomer.56 However, d-Cl-amidine is equally potent in cells as compared to l-Cl-amidine and exhibits better pharmacokinetics, presumably due to decreased proteolysis.55
Since Cl-amidine is a highly hydrophilic compound that readily dissolves in water, Wang and colleagues tried to increase the hydrophobicity and thereby bioavailability of Cl-amidine.57 Specifically, they synthesized a diverse panel of molecules, all comprising the Cl-amidine scaffold, flanked by alternative hydrophobic groups. The most potent inhibitor (16) contained a Cα-amide-methylbenzene as well as an Nα-amide-dimethylnaphthylamine moiety attached to the regular Cl-amidine scaffold. Compound 16 showed similar in vitro inhibition values (IC50 = 1–5 μM) compared to Cl-amidine (IC50 = 5 μM); however, inhibition of cellular proliferation was increased by ∼50-fold, most likely due to an improvement in cell permeability.57
Another interesting Cl-amidine derivative is BB-Cl-amidine (17), which contains a C-terminal benzimidazole and an N-terminal biphenyl moiety. The increased hydrophobicity of this compound also improves its cellular potency, bioavailability, and in vivo half-life.58 BB-Cl-amidine exhibits similar in vitro potencies and selectivities compared to Cl-amidine. However, the cellular potency of BB-Cl-amidine is increased by more than 20-fold; the EC50 value is 8.8 μM versus >200 μM for Cl-amidine when tested against U2OS osteosarcoma cells, a PAD4-expressing cell line.
Projecting forward, with the exception of TDFA and GSK199, most of the currently available compounds are pan PAD inhibitors that block all of the active PAD isozymes with similar potencies. Thus, the identification of isozyme-selective PAD inhibitors remains of crucial importance, and will facilitate the discovery of the individual contributions of PAD isozymes to both cellular physiology and disease.
2.5.3. Chemical Probes for the PADs
Given the high potency and ability to irreversibly modify PAD4, as well as the fact that F-amidine and Cl-amidine selectively modify the active, calcium-bound, form of PAD4, these compounds were adapted for use as ABPP reagents. The first synthesized PAD-selective ABPP was rhodamine-conjugated F-amidine (RFA, 19; Figure 18A).59 This compound retains the F-amidine scaffold but is linked to a fluorescent reporter tag (rhodamine) via a p-benzylic triazole group. Preliminary studies confirmed that this fluorescently tagged PAD4-targeted ABPP preferentially labels the calcium-bound, active, form of PAD4 and does not modify a C645S mutant.59 The probe shows potency equal to that of the non-reporter-tagged F-amidine, indicating that the reporter group does not interfere with enzyme binding. In addition, a biotinylated version of F-amidine (BFA, 20) was synthesized to isolate endogenous PADs.60 The BFA probe further contains a TEV (tobacco etch virus) cleavage site to release bound PAD4 under gentle conditions. This probe was used to coisolate PAD4-interacting proteins from MCF7 cells. While the probe selectively targets endogenous PADs, it was also able to copurify several known PAD4-associated proteins, including p53, HDAC1, and histone H3, indicating that this approach might be used to identify novel PAD4-binding proteins.60 Therefore, these PAD-specific ABPPs represent valuable tools that can be used to label PAD4 in cells, as well as to enrich active PAD4- and PAD4-interacting proteins.
Figure 18.

ABPP probes for PADs. (A) Structures of PAD-selective probes. The amidine group is highlighted in blue, whereas the reporter tags (rhodamine or biotin) are marked in red. (B) Schematic overview of fluorescence polarization assay using the PAD4-specific RFA probe.
In addition to their potential use in targeted proteomic studies and activity-based protein profiling applications, these probes have been used as the basis for developing a number of inhibitor screening platforms. To this end, the Thompson group developed a screening assay that relies on RFA to identify novel PAD inhibitors from diverse chemical libraries in a gel-based format.45 RFA can also be used to measure changes in PAD activity as a function of added inhibitor in a plate-based assay that is compatible with large compound libraries used in high-throughput screening (HTS) approaches by monitoring the changes in fluorescence polarization evoked by probe labeling of the enzyme (Figure 18B). The basis of this assay comes from the fact that when the fluorescent group is excited with polarized light, the RFA–PAD4 complex will rotate slowly and therefore emit highly polarized light. Conversely, free RFA rotates faster and emits nonpolarized light. If an inhibitor is bound to PAD4, it will compete with RFA for enzyme interaction, thereby yielding a low fluorescence polarization signal. Using this fluorescence polarization activity-based protein profiling (fluopol-ABPP)-based HTS assay, the Thompson group screened the NIH validation set, comprising 2000 compounds, and identified streptonigrin (18) as a potent and selective (>35-fold selective for PAD4) PAD4 inactivator.61 This compound shows time-dependent enzyme inactivation and acts as an irreversible PAD4 inhibitor (kinact/KI = 4.4 × 105 min–1 M–1). The detailed mode of inactivation is still, however, unknown.62 In addition to its in vitro activity, streptonigrin also inhibits the histone citrullination activity of PAD4 in HL-60 granulocytes and MCF7 cells.61 Unfortunately, however, streptonigrin has a number of off targets leading to pleiotropic effects on cell viability and signaling, thereby limiting its utility as a probe of PAD4 activity.
More recently, the Thompson group adapted this fluopol-ABPP-based HTS approach to identify PAD2 inhibitors. Here, the authors hypothesized that by lowering the concentration of calcium in the reaction mixture they might identify compounds that specifically bind to the apo, calcium-free form of the enzyme. Using this biased assay format, Lewallen et al. screened the LOPAC collection of pharmacologically active compounds and identified ruthenium red as the first calcium competitive inhibitor for the PADs.49 Although this compound shows limited utility as a cellular probe of PAD activity, its discovery does demonstrate that it is possible to identify potent non-active-site-targeted reversible PAD inhibitors.
2.6. Physiological Roles of Histone Citrullination
2.6.1. Epigenetic Effects of Histone Citrullination
Currently, the known sites of histone citrullination have been mapped to H2AR3,63 H3R2, H3R8, and H3R17,41 H3R26,41,64 and H4R338 (Figure 19). In addition, H4R17, H4R19, and H4R23 can be citrullinated by PAD4 in vitro but have not been found to be a target of citrullination in vivo.31 Histone citrullination is associated with both transcriptional repression and activation.38,41,64 For example, the citrullination of H3R17 by PAD4 at the estrogen receptor α (ERα)-regulated pS2 promoter was shown to correlate with transcriptional repression by interfering with activating, PRMT4-mediated, arginine methylation events.38,41 Moreover, p53-dependent recruitment of PAD4 to the p21 promoter resulted in citrullination of histone H3 and inhibition of gene transcription.65 The function of PAD4 as a p53 corepressor is further enhanced by direct interaction between the histone deacetylase HDAC2, which also represses p53 target genes, and PAD4.66 Exposure to genotoxic stress induced the release of PAD4 from the p21 promoter, which is subsequently derepressed by activating methylation of histone H3R17 by PRMT4.65 PAD4 also mediates histone H3 citrullination on the promoter of the OKL38 gene, thereby repressing the expression of this pro-apoptotic tumor suppressor gene.67 However, following DNA damage, increased p53 binding and histone arginine methylation, as well as a decrease in histone citrullination on the OKL38 promoter, accompany the activation of OKL38, suggesting a direct role of PAD4 and p53 in the expression of OKL38.67
Figure 19.
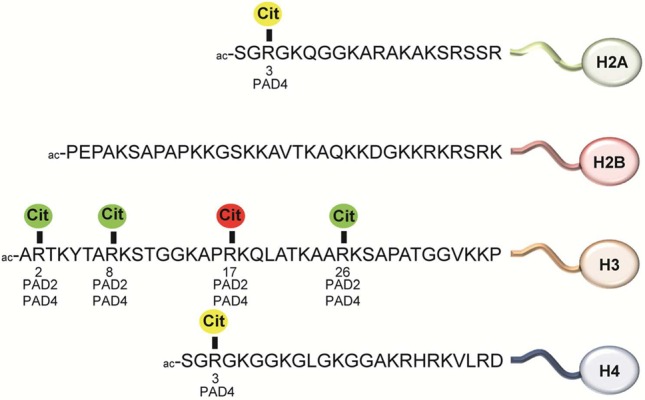
Citrullination sites in histone proteins. Color code: green, gene activation; red, gene repression; yellow, gene activation or repression, or unknown.
Citrullination of H3R8 following estrogen-induced activation of PAD4 also correlates with target gene activation by abolishing the H3K9me3-directed recruitment of HP1α to ERα-dependent promoters.68 HP1α is a chromatin-binding protein that specifically interacts with the H3K9me3 mark to repress gene expression by inducing a heterochromatin-like state that is refractory to high-level transcription. It was further shown that methylation of H3R8 slightly reduces the binding of the transcriptional repressor HP1α to H3K9me3.68 This example of histone PTM crosstalk raises the interesting possibility that differences in HP1α-binding affinity to H3K9me3 are caused by H3R8 methylation or citrullination, thereby regulating the gradual activation of HP1α target genes upon estrogen stimulation.
Increased citrullination at H3R8 in peripheral blood mononuclear cells also results in the activation of downstream genes such as the cytokines TNFα and IL8, which ultimately leads to inappropriate T-lymphocyte activation and uncontrolled immune response in multiple sclerosis.68 More recently, PAD4 was shown to interact with TAL1, a transcription factor that is essential for the generation of embryonic hematopoietic stem cells.69 There, it was demonstrated that TAL1-bound PAD4 acts as an epigenetic coactivator by competing with PRMT6-mediated methylation of the repressive H3R2me2a mark and thus increases IL6ST expression. Alternatively, TAL1-recruited PAD4 can function as a corepressor by counteracting the activating H3R17me2a mark by PRMT4, thereby inhibiting the expression of the CTCF-encoding gene.
More recently, it was shown that PAD4 citrullinates a single arginine residue, H1R54, within the DNA-binding site of histone H1. Modification of this residue results in H1 displacement from chromatin, thereby inducing global chromatin decondensation in pluripotent cells.70 It was also shown that PAD4 is expressed and active in murine embryonic stem (ES) cells as well as reprogrammed induced pluripotent stem (iPS) cells. Interestingly, the expression of PAD1, PAD2, and PAD3, but not that of PAD6, is also observed in pluripotent cells, indicating a potential function for these PADs in pluripotency or cell differentiation as well.70 In these cells, PAD4 plays a critical role in the pluripotency transcriptional network by enhancing the expression of genes involved in stem cell development and maintenance that can be inhibited by addition of Cl-amidine. For example, PAD4 binds to the promoter region of key stem cell genes such as Klf2, Tcl1, Tcfap2c, Kit, and Nanog, thereby activating their expression.70 Chromatin immunoprecipitation–quantitative polymerase chain reaction (ChIP–qPCR) analyses revealed that the association of H1 with chromatin at the regulatory regions of Tcl1 and Nanog is low in pluripotent cells; however, upon Pad4 knockdown, it is significantly enhanced. Moreover, mutation of H1R54 to alanine impairs its interaction with nucleosomes, supporting the critical role of this residue in nucleosome binding. The citrullination of H1 induces chromatin decompaction and may enhance the accessibility of RNA polymerase, transcription factors, and further histone-modifying enzymes. The PAD4-induced open chromatin architecture is also important for stem cell pluripotency during early mouse embryogenesis and can be impaired by Cl-amidine and TDFA treatment.70 The overexpression of PADs in multiple cancers (see below) might induce a similar stem-cell-like state, containing decondensed chromatin, and thereby promote uncontrolled cell growth.71 Interestingly, Dwivedi and colleagues recently observed that the citrullination of histone H1 at arginine 54 is also critical for neutrophil extracellular trap (NET) formation (see below) and represents an autoantibody epitope in sera from patients with systemic lupus erythematosus and Sjögren’s syndrome.72
PAD4 was long thought to be the only nuclear PAD and as such was assumed to be responsible for all nuclear histone citrullination events. Recent studies, however, indicate that this is not the case and that PAD2 is also capable of citrullinating histones.17b,64 For example, PAD2 expression was shown to be upregulated by epidermal growth factor (EGF) stimulation in mammary epithelial cells.17b There, nuclear PAD2 citrullinates histone H3 at R2, R8, and R17. It was proposed that PAD2-induced histone citrullination may play a regulatory role in the expression of lactation-related genes during the diestrus phase of the estrous cycle.17b
Moreover, citrullination of H3R26 by PAD2 at ERα target genes has been linked to transcriptional activation of more than 200 genes.64 The presence of H3R26 Cit destabilizes the nucleosome structure to allow for efficient ER binding to nucleosomal DNA.73 The altered nucleosome structure directly correlates with estradiol administration. Hence, it was proposed that, following estradiol exposure, ER directly or indirectly recruits PAD2 to ER target genes where PAD2 then citrullinates H3R26. The citrullinated H3 was postulated to induce an altered conformation of the nucleosome, manifested by core nucleosome particle protection size shifts from 149 to 125 bp upon H3R26 deimination, which allows for a more stable interaction between ER and its nucleosomal ER-binding sites.73 Interestingly, this arginine residue can also be methylated, and these two modifications are inversely correlated.41 It is also interesting to note that citrullination at H3R26 strongly colocalizes with H3K27 acetylation in MCF-7 cells, thereby raising the possibility for crosstalk between these two modifications.64
2.6.2. Epigenetic Effects of Nonhistone Citrullination
Apart from direct histone citrullination, PADs can also act as direct coactivators of specific transcription factors to induce other histone modifications that affect gene expression. As such, it was shown that PAD4 associates with several transcriptionally active promoters and functions as an activator of c-Fos via a mechanism that involves facilitated phosphorylation of the ETS-domain protein Elk-1.74 EGF-induced activation of PAD4 results in the direct targeting of Elk-1 for citrullination, thereby increasing ERK-mediated-phosphorylation-induced activation of Elk-1. Activated Elk-1 exhibits enhanced association with the histone acetyltransferase p300, which ultimately induces histone H4K5 acetylation and concomitant increased gene transcription.
In addition to the citrullination of histones and transcription factors, the PADs citrullinate a number of other proteins, including themselves. For example, PAD4 is known to autocitrullinate at numerous sites in vitro and in vivo.30b,75 There have been some conflicting observations regarding the functional impact of PAD4 autocitrullination. One report claimed that autocitrullination reduces PAD4 activity.75 By contrast, another study did not detect any significant influence on catalytic activity.30b In addition, it was shown that PAD4 autocitrullination alters protein–protein interactions and is thought to weaken the interaction between PAD4 and citrullinated H3 as well as PRMT1 and the histone deacetylase HDAC1.30b Autocitrullination of PAD4 was also proposed to destabilize a corepressor complex consisting of PAD4 and HDAC1, thereby providing a potential mechanism for decreasing the corepressor activity of this complex.30b
2.6.3. Nonepigenetic Effects of Histone Citrullination
It was postulated that PAD4-mediated citrullination at H4R3 may represent an “apoptotic histone code” to detect damaged cells and induce nuclear fragmentation.76 DNA damage induces PAD4 expression and the concomitant citrullination of various proteins.77 In this respect, PAD4 was shown to citrullinate H4 at arginine 3 and that this activity was blocked by small interfering RNAs (siRNAs) against p53 or PAD4.76 In addition, the presence of H4R3 Cit correlates with the level of apoptosis induction in DNA-damage-exposed U2OS cells. Citrullination of H4R3 was proposed to enhance accessibility of nucleosomal DNA, thereby promoting its apoptotic fragmentation.76 Moreover, PAD4 was shown to citrullinate the nuclear lamina protein lamin C, suggesting the involvement of lamin C citrullination in nuclear fragmentation during apoptosis.76
Histone citrullination is also implicated in innate immunity, where PAD4-mediated histone citrullination is involved in the formation of NETs (Figure 20).78 NETs, which were first identified in 2004 by the Zychlinsky group, are composed of nuclear DNA and associated proteins that are ejected by neutrophils in response to an infection.79 Although the physiological function of NET formation is incompletely understood, NETs are thought to function as a pro-inflammatory form of cell death, known as NETosis, that links the innate and adaptive immune responses. Specifically, NETs are formed in response to a number of stimuli of both bacterial and human host origin that trigger the release of chromatin to form a weblike structure that can trap pathogens and prevent them from spreading throughout the body.
Figure 20.
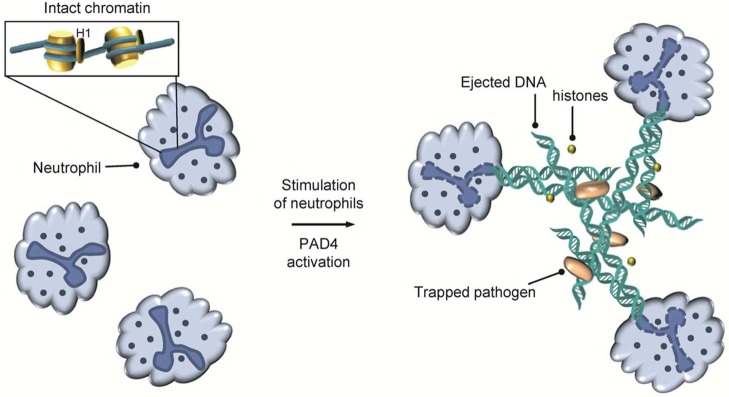
NET formation in neutrophils.
NETs also increase the local concentration of coextruded antimicrobial agents, including histones, myeloperoxidase (MPO), and proteases. Notably, several decades earlier, it had already been shown that histones, in particular H3 and H4, possess bactericidal activity,80 thereby providing at least a partial explanation for the release of histones during NETosis. NET-forming stimuli include lipopolysaccharide (LPS), N-formyl-methionine-leucine-phenylalanine (f-MLP), lipoteichoic acid (LTA), tumor necrosis factor (TNF), interleukin-8 (IL-8), and hydrogen peroxide.78b,78d Although the specific cellular pathways that trigger NET formation are an area of intense investigation, they are currently incompletely understood. Nonetheless, there appears to be a requirement for the generation of reactive oxygen species (ROS), since neutrophils from patients with chronic granulomatous disease, which is due to mutations in the ROS-generating enzyme nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, do not form NETs. In addition, PAD4 activity is a prerequisite and likely the terminal point in this signal transduction cascade because genetic deletion or chemical inhibition of PAD4 results in mouse neutrophils that are unable to citrullinate histones and do not form NETs.78a,78c,81 Thus, PAD4 is a crucial component of the innate immune system in mammals.
2.7. Histone Citrullination in Disease
Dysregulated PAD expression and aberrant protein citrullination have been implicated in numerous human diseases as summarized in several excellent reviews.13,82 Here, we focus on diseases where a direct link between histone citrullination and disease has been established by discussing the involvement of histone citrullination in cancer and inflammatory diseases.
2.7.1. Histone Citrullination in Cancer
Histone modifications play an important role in tumor development and cancer.83 In this respect, PAD4 is overexpressed in various malignant tumor tissues, including osteosarcoma, several adenocarcinomas affecting the colon, esophagus, ovaries, pancreas, and stomach, and carcinomas of the breast, bladder, endometrium, and liver, suggesting that it might be involved in tumors derived from multiple tissue origins.84 It was proposed that aberrant histone modifications can induce tumor suppressor gene silencing, thereby promoting tumorigenesis.83b A key regulator of cell cycle arrest, and programmed cell death, is the tumor suppressor p53, which is mutated in about half of cancers and thereby represents the most frequently altered gene in human cancers.85 p53 is a DNA-binding protein that is responsible for the integration of diverse signals, such as starvation, DNA damage, and various stress signals, by regulating numerous downstream genes that help to cope with stress and control cell fate. As described above, PAD4 functions as a corepressor of p53 to repress its downstream tumor suppressor genes such as p21, GADD45, and PUMA.65,66 Notably, both PAD4 knockdown and inhibition with Cl-amidine result in increased expression of these p53 target genes and increased cell death.65,66 In addition, in osteosarcoma U2OS cancer cells, PAD4 represses the expression of the p53 target gene SESN2, which encodes an upstream inhibitor of the mammalian target of the rapamycin complex 1 (mTORC1) signaling pathway to regulate cellular autophagy.57 Consistently, the PAD inhibitor YW3-56 (14) induces autophagy in U2OS cells.57 In summary, PAD4 might induce tumorigenesis through multiple mechanisms, including chromatin decondensation resulting in a stem-cell-like state, inhibition of tumor suppressor genes by corepressing p53 target genes, and inhibition of autophagy and the concomitant increase in protein synthesis and cell growth.
Interestingly, Tanikawa and colleagues observed that the expression of PAD4 is activated by p53, which binds to the p53-response element p53BS-A in intron 1 of the PAD4 gene. These data indicate that PAD4 may form part of a negative feedback loop to regulate p53 activity.76,77 These data further imply that aberrant histone citrullination caused by dysregulated PAD enzyme activity is actively involved in cancer progression. Given the close link between PADs and cancer, PAD4 represents a suitable target for cancer drug development. In this respect, the PAD inhibitors Cl- and F-amidine both exhibit cytotoxic effects toward several cancerous cell lines such as HL-60, MCF7, and HT-2, whereas no effect was observed in noncancerous lines such as NIH 3T3 and HL-60 granulocytes.86 In addition, these PAD inactivators also potentiate the cytotoxicity of the commonly used anticancer drug doxorubicin.86 Moreover, the Coonrod group showed that the level and activity of PAD2 increase during the transition of normal mammary epithelium to fully malignant breast carcinomas and coincides with HER2/ERBB2 upregulation.87 In addition, when treating MCF10DCIS monolayer carcinoma cells with Cl-amidine, they observed a strong suppression of cell growth in culture, which is induced by cell cycle arrest in the S-phase, followed by apoptosis. Moreover, administration of Cl-amidine to mice containing MCF10DCIS-injected tumor xenografts, a preclinical model of breast cancer, suppresses tumor growth.87
Although high-level PAD expression and histone citrullination are generally considered to be characteristic features of cancer cell proliferation, recently, however, the Coonrod group identified a strong correlation between high PAD2 expression and H3R26 Cit with increased survival in estrogen receptor positive (ER+) tumor patients.73 The authors further suggest that histone citrullination might be a critical prognostic for ER+ tumor development and is thus suited to stratify ER+ tumors into clinically relevant subsets. This observation also raises the intriguing question of whether histone citrullination may act as either a tumor promoter or a suppressor mark in a context and or isozyme-dependent manner. Therefore, further studies are necessary to clarify the detailed roles of individual PADs during tumor development and to understand whether they function as tumor suppressors or oncogenes.
2.7.2. Histone Citrullination in Inflammatory Diseases
Altered histone citrullination is also observed in a range of inflammatory diseases such as RA, lupus, ulcerative colitis, Alzheimer’s disease, and multiple sclerosis.82d Despite their great diversity, a common molecular feature of at least a subset of these diseases is aberrantly upregulated NET formation due to the presence of activated immune cells. As such, progression of these inflammatory diseases may result from the inappropriate and exaggerated induction of NET formation. For example, neutrophils obtained from RA patients are more likely than control neutrophils to spontaneously release NETs.78e Lupus neutrophils possess a similar phenotype, and it is notable that hallmarks of RA and lupus include autoantibodies that bind specifically to citrullinated proteins (RA) and double-stranded DNA (lupus), which are both released from neutrophils during NETosis. Consistent with a role for PAD4 in this process is the fact that both Cl-amidine and GSK199 inhibit NET formation.50,58 Additionally, neutrophils isolated from PAD4(−/−) mice cannot form NETs after stimulation with chemokines or incubation with bacteria, highlighting that the PAD4 function is critical in diseases associated with aberrant NET formation.78c With respect to RA, the links between dysregulated PAD4 activity and disease onset are extremely strong because, in addition to its important role in NET formation, a genome-wide haplotype study identified four single-nucleotide polymorphisms (SNPs) in PAD4 that are associated with an increased risk of developing RA.88 Additionally, the most specific diagnostic for RA is the presence of antibodies to citrullinated proteins, the product of the PAD reaction, and these antibodies now form part of the clinical diagnostic criteria.89 Furthermore, these autoantibodies are present before clinical disease and are predictive of a more severe and erosive form of the disease.90
Notably, it was demonstrated that treatment of mice suffering from collagen-induced arthritis (CIA) with Cl-amidine reduces disease severity, joint inflammation, and joint damage in a dose-dependent manner without apparent signs of cytotoxic effects.91 Moreover, Cl-amidine was also effective in a mouse model of ulcerative colitis where its oral or intraperitoneal administration increased the colon length, as well as mouse mobility and activity, and reduced the disease severity.92 Recently it was shown that PAD inhibition using BB-Cl-amidine and Cl-amidine mitigates vascular, kidney, and skin disease in an MRL/lpr mouse model of lupus.58 Specifically, these PAD inhibitors not only reduce NET formation and interferon (IFN) production, which has been associated with the development of endothelial dysfunction in lupus,93 but also decrease immune complex deposition in kidneys and reduce proteinuria, which constitute major characteristics of this disease. Taken together, inhibition of NET formation by PAD-specific inhibitors represents a promising therapeutic strategy to combat different inflammatory diseases.
Since aberrant NET formation including concomitant increased levels of citrullinated histones is also present in deep vein thrombosis and myocardial infarct formation, inhibition of PAD4 activity also represents a suitable target to interfere with these serious cardiovascular diseases.94 Indeed, recent data with both Cl-amidine and BB-Cl-amidine support this hypothesis.58,81 Apart from PAD-mediated citrullination of histone proteins and NET formation, PAD4 and PAD2 were also shown to hypercitrullinate myelin basic protein (MBP), resulting in the demyelination of the myelin sheath and affecting nerve cell signal transduction, thereby promoting the development of multiple sclerosis (MS).95 In this respect, the PAD inhibitor 2-chloroacetamidine has shown efficacy in multiple preclinical models of MS, indicating that PAD enzymes may also represent a therapeutic target for MS.96
2.8. Future Areas of Protein Citrullination Research
Although most studies dealing with histone citrullination focus on its nuclear effects regarding the modulation of gene expression, it is evident that protein citrullination, even on histone proteins, also exists in the extracellular environment of human sera. The presence of hypercitrullinated proteins, including histones, is a well-documented highly specific marker for rheumatoid arthritis that has diagnostic and prognostic value as well as potential therapeutic implications.97 Since PAD inhibitors can block the accumulation of these hypercitrullinated (histone) proteins and the incidence of anticitrulline antibodies as well as aberrant NET formation, inhibition of PAD function represents a promising target to treat a number of different inflammatory diseases which are linked to hypercitrullination and abnormal NET formation.
Regarding the function of protein citrullination in epigenetic regulation, it will be of great interest to screen for enzymes that can reverse protein citrullination as well as to identify potential citrullination reader proteins that may recognize and integrate this emerging epigenetic mark into the cellular interpretation of the histone code. Since PADs are calcium-dependent enzymes that require near millimolar concentrations of calcium to efficiently citrullinate its protein substrates in vitro, it also remains a topic of future research to elucidate how the PADs get activated in vivo, as cellular calcium concentrations typically remain at low micromolar levels. Another critical aspect to further advance the current knowledge of PAD biology is the generation of isozyme-selective inhibitors to ascertain the physiological contribution of the individual enzymes in health and diseases. In addition, the development of next-generation PAD inhibitors with enhanced selectivity, bioavailability, and pharmacokinetic stability as well as preferentially reversible inhibitors to minimize potential off-target effects encountered by irreversible inhibitors is highly desirable.
3. Histone Arginine Methylation
3.1. Overview of Protein Arginine Methylation
Protein arginine methylation is a common post-translational modification that regulates numerous cellular processes, including gene transcription, mRNA splicing, DNA repair, protein cellular localization, cell fate determination, and signaling.98 Notably, it was shown that about 2% of the total arginine residues isolated from rat liver nuclei are dimethylated.99 The formation of this PTM is catalyzed by the PRMT family of methyltransferases. Currently, there are nine PRMTs annotated in the human genome (Figure 21).100 In addition, two distantly related proteins, FBXO10 and FBXO11, show a low degree of sequence homology to some PRMT motifs, but lack the important substrate-binding double E-loop and THW loop.101 Although the human flag-tagged FBXO11 protein was proposed to harbor arginine methyltransferase activity, the human HA-tagged version of the protein and the Caenorhabditis elegans FBXO11 orthologue DRE-1 did not show any methyltransferase activity in a subsequent study.101a,102 Therefore, FBXO10 and FBXO11 are not considered as true PRMTs.103 Notably, chemogenetic analyses suggest that there may be as many as 44 PRMTs in the human proteome.101c Whether all of these enzymes represent bona fide PRMTs has yet to be proven definitively.
Figure 21.

