

Abstract
Background
Genetic variants in DAT1, the gene encoding the dopamine transporter protein (DAT), have been implicated in many brain disorders. In a recent case-control study of Alzheimer’s disease (AD), a regulatory polymorphism in DAT1 showed a significant association with the clinical stages of dementia.
Methods
We tested whether this variant was associated with increased AD risk, and with measures of cognitive decline and longitudinal ventricular expansion, in a large sample of elderly participants with genetic, neurocognitive, and neuroimaging data from the Alzheimer’s Disease Neuroimaging Initiative.
Results
The minor allele – previously linked with increased DAT expression in vitro – was More common in AD patients than in both individuals with mild cognitive impairment and Healthy elderly controls. The same allele was also associated with poorer cognitive performance and faster ventricular expansion, independently of diagnosis.
Conclusion
These results may be due to reduced dopaminergic transmission in carriers of the DAT1 mutation.
Keywords: neuroimaging genetics, ventricular expansion, dopamine transporter, dementia, DAT1
1. INTRODUCTION
Dopamine (DA) is a powerful regulator of many aspects of brain function, and altered DA transmission can contribute to cognitive impairment [1]. Common variants in DA-related genes have been implicated in cognitive function, age-related cognitive decline, and dementia severity [2,3]. The dopamine transporter protein (DAT) regulates neurotransmission by terminating DA signaling at the synapse, through high-affinity reuptake of DA into presynaptic terminals [4]. The DAT protein limits the activation of DA receptors [5], and changes in DAT expression directly affect the concentration of synaptic DA and the kinetics of reuptake [6,7]. This protein is encoded by the DAT1 (or SLC6A3) gene [8], and DAT1 variants may be related to various brain disorders [9].
A recent Taiwanese study reported an association between the major T allele at rs6347 of DAT1 and moderate dementia [3]. In other words, among demented participants, the minor C allele was significantly more prevalent in patients with severe dementia than in individuals with moderate dementia [3]. Here, we sought to replicate this association in Caucasians, and hypothesized that the minor C allele at this locus would be more common in elderly individuals with Alzheimer’s disease (AD) than in both subjects with mild cognitive impairment (MCI) and healthy elderly controls (CON).
The rs6347 single nucleotide polymorphism (SNP) is a common synonymous variant (T>C, Minor Allele Frequency = 0.299) in exon 9 of DAT1 [10]. It does not affect the amino-acid sequence, but may be a regulatory variant [8]. As DA has a crucial role in cognition [1], and age-related cognitive decline [2], we also predicted that the same allele would be associated with poorer cognitive performance, independently of disease status.
DA also regulates the formation of neurotoxic amyloid beta (Aβ) oligomers [11], and lateral ventricular enlargement indicates an accumulation of brain tissue loss [12]. We therefore hypothesized that carriers of the minor allele at rs6347 would show faster expansion of the lateral ventricles, independently of their dementia status. We tested these predictions in a large elderly cohort (N=738), with genetic, neurocognitive, and neuroimaging data.
2. METHODS
2.1 Subjects
Data used in this study were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database. Written informed consent was obtained from all participants. To avoid the known effects of population stratification on genetic analysis, we included only non-Hispanic Caucasian subjects [13]. Our final analysis comprised 738 individuals (average age 75.52 ± 6.78 years; 438 men/300 women) including 173 AD, 359 MCI, and 206 CON at baseline.
2.2 Cognitive testing and genotyping
All subjects completed detailed cognitive assessments including the Mini-Mental State Examination (MMSE) [14]. Participants were genotyped using the Illumina 610-Quad BeadChip. ApoE genotyping was performed separately, using an ApoE genotyping kit, as described in http://www.adni-info.org/Scientists/Pdfs/adniproceduresmanual12.pdf.
2.3 Statistical analyses of allele frequency, and associations of rs6347 genotype with MMSE scores
The distributions of allele frequencies for rs6347 were evaluated by χ2 tests using contingency tables in SPSS 21.0. Statistical analyses of the odds ratio (OR) and 95% confidence interval (CI) were conducted based on the presence of the minor C allele. We then used the number of minor C alleles at rs6347 to predict baseline MMSE scores, assuming an additive model for allele effects.
2.4 Image acquisition, correction, and pre-processing
Participants were scanned with a standardized MRI protocol developed for this cohort [15,16]. Briefly, high-resolution structural brain MRI scans were acquired at 58 sites across North America, using 1.5 Tesla MRI scanners. A sagittal 3D MP-RAGE sequence was used, and optimized for consistency across sites [16] (TR/TE = 2400/1000 ms; flip angle = 8°; FOV = 24 cm; final reconstructed voxel resolution = 0.9375 × 0.9375 × 1.2 mm3). Image quality control procedures and post-acquisition correction of various image artifacts were performed at a single site (Mayo clinic) [16].