Schematic depiction of the human PRMT family. The SAM-binding methyltransferase region is highlighted in olive green. All family members contain the methyltransferase signature motifs I, post-I, II, and III and the conserved THW loop, labeled as red bars, respectively. Sequence motifs with low or no sequence similarity are depicted in light red. Abbreviations: SH3, SH3 domain; Zn, zinc finger motif; TPR, tetratricopeptide repeat.
The nine S-adenosyl-l-methionine (SAM or AdoMet)-dependent enzymes can be further classified into three types according to their preferred methylation products as the terminal amine(s) of the arginine guanidinium group may be monomethylated or symmetrically or asymmetrically dimethylated to form monomethylarginine (MMA), symmetric dimethylarginine (SDMA), and asymmetric dimethylarginine (ADMA), respectively (Figure 22). Therefore, PRMT-mediated arginine modifications add either ∼14 Da (MMA) or ∼28 Da (SDMA or ADMA) to the overall mass of the histone protein. However, as can be seen in Figure 23A, methylation does not perturb the overall positive charge of the arginine guanidinium group, but changes potential hydrogen bond interactions, since the number of added methyl groups reduces the hydrogen bond donor sites accordingly. On the basis of experimental studies performed with guanidine and a diverse series of methylated guanidine derivatives, the pKa of guanidine is 13.6, whereas the pKa values for N-methylguanidine and N,N-dimethylguanidine are 13.4.104 Interestingly, the pKa value for the N,N′-dimethylguanidine is 13.6, indicating that monomethylated and asymmetrically dimethylated guanidines are only slightly weaker bases than symmetrically dimethylated guanidines. Thus, the major effects of this modification are steric effects as well as changes in hydrogen bond interactions as opposed to the electronic effects observed with citrullination.
Figure 22.

PRMTs are SAM-dependent enzymes that catalyze the transfer of methyl groups onto peptidyl arginine residues. There are three types of PRMTs that are classified according to the site of modification. Type I enzymes generate asymmetric dimethylations, type II enzymes form symmetric dimethylations, and the type III enzyme PRMT7 only catalyzes monomethylation reactions.
Figure 23.

(A) Electrostatic surface potential and hydrogen-bonding donor sites of the side chain of arginine, and the methylated arginine side chains of MMA, ADMA, and SDMA. Cα denotes the α-carbon. Charge potentials were rendered by using SPARTAN (Wavefunction Inc.), with negative electrostatic charges shown in red, positive charges in blue, and neutral charges in green. (B) Distinct stereoisomers for MMA and SDMA. Methyl groups are highlighted in yellow, whereas hydrogen-bonding donor sites are marked in red. Stereoisomers emerging from rotation around the central Cζ–Nε bond are omitted for simplicity.
Moreover, the addition of methyl groups alters the shape of the arginine side chain. Due to the electron delocalization in the arginine guanidinium group and possible rotations around the central carbon–nitrogen bonds, distinct stereoisomers can occur in MMA and SDMA (Figure 23B). On the basis of density functional theory (DFT) calculations, the anti–syn conformation was shown to be the most favorable SDMA form, highlighted by a difference in the ground-state energies for the anti–anti and anti–syn SDMA conformations of 2.8 kcal mol–1.105 A plausible reason for this difference may be steric effects that would disfavor the close proximity between both methyl groups in the anti–anti conformation. However, the activation energy for rotation around the bond between the central guanidinium carbon, Cζ, and one of the terminal nitrogens, Nω, is 14 kcal mol–1, indicating that conversion of the different conformations can occur at room temperature.105,106 In addition, the different stereoisomers represent different hydrogen-bonding patterns that might be harnessed for specific protein interactions. For instance, the anti–anti SDMA conformation was shown to be preferentially bound to the Tudor domains of the human SMN and SPF30 proteins, whereas the extended Tudor domains of the Drosophila TUDOR protein and the human SND1 were shown to bind SDMA in the anti–syn conformation.105,107
Most PRMTs generate ADMA and are classified as type I enzymes (PRMT1–4, PRMT6, PRMT8), with PRMT1 accounting for >50% of the normal steady-state levels of ADMA.108 Of the remaining enzymes, PRMT5 and PRMT9 are the only known type II enzymes that catalyze the formation of SDMA, whereas PRMT7 is a type III enzyme that can only generate MMA on its substrates (Figure 22).109 In addition to these human PRMT orthologues, yeast encodes a PRMT that catalyzes the monomethylation of the internal (Nε) guanidinium nitrogen atom.110 Given the unique specificity of this enzyme, it has been classified as a type IV enzyme. Notably, however, sequence analyses indicate that there is no homologue of this type IV enzyme in higher eukaryotic organisms.
3.2. Structure–Mechanism of PRMTs
All PRMTs contain a conserved catalytic core region of approximately 310 amino acids, and several PRMTs possess additional domains (e.g., SH3, Zn finger, TIM barrel, and TPR) that have been suggested to diversify the substrate specificity of the enzymes and to regulate their activity (Figure 21).98d Typically, PRMTs possess a single catalytic core region. PRMT7 and PRMT9, however, harbor two consecutive methyltransferase domains that may have arisen by gene duplication.111 The catalytic core region comprises five highly characteristic signature motifs (Figures 19 and 22), including (i) motif I (VLD/EVGXGXG), which forms the base of the SAM-binding site and is structurally homologous to sequences found in other nucleotide-binding proteins, (ii) post I (L/V/IXG/AXD/E), which is important for hydrogen bond formation to each hydroxyl of the ribose part of SAM via the carboxylate of the acidic residue, (iii) motif II (F/I/VDI/L/K), which stabilizes motif I by the formation of a parallel β-sheet, (iv) motif III (LR/KXXG), which forms a parallel β-sheet with motif II, and (v) the THW loop, which is close to the active site cavity and helps stabilize the N-terminal helix, which is important for substrate recognition.100
3.2.1. Structure of PRMTs
The crystal structures of several PRMTs revealed that these proteins mainly exist as homodimeric head-to-tail protein complexes (Figure 25A).112 It was proposed that the dimer is critical for proper substrate binding and therefore is required for activity.112c By contrast, PRMT7, the only known type III methyltransferase, is unusual in that it contains two PRMT core units arranged in tandem (Figure 21). Gel filtration profiles and small-angle X-ray scattering (SAXS) experiments indicated that the PRMT7 orthologue from C. elegans exists as a monomer in solution, even in the presence of SAM.113 Notably, the recent crystal structures of mouse and C. elegans PRMT7 revealed that the second methyltransferase domain folds back onto the first catalytically active domain, and thereby forms a pseudodimeric form of the enzyme (Figure 26).113,114
Figure 25.
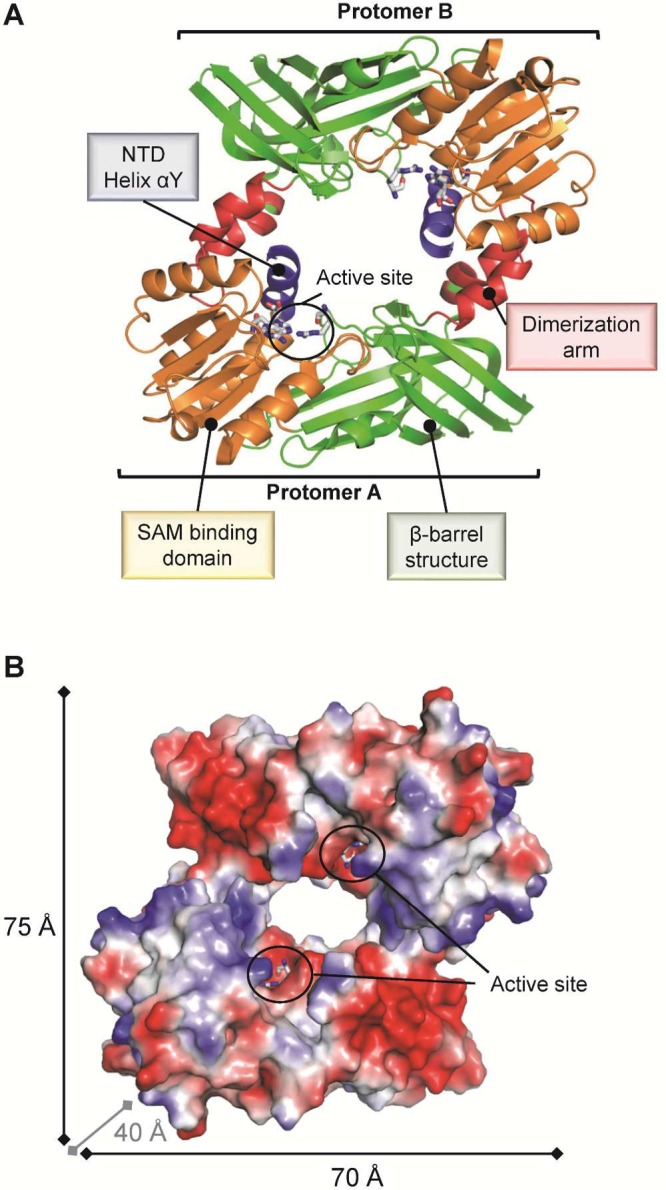
(A) PRMT1 exists as a head-to-tail dimeric protein that comprises four characteristic functional regions, as indicated (PDB code 1OR8). (B) Surface representation of dimeric PRMT1 colored according to its electrostatic surface potential. The catalytic sites of both protomers are located on the same dimer side and are facing each other, separated by ∼30 Å.
Figure 26.

Homodimeric PRMT1 contains two active sites and a central hole (PDB code 1OR8). Monomeric PRMT7 from Mus musculus harbors only one active site and does not possess a central hole (PDB code 4C4A). Homodimeric PRMT7 from T. brucei contains two active sites and does not possess a central hole (PDB code 4M37).
In mouse PRMT7, the two PRMT modules are bridged by a 19-residue linker. Overall, the general architecture is very similar to that of other known PRMT dimer structures. However, only the first (N-terminal) module is active, since the second PRMT module contains several mutations that impair SAM cofactor binding and does not contain a proper double E-loop, thereby rendering this module catalytically nonproductive.114 Moreover, the crystal structures revealed a tight interaction between both modules, which are further stabilized by a zinc finger.114 Interestingly, the interface between both PRMT modules does not contain the typical hole observed in the other PRMT dimer structures and might therefore restrict the flexibility and orientation of peptidyl substrates (Figure 26).114 Notably, PRMT7 derived from plants and Trypanosama are composed of a single PRMT module. Wang et al., however, observed that PRMT7 derived from Trypanosoma brucei exists as a dimer on the basis of SAXS analysis, and also the available crystal structure of T. brucei PRMT7 (TbPRMT7) confirms its dimeric organization (Figure 26).115 Although TbPRMT7 is active as a homodimer, it strictly generates monomethylated arginine residues, indicating that the presence of tandemly arranged PRMT modules versus two active PRMT modules in trans orientation are not critical for determining product specificity.114,115
On the basis of the available crystal structures, the catalytic core region consists of three structurally and functionally distinguishable regions as exemplified by the structure of PRMT1 (Figure 25A). The most critical is the SAM-binding domain, which is highly conserved in other SAM-dependent methyltransferases.116 This SAM-binding domain adopts a typical Rossmann fold and is followed by the β-barrel domain, which is quite unique to the PRMT family and is thought to be important for substrate binding.117 Moreover, the β-barrel domain contains an α-helical insertion that acts as a dimerization arm. Despite the variation in amino acid sequences (Figure 24), the crystal structures of several PRMTs reveal highly similar general folds. In addition, key structural features such as the active site double E-loop, which is critical for guanidinium binding, as well as the SAM-binding residues and several β-strand-forming signature motifs, are conserved among all PRMTs.
Figure 24.

Sequence alignment of the SAM-binding methyltransferase region of human PRMT family members. The product specificity-determining residue is highlighted with a blue asterisk below the alignment. Catalytic residues located on the double E-loop are highlighted with red asterisks below the alignment. The sequence alignment was generated using Clustal Omega and visualized using Espript 3.0.229 The relative accessibility of each residue is depicted below the consensus motif: blue indicates accessible residues, cyan marks intermediately accessible residues, white stands for buried residues, and red indicates that the accessibility is not predicted.
Within the active site are a number of conserved residues that are important for SAM binding, catalysis, and maintaining the overall architecture of the PRMT1 active site (Figure 27).112c These residues include E129 and V128, which are both located on a loop preceding motif II and directly interact with the SAM adenine ring and E100, which forms a bidentate hydrogen bond to the ribose moiety. The side chain of H45 projecting from an N-terminal helix, denoted αY, also hydrogen bonds to a ribose hydroxyl group. Moreover, proper positioning of the SAM cofactor is further mediated by the side chain of M155. The methionine portion of SAM is bound to the side chains of D76, recognizing the free α-amine, whereas R54 forms a bidentate interaction with the carboxylate group. R54 also hydrogen bonds with the side chain of E144 to orient the γ-carboxylate of this residue for optimal electrostatic and hydrogen bond interactions with the ω-nitrogen atom of a substrate arginine. E144 is part of the substrate-binding double E-loop, which also harbors E153. The side chain carboxylates of both of these residues are thought to recognize and align the arginine guanidinium substrate for proper catalysis to occur. Notably, in the structure of PRMT1, the position of this residue does not appear to be catalytically competent since the orientation of the E153 side chain is out of the active site.112c
Figure 27.
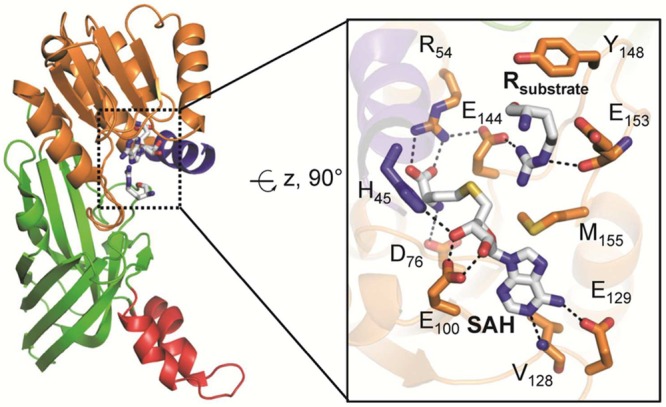
Active site architecture of PRMT1 bound to arginine (PDB code 1OR8). The structural elements are color coded according to Figure 25. The image on the right depicts details of the PRMT1 active site, highlighting critical residues for substrate binding and catalysis. Polar contacts of <3.5 Å are represented as dashed lines.
3.2.2. PRMT Substrate Recognition
On the basis of sequence motif analysis, there are no conserved residues surrounding the sites of arginine methylation.118 The only exception is glycine, which is slightly enriched, especially at positions +1 and +2 following the arginine substrate. However, arginine methylation is often found in unstructured protein regions, including loops, as well as N- or C-terminal regions.119 The lack of structured segments can be rationalized by the deep active site cavity of the PRMTs, which only allows arginine residues present on kinked loops to enter. This structural restraint was highlighted by the peptide-bound PRMT5·MEP50 complex, which revealed the presence of a characteristic β-turn in the peptide substrate (Figure 28).120
Figure 28.

PRMT5·MEP50 (pink) complex bound to histone H4 peptide (gray) (PDB code 4GQB). Polar contacts of <3.5 Å are represented as dashed lines. The highly conserved active site glutamate residues Glu435 and Glu444, forming the double E-loop, bind to the substrate guanidinium group. The hydrogen bond (highlighted in yellow dashed lines) between the carbonyl oxygen of S1 and the amide nitrogen of G4 stabilizes the β-turn conformation. Abbreviation: ac, N-terminal acetylation.
Specifically, the substrate arginine is located at the tip of the β-turn, which is stabilized by a hydrogen bond between the main chain carbonyl oxygen of S1 and the backbone amide nitrogen of G4. In addition, the carbonyl oxygen and the amide nitrogen of S1 form a bidentate hydrogen bond to the Q309 amide side chain group of PRMT5. Similar to the PAD4–substrate peptide interaction, the majority of the interactions between the substrate peptide and PRMT5 are mediated by peptide backbone interactions, rationalizing the lack of strict sequence specificity. The only exception represents a direct hydrogen bond between the side chain of K5 and the carbonyl oxygen of PRMT5 P311. Interestingly, PRMT5 also utilizes several backbone hydrogen bond interactions originating from the main chain of S310, P311, L312, and F580. This observation may explain the lack of sequence conservation of these residues among different PRMT members. Moreover, the hydroxyl group of Y307 hydrogen bonds to the backbone carbonyl oxygen and amide nitrogen of substrate residues G6 and K8, respectively. Thus, the fact that glycine-rich sequences are preferentially targeted is consistent with their ability to endow the polypeptide chain with enhanced conformational freedom, which would facilitate the formation of such β-turn structures. Thus, the context in which an arginine is placed is important for substrate recognition. Moreover, the preference for peptide stretches that can adopt β-turn-like structures coincides with the substrate specificity of PADs, implying that PRMTs and PADs might compete for similar substrates. This appears to be the case because, as noted above, in several instances the methylation and citrullination status of specific arginines in histones are inversely correlated and possess distinct and opposing effects on transcription (see section 2.6.1).
In addition to these structural constraints, local sequences next to the modified arginine are also important for substrate recognition by some PRMTs. As mentioned above, PRMTs, such as PRMT1, PRMT3, PRMT5, PRMT6, and PRMT8, typically target glycine-rich sequences.98d,121 Contrarily, PRMT4 prefers to methylate arginines embedded within proline-, glycine-, and methionine-rich motifs.122 Moreover, crosstalk with lysine acetylation was shown to be critical to enhance H3 peptide substrate methylation by PRMT4 at H3R17, as kinetic studies revealed that PRMT4 has a 5-fold higher activity toward an H3K18-acetylated peptide than the unmodified peptide.112d
Interestingly, the only type III enzyme, PRMT7, specifically recognizes arginines within an RXR motif present in H2B and H4 (see section 3.6).123 There, it was shown that PRMT7 preferentially modifies the N-terminal arginine of the RXR motif (i.e., H2BR29, H2BR31, and H4R17). Notably, efficient catalysis depends on the presence of the second arginine since the replacement of the second arginine by lysine leads to a significant reduction in the methylation signal.123 Although the cocrystal structure of TbPRMT7 bound to a 21-residue histone H4 peptide was recently solved, only the first four residues (SGRG) of this peptide could be observed.115 This structure revealed that the peptide substrate forms a wide turn on the surface of the active site, but not a characteristic β-turn (Figure 29). The side chain guanidinium of the arginine substrate forms five hydrogen bonds to both double E-loop glutamate residues (E172 and E181) and glutamine Q329, which occupies the same position as the central histidine in the THW loop. Mutagenesis studies further revealed that the glutamine residue Q329 can be substituted by a histidine without loss of activity.115 Similar to that in PRMT5, the peptide substrate in PRMT7 is also recognized by several hydrogen bond interactions with the substrate amide backbone. Specifically, the carboxylate of D70, originating from helix αY, makes hydrogen bonds with the main chain amides of residues S1 and G2, and T176, situated on the double E-loop, hydrogen bonds to the backbone carbonyl and the amide of the substrate residue R3. These data indicate that main chain interactions are critical for substrate binding, and the lack of a β-turn in the peptide substrate indicates that PRMT7 may accommodate a wider range of peptide sequences apart from glycine-rich elements.
Figure 29.

PRMT7 (purple) from T. brucei bound to histone H4 peptide (gray) (PDB code 4M38). Polar contacts of <3.5 Å are represented as dashed lines.
For most PRMTs, substrate recognition relies to a great extent on remote sequences (>14 residues distant from the arginine).124 This stands in contrast to the PADs, where long-range interactions are unimportant.19 These distal elements are typically positively charged and are thought to interact via ionic interactions with several negatively charged patches found on PRMT1 (Figure 25B).112c In this respect, mutation of acidic residues in PRMT1 leads to compromised enzyme activity or altered methyltransferase substrate specificity.125 Notably, efficient PRMT-mediated methylation reactions require long peptide substrates, i.e., 21 residues of the N-terminal tail of histone H4 (acH4-21), to achieve H4R3 methylation kinetics comparable to that of full-length H4.124b N-terminal truncation of the two residues preceding the methylated arginine residue (H4–21Δ(1–2)) decreased the methylation efficiency by ∼104-fold, thereby indicating that interactions between the enzyme and the backbone amide N-terminal to the site of methylation are critical for substrate recognition as confirmed by structural analysis. A similar approach employing C-terminal truncation peptides revealed that removal of three residues (AcH4-18) decreased arginine methylation by 150-fold in human PRMT1, while further truncation by three additional residues (AcH4-15) further diminished activity by 860-fold.124b These data clearly indicate that remote sequences are critical for efficient substrate capture, by mediating strong charge–charge interactions between the acidic patches in the SAM-binding domain as well as the β-barrel domain of PRMT1 (as described above) and positively charged residues such as K16, R17, R19, and K20 in the distal portion of the H4 tails.112c,124b,125 Since the great majority of PRMTs form dimers, it is conceivable that this increased amount of distal substrate-binding sites, represented by acidic patches in PRMT1, facilitates the processive dimethylation of Arg residues by allowing the product of the first methylation reaction, monomethylarginine, to enter the active site of the second molecule of the dimer without releasing the substrate from the homodimer. This model is further supported by the high affinity, also reflected by a low KM value, between substrates and the PRMT dimer and the loss of activity of engineered PRMT monomers.112b,112c
Osborne and colleagues further revealed that PRMT1 employs a partially or semiprocessive mechanism, indicating that the substrate stays bound to the enzyme for two consecutive methylation reactions, and the major substrate released is ADMA.124b Similar results have been obtained for PRMT6 as this enzyme has also been suggested to show some processivity.126 By performing double-turnover reaction experiments, the Hevel group observed both MMA and ADMA formation, confirming a general semiprocessive mechanism of PRMT1 catalysis.127 Interestingly, the proportions of MMA and ADMA generated are different among distinct peptide substrates. For example, the N-terminal H4 peptide SGRGKGGKGLGKGGAKR is preferentially processed through a processive mechanism, leading to a dimethylated product, while other sequences such as the fibrillarin-based RKK peptide GGRGGFGGKGGFGGKW partition more frequently through a distributive mechanism where both monomethylated and dimethylated products are ultimately generated. These observations clearly indicate that the degree of processivity is controlled in a substrate-dependent manner and thus distinct patterns of methylation can be deposited by the same PRMT enzyme.127 Thus, conflicting observations regarding the processive or distributive nature of PRMT1 activity may be partially due to the different substrates used and hence changes in the substrate-induced processivity rate. By contrast, recent data indicate that PRMT5 uses a distributive mechanism to catalyze the symmetric dimethylation of histone H4 and its N-terminal peptide fragment.120,124c,128
3.2.3. PRMT Product Selectivity
Structural comparison of several PRMTs reveals similar active site architectures, thereby suggesting that these enzymes employ a similar catalytic mechanism to effect substrate methylation (Figure 30).112a,112c,112e,114 Interestingly, the catalytic domain of PRMT5, which catalyzes SDMA formation, is also highly similar to that of the ADMA-generating type I enzymes as judged by the very high degree of consersation between the available crystal structures (Figure 31).120,129 However, the molecular basis for their distinct product formation paths is largely unknown. Recent reports proposed that a conserved phenylalanine (F327) in the active site of PRMT5 is important for directing symmetric dimethylation, while a methionine (M48) residue in PRMT1 confers specificity toward asymmetric dimethylation.129,130 These observations imply that the generation of symmetrically and asymmetrically dimethylated arginine residues share a common catalytic mechanism, as type I and type II mutant enzymes are capable of performing both of these reactions. Notably, on the basis of the PRMT5 structure, F327, which has been shown to be important for specifying the symmetric dimethylating activity of PRMT5, interacts with the substrate guanidinium group, thereby orienting it for methyl transfer (Figure 31).120 Quantum mechanical models further revealed that the free energy of activation, ΔG⧧, for SDMA formation is 13.4 kcal/mol, and therefore, SDMA formation is more energetically costly than ADMA formation, for which a ΔG⧧ of 10.2 kcal/mol was calculated.130 The higher energy barrier for forming SDMA over ADMA presumably explains the low amount of SDMA formed by a PRMT1M48F mutant and is also reflected by the 160-fold slower rate for symmetric dimethylation, using monomethylated arginine as the substrate, compared to the monomethylation rate observed with PRMT5.131 By contrast, the PRMT1-catalyzed rate of dimethylation is only 2–4-fold slower than the rate of the monomethylation reaction.124b Moreover, introduction of an F379M substitution into PRMT5 partially shifts the product formation specificity such that this enzyme can now generate ADMA, albeit with reduced efficiency compared to that of PRMT1.129 However, it still remains to be determined what other structural features, apart from the mentioned phenylalanine to methionine switch, contribute to product specificity, especially since PRMT9, the other type II enzyme, does not contain a phenylalanine but a methionine at this position. As such, it was recently proposed that subtle differences in the size of the arginine-binding pocket, mainly due to alterations of the THW loop, may be important for controlling the product selectivity of the PRMTs.113,114 In this respect, it is interesting to note that all PRMTs, except the type II enzymes PRMT5 and PRMT9, contain a histidine in the THW loop. PRMT5 and PRMT9 possess serine and cysteine residues at the corresponding site, respectively. It is tempting to speculate that the bulky side chain of histidine impairs binding of MMA, whereas the much smaller side chains of serine and cysteine allow for proper accommodation of a symmetrically dimethylated guanidinium group in the active site pocket (Figure 32).
Figure 30.
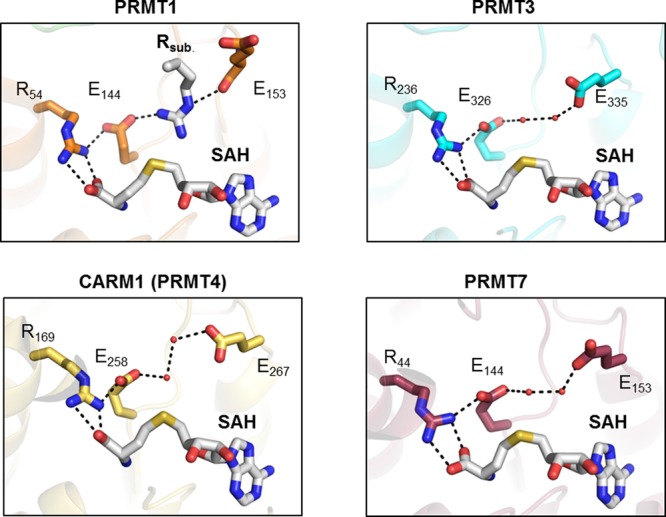
Structural representation of the active site of rat PRMT1 (PDB code 1OR8), rat PRMT3 (PDB code 1F3L), rat PRMT4 (PDB code 3B3F), and mouse PRMT7 (PDB code 4C4A). All structures contain a bound cofactor (SAH, highlighted in gray). Abbreviation: Rsub, substrate arginine.
Figure 31.

Structural comparison of the active site of rat PRMT1 bound to SAH and substrate arginine (PDB code 1OR8) and human PRMT5 bound to sinefungin and histone H4 peptide substrate (PDB code 4GQB).
Figure 32.

Structural comparison of the active site pocket of C. elegans PRMT5 bound to SAH (PDB code 3UA3) and rat PRMT4 (CARM1) bound to SAH (PDB code 3B3F). The lower images depict the putative model of the enzyme active site pockets bound to its products, represented by SAH and SDMA in the case of PRMT5 or SAH and ADMA for PRMT4.
3.2.4. Proposed Catalytic Mechanism of PRMTs
PRMTs employ a bisubstrate mechanism, transferring the methyl group of SAM to specific arginine residues in histone and nonhistone protein substrates, resulting in mono- and dimethylated arginine residues and the byproduct S-adenosyl-l-homocysteine (SAH or AdoHcy). According to the enzyme classification nomenclature, PRMTs belong to the class of transferases that transfer one-carbon groups (EC 2.1.1.125). Notably, PRMT1, PRMT5, and PRMT6 employ rapid equilibrium random kinetic mechanisms wherein substrate binding and product release occur in a random fashion,131,132 although it should be noted that prior work with PRMT6 suggested that this enzyme utilized an ordered kinetic mechanism in which SAM binds first and SAH dissociates last from the enzyme.133 However, this study only used product inhibition patterns to assign the order of substrate binding and product release, and the overall quality of the observed inhibition patterns is quite poor. Thus, PRMT6, like PRMT1 and PRMT5, most likely binds its substrates in a random fashion. Notably, though, the PRMT4-catalyzed reaction has also been suggested to proceed via an ordered sequential mechanism where SAM binding is the first step and SAH is the last product to leave the enzyme.112d
The PRMT1 catalytic mechanism proceeds via a bimolecular nucleophilic substitution (SN2) methyl transfer reaction (Figure 33). The two invariant glutamate residues E144 and E153 are hypothesized to localize the positive charge of the guanidinium group to one ω-nitrogen atom, thereby leaving a lone pair of electrons on the other terminal nitrogen to attack the methylsulfonium group of SAM. Originally, it was proposed that the substrate guanidinium is deprotonated and thereby activated by the carboxylate of E144. However, using solvent isotope effect experiments, Rust et al. suggested that general-acid/base catalysis is not important for promoting methyl transfer in PRMT1.134 Instead they proposed that the PRMT1-catalyzed reaction is primarily driven by proper substrate guanidinium alignment by E144 and E153 with respect to the S-methyl group of SAM and that the prior deprotonation of the substrate guanidinium group is not required for methyl transfer. Subsequent quantum mechanical (QM) calculations indicated that E144 abstracts a proton from the reacting arginine immediately after methyl transfer, consistent with the mechanism proposed by Rust et al.135 These QM studies also suggested that the guanidinium loses planarity in the transfer state, as predicted by the observed inverse solvent isotope effect.134,135 The positioning of the ω-nitrogen of the guanidinium group to attack the SAM methyl group ultimately results in the arginine N-methylation and generation of the byproduct SAH.
Figure 33.
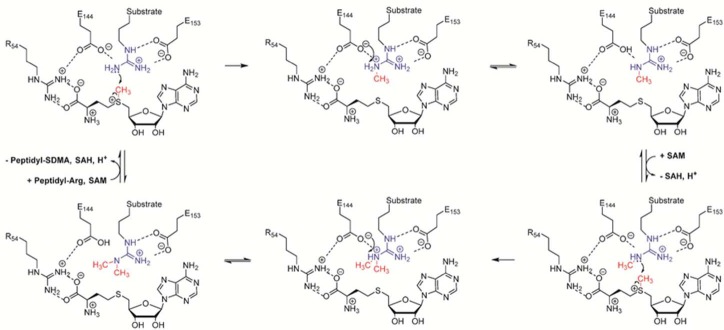
Proposed catalytic mechanism for type I PRMT enzymes, exemplified by PRMT1.
3.3. Is Protein Arginine Methylation a Reversible Modification?
Several PTM regulatory systems such as phosphorylation, ubiquitination, or lysine acetylation are reversibly regulated; however, for arginine methylation, PRMTs act as writers, but the occurrence of a corresponding eraser that clips off the methyl group is still controversial. There was a report claiming that the iron- and α-ketoglutarate-dependent dioxygenase Jumonji domain 6 (Jmjd6) protein acts as an arginine demethylase (eraser).136 However, subsequent detailed analyses revealed that this enzyme is in fact a lysine hydroxylase and does not erase the methyl mark from methylated arginine residues.137 As described above, PAD4 was also suggested to convert methylated arginines to citrulline,38 but this activity is unlikely physiologically relevant due to the extraordinarily low activity.19,39b Therefore, PAD activity antagonizes arginine methylation by competition with PRMTs but does not directly convert methylarginines into citrulline.19,28,41 Nonetheless, due to the dynamic appearance and disappearance of methylarginine marks,41,138 the existence of an arginine demethylase is very likely.100 Examples for aminomethyl demethylases are found in nature, and two separate classes of histone lysine demethylases are known to exist.139 The first class was discovered in 2004 and consists of the lysine-specific demethylases (LSDs), which are flavin adenine dinucleotide (FAD)-dependent amine oxidases that remove mono- and dimethylation marks (Figure 34A).140 The FAD cofactor oxidizes the methyllysine to form an imine intermediate, which is then hydrolyzed to yield unmodified lysine and formaldehyde. The resulting reduced FADH2 is reoxidized by molecular oxygen, thereby forming hydrogen peroxide as a byproduct. The second group comprises the Jumonji C-terminal domain (JmjC) family of histone demethylases, which use iron and α-ketoglutarate as cofactors, thereby acting as oxygenase enzymes to remove mono-, di-, and trimethyl groups from methylated lysine residues (Figure 34B).141 The JmjC-catalyzed demethylation reaction involves oxidative decarboxylation of α-ketoglutarate, coupled to hydroxylation of the methyl group, generating an unstable hydroxymethylammonium intermediate, which is released as formaldehyde.
Figure 34.

Mechanisms of lysine demethylation. (A) Active site of human LSD1 with bound FAD attached to the mechanism-based histone 3 peptide inhibitor N-methylpropargyl-K4 H3 (PDB 2UXN)230 and proposed mechanism. Note that the covalent inhibitor was further reduced using NaBH4. (B) Active site of the JMJD2A with bound H3K9me3, nickel, and N-oxalylglycine that both mimic the actual iron and α-ketoglutarate cofactors, highlighted in green and gray, respectively (PDB 2OQ6),231 and proposed mechanism. Green dashed lines represent CH···O hydrogen bonds. Abbreviations: FAD, flavin adenine dinucleotide; FADH, reduced flavin adenine dinucleotide; FA, formic acid; aKG, α-ketoglutarate; OGA, N-oxalylglycine.
3.4. Methylarginine-Binding Proteins
In contrast to the identification of an arginine demethylase, there is strong structural and biochemical evidence to support the existence of methylarginine readers (Figure 35).107,142 For example, several members of the Tudor protein family specifically recognize methylated arginine residues. These proteins contain a conserved Tudor domain, which is responsible for either methylarginine or methyllysine binding.142c On the basis of sequence analysis, however, it is not possible to unequivocally predict the binding specificity of individual Tudor domains. Structural studies of Tudor domains revealed that an aromatic cage surrounds the methylarginine, thereby forming extensive cation−π and hydrophobic interactions with the bound ligand (Figure 36).105 In addition, it was shown that the hydrophobic cage for methylarginine recognition is much narrower (diameter ∼7 Å) than the cage for methyllysine (diameter >8 Å), thus favoring binding of the planar guanidinium group.105 The structure of ADMA-bound Tudor domains is a good example of how nature utilizes noncovalent cation−π interactions to recognize the arginine guanidinium group.143 In this respect, it was shown that methylation of arginine residues increases the strength of cation−π interactions compared to that for unmodified arginine.144 Notably, the high affinity and specificity in the binding of trimethylated lysine to chromodomains that harbor similar aromatic cage structure also depend on favorable cation−π interactions.145 The strong contribution of multiple cation−π interactions might also provide a plausible mechanism to exclude an uncharged peptidyl citrulline from binding to Tudor domains as it cannot form this type of interaction, although it should be noted that this has not been systematically tested.
Figure 35.

Schematic overview of writers and readers of histone arginine methylation.
Figure 36.
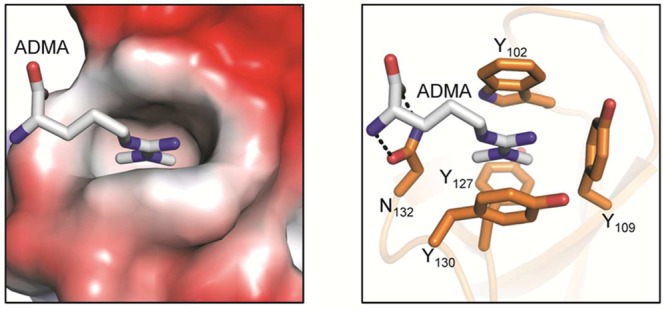
SMN Tudor domain bound to the asymmetric dimethylated arginine residue (PDB code 4A4G). The left panel illustrates the Tudor domain colored according to its electrostatic surface potential. The image on the right highlights the ADMA-interacting residues that form a hydrophobic cage around the methylated guanidinium group.
Recently, it was shown that the TDRD3 protein recognizes ADMA-methylated histone H4 tails via its Tudor domain, thereby activating transcription.146 Although most of the Tudor domains prefer SDMA, it was recently shown that TDRD3 also recognizes ADMA in H3 (H3R17me2a) and H4 (H4R3me2a).146,147 Moreover, it was shown that the PHD domain of the DNA methyltransferase DNMT3A can bind to the H4R3me2s suppressive gene expression mark.148 There, it was proposed that PRMT5 generates H4R3me2s on targeted promoters, which are recognized by DNMT3A. The recruited DNMT3A promotes DNA methylation, thereby inducing gene silencing. However, a subsequent study by Otani et al. could not confirm any interaction between the DNMT3A PHD domain and an H4R3me2s peptide.149
3.5. Chemical Probes–Inhibitors for PRMTs
3.5.1. Inhibitors of PRMTs
Since PRMTs affect a plethora of different target genes, it is unsurprising that, when dysregulated, they play a role in human disease. In fact, dysregulated PRMT activity has been causally linked to the development and progression of numerous cancers, as well as to viral replication and cardiovascular disease. Therefore, the PRMTs constitute promising targets for drug discovery, and inhibitor development is at the frontline of current PRMT research.8b
One of the earliest described PRMT inhibitors to be discovered is autogenerated by the enzyme during catalysis. As described above, the methyl donor substrate SAM is converted to SAH (21) (Figure 37), which represents a potent feedback inhibitor of PRMT activity, and accumulates as an inevitable byproduct of protein methylation. In cells, SAH levels can be raised by blocking its degradation via SAH hydrolase. In fact, adenosine dialdehyde (22) is a potent SAH hydrolase inhibitor that is often used in cell studies to increase the amount of intracellular SAH. Higher SAH levels in turn result in feedback inhibition of most SAM-dependent methylation reactions including PRMT activity. SAM analogues, such as methylthioadenosine (MTA, 23) and sinefungin (24), also function as general PRMT inhibitors (Figure 37); however, they exhibit limited specificity, due to their structural homology to SAM. Thus, they inhibit numerous other SAM-dependent methyltransferases, thereby affecting the cellular methylation of phospholipids, proteins, DNA, and RNA as well. As such, SAM-mimicking methyltransferase inhibitors display limited specificity by indiscriminately inhibiting all SAM-utilizing enzymes.
Figure 37.

Nonselective PRMT inhibitors. Note that adenosine dialdehyde (22) and AMI-1 (25) are both not direct PRMT inhibitors. Adenosine dialdehyde blocks the activity of SAH hydrolase, which induces an increase in SAH levels, thereby inhibiting PRMT activity. AMI-1 binds to the histone substrates and prevents recognition by the PRMT enzyme.
To obtain PRMT-selective inhibitors, several approaches, including both virtual and high-throughput screens, as well as substrate analogue inhibitor design were conducted. One of the first high-throughput screens directed against the yeast type I PRMT, Hmt1p, resulted in the identification of several arginine methyltransferase inhibitors, denoted as AMI.150 Most of these inhibitors were highly nonspecific and also blocked the activity of the protein lysine methyltransferases. AMI-1 (25), however, a symmetric naphthalenesulfonate molecule, inhibited PRMT1 with an IC50 of 8.8 μM but did not diminish the activity of distinct lysine methyltransferases. In addition, AMI-1 blocked the activity of PRMT3, PRMT4, and PRMT6. It was also shown that AMI-1 is cell-permeable and that it inhibits endogenous PRMT1 activity in a concentration-dependent manner.150 However, on the basis of circular dichroism, fluorescence, and absorption spectral analysis, Feng et al. unequivocally determined that AMI-1, and related naphthalenesulfonate derivatives, target the substrate instead of the PRMT enzyme and are therefore not direct PRMT inhibitors.151 Notably, AMI-1 forms a complex with an H4 peptide substrate, most likely via bidentate electrostatic interactions between its sulfonate groups and the arginine guanidinium groups present in the peptide substrate, thereby blocking substrate access to the PRMTs.151
Fragment-based virtual screening identified RM65 (26) as a cell-permeable PRMT1 inhibitor.152 Docking studies suggest that 26 is a competitive inhibitor, occupying both the SAM-binding and the substrate arginine-binding sites. Compound 26 was also shown to reduce histone H4R3 methylation in HepG2 cancer cells at concentrations above 100 μM. Using a similar virtual docking and pharmacophore-based filtering approach, the Jung group identified the diamidine stilbamidine (27) and allantodapsone (28) as PRMT1 inhibitors.153 Both compounds are competitive for the protein substrate, but not for the cofactor SAM. Moreover, these inhibitors are active in a functional assay of estrogen receptor activation and also reduced the cellular methylation of R3 in histone H4 at concentrations below 50 μM while having minor effects on the lysine methylation at H3K4.153 Further optimization resulted in the generation of even more potent inhibitors, such as the dapsone derivative 29, which inhibits PRMT1 with an IC50 of 1.5 μM.154
The Thompson group observed that the SAM congener 5′-(diaminobutyric acid)-N-(iodoethyl)-5′-deoxyadenosine ammonium hydrochloride (AAI, 30) blocks PRMT1 activity with an IC50 of 18.5 μM and a 4.4-fold preference for PRMT1 over CARM1 (Figure 38)155 This compound is thought to form a reactive aziridinium moiety that is susceptible to nucleophilic attack.156 Interestingly, upon incubation with PRMT1 and H4 peptide substrate, this compound reacts with the incoming arginine substrate (H4R3) in situ in an enzyme-dependent manner, thereby autogenerating an effective bisubstrate inhibitor within the enzyme active site (Figure 38). The ability of PRMT1 to chemoenzymatically generate an effective bisubstrate analogue inspired the development of defined bisubstrate derivatives. In this respect, the partial-bisubstrate analogue 31, which comprises an arginine-containing peptide fragment that was linked to the amino acid moiety of SAM, was shown to block PRMT1 activity with an IC50 of 14 μM (Figure 39).157 This compound shows limited PRMT selectivity and also blocks PRMT4 and PRMT6 with similar efficiency. In addition, Dowden and colleagues reported the development of SAM derivatives conjugated to a guanidinium group via varying carbon linkers (32–34).158 Comparison of the generated derivatives revealed that a four-carbon spacer, present in 33 between the guanidinium group and the SAM analogue, is most suited for efficient PRMT inhibition (IC50 = 2.9 μM). Though the selectivity of this inhibitor for other PRMTs was not evaluated, it did not show substantial inhibitory activity against the lysine methyltransferase SET7.158 The Martin group recently developed a partial bisubstrate inhibitor where the SAM adenosine moiety is connected to the guanidinium group (35).159 Interestingly, 35 was more potent than the previously described (partial) bisubstrate inhibitors with IC50 values of 1.3 μM, 560 nM, and 720 nM for PRMT1, PRMT4, and PRMT6, respectively. Moreover, this compound did not display any measurable inhibitory effect on the lysine methyltransferase G9a; however, it also did not show any inhibitory effect on cell proliferation using MCF7 and Caco2 cells.159 Although bisubstrate analogues represent interesting tools to analyze PRMT activity, they comprise several limitations, including lack of selectivity, within the PRMT family, and, with respect to peptide-based bisubstrate inhibitors, undesirable pharmacological properties. Therefore, recently, great efforts have been taken to yield potent and isozyme-selective PRMT inhibitors.
Figure 38.
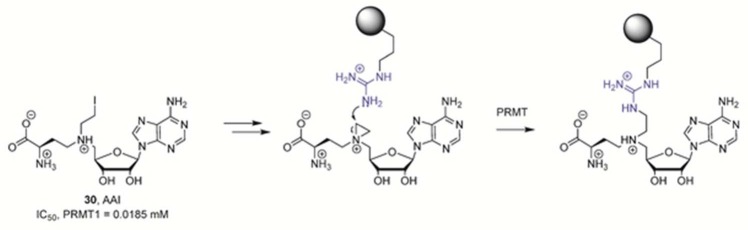
SAM derivative AAI is transformed in situ to generate a bisubstrate PRMT inhibitor. The gray sphere denotes a peptidyl arginine substrate.
Figure 39.
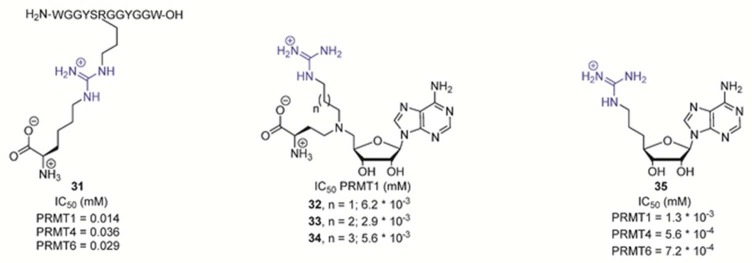
Bisubstrate-based PRMT inhibitors.
The stilbamidine derivative furamidine 36 contains two amidine moieties and is specific for PRMT1 with an IC50 of 9.4 μM (Figure 40).160 Another potent PRMT1 inhibitor is a peptide-based haloacetamidine-containing compound dubbed C21 (37).161 This compound is derived from the N-terminal sequence of histone H4 with a chloroacetamidine-modified residue in place of H4R3 to serve as a reactive warhead. In contrast to most other PRMT inhibitors, C21 acts as an irreversible inhibitor that forms a covalent bond with a hyper-reactive cysteine residue, C101,162 present in the active site of the enzyme.163 In addition, despite the presence of a chloroacetamidine warhead, which is also present in the most potent PAD inhibitors (see above), C21 is selective for PRMT1 (IC50 = 1.8 μM) and displays poor inhibitory activity toward the PAD enzymes (IC50 for PAD4 = 145 μM).161 Notably, C21 selectively inhibits cellular PRMT1 activity over PRMT4 when delivered into cells with peptide transfection reagents.161 However, C21 is only ∼5-fold more selective for PRMT1 over PRMT6. To screen for more selective PRMT1 inhibitors, Bicker and colleagues employed a combinatorial peptide library approach.164 The identified hit, denoted C21-1F (38), contains a phenylalanine instead of a glycine at the R-1 position (Figure 40). Although C21-1F is slightly less potent than C21, it is 3 times more selective for PRMT1 over PRMT6, indicating that residues around the substrate arginine might be exchanged or modified to develop isozyme-selective PRMT inhibitors.
Figure 40.
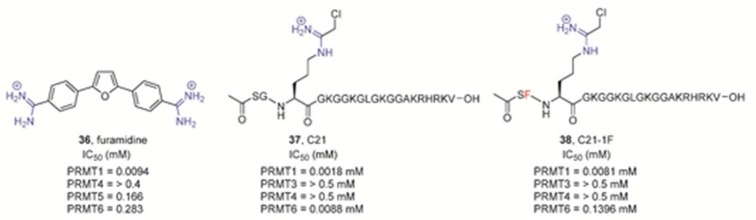
PRMT1-selective inhibitors.
With respect to PRMT3, Siarheyeva et al. employed a library screening approach to identify selective inhibitors targeting this enzyme.165 Optimization of the initial hit compounds revealed inhibitor 39.166 Notably, this inhibitor is noncompetitive with respect to both SAM and the peptide substrate and binds to an allosteric site present in PRMT3 (Figure 41). This compound is highly selective for PRMT3, and detailed structural analyses showed that 39 binds to an allosteric pocket at the base of the dimerization arm between two PRMT3 subunits. There, 39 interacts with and distorts the activation helix, which is critical for proper SAM binding. It is thought that 39 induces conformational constraints on this α-helix that prevent formation of a catalytically competent state.166 The isoquinoline moiety of 39 forms a buried hydrogen bond with T466, the urea group forms hydrogen bonds with the side chains of E422 and R39, and the pyrrolidine amide pushes against the α-helix (Figure 41). To test the in vivo efficacy, 39 was evaluated for target engagement using an InCELL Hunter assay. There, it was shown that 39 stabilized PRMT3 in HEK293 cells with an EC50 value of 1.3 μM. Moreover, 39 can block the PRMT3-dependent dimethylation of endogenous histone 4 at H4R3 with an IC50 of 225 nM. These data demonstrate that 39 is a potent, selective, and cell-active allosteric inhibitor of PRMT3 that is suitable for further cell or animal studies.
Figure 41.
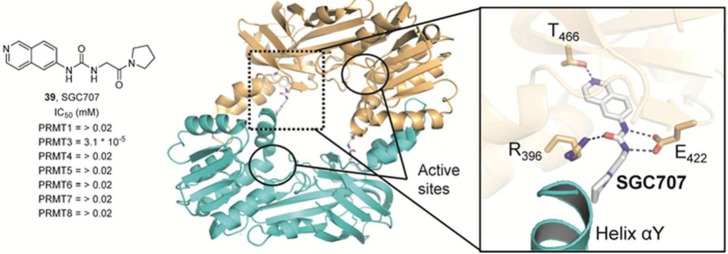
Allosteric PRMT3 inhibitor SGC707. Crystal structure of dimeric PRMT3 bound to inhibitor 39 (PDB code 4RYL). SGC707 (gray) binds an allosteric site located at the interface between two PRMT3 protomers. Hydrogen bonds of <3.5 Å are represented as dashed black lines.
High-throughput screening efforts also led to the identification of pyrazole and benzimidazole derivatives as potent PRMT4 inhibitors.167 Subsequent optimization of the initial hit compounds resulted in nanomolar inhibitors such as the indole derivative 40 and the pyrazole derivative 41, which possess IC50 values of 30 and 27 nM, respectively (Figure 42).168 Interestingly, both of these inhibitors bind to the substrate arginine-binding cavity of PRMT4 and require the presence of bound cofactor SAH (Figure 42).168 Structural studies of PRMT4 in complex with sinefungin and 40 revealed that the N-methylethanamine moiety of the inhibitor is directed toward the bottom of the arginine-binding cavity and directly interacts with the active site residue E258. The piperidine group is positioned at the entrance of the active site cavity and hydrogen bonds to H415 of the THW loop, whereas the indole moiety makes hydrophobic interactions with several aromatic side chains of PRMT4 and forms a water-mediated hydrogen bond to the main chain carbonyl of K471 and the side chain of N266. The crystal structure of PRMT4 bound to 41 revealed that the terminal l-alaninamide moiety mimics the arginine guanidinium group and makes several polar interactions with PRMT4, including the carboxyl groups of the double E-loop residues E258 and E267. In addition, the alanylmethyl group of 41 forms CH···O hydrogen bonds to the hydroxyl oxygen of Y154 and the backbone carbonyl oxygen of M260, while the carbonyl oxygen of the l-alaninamide hydrogen bonds with one of the imidazole nitrogens of H415. Notably, the (trifluoromethyl)pyrazole, 1,3,4-oxadiazole, and indole scaffolds are thought to mainly interact with PRMT4 via shape complementarity rather than by polar interactions, except for two hydrogen bonds formed between the side chain of Y262 and the oxadiazole oxygen atom and between the hydroxyl og Y477 and a fluorine of the trifluoromethyl group.
Figure 42.