2.5 Segmentation of the lateral ventricles
Raw MRI scans were pre-processed to reduce signal inhomogeneity and linearly registered to a template (using 9 parameter registration). Prior methods for ventricular segmentation have used semi-automated, automated [17], and single-atlas or multi-atlas methods [18]. Here we segmented the ventricles with our modified multi-atlas approach described previously [19]. An inverse-consistent fluid registration with a mutual information fidelity term aligned a set of hand-labeled ventricular templates to each scan [20]. The template surfaces were registered into homologous point-to-point correspondence as a group using medial-spherical registration [21]. This approach is very similar to that of [22], except ours is based on surface geometry rather than image voxels. 1 subject whose meshes deviated by several millimeters from the actual periventricular boundaries was excluded. Our final analysis included 737 ADNI subjects at baseline, 623 at 12-month follow-up, and 481 at 24-month follow-up.
2.6 Statistical associations of rs6347 genotype with ventricular volumes
We first determined if genotype at the rs6347 locus might be associated with baseline ventricular volumes, after adjusting for age, sex, and diagnosis, testing both recessive and additive models of minor C allele effects. As we did not detect an association, we then used generalized linear mixed models (GLMMs) to determine if genotype at the rs6347 locus predicted ventricular expansion over a period of two years, also testing both recessive and additive models of minor allele effects. We used changes in volume of the lateral ventricles (in cubic mm) as dependent variables, controlling for age, sex, diagnosis, and ApoE genotype, with subjects included as a random factor and time point (i.e., change in volume at 12 months and 24 months) as a repeated measure.
3. RESULTS
3.1 Primary analyses
3.1.1 Allele frequency
Allele frequency was computed from genotype frequency in baseline participants (N=738) and significantly differed across the 3 diagnostic groups (p=0.018, Table 1). The minor C allele was more prevalent in AD than in both CON (p=0.006, Table 2) and MCI (p=0.039, Table 3), but did not significantly differ between MCI and CON (Table 4).
Table 1.
Genotype and allele frequency by diagnostic group (baseline subjects)
| CON | MCI | AD | Pearson Chi-Square Test | ||
|---|---|---|---|---|---|
| Total | N=738 | 206 | 359 | 173 | |
| Genotype Frequency | TT | 126 (61%) | 203 (57%) | 85 (49%) | p = 0.091 |
| TC | 68 (33%) | 131 (36%) | 68 (39%) | ||
| CC | 12 (6%) | 25 (7%) | 20 (12%) | ||
| Allele Frequency | T | 320 (78%) | 537 (75%) | 238 (69%) | p = 0.018 |
| C | 92 (22%) | 181 (25%) | 108 (31%) | ||
Table 2.
Allele frequency in the AD versus CON group
| Allele Frequency | Pearson Chi-Square Test | Odds Ratio (Confidence Interval) |
||
|---|---|---|---|---|
| C | T | |||
| AD | 108 | 238 | p = 0.006 | 1.578 (1.141–2.184) |
| CON | 92 | 320 | ||
Table 3.
Allele frequency in the AD versus MCI group
| Allele Frequency | Pearson Chi-Square Test | Odds Ratio (Confidence Interval) |
||
|---|---|---|---|---|
| C | T | |||
| AD | 108 | 238 | p = 0.039 | 1.346 (1.014–1.787) |
| MCI | 181 | 537 | ||
Table 4.
Allele frequency in the MCI versus CON group
| Allele Frequency | Pearson Chi-Square Test | Odds Ratio (Confidence Interval) |
||
|---|---|---|---|---|
| C | T | |||
| MCI | 181 | 537 | p = 0.277 | 1.172 (0.880–1.561) |
| CON | 92 | 320 | ||
3.1.2 Associations of rs6347 genotype with MMSE scores
MMSE scores significantly differed between the 3 genotype groups (p=0.006, Figure 1). Carriers of 2 C alleles performed more poorly than both carriers of 0 (p=0.004) and 1 (p=0.026) C allele, but carriers of 0 and 1 C alleles did not differ, suggesting a recessive model of minor allele effects on cognitive decline (Figure 1). These differences remained significant after controlling for sex, age, diagnosis, and ApoE status (p=0.030, F-ratio=3.524).
Figure 1. Effects of genotype at the rs6347 locus on MMSE scores (N=738).
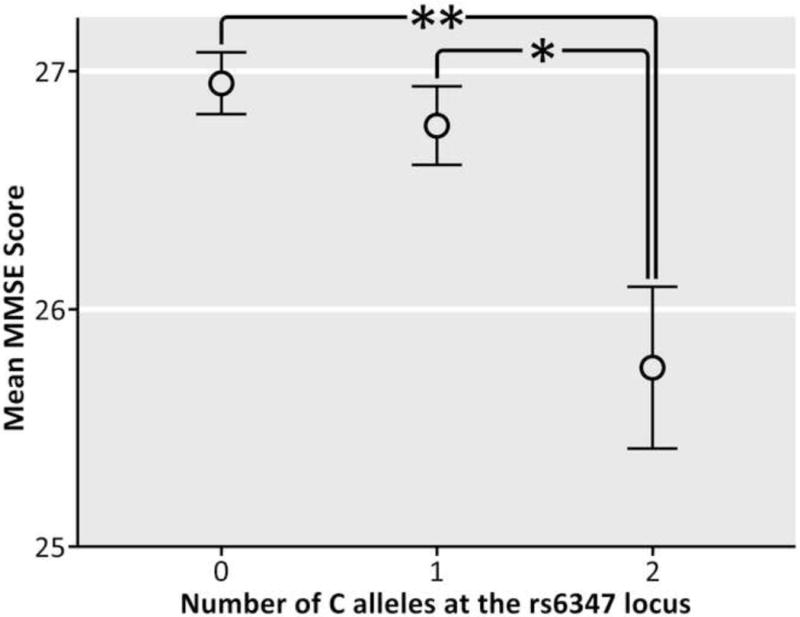
The vertical bars represent the standard error of the mean (SEM) for each genotype group. The asterisks represent a significant difference between genotype groups. (*p=0.026; **p=0.004).