PRMT4 (CARM1)-selective inhibitors. The top panel illustrates the structural characterization of the indole inhibitor 40 bound to PRMT4 (PDB code 2Y1W). The lower image represents the crystal structure of PRMT4 bound to the pyrazole inhibitor 41 (PDB code 2Y1X). Dashed green lines indicate CH···O hydrogen bonds, whereas other polar contacts of <3.5 Å are represented as dashed black lines.
Very recently, researchers from Epizyme developed selective PRMT5 inhibitors containing a di- or tetrahydroisoquinoline–hydroxypropyl–arylcarboxamide scaffold.169 One of the most effective compounds, EPZ015666 (Figure 43), was shown to act as a very potent (Ki = 5 nM), selective (>20000-fold compared to other protein methyltransferases), and orally bioavailable inhibitor of PRMT5.170 Interestingly, the tetrahydroisoquinoline moiety of 42 is thought to directly bind to the characteristic F327 residue that is present in PRMT5 but absent in other PRMTs via π–π stacking interactions.170 In addition, it was demonstrated that 42 reduces cellular SDMA levels in Z-138 cell lines with an IC50 of 44 nM and displays robust antitumor activity in mantle cell lymphoma (MCL) xenograft mouse models. Thus, EPZ015666 has cellular activity and in vivo efficacy and represents a promising lead compound for the development of PRMT5 inhibitors as potential cancer therapeutics.
Figure 43.

PRMT5-selective inhibitors.
In 2015, Alinari and colleagues also reported the discovery of a selective PRMT5 inhibitor employing a structure-based virtual screening approach.171 After initial cell testing assays, CMP5 (43) was identified as the top hit and used for further characterizations. It was shown that 43 is selective for PRMT5 and does not block PRMT1, PRMT4, or PRMT7 at concentrations below 100 μM. Moreover, on the basis of modeling studies, this compound is predicted to occupy the SAM-binding pocket via its carbazole ring, while the pyridine ring is thought to form π-stacking interactions with the characteristic F327 of PRMT5. Inhibitor 43 was selectively toxic to lymphoma cells and killed Pfeiffer cells and SUDHL2 cells with IC50 values of 30 and 35 μM, respectively.171 In addition, CMP5 treatment reduces the cellular level of H4R2me2s as well as H3R8me2s marks, thus highlighting its cellular efficacy.
3.5.2. Chemical Probes for the PRMTs
Since C21 is an irreversible, covalent, and highly selective PRMT1 inhibitor, it was adapted for use as a chemical probe of PRMT1 activity by attaching fluorescein (F-C21, 44) and biotin (B-C21, 45) reporter tags (Figure 44).172 Although F-C21 labels recombinant PRMT1, the fluorescent probe cannot detect cellular PRMT1, likely due to low levels of the active protein. However, B-C21 can efficiently be used to label and isolate endogenous PRMT1 from MCF-7 whole cell extracts. Notably, using B-C21 as an ABPP, it was shown that cellular PRMT1 activity is regulated in response to estrogen. Specifically, the amount of active PRMT1 isolated from nuclear extracts is reduced after estrogen treatment, suggesting that PRMT1 activity is negatively regulated in a manner that ultimately precludes the enzyme from interacting with its substrates (mimicked by B-C21).172 Interestingly, the subcellular localization of the PRMTs also appears to be altered in cancer cells (see below), suggesting that the non-nuclear effects of the PRMTs may be more important than previously thought.
Figure 44.
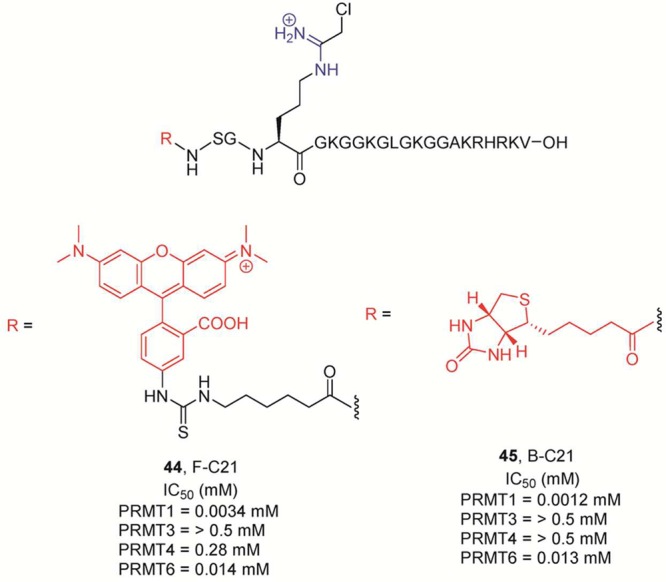
Probes for PRMT1. The amidine group is highlighted in blue, whereas the reporter tags (biotin or fluorescein) are marked in red. IC50 values for the PRMTs were determined after incubation of the enzyme with 15 μM 14C-methyl-SAM for 10 min at 37 °C.
Since the fluorescent conjugate F-C21 acts as a potent PRMT1 inactivator, it could also be used in Fluopol-ABPP-based HTS approaches to identify specific PRMT1 inhibitors in a manner similar to that described for the PAD enzymes (see above). In this respect, Dillon and colleagues adapted a cysteine-reactive maleimide conjugated to AlexaFluor488 as an ABPP to screen for PRMT1 inhibitors.173 This probe, although much less specific than F-C21, can label PRMT1 by forming a covalent bond with a hyper-reactive cysteine residue, C101,162 that is located in the SAM-binding pocket close to the purine ring. Using this Fluopol-ABPP assay, they identified two potent PRMT1 inhibitors, 46 and 47 (Figure 45), that also block PRMT8 activity but not PRMT4 activity.173 Both of these compounds are nitroalkenes and are expected to react with the cysteine residue in PRMT1. Notably, PRMT8 also possesses a cysteine residue in its SAM-binding pocket, whereas PRMT4 does not.
Figure 45.

Nitroalkenes as cysteine-reactive PRMT1 inhibitors.
3.6. Physiological Role of Histone Arginine Methylation
PRMTs methylate numerous cellular protein substrates, including nuclear proteins such as transcription factors, other coregulators, and histones. The importance of this PTM to cellular growth is probably best exemplified by the fact that both PRMT1 and CARM1 mouse knockouts are embryonically lethal. The currently identified sites of histone methylation are H2AR3 and R11, H2BR29, R31, and R33, H3R2, R8, R17, and R26, and H4R3, R17, R19, and R23 (Figure 46). The diversity of arginine methylation sites on histone proteins provides multiple routes to directly link arginine methylation to the epigenetic regulation of gene expression (Figure 46). In addition, the methylation of arginine residues in several transcriptional coactivators (e.g., the histone acetyltransferases p300 and CBP) provides an indirect route to influence the epigenetic state of affected genes. Originally, it was assumed that the methylation of histone arginine residues was associated with gene activation, as the dimethylation of histone H4R3 by PRMT1 facilitates transcriptional activation by a variety of nuclear hormone receptors.174 More recently, however, it became apparent that arginine methylation can be either an activating or a repressing mark that regulates the expression of multiple genes. Below we highlight several examples of PRMT-mediated histone methylation events that are correlated with the activation and repression of gene transcription.
Figure 46.

Arginine methylation sites in histone proteins. Abbreviations: a, asymmetric dimethylation; s, symmestric dimethylation; m, monomethylation. Color code: green, gene activation; red, gene repression; yellow, gene activation or repression, or unknown.
3.6.1. Methylation of H2AR3
H2AR3 methylation can be catalyzed by PRMT1, PRMT5, PRMT6, or PRMT7, generating ADMA (PRMT1 and PRMT6), SDMA (PRMT5), or MMA (PRMT7). The effects of these methylation events are varied. For example, in mouse ES cells, PRMT5 expression and activity are upregulated and the enzyme was shown to symmetrically dimethylate H2AR3. This event helps maintain pluripotency by repressing differentiation genes, including Fgf5, Gata4, Gata6, and HoxD9.175 This report also indicated that PRMT5 is critical for embryonic stem cell (ESC) generation, as no ESCs were generated in Prmt5–/– mouse embryos.175 However, in a separate study investigating human ESCs, PRMT5 knockdown had no effect on pluripotency, as evidenced by the fact that the PRMT5 knockdown cells and wild-type cells had similar RNA levels for genes associated with multiple tissue types.176 PRMT5 knockdown did, however, correlate with the repression of 78 genes. Notably, only two of these genes are known developmental genes, whereas the rest are associated with basic cellular processes.176 These results illustrate the differences between the human and mouse epigenetic landscape.
PRMT7 also methylates H2AR3 in response to DNA damage to repress the transcription of the DNA polymerases POLD1 and POLD2. Importantly, when PRMT7 was knocked down in a cellular model, cells were more resistant to DNA damage due to the derepression of DNA damage response genes such as POLD1 and POLD2.177 Collectively, these results illustrate the importance of isozyme-specific PRMT inhibitors. In this case, if a PRMT inhibitor were used in combination with a DNA-damaging agent, inhibition of PRMT7 could promote tumor resistance to the chemotherapeutic drug.
3.6.2. Methylation of H2AR11 and R29
H2AR3, R11, and R29 were all shown to be methylated in a proteomic study of histones extracted from HeLa cells. Notably, H2AR29 was found to be methylated at sites known to be associated with PRMT6-mediated gene repression, suggesting that PRMT6 is able to asymmetrically dimethylate H2AR29 in vivo.178 These genes include EIF1b, MMP9, THBS1, and TNFRSF11B. THBS1 is of particular interest as it is dysregulated in a variety of cancers, specifically playing a role in angiogenesis.178
3.6.3. Methylation of H3R2
On the basis of current data, histone H3 is the most heavily modified histone, and the methylation of arginine residues in this protein follows this trend. PRMT6 dimethylates H3R2, and this modification alters the binding of several effector proteins that typically bind H3K4Me. These effectors, including JMJD2, and several Tudor- and PHD-domain-containing proteins, play a variety of roles in gene activation. It follows then that H3R2 methylation causes a change in the gene activation profile of the cell. These changes were characterized prominently by investigating the downstream effects of one particular effector protein, WDR5, which is an integral component of the MLL complex that catalyzes the methylation of H3K4. Interestingly, this WD40-repeat protein was shown to have decreased binding upon H3R2 dimethylation, resulting in a reduction in the activation of a number of target genes, including HoxA5 and cyclin D1.179 Interestingly, PRMT6 is able to methylate H3R2 regardless of the methylation state of H3K4, suggesting that the methylation of H3R2 has a dominant effect on H3K4 methylation.
PRMT5 also symmetrically dimethylates H3R2, and this modification promotes chromatin remodeling, exposing a binding site for a transcription factor called CREB (cAMP-response-element-binding protein). CREB is then phosphorylated by PKA, allowing for the activation of target genes involved in glucose metabolism, including G6pc, Pck1, and Ppargc1a.180 Expression of CARM1 is similarly necessary for expression of glucose homeostasis factors, including Gys1, Pgam2, and Pgym. This role, however, was not linked to a specific histone methylation event.181
3.6.4. Methylation of H3R8
PRMT5 has also been implicated in metabolic regulation via the dimethylation of H3R8. In both cell culture and primary adipocytes, ChIP results show that PRMT5 associates with PPARγ2 and PPARγ2-responsive promoter sequences, activating adipogenesis. In type 2 diabetes, PRMT5 regulates metabolic signals during a fasting state by associating with CRTC2, which directs it to target genes.182
3.6.5. Methylation of H3R17 and R26
CARM1 activity on H3R17 also regulates effector binding. In a recent study, asymmetric dimethylation of this site not only blocked association of the TIF family of corepressors, but prevented deacetylation by abrogating the interaction of H3 with the NuRD complex.183 Like PRMT5, CARM1 was also found to maintain pluripotency in mouse ESCs. Notably in this study, ChIP experiments using antibodies for CARM1 as well as for two different methylated H3 substrates, H3R26me2a and H3R17me2a, showed that CARM1 is recruited to the promoters of a variety of genes involved in differentiation. This more precisely implicates not only CARM1 but histone modification in gene activation.184 In another study, CARM1 was shown to be recruited to the creatine kinase promoter during skeletal myogenesis.185 Further studies indicate that CARM1 is specifically expressed during differentiation and recruited to the nucleus and subsequently to chromatin.185 Knockdown studies show that, in the absence of CARM1, the levels of other members of the transcription factor complex associated with creatine kinase activation are not expressed, giving another facet of the role of CARM1 in this process. Together, this study shows an enhancing role for CARM1 in muscle differentiation.185
3.6.6. Methylation of H4R3
H4R3 is methylated by a number of PRMTs, including PRMT1, PRMT5, and PRMT6. The first indication that the methylation of this residue could alter gene transcription began with reports investigating PRMT1 activity on purified H4 with varied levels of lysine acetylation.174 These studies showed that PRMT1 methylates unacetylated histones more efficiently than acetylated histones. This allowed Wang et al. to conclude that asymmetric dimethylation of H4R3 was likely a transcriptional activating event that promoted the acetylation of histone H4 by the histone acetyltransferase p300 to activate gene expression.174 The first report to clearly identify a role for a PRMT in tumorgenesis came from a study showing that PRMT1 methylates H4R3 as a part of the MLL complex in hematopoietic cells.186 With the introduction of the full MLL complex, these cells showed enhanced self-renewal when compared to those with an MLL complex with a catalytically dead PRMT1. This implicates PRMT1, specifically its role in dimethylating H4R3, in cell survival.186
By contrast with the results obtained for PRMT1, the symmetric dimethylation of H4R3 by PRMT5 is generally a repressive mark. For example, in an early study of arginine methylation, PRMT5 was shown to associate with the SWI/SNF (switch/sucrose nonfermentable) chromatin remodeling complex as well as with the Brg1 complex. As a part of this complex, PRMT5-mediated methylation of H4R3 displayed a repressive functionality specifically on the c-Myc target gene Cad.187 This report further implicated PRMT5 as playing a role in oncogenesis as well as being a part of a larger chromatin-modifying complex and thereby working in collaboration with other epigenetic transformations. A later bioinformatics study using ChIP-seq data analysis of histone methylation found PRMT5-mediated H4R3 dimethylation to be the second most repressive mark of the 20 lysine and arginine methylation events tested. Also shown in this study was the dependence of DNMT3A-mediated DNA methylation on the symmetric dimethylation of H4R3.188 DNA methylation by DNMT3A is also known to be a transcriptionally repressive modification.148 H4R3 methylation also affects the binding of the effector proteins SRP68/72 to H4. Interestingly, both asymmetric dimethylation and symmetric dimethylation of H4R3 inhibit the binding of the SRP68/72 heterodimer to chromatin both in vitro and in cells.189
3.7. Nonhistone Methylation in Epigenetic Regulation
PRMT1 also methylates ERα at R260, within its DNA-binding domain. This methylation event occurs in response to estradiol and allows for the association of ERα with PI3K and Src, leading to Akt1 activation. Moreover, in highly malignant ER+ breast cancer samples, ERα was found to be hypermethylated, suggesting a role for PRMT1 in breast cancer.138b BRCA1 is also methylated by PRMT1, which alters its binding to a variety of promoters, leading to increased binding to the APEX, ARHG, and GADD45G promoters, and a decrease in binding to ESR2, SREB, and FGF9 promoters.190
PRMT1 also affects mRNA processing by methylating FUS (fused in sarcoma).191 FUS is an mRNA-binding protein that is important for mRNA processing and shuttling RNAs from the nucleus to the cytoplasm. However, nuclear import of FUS relies upon its binding to transportin 1. The FUS/transportin 1 interaction is abrogated by a PRMT1-mediated methylation event, causing FUS to be trafficked to inclusion bodies that are found in amyotrophic lateral sclerosis (ALS) patients. PRMT1 knockdown increases FUS/transportin 1 binding as well as the nuclear localization of FUS.191
PRMT5-mediated methylation of the tumor suppressor p53 also helps to regulate the expression of p53 target genes. The sites of modification were mapped to R333, R335, and R337, and methylation at these sites induced cell cycle arrest, whereas the deletion of PRMT5 induced apoptosis.192 These data indicate that PRMT5-mediated methylation of p53 causes a change in the response to DNA damage, inducing cell proliferation.192 In a later study, the role of PRMT5 in p53 signaling was further investigated. Here, PRMT5 was found to be essential for p53 stability as well as for the expression of two p53 target genes, MDM2 and p21.193
3.8. Arginine Methylation in Cancer
Dysregulated PRMT expression and activity have been observed in a variety of cancers. Specifically, PRMT1, PRMT2, PRMT3, PRMT4, PRMT5, PRMT6, and PRMT7 have been shown to be overexpressed or otherwise contribute to tumorigenesis, while PRMT8 and PRMT9 have not yet been implicated in oncogenesis.
3.8.1. PRMT1 in Cancer
Recent studies have linked the increased expression of different PRMT1 splice variants (i.e., PRMT1v1 and PRMT1v2) to enhanced malignancy and poor prognosis.194 In one study, based on immunohistochemistry of primary tumor samples, overall PRMT1 expression was linked to the patient’s age, menopausal status, and progesterone receptor status.194a In the same study, low expression levels of PRMT1v1 were associated with increased survival. This research found no link between PRMT1v2 levels and survival rate.194a However, in a cell-based study using MCF7 cells, specific knockdown of PRMT1v2 increased apoptosis. Similarly, induced expression of PRMT1v2 promoted cell invasion in nonaggressive cell lines; this effect was not achieved by overexpressing other splice variants of PRMT1.194b Interestingly, both of these papers stress the importance of PRMT1 as a cytosolic methyltransferase and both report that PRMT1 was shown to be more malignant when expressed in the extranuclear environment. While the role of PRMT1 as a histone arginine methyltransferase has been clearly described in the literature, the effect of arginine methylation on general cell signaling is only beginning to be explored.
Dysregulation of PRMT1 has also been linked to breast cancer and leukemia, again suggesting that it is a potential therapeutic target.186 As mentioned previously, asymmetric dimethylation of H4R3 upregulates the expression of estrogen receptor target genes. While this response was first attributed to PRMT1, the increased expression of estrogen receptor target genes was later found to be promoted by either PRMT1 or CARM1.138a In this report, the protein complexes that associate with the pS2 promoter were identified by ChIP and reChIP analysis. In two of the six complexes, a type 1 PRMT was found, either PRMT1 or CARM1, but never both. In one of these complexes, PRMT1 and CARM1 were interchangeable. This complex, notably, also contains histone acetyltransferases. The other found complex only contained PRMT1 but also SWI/SNF chromatin remodeler proteins Brg1 and Ini1.138a
3.8.2. PRMT2 in Cancer
PRMT2 is also implicated in breast cancer relating to its ability to act as a transcriptional coactivator for ERα.195 In breast cancer cell lines, the levels of PRMT2 and a splice variant, PRMT2L2, were shown to be increased in ER+ lines. Upon overexpression of PRMT2L2, increased expression of ERα target genes was observed.195 It was later observed that different splice variants of PRMT2 have a distinct subcellular localization.
3.8.3. PRMT3 in Cancer
PRMT3 has been shown to be regulated post-transcriptionally by a tumor suppressor protein, DAL-1/4.1B, which inhibits its methyltransferase activity both in vitro and in cell culture.196 Overexpression of DAL-1/4.1B in MCF7 cells reduces PRMT3-catalyzed methylation of a variety of unidentified cellular proteins. As DAL-1/4.1B has been shown to have an antiproliferative role, this activity indirectly implicates PRMT3 in oncogenesis.196
3.8.4. PRMT4/CARM1 in Cancer
CARM1 levels have been shown to be increased in colon, prostate, and breast cancer.197 Like PRMT1, CARM1 plays a role in ER+ breast cancer by activating ERα target genes. Furthermore, CARM1 is necessary for this activation event as knockdown of CARM1 abrogates the estrogen response in mouse embryos.198 CARM1 has also been shown to be a marker for well-differentiated breast cancer, suggesting that CARM1 plays a role in reprogramming the epigenome of breast cancer cells. Overexpression of CARM1 in MCF7 cells causes a change in morphology as well as a change in the estradiol-induced gene signature.197c In another breast cancer study, CARM1 was shown to be necessary for E2F1 expression. E2F1, in turn, is required for cyclin E1 expression. The cyclin E1 promoter has high levels of both H3R17me2a and H3R26me2a, both of which are reduced upon CARM1 knockdown.199
CARM1 is also necessary for NFκB target gene expression as shown in MEF cells by CARM1 knockdown.200 NFκB is a transcription factor responsible for the regulation of genes involved in inflammation and cell survival. The effect of CARM1 was linked to H3R17me2a, though investigation into the potential role of H3R26me2a was not performed. These studies also showed a link between CARM1 and p300 acetyltransferase activity in NFκB recruitment and gene activation. Both enzymes are critical for this event.200
3.8.5. PRMT5 in Cancer
PRMT5 methylates both H4R3 and H3R8 in chronic lymphocytic lymphoma, causing transcriptional silencing of known target genes Rb1, Rbl1, and Rbl2.201 Knockdown of PRMT5 in a B-CLL cell line model decreases H4R3 and H3R8 dimethylation, increases protein expression of RBL2, and inhibits cell proliferation.201 Furthermore, PRMT5 overexpression has been linked to a number of cancers, including colon, lung, astrocytoma, and fibrosarcoma.202 Although it is unclear how PRMT5 promotes tumorigenesis, PRMT5 does regulate eIF4E expression and p53 function as a prosurvival factor.193 PRMT5 also associates with the SWI/SNF chromatin remodeling proteins, and this interaction has been suggested to be causal in dysregulating the expression of tumor suppressor genes, including ST7 and NM23.203 PRMT5 has also been shown to methylate NF-κB, promoting its gene regulation functionality in a colon cancer cell model. By mutating the target arginine on NF-κB to a lysine residue and therein blocking the ability of PRMT5 to methylate NF-κB, a more dramatic dysregulation of gene regulation was observed than by knocking down PRMT5. This indicates the necessity of arginine methylation for NF-κB activity.202a
Many of the functions of PRMT5 have been attributed to cytosolic and other nonhistone targets of arginine methylation. While PRMT5, as with the other isozymes, was initially characterized as a histone-modifying enzyme, the role of PRMT5 in the cellular environment is constantly evolving and appears to reside more frequently in the cytoplasm, not the nucleus. For example, Shilo et al. showed that PRMT5 mRNA and protein levels are dramatically increased in lung carcinoma tissues over normal lung tissues.202b Along with this trend, dimethylation of H4R3 also increases, illustrating the role of PRMT5 in tumor suppression. However, the more striking trend from this report indicates that there is more cytoplasmic PRMT5 present not only when comparing cancerous to normal lung tissues, but also in higher grade tumors with poorer prognosis.202b
3.8.6. PRMT7 in Cancer
As described previously, PRMT7-mediated methylation of H2AR3 affects DNA damage repair. The effects of inhibiting PRMT7 are complex and could result in resistance to certain chemotherapeutics.177 For these reasons, isozyme-specific PRMT inhibitors are essential.
3.9. Arginine Methylation in Atherosclerosis
Nitric oxide (NO) is a regulator of vasodilation known to be integral to cardiac health. The free amino acid l-arginine is the precursor for the generation of NO by the nitric oxide synthase (NOS) family of enzymes. However, when excess ADMA is present in the system due to the breakdown of proteins containing ADMA, this serves as an inhibitor of the NOS enzymes, lowering the production of NO.204 In the absence of l-arginine or presence of ADMA, NOS can also produce superoxide, which can have the opposite physiological effect compared to NO.205 In patients with heart disease, PRMT1 is observed at elevated levels, as well as a lowered expression of DDAH, which metabolizes ADMA. Both of these factors cause increases in ADMA levels and decreased vasodilation.206
4. Noncanonical Histone Arginine Modifications
4.1. Protein Arginine Phosphorylation
The phosphorylation of arginine residues in histone H3 was first reported in 1994.207 Although the underlying kinase could not be identified, using a partially purified kinase fraction derived from nuclear cell extracts of mouse leukemia cells, it was shown that histone H3 can be arginine phosphorylated at R2 as well as R128, R129, and R131 in the C-terminus of the protein.207 This modification introduces significant negative charge into the histone that undoubtedly would influence its DNA-binding ability and thereby chromatin structure. In contrast to other phosphorylated residues that are stable under acidic conditions (e.g., phosphoserine, phosphothreonine, and phosphotyrosine; O-phosphorylations), phosphoarginine, like other N-linked phosphorylation events, is an acid-labile modification. Therefore, current methods to directly detect protein arginine phosphorylation are sparse, and care has to be taken to preserve the acid-labile phospho mark during sample preparation and analysis.208
Although the molecular identity of the responsible eukaryotic protein arginine kinase is still unclear, it was recently demonstrated that protein arginine phosphorylation also occurs in bacteria.208a The underlying protein arginine kinase (PAK) was identified as McsB (EC 2.7.14.1) (Figure 47).208a This enzyme shows close homology to guanidine phosphotransferase enzymes such as creatine kinase and l-arginine kinase. Guanidine phosphotransferases are typically involved in energy homeostasis and shuttle the γ-phosphate from ATP onto small-molecule guanidine-containing compounds that serve as chemical energy storage devices in cells relying on high rates of energy turnover such as muscles and neurons.209
Figure 47.
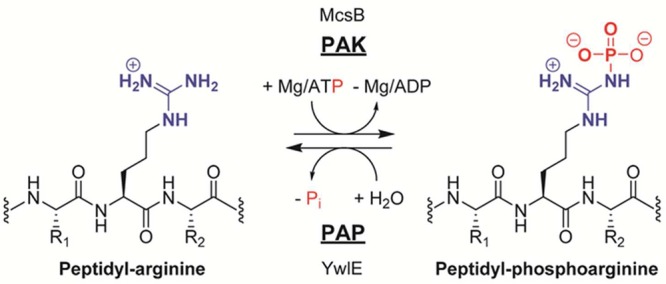
Protein arginine kinase McsB transfers the γ-phosphoryl group from ATP onto the arginine guanidinium group. The generated phosphoarginine residue can be hydrolyzed by the protein arginine phosphatase YwlE.
McsB shares several highly conserved residues which are important for nucleotide and substrate guanidinium binding with other guanidine phosphotransferases. However, it lacks the entire N-terminal domain that is critical for trapping small-molecule substrates, such as the amino acid l-arginine, thereby allowing larger peptidyl substrates to enter the active site.208a Besides McsB’s preference for peptidyl arginine residues, the catalytic mechanism is thought to be similar to that of l-arginine kinases that employ a direct, in-line phosphoryl transfer between ATP and the guanidinium group (Figure 48A). Notably, it was shown that l-arginine kinase binds arginine and ATP randomly, i.e., without a specific order, to the active site and that the resulting products phosphoarginine and ADP are individually released.210 Moreover, structural analysis of arginine kinase bound with a transition-state analogue, composed of nitrate, ADP, and l-arginine, revealed that five arginine residues form a dense network of electrostatic interactions with the phosphate groups and the γ-phosphate mimicking nitrate (Figure 48A).211 In addition, a tightly bound magnesium ion, which is essential for catalysis, is thought to coordinate all three ATP phosphate groups to properly orient the γ-phosphate for the transfer reaction to occur.
Figure 48.

(A) Active site of l-arginine kinase with bound ADP, nitrate, and l-arginine (PDB code 1BG0) and proposed reaction mechanism. Note that several arginine residues were omitted in the proposed reaction scheme for clarity. (B) Active site of YwlE C7S with bound peptidyl arginine and containing a phosphorylated S7 residue, which mimics the thiophosphate reaction intermediate generated after the first SN2 reaction (PDB code 4KK4). Residue R149* originates from a symmetry-equivalent molecule. Polar contacts of <3.5 Å are represented as dashed lines. The proposed catalytic mechanism for the PAP enzyme YwlE is shown on the right side. The guanidinium group of the incoming phosphoarginine substrate is colored in blue, whereas the phosphoryl group is shown in red.
The substrate arginine is aligned between two carboxylate groups originating from E225 and E314 that are both thought to act as general bases. However, mutagenesis studies suggest that base catalysis by these residues may enhance the catalytic rate but is not absolutely essential.212 It was suggested that proper substrate prealignment might be more important than acid/base chemistry, electrostatics, or other potential effects.212 Due to the polarization of the guanidinium group by the two glutamate residues, the substrate arginine Nω atom is highly nucleophilic and predisposed to attack the electrophilic phosphorus of the ATP γ-phosphate in a classical SN2 reaction. In addition, the positive charges of several arginine residues and the magnesium ion pull electrons toward the phosphate oxygens, away from the phosphorus, thereby increasing its electrophilicity.
Recently, a corresponding protein arginine phosphatase (PAP), YwlE (EC 3.9.1.2), was identified that can hydrolyze the phosphoramidate (P–N) bond of phosphoarginine, thereby releasing unmodified arginine (Figure 47).213 Notably, YwlE shares homology with the protein tyrosine phosphatase (PTP) family and utilizes a similar catalytic mechanism. In contrast to other PTP members, YwlE employs a dual size and polarity filter to select phosphoarginine residues.213a The highly polar substrate guanidinium group is sandwiched between the carboxylate of D118 and the hydroxyl of T11 and further stabilized by cation−π interactions with the phenyl ring of F120 (Figure 48B). The molecular mechanism of phosphoarginine hydrolysis consists of a two-step process comprising two consecutive SN2 reactions (Figure 48B).213a The incoming phosphoarginine phosphoryl group forms a bidentate bond with the guanidinium group of R13, whereas the phosphoarginine guanidinium group is clamped between the hydroxyl group of T11 and the carboxyl group of D118. The phosphorus atom of the substrate molecule is attacked by the highly nucleophilic active site cysteine residue C7, forming a thiophosphate reaction intermediate. This reaction is accompanied by the protonation of the arginine leaving group by D118. Subsequently, an incoming water molecule is deprotonated by D118, and the water-derived nucleophilic hydroxyl anion attacks the phosphocysteine residue, performing the second SN2 reaction, thereby releasing the phosphate ion and regenerating the active C7 thiolate anion. The active site cysteine was shown to be essential for catalysis and subject to oxidative regulation by the formation of a disulfide bridge with an adjacent backdoor cysteine.214
4.1.1. Physiological Role of Histone Arginine Phosphorylation
As shown by Wakim et al., histone H3 is subject to arginine phosphorylation by a Ca2+-calmodulin-dependent kinase derived from mouse leukemia cells.207 It was also demonstrated that in vivo 32P incorporation into H3 in rat heart endothelial cells results in phosphorylation of a basic amino acid in quiescent but not in dividing cells.215 This Ca2+-calmodulin-dependent kinase is present in nearly equal amounts in both quiescent and dividing cells; however, the histone H3 phosphorylating activity was 20–100-fold higher in quiescent cells.215 In addition, it was suggested that phosphorylation of histone H3 was involved in cell cycle exit.
The studies by Wakim et al. have, however, not been followed up, and there is still some uncertainty about the existence of a eukaryotic protein arginine kinase. Notably, the phosphorylation of arginine residues was solely derived from indirect methods, including the acid lability of 32P radiolabeling and a missing signal for arginine using Edman sequencing. However, to unequivocally prove the presence of phosphoarginine in histone proteins, direct phosphoarginine detection methods such as recently optimized mass spectrometry techniques or 31P NMR analysis should be employed.208a,216
4.2. Histone Arginine ADP-Ribosylation
ADP-ribosylation is a covalent PTM which is catalyzed by ADP-ribosyltransferases (ARTs) and is involved in several cellular processes such as cell cycle regulation and DNA damage response, replication, or transcription. The generation of ADP-ribosylated proteins requires nicotinamide adenine dinucleotide (NAD+) as a cofactor and leads to the formation of nicotinamide (Figure 49). ADP-ribosylation was shown to be reversibly regulated by the hydrolysis of the ADP-ribose group catalyzed by ADP-ribosyl hydrolase (ARH) enzymes. ARTs can either attach mono-ADP-ribosyl groups (catalyzed by mARTs) or poly-ADP-ribosyl groups (catalyzed by pARTs). In contrast to mARTs that only transfer a single ADP-ribose moiety onto a specific amino acid side chain, pARTs (also known as poly-ADP-ribose polymerases or PARPs), additionally catalyze the elongation and branching of ADP-ribose units on ADP-ribosylated targets.217 ADP-ribose can be linked to either negatively charged glutamates and aspartates via ester bonds that are highly sensitive to hydroxylamine or to positively charged arginine or lysine residues via N-glycosidic bonds that are resistant to hydroxylamine treatment as well as to cysteine and asparagine residues.218 Interestingly, most known mARTs transfer ADP-ribose onto arginine or lysine residues, whereas pARTs mainly target glutamate residues.219 Notably, ARTs are mainly extracellular enzymes that modify integrins and growth factor receptors. The only known intracellular enzymes possessing mono-ADP-ribosyltransferase activity are members of the sirtuin (SIRT) family of NAD+-dependent deacetylases. Sirtuins SIRT1, SIRT2, SIRT4, and SIRT6 harbor weak intrinsic mono-ADP-ribosylation activity, transferring a single ADP-ribose to an arginine residue of specific target proteins.220,221 There are three ARH and one poly-ADP-ribose glycohydrolase (PARG) known in humans. The poly-ADP-ribose polymer can be degraded by PARG and ARH3, which hydrolyze glycosidic bonds between two ADP-ribose units, thus removing ADP-ribose moieties from the polymers. The only enzyme able to release a mono-ADP-ribose moiety from ADP-ribosylated proteins is ARH1, which cleaves off a mono-ADP-ribose from arginine residues.222
Figure 49.
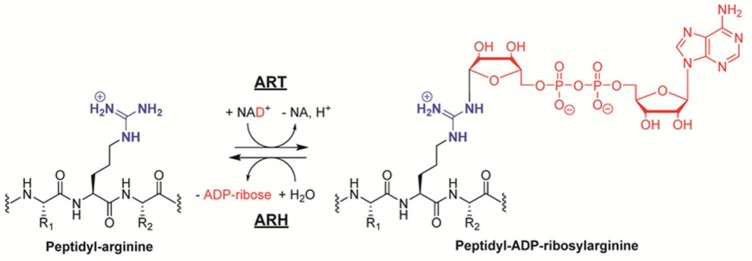
Generation of ADP-ribosylated arginines is catalyzed by ART enzymes, while the hydrolysis of peptidyl ADP-ribosylated arginine residues is mediated by ARH enzymes.
Several decades ago, it was shown that histone proteins can be ADP-ribosylated.223 There, it was reported that ADP-ribosylated histone proteins contain primarily ADP-ribose monomers or short oligomers rather than long polymers.223b Despite some evidence that histone proteins can be ADP-ribosylated on arginine residues, further studies are necessary to unequivocally prove the presence of ADP-ribosylated histones and to identify the underlying transferase enzyme(s).
4.3. Histone Arginylation
Histone proteins are also subject to protein arginylation, which represents the post-translational addition of an arginine residue. This type of modification affects the proteolytically processed and thereby exposed α-amine of histone H2B type 2-B at Q48 and A59, histone H4 at T136 and V116, and histone H2A.1 at S41, as well as histone H1 at L61.224 Interestingly, arginylation sites can be further modified by arginine methylation. On the basis of structural modeling, arginylation of histone proteins potentially facilitates the interaction of the histones with DNA due to the introduced positive charge of the additional arginine residue.224b Protein arginylation is mediated by the arginyl-tRNA-protein transferase 1 (ATE1), which transfers a single tRNA-bound arginine onto proteins (Figure 50).225 Typically, arginylation occurs at the unprotected N-terminal α-amino groups. However, following proteolytic processing, internal α-amino groups can be arginylated in vivo.224a It was proposed that the main function of protein arginylation is to mark target protein substrates for degradation by the ubiquitin-dependent N-end rule pathway.226 However, the detailed functions and the extent of histone arginylation are currently not known.
Figure 50.

Post-translational addition of arginine residues is catalyzed by ATE1.
5. Concluding Remarks
Epigenetic regulation governed by arginine modification is an emerging hallmark of eukaryotic organisms, and interference with the underlying enzymes holds great promise to intervene in various diseases ranging from cancer to rheumatoid arthritis. The abundance of various histone modifications on nucleosomes implies that crosstalk between these modifications is very likely. Different types of modifications occur on arginine residues, resulting in some form of antagonism since distinct types of modifications on arginines are mutually exclusive. In addition, the introduction of arginine modifications can either create or compromise a substrate recognition site for other histone-modifying enzymes even spanning different histone tails. The molecular details of such communication between modifications are a topic of intense research. In this respect, it is interesting to note that PAD-mediated histone citrullination functions as a general opponent of methylation by PRMTs. Depending on the methylation signal, i.e., activating or inhibiting, PADs might act as a repressor if activating arginine methylation is inhibited or an activator if the balance of PAD activity is shifted toward the suppression of a repressive arginine methylation mark.
Acknowledgments
This work was supported in part by NIH Grants GM079357, GM110394, and CA151304 (to P.R.T.), by an Austrian Science Fund (FWF) fellowship (Grant J 3548-B21) (to J.F.), and by an Eli Lilly LIFA fellowship (to J.W.K.).
Glossary
Abbreviations
- ABPP
activity-based protein profiling
- ADP
adenosine diphosphate
- AMP
adenosine monophosphate
- ARH
ADP-ribosyl hydrolase
- ART
ADP-ribosyltransferase
- ATE
arginyl-tRNA-protein transferase
- ATP
adenosine triphosphate
- ADMA
asymmetric dimethylarginine
- BAA
benzoyl-l-arginine amide
- bp
base pair
- Bz-ADMA
benzoyl-Nω,Nω-dimethylarginine
- CID
collision-induced dissociation
- ChIP
chromatin immunoprecipitation
- DFT
density functional theory
- DTT
dithiothreitol
- ER
estrogen receptor
- ES cells
embryonic stem cells
- ETD
electron-transferring dissociation
- FAD
flavin adenine dinucleotide
- Fluopol-ABPP
fluorescence polarization activity-based protein profiling
- HTS
high-throughput screening
- LSD
lysine-specific demethylases
- MMA
monomethylarginine
- MS
multiple sclerosis
- NAD
nicotinamide adenine dinucleotide
- NET
neutrophil extracellular trap
- NLS
nuclear localization signal
- PAD
protein arginine deiminase
- PAP
protein arginine phosphatase
- PAK
protein arginine kinase
- PCMB
p-chloromercuribenzoate
- PRMT
protein arginine methyltransferase
- PTM
post-translational modification
- PTP
protein tyrosine phosphatase
- QM
quantum mechanical
- RA
rheumatoid arthritis
- SAH
S-adenosyl-l-homocysteine
- SAM
S-adenosyl-l-methionine
- SAXS
small-angle X-ray scattering
- SDMA
symmetric dimethylarginine
- SN2 reaction
bimolecular nucleophilic substitution reaction
Biographies

Jakob Fuhrmann received his masters in molecular biology from the University of Vienna, Austria, in 2005. Afterward, he performed his doctoral studies in molecular/structural biology under the direction of Professor Tim Clausen at the Institute of Molecular Pathology (IMP), Vienna, Austria. His Ph.D. thesis was honored with several awards, including the Award of Excellence by the Austrian Ministry of Science and Research in 2010. In 2011, he joined the group of Professor Paul Thompson at the Scripps Research Institute, Scripps Florida. Jakob is currently a postdoctoral fellow, supported by an Erwin Schroedinger Fellowship. Previously, he also received a fellowship from the European Molecular Biology Organization (EMBO). His current interests cover the area of protein arginine phosphorylation, where he investigates the link between the chemical biology and the physiological role of this novel post-translational modification in diverse organisms.

Kathleen W. Clancy was born in 1985 in Prince Frederick, MD. She received her B.S. in chemistry with a concentration in biochemistry from Haverford College in 2007. While an undergraduate, Kate spent summers performing research under the mentorship of Dr. Solomon Snyder at Johns Hopkins Medical School in the Department of Neuroscience. She then received her Ph.D. in chemistry from Duke University, working under Dr. Dewey McCafferty conducting research on enzymes involved in bacterial pathogenesis and peptidoglycan recycling. Kate then moved to Scripps Florida to begin her postdoctoral research with Dr. Paul Thompson examining post-translational modification crosstalk with an emphasis on arginine functionalization. Kate received the Lilly Innovation Fellowship Award from Eli Lilly and Co. in 2013. She is currently working with Paul Thompson in the Department of Biochemistry and Molecular Pharmacology at the University of Massachusetts Medical School.

Paul R. Thompson is a Professor and the Director of Chemical Biology in the Department of Biochemistry and Molecular Pharmacology at the University of Massachusetts Medical School (UMASS Med) in Worcester, MA, where his research focuses on the development of novel therapeutics for a range of diseases, including cancer, rheumatoid arthritis, inflammatory bowel disease, and lupus. In particular, he is a world leader in the biology and biochemistry of the protein arginine deiminases. Paul received his B.Sc. (Summa Cum Laude) and Ph.D. degrees from McMaster University in Canada before moving to the United States to take a postdoctoral position with Philip Cole at the Johns Hopkins School of Medicine. Paul then moved to the University of South Carolina to begin his independent career before moving to the Department of Chemistry at The Scripps Research Institute, Scripps Florida, in May 2010. Paul subsequently moved to UMASS Med in August 2014. Paul has published more than 100 articles in major scientific journals, including Nature, Cell, Nature Structural and Molecular Biology, and the Journal of the American Chemical Society. Paul has also won a number of awards, including a Canadian Institutes of Health Research Postdoctoral Fellowship and a Camille Dreyfus Teacher-Scholar Award.
Special Issue
This paper is an additional review for Chem. Rev. 2015, 115, issue 6, “Epigenetics”.
The authors declare the following competing financial interest(s): P.R.T. is a cofounder and consultant to Padlock Therapeutics.
Funding Statement
National Institutes of Health, United States
References
- a Jenuwein T.; Allis C. D. Translating the histone code. Science 2001, 29355321074–80. [DOI] [PubMed] [Google Scholar]; b Strahl B. D.; Allis C. D. The language of covalent histone modifications. Nature 2000, 403676541–5. [DOI] [PubMed] [Google Scholar]
- Ruthenburg A. J.; Allis C. D.; Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol. Cell 2007, 25115–30. [DOI] [PubMed] [Google Scholar]
- Kouzarides T., SnapShot: histone-modifying enzymes. Cell 2007, 131 (4). [DOI] [PubMed] [Google Scholar]
- Arnaudo A. M.; Garcia B. A. Proteomic characterization of novel histone post-translational modifications. Epigenet. Chromatin 2013, 6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M.; Luo H.; Lee S.; Jin F.; Yang J. S.; Montellier E.; Buchou T.; Cheng Z.; Rousseaux S.; Rajagopal N.; Lu Z.; Ye Z.; Zhu Q.; Wysocka J.; Ye Y.; Khochbin S.; Ren B.; Zhao Y. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 14661016–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K.; Mader A. W.; Richmond R. K.; Sargent D. F.; Richmond T. J. Crystal structure of the nucleosome core particle at 2.8 angstrom resolution. Nature 1997, 3896648251–260. [DOI] [PubMed] [Google Scholar]
- a Lee W.; Tillo D.; Bray N.; Morse R. H.; Davis R. W.; Hughes T. R.; Nislow C. A high-resolution atlas of nucleosome occupancy in yeast. Nat. Genet. 2007, 39101235–1244. [DOI] [PubMed] [Google Scholar]; b Valouev A.; Johnson S. M.; Boyd S. D.; Smith C. L.; Fire A. Z.; Sidow A. Determinants of nucleosome organization in primary human cells. Nature 2011, 4747352516–U148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pray L. At the flick of a switch: epigenetic drugs. Chem. Biol. 2008, 157640–641. [DOI] [PubMed] [Google Scholar]; b Copeland R. A.; Solomon M. E.; Richon V. M. Protein methyltransferases as a target class for drug discovery. Nat. Rev. Drug Discovery 2009, 89724–732. [DOI] [PubMed] [Google Scholar]; c Khan O.; La Thangue N. B. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol. Cell Biol. 2012, 90185–94. [DOI] [PubMed] [Google Scholar]
- Jencks W. P.; Regenstein J.. Ionization Constants of Acids and Bases. In Handbook of Biochemistry and Molecular Biology, 4th ed.; CRC Press: Boca Raton, FL, 2010; pp 595–635. [Google Scholar]
- a Luscombe N. M.; Laskowski R. A.; Thornton J. M. Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 2001, 29132860–74. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Calnan B. J.; Tidor B.; Biancalana S.; Hudson D.; Frankel A. D. Arginine-mediated RNA recognition: the arginine fork. Science 1991, 25250091167–71. [DOI] [PubMed] [Google Scholar]
- Mandell D. J.; Chorny I.; Groban E. S.; Wong S. E.; Levine E.; Rapp C. S.; Jacobson M. P. Strengths of hydrogen bonds involving phosphorylated amino acid side chains. J. Am. Chem. Soc. 2007, 1294820–7. [DOI] [PubMed] [Google Scholar]
- Bannister A. J.; Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011, 213381–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossenaar E. R.; Zendman A. J.; van Venrooij W. J.; Pruijn G. J. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. BioEssays 2003, 25111106–18. [DOI] [PubMed] [Google Scholar]
- Hirofumi T.; Tomoharu G.; Bryan K.; Paul R T.; Oliver V.; Kazuya H.; Tatsurou M.; Kouichiro S.; Hiroyuki H.; Eiji S. Purification of enzymatically inactive peptidylarginine deiminase type 6 from mouse ovary that reveals hexameric structure different from other dimeric isoforms. Adv. Biosci. Biotechnol. 2011, 24304–310. [Google Scholar]
- Nakashima K.; Hagiwara T.; Ishigami A.; Nagata S.; Asaga H.; Kuramoto M.; Senshu T.; Yamada M. Molecular characterization of peptidylarginine deiminase in HL-60 cells induced by retinoic acid and 1alpha,25-dihydroxyvitamin D(3). J. Biol. Chem. 1999, 2743927786–92. [DOI] [PubMed] [Google Scholar]
- a Guerrin M.; Ishigami A.; Mechin M. C.; Nachat R.; Valmary S.; Sebbag M.; Simon M.; Senshu T.; Serre G. cDNA cloning, gene organization and expression analysis of human peptidylarginine deiminase type I. Biochem. J. 2003, 370, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ishigami A.; Ohsawa T.; Asaga H.; Akiyama K.; Kuramoto M.; Maruyama N. Human peptidylarginine deiminase type II: molecular cloning, gene organization, and expression in human skin. Arch. Biochem. Biophys. 2002, 407125–31. [DOI] [PubMed] [Google Scholar]; c Kanno T.; Kawada A.; Yamanouchi J.; Yosida-Noro C.; Yoshiki A.; Shiraiwa M.; Kusakabe M.; Manabe M.; Tezuka T.; Takahara H. Human peptidylarginine deiminase type III: Molecular cloning and nucleotide sequence of the cDNA, properties of the recombinant enzyme, and immunohistochemical localization in human skin. J. Invest. Dermatol. 2000, 1155813–823. [DOI] [PubMed] [Google Scholar]; d Chavanas S.; Mechin M. C.; Takahara H.; Kawada A.; Nachat R.; Serre G.; Simon M. Comparative analysis of the mouse and human peptidylarginine deiminase gene clusters reveals highly conserved non-coding segments and a new human gene, PADI6. Gene 2004, 330, 19–27. [DOI] [PubMed] [Google Scholar]; e Wright P. W.; Bolling L. C.; Calvert M. E.; Sarmento O. F.; Berkeley E. V.; Shea M. C.; Hao Z. L.; Jayes F. C.; Bush L. A.; Shetty J.; Shore A. N.; Reddi P. P.; Tung K. S.; Samy E.; Allietta M. M.; Sherman N. E.; Herr J. C.; Coonrod S. A. ePAD, an oocyte and early embryo-abundant peptidylarginine deiminase-like protein that localizes to egg cytoplasmic sheets. Dev. Biol. 2003, 256173–88. [DOI] [PubMed] [Google Scholar]
- a Nakashima K.; Hagiwara T.; Yamada M. Nuclear localization of peptidylarginine deiminase V and histone deimination in granulocytes. J. Biol. Chem. 2002, 2775149562–49568. [DOI] [PubMed] [Google Scholar]; b Cherrington B. D.; Morency E.; Struble A. M.; Coonrod S. A.; Wakshlag J. J. Potential Role for Peptidylarginine Deiminase 2 (PAD2) in Citrullination of Canine Mammary Epithelial Cell Histones. PLoS One 2010, 57e11768. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Jang B.; Shin H. Y.; Choi J. K.; Du P. T. N.; Jeong B. H.; Ishigami A.; Maruyama N.; Carp R. I.; Kim Y. S.; Choi E. K. Subcellular Localization of Peptidylarginine Deiminase 2 and Citrullinated Proteins in Brains of Scrapie-Infected Mice: Nuclear Localization of PAD2 and Membrane Fraction-Enriched Citrullinated Proteins. J. Neuropathol. Exp. Neurol. 2011, 702116–124. [DOI] [PubMed] [Google Scholar]
- a Shirai H.; Blundell T. L.; Mizuguchi K. A novel superfamily of enzymes that catalyze the modification of guanidino groups. Trends Biochem. Sci. 2001, 268465–468. [DOI] [PubMed] [Google Scholar]; b Shirai H.; Mokrab Y.; Mizuguchi K. The guanidino-group modifying enzymes: structural basis for their diversity and commonality. Proteins 2006, 6441010–1023. [DOI] [PubMed] [Google Scholar]; c Linsky T.; Fast W. Mechanistic similarity and diversity among the guanidine-modifying members of the pentein superfamily. Biochim. Biophys. Acta, Proteins Proteomics 2010, 1804101943–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney P. L.; Bhatia M.; Jones N. G.; Yuan L.; Glascock M. C.; Catchings K. L.; Yamada M.; Thompson P. R. Kinetic characterization of protein arginine deiminase 4: a transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry 2005, 443110570–10582. [DOI] [PubMed] [Google Scholar]
- Lewis C. A.; Wolfenden R. The Nonenzymatic Decomposition of Guanidines and Amidines. J. Am. Chem. Soc. 2014, 1361130–136. [DOI] [PubMed] [Google Scholar]
- Arita K.; Hashimoto H.; Shimizu T.; Nakashima K.; Yamada M.; Sato M. Structural basis for Ca2+-induced activation of human PAD4. Nat. Struct. Mol. Biol. 2004, 118777–783. [DOI] [PubMed] [Google Scholar]
- Liu Y. L.; Chiang Y. H.; Liu G. Y.; Hung H. C. Functional Role of Dimerization of Human Peptidylarginine Deiminase 4 (PAD4). PLoS One 2011, 66e21314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade D. J.; Fang P.; Dreyton C. J.; Zhang Y.; Fuhrmann J.; Rempel D.; Bax B. D.; Coonrod S. A.; Lewis H. D.; Guo M.; Gross M. L.; Thompson P. R. Protein Arginine Deiminase 2 Binds Calcium in an Ordered Fashion: Implications for Inhibitor Design. ACS Chem. Biol. 2015, 1041043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tour O.; Adams S. R.; Kerr R. A.; Meijer R. M.; Sejnowski T. J.; Tsien R. W.; Tsien R. Y. Calcium Green FlAsH as a genetically targeted small-molecule calcium indicator. Nat. Chem. Biol. 2007, 37423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrah E.; Giles J. T.; Ols M. L.; Bull H. G.; Andrade F.; Rosen A., Erosive Rheumatoid Arthritis Is Associated with Antibodies That Activate PAD4 by Increasing Calcium Sensitivity. Sci. Transl. Med. 2013, 5 (186). [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Murray-Rust J.; Leiper J.; McAlister M.; Phelan J.; Tilley S.; Maria J. S.; Vallance P.; McDonald N. Structural insights into the hydrolysis of cellular nitric oxide synthase inhibitors by dimethylarginine dimethylaminohydrolase. Nat. Struct. Mol. Biol. 2001, 88679–683. [DOI] [PubMed] [Google Scholar]; b Das K.; Butler G. H.; Kwiatkowski V.; Clark A. D.; Yadav P.; Arnold E. Crystal structures of arginine deiminase with covalent reaction intermediates: Implications for catalytic mechanism. Structure 2004, 124657–667. [DOI] [PubMed] [Google Scholar]
- a Linsky T. W.; Fast W., Guanidine-Modifying Enzymes in the Pentein Superfamily. Comprehensive Natural Products Chemistry II, Chemistry and Biology; Elsevier: Amsterdam, 2010; Vol. 8, pp 125–159. [Google Scholar]; b Frey D.; Braun O.; Briand C.; Vasak M.; Grutter M. G. Structure of the mammalian NOS regulator dimethylarginine dimethylaminohydrolase: a basis for the design of specific inhibitors. Structure 2006, 145901–911. [DOI] [PubMed] [Google Scholar]
- Thompson P. R.; Fast W. Histone citrullination by protein arginine deiminase: is arginine methylation a green light or a roadblock?. ACS Chem. Biol. 2006, 17433–441. [DOI] [PubMed] [Google Scholar]
- Teo C. Y.; Shave S.; Chor A. L. T.; Salleh A.; Rahman M. B. B.; Walkinshaw M. D.; Tejo B. A., Discovery of a new class of inhibitors for the protein arginine deiminase type 4 (PAD4) by structure-based virtual screening. BMC Bioinf. 2012, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Causey C. P.; Jones J. E.; Slack J. L.; Kamei D.; Jones L. E.; Subramanian V.; Knuckley B.; Ebrahimi P.; Chumanevich A. A.; Luo Y.; Hashimoto H.; Sato M.; Hofseth L. J.; Thompson P. R. The Development of N-α-(2-Carboxyl)benzoyl-N-5-(2-fluoro-1-iminoethyl)-l-ornithine Amide (o-F-amidine) and N-α-(2-Carboxyl)benzoyl-N-5-(2-chloro-1-iminoethyl)-l-ornithine Amide (o-Cl-amidine) as Second Generation Protein Arginine Deiminase (PAD) Inhibitors. J. Med. Chem. 2011, 54196919–6935. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Slack J. L.; Jones L. E.; Bhatia M. M.; Thompson P. R. Autodeimination of Protein Arginine Deiminase 4 Alters Protein–Protein Interactions but Not Activity. Biochemistry 2011, 50193997–4010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita K.; Shimizu T.; Hashimoto H.; Hidaka Y.; Yamada M.; Sato M. Structural basis for histone N-terminal recognition by human peptidylarginine deiminase 4. Proc. Natl. Acad. Sci. U.S.A. 2006, 103145291–5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khorasanizadeh S. The nucleosome: from genomic organization to genomic regulation. Cell 2004, 1162259–272. [DOI] [PubMed] [Google Scholar]
- a Knuckley B.; Bhatia M.; Thompson P. R. Protein arginine deiminase 4: evidence for a reverse protonation mechanism. Biochemistry 2007, 46226578–87. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Knuckley B.; Causey C. P.; Jones J. E.; Bhatia M.; Dreyton C. J.; Osborne T. C.; Takahara H.; Thompson P. R. Substrate specificity and kinetic studies of PADs 1, 3, and 4 identify potent and selective inhibitors of protein arginine deiminase 3. Biochemistry 2010, 49234852–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisaki M.; Sugawara K. Properties of peptidylarginine deiminase from the epidermis of newborn rats. J. Biochem. 1981, 891257–63. [DOI] [PubMed] [Google Scholar]
- a Luo Y.; Arita K.; Bhatia M.; Knuckley B.; Lee Y. H.; Stallcup M. R.; Sato M.; Thompson P. R. Inhibitors and inactivators of protein arginine deiminase 4: functional and structural characterization. Biochemistry 2006, 453911727–11736. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jones J. E.; Slack J. L.; Fang P. F.; Zhang X. S.; Subramanian V.; Causey C. P.; Coonrod S. A.; Guo M.; Thompson P. R. Synthesis and Screening of a Haloacetamidine Containing Library To Identify PAD4 Selective Inhibitors. ACS Chem. Biol. 2012, 71160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyton C. J.; Knuckley B.; Jones J. E.; Lewallen D. M.; Thompson P. R. Mechanistic Studies of Protein Arginine Deiminase 2: Evidence for a Substrate-Assisted Mechanism. Biochemistry 2014, 53274426–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone E. M.; Costello A. L.; Tierney D. L.; Fast W. Substrate-assisted cysteine deprotonation in the mechanism of dimethylargininase (DDAH) from Pseudomonas aeruginosa. Biochemistry 2006, 45175618–5630. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wysocka J.; Sayegh J.; Lee Y. H.; Perlin J. R.; Leonelli L.; Sonbuchner L. S.; McDonald C. H.; Cook R. G.; Dou Y.; Roeder R. G.; Clarke S.; Stallcup M. R.; Allis C. D.; Coonrod S. A. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 2004, 3065694279–283. [DOI] [PubMed] [Google Scholar]
- a Hidaka Y.; Hagiwara T.; Yamada M. Methylation of the guanidino group of arginine residues prevents citrullination by peptidylarginine deiminase IV. FEBS Lett. 2005, 579194088–4092. [DOI] [PubMed] [Google Scholar]; b Raijmakers R.; Zendman A. J. W.; Egberts W. V.; Vossenaar E. R.; Raats J.; Soede-Huijbregts C.; Rutjes F. P. J. T.; van Veelen P. A.; Drijfhout J. W.; Pruijn G. J. M. Methylation of arginine residues interferes with citrullination by peptidylarginine deiminases in vitro. J. Mol. Biol. 2007, 36741118–1129. [DOI] [PubMed] [Google Scholar]
- Lluis M.; Wang Y.; Monzingo A. F.; Fast W.; Robertus J. D. Characterization of C-Alkyl Amidines as Bioavailable Covalent Reversible Inhibitors of Human DDAH-1. ChemMedChem 2011, 6181–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbert G. L.; Daujat S.; Snowden A. W.; Erdjument-Bromage H.; Hagiwara T.; Yamada M.; Schneider R.; Gregory P. D.; Tempst P.; Bannister A. J.; Kouzarides T. Histone deimination antagonizes arginine methylation. Cell 2004, 1185545–553. [DOI] [PubMed] [Google Scholar]
- a Allis C. D.; Bowen J. K.; Abraham G. N.; Glover C. V. C.; Gorovsky M. A. Proteolytic Processing of Histone-H3 in Chromatin—a Physiologically Regulated Event in Tetrahymena Micronuclei. Cell 1980, 20155–64. [DOI] [PubMed] [Google Scholar]; b Jin J. J.; Cai Y.; Li B.; Conaway R. C.; Workman J. L.; Conaway J. W.; Kusch T. In and out: histone variant exchange in chromatin. Trends Biochem. Sci. 2005, 3012680–687. [DOI] [PubMed] [Google Scholar]; c Bannister A. J.; Schneider R.; Kouzarides T. Histone methylation: dynamic or static?. Cell 2002, 1097801–806. [DOI] [PubMed] [Google Scholar]
- a Wu C. Y.; Lee H. J.; Wu S. H.; Chen S. T.; Chiou S. H.; Chang G. G. Chemical mechanism of the endogenous argininosuccinate lyase activity of duck lens delta2-crystallin. Biochem. J. 1998, 333, 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Goto M.; Omi R.; Miyahara I.; Sugahara M.; Hirotsu K. Structures of argininosuccinate synthetase in enzyme-ATP substrates and enzyme-AMP product forms—stereochemistry of the catalytic reaction. J. Biol. Chem. 2003, 2782522964–22971. [DOI] [PubMed] [Google Scholar]; c Sampaleanu L. M.; Codding P. W.; Lobsanov Y. D.; Tsai M.; Smith G. D.; Horvatin C.; Howell P. L. Structural studies of duck δ2 crystallin mutants provide insight into the role of Thr(161) and the 280s loop in catalysis. Biochem. J. 2004, 384, 437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Morris S. M. Regulation of enzymes of the urea cycle and arginine metabolism. Annu. Rev. Nutr. 2002, 22, 87–105. [DOI] [PubMed] [Google Scholar]
- Pritzker L. B.; Moscarello M. A. A novel microtubule independent effect of paclitaxel: the inhibition of peptidylarginine deiminase from bovine brain. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1998, 13881154–160. [DOI] [PubMed] [Google Scholar]
- Knuckley B.; Luo Y.; Thompson P. R. Profiling protein arginine deiminase 4 (PAD4): a novel screen to identify PAD4 inhibitors. Bioorg. Med. Chem. 2008, 162739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdag M.; Dreker T.; Henry C.; Tosco P.; Vallaro M.; Fruttero R.; Scozzafava A.; Carta F.; Supuran C. T. Novel small molecule protein arginine deiminase 4 (PAD4) inhibitors. Bioorg. Med. Chem. Lett. 2013, 233715–719. [DOI] [PubMed] [Google Scholar]
- Ferretti P.; Pong U. K.; Vagaska B.; Merchant R.; Matthews C. J.; Marson C. M. Discovery of a structurally novel, drug-like and potent inhibitor of peptidylarginine deiminase. MedChemComm 2013, 471109–1113. [Google Scholar]
- Ghorai P.; Kraus A.; Keller M.; Gotte C.; Igel P.; Schneider E.; Schnell D.; Bernhardt G.; Dove S.; Zabel M.; Elz S.; Seifert R.; Buschauer A. Acylguanidines as Bioisosteres of Guanidines: NG-Acylated Imidazolylpropylguanidines, a New Class of Histamine H2 Receptor Agonists. J. Med. Chem. 2008, 51227193–7204. [DOI] [PubMed] [Google Scholar]
- Lewallen D. M.; Bicker K. L.; Madoux F.; Chase P.; Anguish L.; Coonrod S.; Hodder P.; Thompson P. R. A FluoPol-ABPP PAD2 High-Throughput Screen Identifies the First Calcium Site Inhibitor Targeting the PADs. ACS Chem. Biol. 2014, 94913–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis H. D.; Liddle J.; Coote J. E.; Atkinson S. J.; Barker M. D.; Bax B. D.; Bicker K. L.; Bingham R. P.; Campbell M.; Chen Y. H.; Chung C. W.; Craggs P. D.; Davis R. P.; Eberhard D.; Joberty G.; Lind K. E.; Locke K.; Maller C.; Martinod K.; Patten C.; Polyakova O.; Rise C. E.; Rudiger M.; Sheppard R. J.; Slade D. J.; Thomas P.; Thorpe J.; Yao G.; Drewes G.; Wagner D. D.; Thompson P. R.; Prinjha R. K.; Wilson D. M. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 2015, 113189–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone E. M.; Schaller T. H.; Bianchi H.; Person M. D.; Fast W. Inactivation of two diverse enzymes in the amidinotransferase superfamily by 2-chloroacetamidine: dimethylargininase and peptidylarginine deiminase. Biochemistry 2005, 444213744–13752. [DOI] [PubMed] [Google Scholar]
- Luo Y.; Knuckley B.; Lee Y. H.; Stallcup M. R.; Thompson P. R. A fluoroacetamidine-based inactivator of protein arginine deiminase 4: design, synthesis, and in vitro and in vivo evaluation. J. Am. Chem. Soc. 2006, 12841092–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuckley B.; Causey C. P.; Pellechia P. J.; Cook P. F.; Thompson P. R. Haloacetamidine-Based Inactivators of Protein Arginine Deiminase 4 (PAD4): Evidence that General Acid Catalysis Promotes Efficient Inactivation. ChemBioChem 2010, 112161–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.; Liu C.; Lin J. Theoretical study of the mechanism of protein arginine deiminase 4 (PAD4) inhibition by F-amidine. J. Mol. Graphics Modell. 2015, 55, 25–32. [DOI] [PubMed] [Google Scholar]
- Bicker K. L.; Anguish L.; Chumanevich A. A.; Cameron M. D.; Cui X. L.; Witalison E.; Subramanian V.; Zhang X. S.; Chumanevich A. P.; Hofseth L. J.; Coonrod S. A.; Thompson P. R. d-Amino Acid-Based Protein Arginine Deiminase Inhibitors: Synthesis, Pharmacokinetics, and in Cellulo Efficacy. ACS Med. Chem. Lett. 2012, 3121081–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura K. Specificity and Mode of Action of the Muscle-Type Protein-Arginine Deiminase. Arch. Biochem. Biophys. 1992, 2932362–369. [DOI] [PubMed] [Google Scholar]
- Wang Y. J.; Li P. X.; Wang S.; Hu J.; Chen X. A.; Wu J. H.; Fisher M.; Oshaben K.; Zhao N.; Gu Y.; Wang D.; Chen G.; Wang Y. M. Anticancer Peptidylarginine Deiminase (PAD) Inhibitors Regulate the Autophagy Flux and the Mammalian Target of Rapamycin Complex 1 Activity. J. Biol. Chem. 2012, 2873125941–25953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight J. S.; Subramanian V.; O’Dell A. A.; Yalavarthi S.; Zhao W.; Smith C. K.; Hodgin J. B.; Thompson P. R.; Kaplan M. J.. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann. Rheum. Dis. 2014, DOI: 10.1136/annrheumdis-2014-205365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y.; Knuckley B.; Bhatia M.; Pellechia P. J.; Thompson P. R. Activity-based protein profiling reagents for protein arginine deiminase 4 (PAD4): synthesis and in vitro evaluation of a fluorescently labeled probe. J. Am. Chem. Soc. 2006, 1284514468–14469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack J. L.; Causey C. P.; Luo Y.; Thompson P. R. Development and Use of Clickable Activity Based Protein Profiling Agents for Protein Arginine Deiminase 4. ACS Chem. Biol. 2011, 65466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuckley B.; Jones J. E.; Bachovchin D. A.; Slack J.; Causey C. P.; Brown S. J.; Rosen H.; Cravatt B. F.; Thompson P. R.; fluopol-ABPP A. HTS assay to identify PAD inhibitors. Chem. Commun. (Cambridge, U.K.) 2010, 46387175–7177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyton C. J.; Anderson E. D.; Subramanian V.; Boger D. L.; Thompson P. R. Insights into the mechanism of streptonigrin-induced protein arginine deiminase inactivation. Bioorg. Med. Chem. 2014, 2241362–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara T.; Hidaka Y.; Yamada M. Deimination of histone H2A and H4 at arginine 3 in HL-60 granulocytes. Biochemistry 2005, 44155827–5834. [DOI] [PubMed] [Google Scholar]
- Zhang X. S.; Bolt M.; Guertin M. J.; Chen W.; Zhang S.; Cherrington B. D.; Slade D. J.; Dreyton C. J.; Subramanian V.; Bicker K. L.; Thompson P. R.; Mancini M. A.; Lis J. T.; Coonrod S. A. Peptidylarginine deiminase 2-catalyzed histone H3 arginine 26 citrullination facilitates estrogen receptor alpha target gene activation. Proc. Natl. Acad. Sci. U.S.A. 2012, 1093313331–13336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P. X.; Yao H. J.; Zhang Z. Q.; Li M.; Luo Y.; Thompson P. R.; Gilmour D. S.; Wang Y. M. Regulation of p53 target gene expression by peptidylarginine deiminase 4. Mol. Cell. Biol. 2008, 28154745–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.; Wang D.; Yao H.; Doret P.; Hao G.; Shen Q.; Qiu H.; Zhang X.; Wang Y.; Chen G.; Wang Y. Coordination of PAD4 and HDAC2 in the regulation of p53-target gene expression. Oncogene 2010, 29213153–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H.; Li P.; Venters B. J.; Zheng S.; Thompson P. R.; Pugh B. F.; Wang Y. Histone Arg modifications and p53 regulate the expression of OKL38, a mediator of apoptosis. J. Biol. Chem. 2008, 2832920060–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P.; Azebi S.; England P.; Christensen T.; M?ller-Larsen A.; Petersen T.; Batsche E.; Muchardt C., Citrullination of Histone H3 Interferes with HP1-Mediated Transcriptional Repression. PLoS Genet. 2012, 8 (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodziej S.; Kuvardina O. N.; Oellerich T.; Herglotz J.; Backert I.; Kohrs N.; Buscato E.; Wittmann S. K.; Salinas-Riester G.; Bonig H.; Karas M.; Serve H.; Proschak E.; Lausen J., PADI4 acts as a coactivator of Tal1 by counteracting repressive histone arginine methylation. Nat. Commun. 2014, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophorou M. A.; Castelo-Branco G.; Halley-Stott R. P.; Oliveira C. S.; Loos R.; Radzisheuskaya A.; Mowen K. A.; Bertone P.; Silva J. C. R.; Zernicka-Goetz M.; Nielsen M. L.; Gurdon J. B.; Kouzarides T. Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature 2014, 5077490104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slade D. J.; Subramanian V.; Thompson P. R. Pluripotency: citrullination unravels stem cells. Nat. Chem. Biol. 2014, 105327–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi N.; Neeli I.; Schall N.; Wan H.; Desiderio D. M.; Csernok E.; Thompson P. R.; Dali H.; Briand J. P.; Muller S.; Radic M. Deimination of linker histones links neutrophil extracellular trap release with autoantibodies in systemic autoimmunity. FASEB J. 2014, 2872840–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin M. J.; Zhang X.; Anguish L.; Kim S.; Varticovski L.; Lis J. T.; Hager G. L.; Coonrod S. A. Targeted H3R26 deimination specifically facilitates estrogen receptor binding by modifying nucleosome structure. PLoS Genet. 2014, 109e1004613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X. S.; Gamble M. J.; Stadler S.; Cherrington B. D.; Causey C. P.; Thompson P. R.; Roberson M. S.; Kraus W. L.; Coonrod S. A., Genome-Wide Analysis Reveals PADI4 Cooperates with Elk-1 To Activate c-Fos Expression in Breast Cancer Cells. PLoS Genet. 2011, 7 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade F.; Darrah E.; Gucek M.; Cole R. N.; Rosen A.; Zhu X. M. Autocitrullination of Human Peptidyl Arginine Deiminase Type 4 Regulates Protein Citrullination during Cell Activation. Arthritis Rheum. 2010, 6261630–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanikawa C.; Espinosa M.; Suzuki A.; Masuda K.; Yamamoto K.; Tsuchiya E.; Ueda K.; Daigo Y.; Nakamura Y.; Matsuda K.. Regulation of histone modification and chromatin structure by the p53-PADI4 pathway. Nat. Commun. 2012, 3, No. 676. [DOI] [PubMed] [Google Scholar]
- Tanikawa C.; Ueda K.; Nakagawa H.; Yoshida N.; Nakamura Y.; Matsuda K. Regulation of Protein Citrullination through p53/PAD14 Network in DNA Damage Response. Cancer Res. 2009, 69228761–8769. [DOI] [PubMed] [Google Scholar]
- a Hemmers S.; Teijaro J. R.; Arandjelovic S.; Mowen K. A. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS One 2011, 67e22043. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Neeli I.; Khan S. N.; Radic M. Histone deimination as a response to inflammatory stimuli in neutrophils. J. Immunol. 2008, 18031895–1902. [DOI] [PubMed] [Google Scholar]; c Li P. X.; Li M.; Lindberg M. R.; Kennett M. J.; Xiong N.; Wang Y. M. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 20791853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wang Y. M.; Li M.; Stadler S.; Correll S.; Li P. X.; Wang D. C.; Hayama R.; Leonelli L.; Han H.; Grigoryev S. A.; Allis C. D.; Coonrod S. A. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 1842205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Khandpur R.; Carmona-Rivera C.; Vivekanandan-Giri A.; Gizinski A.; Yalavarthi S.; Knight J. S.; Friday S.; Li S.; Patel R. M.; Subramanian V.; Thompson P.; Chen P. J.; Fox D. A.; Pennathur S.; Kaplan M. J., NETs Are a Source of Citrullinated Autoantigens and Stimulate Inflammatory Responses in Rheumatoid Arthritis. Sci. Transl. Med. 2013, 5 (178). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V.; Reichard U.; Goosmann C.; Fauler B.; Uhlemann Y.; Weiss D. S.; Weinrauch Y.; Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science 2004, 30356631532–1535. [DOI] [PubMed] [Google Scholar]
- Hirsch J. G. Bactericidal Action of Histone. J. Exp. Med. 1958, 1086925–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Knight J. S.; Zhao W. P.; Luo W.; Subramanian V.; O’Dell A. A.; Yalavarthi S.; Hodgin J. B.; Eitzman D. T.; Thompson P. R.; Kaplan M. J. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J. Clin. Invest. 2013, 12372981–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Knight J. S.; Luo W.; O’Dell A. A.; Yalavarthi S.; Zhao W. P.; Subramanian V.; Guo C. A.; Grenn R. C.; Thompson P. R.; Eitzman D. T.; Kaplan M. J. Peptidylarginine Deiminase Inhibition Reduces Vascular Damage and Modulates Innate Immune Responses in Murine Models of Atherosclerosis. Circ. Res. 2014, 1146947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bicker K. L.; Thompson P. R. The protein arginine deiminases: structure, function, inhibition, and disease. Biopolymers 2013, 992155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang S.; Wang Y. M. Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim. Biophys. Acta, Gene Regul. Mech. 2013, 1829101126–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gyorgy B.; Toth E.; Tarcsa E.; Falus A.; Buzas E. I. Citrullination: a posttranslational modification in health and disease. Int. J. Biochem. Cell Biol. 2006, 38101662–1677. [DOI] [PubMed] [Google Scholar]; d Jones J. E.; Causey C. P.; Knuckley B.; Slack-Noyes J. L.; Thompson P. R. Protein arginine deiminase 4 (PAD4): current understanding and future therapeutic potential. Curr. Opin. Drug Discovery Dev. 2009, 125616–627. [PMC free article] [PubMed] [Google Scholar]
- a Esteller M. Molecular origins of cancer: epigenetics in cancer. N. Engl. J. Med. 2008, 358111148–1159. [DOI] [PubMed] [Google Scholar]; b Chi P.; Allis C. D.; Wang G. G. Covalent histone modifications—miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 2010, 107457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chang X. T.; Han J. X. Expression of peptidylarginine deiminase type 4 (PAD4) in various tumors. Mol. Carcinog. 2006, 453183–196. [DOI] [PubMed] [Google Scholar]; b Chang X. T.; Han J. X.; Pang L.; Zhao Y.; Yang Y.; Shen Z. L., Increased PADI4 expression in blood and tissues of patients with malignant tumors. BMC Cancer 2009, 9;. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wang L.; Chang X. T.; Yuan G. Y.; Zhao Y.; Wang P. C. Expression of Peptidylarginine Deiminase Type 4 in Ovarian Tumors. Int. J. Biol. Sci. 2010, 65454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hollstein M.; Sidransky D.; Vogelstein B.; Harris C. C. P53 Mutations in Human Cancers. Science 1991, 253501549–53. [DOI] [PubMed] [Google Scholar]; b Levine A. J.; Momand J.; Finlay C. A. The P53 Tumor Suppressor Gene. Nature 1991, 3516326453–456. [DOI] [PubMed] [Google Scholar]; c Vogelstein B.; Lane D.; Levine A. J. Surfing the p53 network. Nature 2000, 4086810307–310. [DOI] [PubMed] [Google Scholar]
- Slack J. L.; Causey C. P.; Thompson P. R. Protein arginine deiminase 4: a target for an epigenetic cancer therapy. Cell. Mol. Life Sci. 2011, 684709–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElwee J. L.; Mohanan S.; Griffith O. L.; Breuer H. C.; Anguish L. J.; Cherrington B. D.; Palmer A. M.; Howe L. R.; Subramanian V.; Causey C. P.; Thompson P. R.; Gray J. W.; Coonrod S. A.. Identification of PADI2 as a potential breast cancer biomarker and therapeutic target. BMC Cancer 2012, 12, No. 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A.; Yamada R.; Chang X.; Tokuhiro S.; Sawada T.; Suzuki M.; Nagasaki M.; Nakayama-Hamada M.; Kawaida R.; Ono M.; Ohtsuki M.; Furukawa H.; Yoshino S.; Yukioka M.; Tohma S.; Matsubara T.; Wakitani S.; Teshima R.; Nishioka Y.; Sekine A.; Iida A.; Takahashi A.; Tsunoda T.; Nakamura Y.; Yamamoto K. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat. Genet. 2003, 344395–402. [DOI] [PubMed] [Google Scholar]
- Aletaha D.; Neogi T.; Silman A. J.; Funovits J.; Felson D. T.; Bingham C. O.; Birnbaum N. S.; Burmester G. R.; Bykerk V. P.; Cohen M. D.; Combe B.; Costenbader K. H.; Dougados M.; Emery P.; Ferraccioli G.; Hazes J. M. W.; Hobbs K.; Huizinga T. W. J.; Kavanaugh A.; Kay J.; Kvien T. K.; Laing T.; Mease P.; Menard H. A.; Moreland L. W.; Naden R. L.; Pincus T.; Smolen J. S.; Stanislawska-Biernat E.; Symmons D.; Tak P. P.; Upchurch K. S.; Vencovsky J.; Wolfe F.; Hawker G. 2010 Rheumatoid Arthritis Classification Criteria: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. Arthritis Rheum. 2010, 6292569–2581. [DOI] [PubMed] [Google Scholar]
- a Schellekens G. A.; Visser H.; de Jong B. A. W.; Van den Hoogen F. H. J.; Hazes J. M. W.; Breedveld F. C.; van Venrooij W. J. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000, 431155–163. [DOI] [PubMed] [Google Scholar]; b Machold K. P.; Stamm T. A.; Nell V. P. K.; Pflugbeil S.; Aletaha D.; Steiner G.; Uffmann M.; Smolen J. S. Very recent onset rheumatoid arthritis: clinical and serological patient characteristics associated with radiographic progression over the first years of disease. Rheumatology 2007, 462342–349. [DOI] [PubMed] [Google Scholar]
- Willis V.; Gizinski A. M.; Banda N. K.; Causey C. P.; Knuckley B.; Cordova K. N.; Luo Y. A.; Levitt B.; Glogowska M.; Chandra P.; Kulik L.; Robinson W. H.; Arend W. P.; Thompson P. R.; Holers V. M. N-α-Benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine Amide, a Protein Arginine Deiminase Inhibitor, Reduces the Severity of Murine Collagen-Induced Arthritis. J. Immunol. 2011, 18674396–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chumanevich A. A.; Causey C. P.; Knuckley B. A.; Jones J. E.; Poudyal D.; Chumanevich A. P.; Davis T.; Matesic L. E.; Thompson P. R.; Hofseth L. J. Suppression of colitis in mice by Cl-amidine: a novel peptidylarginine deiminase inhibitor. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 3006G929–G938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkon K. B.; Wiedeman A. Type I IFN system in the development and manifestations of SLE. Curr. Opin. Rheumatol. 2012, 245499–505. [DOI] [PubMed] [Google Scholar]
- a Martinod K.; Demers M.; Fuchs T. A.; Wong S. L.; Brill A.; Gallant M.; Hu J.; Wang Y. M.; Wagner D. D. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl. Acad. Sci. U.S.A. 2013, 110218674–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Savchenko A. S.; Borissoff J. I.; Martinod K.; De Meyer S. F.; Gallant M.; Erpenbeck L.; Brill A.; Wang Y. M.; Wagner D. D. VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood 2014, 1231141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zheng X. L. DNase, ADAMTS13, and iPAD4: good for the heart. Blood 2014, 123110–11. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Martinod K.; Wagner D. D. Thrombosis: tangled up in NETs. Blood 2014, 123182768–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wood D. D.; Bilbao J. M.; OConnors P.; Moscarello M. A. Acute multiple sclerosis (Marburg type) is associated with developmentally immature myelin basic protein. Ann. Neurol. 1996, 40118–24. [DOI] [PubMed] [Google Scholar]; b Mastronardi F. G.; Mak B.; Ackerley C. A.; Roots B. I.; Moscarello M. A. Modifications of myelin basic protein in DM20 transgenic mice are similar to those in myelin basic protein from multiple sclerosis. J. Clin. Invest. 1996, 972349–358. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Moscarello M. A.; Pritzker L.; Mastronardi F. G.; Wood D. D. Peptidylarginine deiminase: a candidate factor in demyelinating disease. J. Neurochem. 2002, 812335–343. [DOI] [PubMed] [Google Scholar]; d Musse A. A.; Li Z.; Ackerley C. A.; Bienzle D.; Lei H.; Poma R.; Harauz G.; Moscarello M. A.; Mastronardi F. G. Peptidylarginine deiminase 2 (PAD2) overexpression in transgenic mice leads to myelin loss in the central nervous system. Dis. Models Mech. 2008, 14–5229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscarello M. A.; Lei H.; Mastronardi F. G.; Winer S.; Tsui H.; Li Z.; Ackerley C.; Zhang L.; Raijmakers R.; Wood D. D. Inhibition of peptidyl-arginine deiminases reverses protein-hypercitrullination and disease in mouse models of multiple sclerosis. Dis. Models Mech. 2013, 62467–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a van Boekel M. A. M.; Vossenaar E. R.; van den Hoogen F. H. J.; van Venrooij W. J. Autoantibody systems in rheumatoid arthritis: specificity, sensitivity and diagnostic value. Arthritis Res. 2002, 4287–93. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Steiner G.; Smolen J., Autoantibodies in rheumatoid arthritis and their clinical significance. Arthritis Res. Ther. 2002, 4;. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Suzuki A.; Yamada R.; Yamamoto K. Citrullination by peptidylarginine deiminase in rheumatoid arthritis. Ann. N.Y. Acad. Sci. 2007, 1108, 323–339. [DOI] [PubMed] [Google Scholar]; d Wegner N.; Lundberg K.; Kinloch A.; Fisher B.; Malmstrom V.; Feldmann M.; Venables P. J. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol. Rev. 2010, 233, 34–54. [DOI] [PubMed] [Google Scholar]; e Bicker K. L.; Subramanian V.; Chumanevich A. A.; Hofseth L. J.; Thompson P. R. Seeing Citrulline: Development of a Phenylglyoxal-Based Probe To Visualize Protein Citrullination. J. Am. Chem. Soc. 2012, 1344117015–17018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Chen D. G.; Ma H.; Hong H.; Koh S. S.; Huang S. M.; Schurter B. T.; Aswad D. W.; Stallcup M. R. Regulation of transcription by a protein methyltransferase. Science 1999, 28454232174–2177. [DOI] [PubMed] [Google Scholar]; b Bedford M. T.; Richard S. Arginine methylation: an emerging regulator of protein function. Mol. Cell 2005, 183263–272. [DOI] [PubMed] [Google Scholar]; c Wysocka J.; Allis C. D.; Coonrod S. Histone arginine methylation and its dynamic regulation. Front. Biosci. 2006, 11, 344–355. [DOI] [PubMed] [Google Scholar]; d Bedford M. T.; Clarke S. G. Protein Arginine Methylation in Mammals: Who, What, and Why. Mol. Cell 2009, 3311–13. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Di Lorenzo A.; Bedford M. T. Histone arginine methylation. FEBS Lett. 2011, 585132024–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Torres-Padilla M. E.; Parfitt D. E.; Kouzarides T.; Zernicka-Goetz M. Histone arginine methylation regulates pluripotency in the early mouse embryo. Nature 2007, 4457124214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boffa L. C.; Karn J.; Vidali G.; Allfrey V. G. Distribution of NG,NG-Dimethylarginine in Nuclear Protein-Fractions. Biochem. Biophys. Res. Commun. 1977, 743969–976. [DOI] [PubMed] [Google Scholar]
- Yang Y. Z.; Bedford M. T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13137–50. [DOI] [PubMed] [Google Scholar]
- a Cook J. R.; Lee J. H.; Yang Z. H.; Krause C. D.; Herth N.; Hoffmann R.; Pestka S. FBXO11/PRMT9, a new protein arginine methyltransferase, symmetrically dimethylates arginine residues. Biochem. Biophys. Res. Commun. 2006, 3422472–481. [DOI] [PubMed] [Google Scholar]; b Krause C. D.; Yang Z. H.; Kim Y. S.; Lee J. H.; Cook J. R.; Pestka S. Protein arginine methyltransferases: evolution and assessment of their pharmacological and therapeutic potential. Pharmacol. Ther. 2007, 113150–87. [DOI] [PubMed] [Google Scholar]; c Richon V. M.; Johnston D.; Sneeringer C. J.; Jin L.; Majer C. R.; Elliston K.; Jerva L. F.; Scott M. P.; Copeland R. A. Chemogenetic Analysis of Human Protein Methyltransferases. Chem. Biol. Drug Des. 2011, 782199–210. [DOI] [PubMed] [Google Scholar]
- Fielenbach N.; Guardavaccaro D.; Neubert K.; Chan T.; Li D. L.; Feng Q.; Hutter H.; Pagano M.; Antebi A. DRE-1: an evolutionarily conserved F box protein that regulates C-elegans developmental age. Dev. Cell 2007, 123443–455. [DOI] [PubMed] [Google Scholar]
- Wang Y. C.; Li C. Evolutionarily conserved protein arginine methyltransferases in non-mammalian animal systems. FEBS J. 2012, 2796932–45. [DOI] [PubMed] [Google Scholar]
- Angyal S. J.; Warburton W. K., The Basic Strengths of Methylated Guanidines. J. Chem. Soc. 1951. (Sept), 2492–2494. [Google Scholar]
- Tripsianes K.; Madl T.; Machyna M.; Fessas D.; Englbrecht C.; Fischer U.; Neugebauer K. M.; Sattler M. Structural basis for dimethylarginine recognition by the Tudor domains of human SMN and SPF30 proteins. Nat. Struct. Mol. Biol. 2011, 18121414–U136. [DOI] [PubMed] [Google Scholar]
- Kessler H. Detection of Intramolecular Mobility by NMR Spectroscopy 0.13. Detection of Hindered Rotation and Inversion by NMR Spectroscopy. Angew. Chem., Int. Ed. Engl. 1970, 93219–&. [Google Scholar]
- a Liu H. P.; Wang J. Y. S.; Huang Y.; Li Z. Z.; Gong W. M.; Lehmann R.; Xu R. M. Structural basis for methylarginine-dependent recognition of Aubergine by Tudor. Genes Dev. 2010, 24171876–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Liu K.; Chen C.; Guo Y. H.; Lam R.; Bian C. B.; Xu C.; Zhao D. Y.; Jin J.; MacKenzie F.; Pawson T.; Min J. R. Structural basis for recognition of arginine methylated Piwi proteins by the extended Tudor domain. Proc. Natl. Acad. Sci. U.S.A. 2010, 1074318398–18403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tang J.; Frankel A.; Cook R. J.; Kim S.; Paik W. K.; Williams K. R.; Clarke S.; Herschman H. R. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J. Biol. Chem. 2000, 275117723–7730. [DOI] [PubMed] [Google Scholar]; b Pawlak M. R.; Scherer C. A.; Chen J.; Roshon M. J.; Ruley H. E. Arginine N-methyltransferase 1 is required for early postimplantation mouse development, but cells deficient in the enzyme are viable. Mol. Cell. Biol. 2000, 20134859–4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Branscombe T. L.; Frankel A.; Lee J. H.; Cook J. R.; Yang Z. H.; Pestka S.; Clarke S. PRMT5 (Janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J. Biol. Chem. 2001, 2763532971–32976. [DOI] [PubMed] [Google Scholar]; b Zurita-Lopez C. I.; Sandberg T.; Kelly R.; Clarke S. G. Human Protein Arginine Methyltransferase 7 (PRMT7) Is a Type III Enzyme Forming ω-NG-Monomethylated Arginine Residues. J. Biol. Chem. 2012, 287117859–7870. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yang Y.; Hadjikyriacou A.; Xia Z.; Gayatri S.; Kim D.; Zurita-Lopez C.; Kelly R.; Guo A.; Li W.; Clarke S. G.; Bedford M. T. PRMT9 is a type II methyltransferase that methylates the splicing factor SAP145. Nat. Commun. 2015, 6, 6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zobel-Thropp P.; Gary J. D.; Clarke S. δ-N-Methylarginine is a novel posttranslational modification of arginine residues in yeast proteins. J. Biol. Chem. 1998, 2734529283–29286. [DOI] [PubMed] [Google Scholar]
- Miranda T. B.; Miranda M.; Frankel A.; Clarke S. PRMT7 is a member of the protein arginine methyltransferase family with a distinct substrate specificity. J. Biol. Chem. 2004, 2792222902–22907. [DOI] [PubMed] [Google Scholar]
- a Zhang X.; Zhou L.; Cheng X. D. Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. EMBO J. 2000, 19143509–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Weiss V. H.; McBride A. E.; Soriano M. A.; Filman D. J.; Silver P. A.; Hogle J. M. The structure and oligomerization of the yeast arginine methyltransferase, Hmt1. Nat. Struct. Mol. Biol. 2000, 7121165–1171. [DOI] [PubMed] [Google Scholar]; c Zhang X.; Cheng X. D. Structure of the predominant protein arginine methyltransferase PRMT1 and analysis of its binding to substrate peptides. Structure 2003, 115509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Yue W. W.; Hassler M.; Roe S. M.; Thompson-Vale V.; Pearl L. H. Insights into histone code syntax from structural and biochemical studies of CARM1 methyltransferase. EMBO J. 2007, 26204402–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Troffer-Charlier N.; Cura V.; Hassenboehler P.; Moras D.; Cavarelli J. Functional insights from structures of coactivator-associated arginine methyltransferase 1 domains. EMBO J. 2007, 26204391–4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M.; Toma-Fukai S.; Kim J. D.; Fukamizu A.; Shimizu T. Protein arginine methyltransferase 7 has a novel homodimer-like structure formed by tandem repeats. FEBS Lett. 2014, 588101942–1948. [DOI] [PubMed] [Google Scholar]
- Cura V.; Troffer-Charlier N.; Wurtz J. M.; Bonnefond L.; Cavarelli J. Structural insight into arginine methylation by the mouse protein arginine methyltransferase 7: a zinc finger freezes the mimic of the dimeric state into a single active site. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2014, 70, 2401–2412. [DOI] [PubMed] [Google Scholar]
- Wang C. Y.; Zhu Y. W.; Caceres T. B.; Liu L.; Peng J. H.; Wang J. C.; Chen J. J.; Chen X. W.; Zhang Z. Y.; Zuo X. B.; Gong Q. G.; Teng M. K.; Hevel J. M.; Wu J. H.; Shi Y. Y. Structural Determinants for the Strict Monomethylation Activity by Trypanosoma brucei Protein Arginine Methyltransferase 7. Structure 2014, 225756–768. [DOI] [PubMed] [Google Scholar]
- Schubert H. L.; Blumenthal R. M.; Cheng X. D. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 2003, 286329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X. D.; Collins R. E.; Zhang X. Structural and sequence motifs of protein (histone) methylation enzymes. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 267–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shien D. M.; Lee T. Y.; Chang W. C.; Hsu J. B. K.; Horng J. T.; Hsu P. C.; Wang T. Y.; Huang H. D. Incorporating Structural Characteristics for Identification of Protein Methylation Sites. J. Comput. Chem. 2009, 3091532–1543. [DOI] [PubMed] [Google Scholar]
- Kolbel K.; Ihling C.; Kuhn U.; Neundorf I.; Otto S.; Stichel J.; Robaa D.; Beck-Sickinger A. G.; Sinz A.; Wahle E. Peptide Backbone Conformation Affects the Substrate Preference of Protein Arginine Methyltransferase I. Biochemistry 2012, 51275463–5475. [DOI] [PubMed] [Google Scholar]
- Antonysamy S.; Bonday Z.; Campbell R. M.; Doyle B.; Druzina Z.; Gheyi T.; Han B.; Jungheim L. N.; Qian Y. W.; Rauch C.; Russell M.; Sauder J. M.; Wasserman S. R.; Weichert K.; Willard F. S.; Zhang A. P.; Emtage S. Crystal structure of the human PRMT5:MEP50 complex. Proc. Natl. Acad. Sci. U.S.A. 2012, 1094417960–17965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thandapani P.; O’Connor T. R.; Bailey T. L.; Richard S. Defining the RGG/RG Motif. Mol. Cell 2013, 505613–623. [DOI] [PubMed] [Google Scholar]
- Cheng D. H.; Cote J.; Shaaban S.; Bedford M. T. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol. Cell 2007, 25171–83. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Maity R.; Whitelegge J. P.; Hadjikyriacou A.; Li Z. W.; Zurita-Lopez C.; Al-Hadid Q.; Clark A. T.; Bedford M. T.; Masson J. Y.; Clarke S. G. Mammalian Protein Arginine Methyltransferase 7 (PRMT7) Specifically Targets RXR Sites in Lysine- and Arginine-Rich Regions. J. Biol. Chem. 2013, 2885237010–37025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wooderchak W. L.; Zang T. Z.; Zhou Z. S.; Acuna M.; Tahara S. M.; Hevel J. M. Substrate profiling of PRMT1 reveals amino acid sequences that extend beyond the “RGG” paradigm. Biochemistry 2008, 47369456–9466. [DOI] [PubMed] [Google Scholar]; b Osborne T. C.; Obianyo O.; Zhang X.; Cheng X.; Thompson P. R. Protein arginine methyltransferase 1: positively charged residues in substrate peptides distal to the site of methylation are important for substrate binding and catalysis. Biochemistry 2007, 464613370–13381. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wang M.; Fuhrmann J.; Thompson P. R. Protein Arginine Methyltransferase 5 Catalyzes Substrate Dimethylation in a Distributive Fashion. Biochemistry 2014, 53507884–7892. [DOI] [PubMed] [Google Scholar]
- Lee D. Y.; Ianculescu I.; Purcell D.; Zhang X.; Cheng X. D.; Stallcup M. R. Surface-scanning mutational analysis of protein arginine methyltransferase 1: roles of specific amino acids in methyltransferase substrate specificity, oligomerization, and coactivator function. Mol. Endocrinol. 2007, 2161381–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel A.; Yadav N.; Lee J. H.; Branscombe T. L.; Clarke S.; Bedford M. T. The novel human protein arginine N-methyltransferase PRMT6 is a nuclear enzyme displaying unique substrate specificity. J. Biol. Chem. 2002, 27753537–3543. [DOI] [PubMed] [Google Scholar]
- Gui S. Y.; Wooderchak-Donahue W. L.; Zang T. Z.; Chen D.; Daly M. P.; Zhou Z. S.; Hevel J. M. Substrate-Induced Control of Product Formation by Protein Arginine Methyltransferase 1. Biochemistry 2013, 521199–209. [DOI] [PubMed] [Google Scholar]
- Burgos E. S.; Wilczek C.; Onikubo T.; Bonanno J. B.; Jansong J.; Reimer U.; Shechter D.. Histone H2A and H4 N-Terminal Tails Are Positioned by the MEP50 WD-Repeat Protein for Efficient Methylation by the PRMT5 Arginine Methyltransferase J. Biol. Chem. 2015, DOI: 10.1074/jbc.M115.636894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L. T.; Wang M. Z.; Lv Z. Y.; Yang N.; Liu Y. F.; Bao S. L.; Gong W. M.; Xu R. M. Structural insights into protein arginine symmetric dimethylation by PRMT5. Proc. Natl. Acad. Sci. U.S.A. 2011, 1085120538–20543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui S.; Gathiaka S.; Li J.; Qu J.; Acevedo O.; Hevel J. M. A remodeled protein arginine methyltransferase 1 (PRMT1) generates symmetric dimethylarginine. J. Biol. Chem. 2014, 289139320–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Xu R. M.; Thompson P. R. Substrate Specificity, Processivity, and Kinetic Mechanism of Protein Arginine Methyltransferase 5. Biochemistry 2013, 52325430–5440. [DOI] [PubMed] [Google Scholar]
- a Obianyo O.; Osborne T. C.; Thompson P. R. Kinetic mechanism of protein arginine methyltransferase 1. Biochemistry 2008, 473910420–10427. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Obianyo O.; Thompson P. R. Kinetic mechanism of protein arginine methyltransferase 6 (PRMT6). J. Biol. Chem. 2012, 28786062–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakowski T. M.; Frankel A. A kinetic study of human protein arginine N-methyltransferase 6 reveals a distributive mechanism. J. Biol. Chem. 2008, 2831510015–10025. [DOI] [PubMed] [Google Scholar]
- Rust H. L.; Zurita-Lopez C. I.; Clarke S.; Thompson P. R. Mechanistic Studies on Transcriptional Coactivator Protein Arginine Methyltransferase 1. Biochemistry 2011, 50163332–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R. H.; Li X.; Liang Z. J.; Zhu K. K.; Lu J. Y.; Kong X. Q.; Ouyang S. S.; Li L.; Zheng Y. G.; Luo C., Theoretical Insights into Catalytic Mechanism of Protein Arginine Methyltransferase 1. PLoS One 2013, 8 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang B. S.; Chen Y.; Zhao Y. M.; Bruick R. K. JMJD6 is a histone arginine demethylase. Science 2007, 3185849444–447. [DOI] [PubMed] [Google Scholar]
- a Webby C. J.; Wolf A.; Gromak N.; Dreger M.; Kramer H.; Kessler B.; Nielsen M. L.; Schmitz C.; Butler D. S.; Yates J. R.; Delahunty C. M.; Hahn P.; Lengeling A.; Mann M.; Proudfoot N. J.; Schofield C. J.; Bottger A. Jmjd6 Catalyses Lysyl-Hydroxylation of U2AF65, a Protein Associated with RNA Splicing. Science 2009, 325593690–93. [DOI] [PubMed] [Google Scholar]; b Hahn P.; Wegener I.; Burrells A.; Bose J.; Wolf A.; Erck C.; Butler D.; Schofield C. J.; Bottger A.; Lengeling A., Analysis of Jmjd6 Cellular Localization and Testing for Its Involvement in Histone Demethylation. PLoS One 2010, 5 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Metivier R.; Penot G.; Hubner M. R.; Reid G.; Brand H.; Kos M.; Gannon F. Estrogen receptor-α directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 2003, 1156751–763. [DOI] [PubMed] [Google Scholar]; b Le Romancer M.; Treilleux I.; Leconte N.; Robin-Lespinasse Y.; Sentis S.; Bouchekioua-Bouzaghou K.; Goddard S.; Gobert-Gosse S.; Corbo L. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol. Cell 2008, 312212–221. [DOI] [PubMed] [Google Scholar]
- Hou H. F.; Yu H. T. Structural insights into histone lysine demethylation. Curr. Opin. Struct. Biol. 2010, 206739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.; Lan F.; Matson C.; Mulligan P.; Whetstine J. R.; Cole P. A.; Casero R. A.; Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 1197941–53. [DOI] [PubMed] [Google Scholar]
- Tsukada Y.; Fang J.; Erdjument-Bromage H.; Warren M. E.; Borchers C. H.; Tempst P.; Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 4397078811–6. [DOI] [PubMed] [Google Scholar]
- a Friesen W. J.; Massenet S.; Paushkin S.; Wyce A.; Dreyfuss G. SMN, the product of the spinal muscular atrophy gene, binds preferentially to dimethylarginine-containing protein targets. Mol. Cell 2001, 751111–1117. [DOI] [PubMed] [Google Scholar]; b Sprangers R.; Groves M. R.; Sinning L.; Sattler M. High-resolution X-ray and NMR structures of the SMN tudor domain: conformational variation in the binding site for symmetrically dimethylated arginine residues. J. Mol. Biol. 2003, 3272507–520. [DOI] [PubMed] [Google Scholar]; c Cote J.; Richard S. Tudor domains bind symmetrical dimethylated arginines. J. Biol. Chem. 2005, 2803128476–28483. [DOI] [PubMed] [Google Scholar]
- Ma J. C.; Dougherty D. A. The cation−π interaction. Chem. Rev. (Washington, DC, U.S.) 1997, 9751303–1324. [DOI] [PubMed] [Google Scholar]
- Hughes R. M.; Waters M. L. Arginine methylation in a β-hairpin peptide: implications for Arg−π interactions, ΔCp°, and the cold denatured state. J. Am. Chem. Soc. 2006, 1283912735–42. [DOI] [PubMed] [Google Scholar]
- Hughes R. M.; Wiggins K. R.; Khorasanizadeh S.; Waters M. L. Recognition of trimethyllysine by a chromodomain is not driven by the hydrophobic effect. Proc. Natl. Acad. Sci. U.S.A. 2007, 1042711184–11188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. Z.; Lu Y.; Espejo A.; Wu J. C.; Xu W.; Liang S. D.; Bedford M. T. TDRD3 Is an Effector Molecule for Arginine-Methylated Histone Marks. Mol. Cell 2010, 4061016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.; Nott T. J.; Jin J.; Pawson T. Deciphering arginine methylation: Tudor tells the tale. Nat. Rev. Mol. Cell Biol. 2011, 1210629–642. [DOI] [PubMed] [Google Scholar]
- Zhao Q.; Rank G.; Tan Y. T.; Li H. T.; Moritz R. L.; Simpson R. J.; Cerruti L.; Curtis D. J.; Patel D. J.; Allis C. D.; Cunningham J. M.; Jane S. M. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat. Struct. Mol. Biol. 2009, 163304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani J.; Nankumo T.; Arita K.; Inamoto S.; Ariyoshi M.; Shirakawa M. Structural basis for recognition of H3K4 methylation status by the DNA methyltransferase 3A ATRX-DNMT3-DNMT3L domain. EMBO Rep. 2009, 10111235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng D.; Yadav N.; King R. W.; Swanson M. S.; Weinstein E. J.; Bedford M. T. Small molecule regulators of protein arginine methyltransferases. J. Biol. Chem. 2004, 2792323892–9. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Li M. Y.; Wang B. H.; Zheng Y. G. Discovery and Mechanistic Study of a Class of Protein Arginine Methylation Inhibitors. J. Med. Chem. 2010, 53166028–6039. [DOI] [PubMed] [Google Scholar]
- Spannhoff A.; Machmur R.; Heinke R.; Trojer P.; Bauer I.; Brosch G.; Schule R.; Hanefeld W.; Sippl W.; Jung M. A novel arginine methyltransferase inhibitor with cellular activity. Bioorg. Med. Chem. Lett. 2007, 17154150–4153. [DOI] [PubMed] [Google Scholar]
- Spannhoff A.; Heinke R.; Bauer I.; Trojer P.; Metzger E.; Gust R.; Schule R.; Brosch G.; Sippl W.; Jung M. Target-based approach to inhibitors of histone arginine methyltransferases. J. Med. Chem. 2007, 50102319–2325. [DOI] [PubMed] [Google Scholar]
- Bissinger E. M.; Heinke R.; Spannhoff A.; Eberlin A.; Metzger E.; Cura V.; Hassenboehler P.; Cavarelli J.; Schule R.; Bedford M. T.; Sippl W.; Jung M. Acyl derivatives of p-aminosulfonamides and dapsone as new inhibitors of the arginine methyltransferase hPRMT1. Bioorg. Med. Chem. 2011, 19123717–3731. [DOI] [PubMed] [Google Scholar]
- Osborne T.; Roska R. L.; Rajski S. R.; Thompson P. R. In situ generation of a bisubstrate analogue for protein arginine methyltransferase 1. J. Am. Chem. Soc. 2008, 130144574–5. [DOI] [PubMed] [Google Scholar]
- Weller R. L.; Rajski S. R. Design, synthesis, and preliminary biological evaluation of a DNA methyltransferase-directed alkylating agent. ChemBioChem 2006, 72243–5. [DOI] [PubMed] [Google Scholar]
- ’T Hart P.; Lakowski T. M.; Thomas D.; Frankel A.; Martin N. I. Peptidic Partial Bisubstrates as Inhibitors of the Protein Arginine N-Methyltransferases. ChemBioChem 2011, 1291427–1432. [DOI] [PubMed] [Google Scholar]
- Dowden J.; Hong W.; Parry R. V.; Pike R. A.; Ward S. G. Toward the development of potent and selective bisubstrate inhibitors of protein arginine methyltransferases. Bioorg. Med. Chem. Lett. 2010, 2072103–5. [DOI] [PubMed] [Google Scholar]
- van Haren M.; van Ufford L. Q.; Moret E. E.; Martin N. I. Synthesis and evaluation of protein arginine N-methyltransferase inhibitors designed to simultaneously occupy both substrate binding sites. Org. Biomol. Chem. 2015, 132549–60. [DOI] [PubMed] [Google Scholar]
- Yan L. L.; Yan C. L.; Qian K.; Su H. R.; Kofsky-Wofford S. A.; Lee W. C.; Zhao X. Y.; Ho M. C.; Ivanov I.; Zheng Y. G. Diamidine Compounds for Selective Inhibition of Protein Arginine Methyltransferase 1. J. Med. Chem. 2014, 5762611–2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obianyo O.; Causey C. P.; Osborne T. C.; Jones J. E.; Lee Y. H.; Stallcup M. R.; Thompson P. R. A Chloroacetamidine-Based Inactivator of Protein Arginine Methyltransferase 1: Design, Synthesis, and in Vitro and in Vivo Evaluation. ChemBioChem 2010, 1191219–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerapana E.; Wang C.; Simon G. M.; Richter F.; Khare S.; Dillon M. B. D.; Bachovchin D. A.; Mowen K.; Baker D.; Cravatt B. F. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 2010, 4687325790–U79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust H. L.Decoding PRMT1: Studies On the Catalytic Mechanism, Regulation, Inhibition, and Crosstalk of PRMT1-Dependent Methylation. Ph.D. Thesis, University of South Carolina, 2013. [Google Scholar]
- Bicker K. L.; Obianyo O.; Rust H. L.; Thompson P. R. A combinatorial approach to characterize the substrate specificity of protein arginine methyltransferase 1. Mol. BioSyst. 2011, 7148–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siarheyeva A.; Senisterra G.; Allali-Hassani A.; Dong A.; Dobrovetsky E.; Wasney G. A.; Chau I.; Marcellus R.; Hajian T.; Liu F.; Korboukh I.; Smil D.; Bolshan Y.; Min J.; Wu H.; Zeng H.; Loppnau P.; Poda G.; Griffin C.; Aman A.; Brown P. J.; Jin J.; Al-Awar R.; Arrowsmith C. H.; Schapira M.; Vedadi M. An allosteric inhibitor of protein arginine methyltransferase 3. Structure 2012, 2081425–35. [DOI] [PubMed] [Google Scholar]
- Kaniskan H. U.; Szewczyk M. M.; Yu Z.; Eram M. S.; Yang X.; Schmidt K.; Luo X.; Dai M.; He F.; Zang I.; Lin Y.; Kennedy S.; Li F.; Dobrovetsky E.; Dong A.; Smil D.; Min S. J.; Landon M.; Lin-Jones J.; Huang X. P.; Roth B. L.; Schapira M.; Atadja P.; Barsyte-Lovejoy D.; Arrowsmith C. H.; Brown P. J.; Zhao K.; Jin J.; Vedadi M. A Potent, Selective and Cell-Active Allosteric Inhibitor of Protein Arginine Methyltransferase 3 (PRMT3). Angew. Chem., Int. Ed. 2015, 54175166–5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Purandare A. V.; Chen Z.; Huynh T.; Pang S.; Geng J.; Vaccaro W.; Poss M. A.; Oconnell J.; Nowak K.; Jayaraman L. Pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1). Bioorg. Med. Chem. Lett. 2008, 18154438–4441. [DOI] [PubMed] [Google Scholar]; b Wan H.; Huynh T.; Pang S.; Geng J.; Vaccaro W.; Poss M. A.; Trainor G. L.; Lorenzi M. V.; Gottardis M.; Jayaraman L.; Purandare A. V. Benzo[d]imidazole inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1)—Hit to Lead Studies. Bioorg. Med. Chem. Lett. 2009, 19175063–6. [DOI] [PubMed] [Google Scholar]
- Sack J. S.; Thieffine S.; Bandiera T.; Fasolini M.; Duke G. J.; Jayaraman L.; Kish K. F.; Klei H. E.; Purandare A. V.; Rosettani P.; Troiani S.; Xie D. L.; Bertrand J. A. Structural basis for CARM1 inhibition by indole and pyrazole inhibitors. Biochem. J. 2011, 436, 331–339. [DOI] [PubMed] [Google Scholar]
- a Duncan K. W.; Chesworth R.; Boriack-Sjodin P. A.; Jin L.. Methods of inhibiting PRMT5. WO2014100764A2, 2014.; b Duncan K. W.; Chesworth R.; Munchhof M. J.. Preparation of PRMT5 inhibitors useful in the treatment of cancer and metabolic disorder. WO2014100716A1, 2014.; c Duncan K. W.; Chesworth R.; Boriack-Sjodin P. A.; Munchhof M. J.; Jin L.. Preparation of N-[3-(3,4-Dihydroisoquinolin-2(1H)-yl)-2-hydroxypropyl]arylcarboxamide as PRMT5 inhibitors. WO2014100719A2, 2014.
- Penebre E.; Kuplast K. G.; Majer C. R.; Johnston L. D.; Rioux N.; Munchhof M.; Jin L.; Boriak-Sjodin P. A.; Wigle T. J.; Jacques S. L.; West K. A.; Lingaraj T.; Stickland K.; Ribich S. A.; Raimondi A.; Porter-Scott M.; Waters N. J.; Pollock R. M.; Smith J. J.; Barbash O.; Kruger R. G.; Copeland R. A.; Moyer M. P.; Chesworth R.; Duncan K. W.. Identification of a First-in-Class PRMT5 Inhibitor with Potent in Vitro and in Vivo Activity in Preclinical Models of Mantle Cell Lymphoma, 56th ASH Annual Meeting; American Society of Hematology: Washington, DC, 2014; p 438. [Google Scholar]
- Alinari L.; Mahasenan K. V.; Yan F.; Karkhanis V.; Chung J. H.; Smith E. M.; Quinion C.; Smith P. L.; Kim L.; Patton J. T.; Lapalombella R.; Yu B.; Wu Y.; Roy S.; De Leo A.; Pileri S.; Agostinelli C.; Ayers L.; Bradner J. E.; Chen-Kiang S.; Elemento O.; Motiwala T.; Majumder S.; Byrd J. C.; Jacob S.; Sif S.; Li C.; Baiocchi R. A. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood 2015, 125162530–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obianyo O.; Causey C. P.; Jones J. E.; Thompson P. R. Activity-Based Protein Profiling of Protein Arginine Methyltransferase 1. ACS Chem. Biol. 2011, 6101127–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon M. B. C.; Bachovchin D. A.; Brown S. J.; Finn M. G.; Rosen H.; Cravatt B. F.; Mowen K. A. Novel Inhibitors for PRMT1 Discovered by High-Throughput Screening Using Activity-Based Fluorescence Polarization. ACS Chem. Biol. 2012, 771198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. B.; Huang Z. Q.; Xia L.; Feng Q.; Erdjument-Bromage H.; Strahl B. D.; Briggs S. D.; Allis C. D.; Wong J. M.; Tempst P.; Zhang Y. Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science 2001, 2935531853–857. [DOI] [PubMed] [Google Scholar]
- Tee W. W.; Pardo M.; Theunissen T. W.; Yu L.; Choudhary J. S.; Hajkova P.; Surani M. A. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 2010, 24242772–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkountela S.; Li Z. W.; Chin C. J.; Lee S. A.; Clark A. T. PRMT5 Is Required for Human Embryonic Stem Cell Proliferation but Not Pluripotency. Stem Cell Rev. Rep. 2014, 102230–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkhanis V.; Wang L.; Tae S.; Hu Y. J.; Imbalzano A. N.; Sif S. Protein Arginine Methyltransferase 7 Regulates Cellular Response to DNA Damage by Methylating Promoter Histones H2A and H4 of the Polymerase δ Catalytic Subunit Gene, POLD1. J. Biol. Chem. 2012, 2873529801–29814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann T.; Izzo A.; Kamieniarz K.; Richter F.; Vogler C.; Sarg B.; Lindner H.; Young N. L.; Mittler G.; Garcia B. A.; Schneider R., Methylation of H2AR29 is a novel repressive PRMT6 target. Epigenet. Chromatin 2011, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iberg A. N.; Espejo A.; Cheng D.; Kim D.; Michaud-Levesque J.; Richard S.; Bedford M. T. Arginine methylation of the histone H3 tail impedes effector binding. J. Biol. Chem. 2008, 28363006–3010. [DOI] [PubMed] [Google Scholar]
- Tsai W. W.; Niessen S.; Goebel N.; Yates J. R.; Guccione E.; Montminy M. PRMT5 modulates the metabolic response to fasting signals. Proc. Natl. Acad. Sci. U.S.A. 2013, 110228870–8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. C. M.; Dowhan D. H.; Eriksson N. A.; Muscat G. E. O. CARM1/PRMT4 is necessary for the glycogen gene expression programme in skeletal muscle cells. Biochem. J. 2012, 444, 323–331. [DOI] [PubMed] [Google Scholar]
- LeBlanc S. E.; Konda S.; Wu Q.; Hu Y. J.; Oslowski C. M.; Sif S.; Imbalzano A. N. Protein arginine methyltransferase 5 (Prmt5) promotes gene expression of peroxisome proliferator-activated receptor γ2 (PPARγ2) and its target genes during adipogenesis. Mol. Endocrinol. 2012, 264583–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.; Cui N.; Wang R.; Li J. W.; Wong J. M., A Role for CARM1-Mediated Histone H3 Arginine Methylation in Protecting Histone Acetylation by Releasing Corepressors from Chromatin. PLoS One 2012, 7 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q.; Bruce A. W.; Jedrusik A.; Ellis P. D.; Andrews R. M.; Langford C. F.; Glover D. M.; Zernicka-Goetz M. CARM1 Is Required in Embryonic Stem Cells To Maintain Pluripotency and Resist Differentiation. Stem Cells 2009, 27112637–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. L.; Loffler K. A.; Chen D. G.; Stallcup M. R.; Muscat G. E. O. The coactivator-associated arginine methyltransferase is necessary for muscle differentiation—CARM1 coactivates myocyte enhancer factor-2. J. Biol. Chem. 2002, 27764324–4333. [DOI] [PubMed] [Google Scholar]
- Cheung N.; Chan L. C.; Thompson A.; Cleary M. L.; So C. W. E. Protein arginine-methyltransferase-dependent oncogenesis. Nat. Cell Biol. 2007, 9101208–1215. [DOI] [PubMed] [Google Scholar]
- Pal S.; Yun R.; Datta A.; Lacomis L.; Erdjument-Bromage H.; Kumar J.; Tempst P.; Sif S. mSin3A/histone deacetylase 2- and PRMT5-containing Brg1 complex is involved in transcriptional repression of the Myc target gene cad. Mol. Cell. Biol. 2003, 23217475–7487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X. J.; Hoang S.; Mayo M. W.; Bekiranov S., Application of machine learning methods to histone methylation ChIP-Seq data reveals H4R3me2 globally represses gene expression. BMC Bioinf. 2010, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. J.; Zhou F.; Zhan D. G.; Gao Q. Q.; Cui N.; Li J. W.; Iakhiaeva E.; Zwieb C.; Lin B. Y.; Wong J. M. A Novel Histone H4 Arginine 3 Methylation-Sensitive Histone H4 Binding Activity and Transcriptional Regulatory Function for Signal Recognition Particle Subunits SRP68 and SRP72. J. Biol. Chem. 2012, 2874840641–40651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guendel I.; Carpio L.; Pedati C.; Schwartz A.; Teal C.; Kashanchi F.; Kehn-Hall K., Methylation of the Tumor Suppressor Protein, BRCA1, Influences Its Transcriptional Cofactor Function. PLoS One 2010, 5 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D.; Madl T.; Valori C. F.; Bentmann E.; Tahirovic S.; Abou-Ajram C.; Kremmer E.; Ansorge O.; Mackenzie I. R. A.; Neumann M.; Haass C. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012, 31224258–4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson M.; Durant S. T.; Cho E. C.; Sheahan S.; Edelmann M.; Kessler B.; La Thangue N. B. Arginine methylation regulates the p53 response. Nat. Cell Biol. 2008, 10121431–U122. [DOI] [PubMed] [Google Scholar]
- Scoumanne A.; Zhang J.; Chen X. PRMT5 is required for cell-cycle progression and p53 tumor suppressor function. Nucleic Acids Res. 2009, 37154965–4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mathioudaki K.; Scorilas A.; Ardavanis A.; Lymberi P.; Tsiambas E.; Devetzi M.; Apostolaki A.; Talieri M. Clinical evaluation of PRMT1 gene expression in breast cancer. Tumor Biol. 2011, 323575–582. [DOI] [PubMed] [Google Scholar]; b Baldwin R. M.; Morettin A.; Paris G.; Goulet I.; Cote J. Alternatively spliced protein arginine methyltransferase 1 isoform PRMT1v2 promotes the survival and invasiveness of breast cancer cells. Cell Cycle 2012, 11244597–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J.; Cao R. X.; Hong T.; Yang J.; Zu X. Y.; Xiao X. H.; Liu J. H.; Wen G. B. Identification and expression analysis of a novel transcript of the human PRMT2 gene resulted from alternative polyadenylation in breast cancer. Gene 2011, 48711–9. [DOI] [PubMed] [Google Scholar]
- Singh V.; Miranda T. B.; Jiang W.; Frankel A.; Roemer M. E.; Robb V. A.; Gutmann D. H.; Herschman H. R.; Clarke S.; Newsham I. F. DAL-1/4.1B tumor suppressor interacts with protein arginine N-methyltransferase 3 (PRMT3) and inhibits its ability to methylate substrates in vitro and in vivo. Oncogene 2004, 23477761–7771. [DOI] [PubMed] [Google Scholar]
- a Kim Y. R.; Lee B. K.; Park R. Y.; Nguyen N. T. X.; Bae J. A.; Kwon D. D.; Jung C.. Differential CARM1 expression in prostate and colorectal cancers. BMC Cancer 2010, 10, No. 197. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Hong H.; Kao C. H.; Jeng M. H.; Eble J. N.; Koch M. O.; Gardner T. A.; Zhang S. B.; Li L.; Pan C. X.; Hu Z. Q.; MacLennan G. T.; Cheng L. Aberrant expression of CARM1, a transcriptional coactivator of androgen receptor, in the development of prostate carcinoma and androgen-independent status. Cancer 2004, 101183–89. [DOI] [PubMed] [Google Scholar]; c Al-Dhaheri M.; Wu J. C.; Skliris G. P.; Li J.; Higashimato K.; Wang Y. D.; White K. P.; Lambert P.; Zhu Y. R.; Murphy L.; Xu W. CARM1 Is an Important Determinant of ERα-Dependent Breast Cancer Cell Differentiation and Proliferation in Breast Cancer Cells. Cancer Res. 2011, 7162118–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav N.; Lee J.; Kim J.; Shen J.; Hu M. C.; Aldaz C. M.; Bedford M. T. Specific protein methylation defects and gene expression perturbations in coactivator-associated arginine methyltransferase 1-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 2003, 100116464–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Messaoudi S.; Fabbrizio E.; Rodriguez C.; Chuchana P.; Fauquier L.; Cheng D. H.; Theillet C.; Vandel L.; Bedford M. T.; Sardet C. Coactivator-associated arginine methyltransferase 1 (CARM1) is a positive regulator of the Cyclin E1 gene. Proc. Natl. Acad. Sci. U.S.A. 2006, 1033613351–13356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covic M.; Hassa P. O.; Saccani S.; Buerki C.; Meier N. I.; Lombardi C.; Imhof R.; Bedford M. T.; Natoli G.; Hottiger M. O. Arginine methyltransferase CARM1 is a promoter-specific regulator of NF-κB-dependent gene expression. EMBO J. 2005, 24185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Pal S.; Sif S. Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol. Cell. Biol. 2008, 28206262–6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wei H.; Wang B. L.; Miyagi M.; She Y.; Gopalan B.; Huang D. B.; Ghosh G.; Stark G. R.; Lu T. PRMT5 dimethylates R30 of the p65 subunit to activate NF-κB. Proc. Natl. Acad. Sci. U.S.A. 2013, 1103313516–13521. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Shilo K.; Wu X.; Sharma S.; Welliver M.; Duan W. R.; Villalona-Calero M.; Fukuoka J.; Sif S.; Baiocchi R.; Hitchcock C. L.; Zhao W. Q.; Otterson G. A.. Cellular localization of protein arginine methyltransferase-5 correlates with grade of lung tumors. Diagn. Pathol. 2013, 8, No. 201. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yan F. T.; Alinari L.; Lustberg M. E.; Martin L. K.; Cordero-Nieves H. M.; Banasavadi-Siddegowda Y.; Virk S.; Barnholtz-Sloan J.; Bell E. H.; Wojton J.; Jacob N. K.; Chakravarti A.; Nowicki M. O.; Wu X.; Lapalombella R.; Datta J.; Yu B.; Gordon K.; Haseley A.; Patton J. T.; Smith P. L.; Ryu J.; Zhang X. L.; Mo X. K.; Marcucci G.; Nuovo G.; Kwon C. H.; Byrd J. C.; Chiocca E. A.; Li C. L.; Sif S.; Jacob S.; Lawler S.; Kaur B.; Baiocchi R. A. Genetic Validation of the Protein Arginine Methyltransferase PRMT5 as a Candidate Therapeutic Target in Glioblastoma. Cancer Res. 2014, 7461752–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S.; Vishwanath S. N.; Erdjument-Bromage H.; Tempst P.; Sif S. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol. Cell. Biol. 2004, 24219630–9645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger R. H.; Vallance P.; Cooke J. P. Asymmetric dimethylarginine (ADMA): a key regulator of nitric oxide synthase. Atheroscler. Suppl. 2003, 441–3. [DOI] [PubMed] [Google Scholar]
- Pou S.; Keaton L.; Surichamorn W.; Rosen G. M. Mechanism of superoxide generation by neuronal nitric-oxide synthase. J. Biol. Chem. 1999, 274149573–9580. [DOI] [PubMed] [Google Scholar]
- Jacobi J.; Tsao P. S. Asymmetrical dimethylarginine in renal disease: limits of variation or variation limits?. Am. J. Nephrol. 2008, 282224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakim B. T.; Aswad G. D. Ca2+-calmodulin-dependent phosphorylation of arginine in histone 3 by a nuclear kinase from mouse leukemia cells. J. Biol. Chem. 1994, 26942722–7. [PubMed] [Google Scholar]
- a Fuhrmann J.; Schmidt A.; Spiess S.; Lehner A.; Turgay K.; Mechtler K.; Charpentier E.; Clausen T. McsB Is a Protein Arginine Kinase That Phosphorylates and Inhibits the Heat-Shock Regulator CtsR. Science 2009, 32459321323–1327. [DOI] [PubMed] [Google Scholar]; b Slade D. J.; Subramanian V.; Fuhrmann J.; Thompson P. R. Chemical and Biological Methods To Detect Post-Translational Modifications of Arginine. Biopolymers 2014, 1012133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington W. R. Evolution and physiological roles of phosphagen systems. Annu. Rev. Physiol. 2001, 63, 289–325. [DOI] [PubMed] [Google Scholar]
- Blethen S. L. Kinetic Properties of Arginine Kinase Isoenzymes of Limulus Polyphemus. Arch. Biochem. Biophys. 1972, 1491244–&. [DOI] [PubMed] [Google Scholar]
- Zhou G. F.; Somasundaram T.; Blanc E.; Parthasarathy G.; Ellington W. R.; Chapman M. S. Transition state structure of arginine kinase: implications for catalysis of bimolecular reactions. Proc. Natl. Acad. Sci. U.S.A. 1998, 95158449–8454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruett P. S.; Azzi A.; Clark S. A.; Yousef M. S.; Gattis J. L.; Somasundaram T.; Ellington W. R.; Chapman M. S. The putative catalytic bases have, at most, an accessory role in the mechanism of arginine kinase. J. Biol. Chem. 2003, 2782926952–26957. [DOI] [PubMed] [Google Scholar]
- a Fuhrmann J.; Mierzwa B.; Trentini D. B.; Spiess S.; Lehner A.; Charpentier E.; Clausen T. Structural Basis for Recognizing Phosphoarginine and Evolving Residue-Specific Protein Phosphatases in Gram-Positive Bacteria. Cell Rep. 2013, 361832–1839. [DOI] [PubMed] [Google Scholar]; b Elsholz A. K. W.; Turgay K.; Michalik S.; Hessling B.; Gronau K.; Oertel D.; Mader U.; Bernhardt J.; Becher D.; Hecker M.; Gerth U. Global impact of protein arginine phosphorylation on the physiology of Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 2012, 109197451–7456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann J.; Subramanian V.; Thompson P. R. Targeting the Arginine Phosphatase YwIE with a Catalytic Redox-Based Inhibitor. ACS Chem. Biol. 2013, 892024–2032. [DOI] [PubMed] [Google Scholar]
- Wakim B. T.; Grutkoski P. S.; Vaughan A. T.; Engelmann G. L. Stimulation of a Ca2+-calmodulin-activated histone 3 arginine kinase in quiescent rat heart endothelial cells compared to actively dividing cells. J. Biol. Chem. 1995, 2703923155–8. [DOI] [PubMed] [Google Scholar]
- Schmidt A.; Ammerer G.; Mechtler K. Studying the fragmentation behavior of peptides with arginine phosphorylation and its influence on phospho-site localization. Proteomics 2013, 136945–954. [DOI] [PubMed] [Google Scholar]
- Glowacki G.; Braren R.; Firner K.; Nissen M.; Kuhl M.; Reche P.; Bazan F.; Cetkovic-Cvrlje M.; Leiter E.; Haag F.; Koch-Nolte F. The family of toxin-related ecto-ADP-ribosyltransferases in humans and the mouse. Protein Sci. 2002, 1171657–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottiger M. O. ADP-ribosylation of histones by ARTD1: an additional module of the histone code?. FEBS Lett. 2011, 585111595–1599. [DOI] [PubMed] [Google Scholar]
- Smith S. The world according to PARP. Trends Biochem. Sci. 2001, 263174–179. [DOI] [PubMed] [Google Scholar]
- Frye R. A. Characterization of five human cDNAs with homology to the yeast SIR2 gene: Sir2-like proteins (sirtuins) metabolize NAD and may have protein ADP-ribosyltransferase activity. Biochem. Biophys. Res. Commun. 1999, 2601273–279. [DOI] [PubMed] [Google Scholar]
- Hawse W. F.; Wolberger C. Structure-Based Mechanism of ADP-Ribosylation by Sirtuins. J. Biol. Chem. 2009, 2844833654–33661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss J.; Stanley S. J.; Nightingale M. S.; Murtagh J. J.; Monaco L.; Mishima K.; Chen H. C.; Williamson K. C.; Tsai S. C. Molecular and Immunological Characterization of Adp-Ribosylarginine Hydrolases. J. Biol. Chem. 1992, 2671510481–10488. [PubMed] [Google Scholar]
- a Stone P. R.; Lorimer W. S.; Kidwell W. R. Properties of Complex between Histone H-1 and Poly(Adp-Ribose) Synthesized in Hela-Cell Nuclei. Eur. J. Biochem. 1977, 8119–18. [DOI] [PubMed] [Google Scholar]; b Burzio L. O.; Riquelme P. T.; Koide S. S. Adp Ribosylation of Rat-Liver Nucleosomal Core Histones. J. Biol. Chem. 1979, 25483029–3037. [PubMed] [Google Scholar]; c Golderer G.; Grobner P. Adp-Ribosylation of Core Histones and Their Acetylated Subspecies. Biochem. J. 1991, 277, 607–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wong C. C. L.; Xu T.; Rai R.; Bailey A. O.; Yates J. R.; Wolf Y. I.; Zebroski H.; Kashina A. Global analysis of posttranslational protein arginylation. PLoS Biol. 2007, 5102231–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Saha S.; Wong C. C. L.; Xu T.; Namgoong S.; Zebroski H.; Yates J. R.; Kashina A. Arginylation and Methylation Double Up To Regulate Nuclear Proteins and Nuclear Architecture in Vivo. Chem. Biol. 2011, 18111369–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzi E.; Choder M.; Chen W. N.; Varshavsky A.; Goffeau A. Cloning and Functional-Analysis of the Arginyl-Transfer RNA-Protein Transferase Gene Ate1 of Saccharomyces cerevisiae. J. Biol. Chem. 1990, 265137464–7471. [PubMed] [Google Scholar]
- a Bachmair A.; Finley D.; Varshavsky A. In Vivo Half-Life of a Protein Is a Function of Its Amino-Terminal Residue. Science 1986, 2344773179–186. [DOI] [PubMed] [Google Scholar]; b Lee M. J.; Tasaki T.; Moroi K.; An J. Y.; Kimura S.; Davydov I. V.; Kwon Y. T. RGS4 and RGS5 are in vivo substrates of the N-end rule pathway. Proc. Natl. Acad. Sci. U.S.A. 2005, 1024215030–15035. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Hu R. G.; Sheng J.; Qi X.; Xu Z. M.; Takahashi T. T.; Varshavsky A. The N-end rule pathway as a nitric oxide sensor controlling the levels of multiple regulators. Nature 2005, 4377061981–986. [DOI] [PubMed] [Google Scholar]
- Kaneko T.; Huang H. M.; Cao X.; Li X.; Li C. J.; Voss C.; Sidhu S. S.; Li S. S. C. Superbinder SH2 Domains Act as Antagonists of Cell Signaling. Sci. Signaling 2012, 5243ra68. [DOI] [PubMed] [Google Scholar]
- Emamzadah S.; Tropia L.; Halazonetis T. D. Crystal Structure of a Multidomain Human p53 Tetramer Bound to the Natural CDKN1A (p21) p53-Response Element. Mol. Cancer Res. 2011, 9111493–1499. [DOI] [PubMed] [Google Scholar]
- a Sievers F.; Wilm A.; Dineen D.; Gibson T. J.; Karplus K.; Li W. Z.; Lopez R.; McWilliam H.; Remmert M.; Soding J.; Thompson J. D.; Higgins D. G., Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7;. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Robert X.; Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42W1W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M. J.; Culhane J. C.; Szewczuk L. M.; Gocke C. B.; Brautigam C. A.; Tomchick D. R.; Machius M.; Cole P. A.; Yu H. T. Structural basis of histone demethylation by LSD1 revealed by suicide inactivation. Nat. Struct. Mol. Biol. 2007, 146535–539. [DOI] [PubMed] [Google Scholar]
- Ng S. S.; Kavanagh K. L.; McDonough M. A.; Butler D.; Pilka E. S.; Lienard B. M. R.; Bray J. E.; Savitsky P.; Gileadi O.; von Delft F.; Rose N. R.; Offer J.; Scheinost J. C.; Borowski T.; Sundstrom M.; Schofield C. J.; Oppermann U. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature 2007, 448714987–91. [DOI] [PubMed] [Google Scholar]