3.1.3 Associations of rs6347 genotype with ventricular expansion
Baseline ventricular volumes, as well as volume loss over a period of 2 years (in cubic mm) are reported in Table 5, by diagnostic and genotype groups. Genotype at the rs6347 locus was not significantly related to total ventricular volume at baseline (p=0.474, F-ratio=0.514 and p=0.612, F-ratio=0.491 for the recessive and additive models, respectively), after controlling for sex, age, and diagnosis. However, carrying the minor C allele was associated with greater overall ventricular expansion over a period of 2 years (p=0.017 and p=0.037 for the recessive and additive models, respectively), after controlling for sex, age, and dementia status (Table 6). These results remained significant after introducing ApoE genotype as an additional covariate (Table 6).
Table 5.
Total baseline ventricular volumes (VB) and total volume loss (VL) over a period of 2 years (in cubic mm) by diagnostic and genotype groups: Mean +/− SEM
| CON | MCI | AD | Total | ||
|---|---|---|---|---|---|
| TT | VB | 49,041 (±1,562) | 49,253 (±1,197) | 47,052 (±1,454) | 48,735 (± 811) |
| VL | 3,552 (±292) | 6,482 (±499) | 11,363 (±1,060) | 6,364 (± 361) | |
| TC | VB | 47,605 (±2,133) | 48,629 (±1,341) | 45,798 (± 1,972) | 47,647 (± 989) |
| VL | 3,627 (±454) | 5,951 (±512) | 10,572 (± 1,270) | 6,252 (± 434) | |
| CC | VB | 42,169 (±4,503) | 47,680 (±3,505) | 46,775 (± 3,511) | 46,202 (± 2,167) |
| VL | 2,352 (±1,085) | 6,923 (±1,769) | 12,660 (± 1,729) | 8,039 (± 1,197) | |
| Total | VB | 48,167(±1,215) | 48,915 (±868) | 46,527 (± 1,124) | 48,145 (± 602) |
| VL | 3,525 (±242) | 6,304 (±355) | 11,205 (± 744) | 6,437 (± 271) | |
Table 6.
Results of multiple regression analyses: associations between rs6347 genotype and ventricular expansion (Recessive and Additive Models)
| Recessive Model | Genotype effects (3 covariates)1 |
Genotype effects (4 covariates)2 |
Genotype by diagnosis interaction3 |
|---|---|---|---|
| Total expansion (cubic mm) |
4,5F-ratio=5.710 p=0.017 |
F-ratio=6.198 p=0.013 |
F-ratio=0.134 p=0.715 |
| Left expansion (cubic mm) |
F-ratio=6.230 p=0.013 |
F-ratio=5.817 p=0.016 |
F-ratio=0.075 p=0.784 |
| Right expansion (cubic mm) |
F-ratio=4.460 p=0.035 |
F-ratio=4.684 p=0.031 |
F-ratio=0.123 p=0.726 |
| Additive Model |
Genotype effects (3 covariates)1 |
Genotype effects (4 covariates)2 |
Genotype by Diagnosis interaction3 |
| Total expansion (cubic mm) |
F-ratio=3.330 p=0.037 |
F-ratio=3.873 p=0.021 |
F-ratio=1.142 p=0.320 |
| Left expansion (cubic mm) |
F-ratio=3.384 p=0.035 |
F-ratio=3.630 p=0.027 |
F-ratio=0.390 p=0.677 |
| Right expansion (cubic mm) |
F-ratio=3.407 p=0.034 |
F-ratio=3.744 p=0.024 |
F-ratio=2.216 p=0.110 |
rs6347 genotype (top: recessive model; bottom: additive model) was used to predict variations in ventricular expansion, with age, sex, and diagnosis regressed out.
rs6347 genotype (top: recessive model; bottom: additive model) was used to predict variations in ventricular expansion, with age, sex, diagnosis, and ApoE status regressed out.
Significance of the genotype by diagnosis interaction term using the following equation: expansion measure = constant + genotype + age + sex + diagnosis + genotype*diagnosis.
In multiple regressions, the F-ratio is used to test the hypothesis that the slopes of the regression lines are 0. The F is large when the independent variable helps to explain the variation in the dependent variable, independently of the other explanatory variables that are regressed out. For instance, here we reject the hypothesis that the slope of the regression line is 0 (F-ratio=5.710, p=0.017), meaning that there is a significant linear relation between rs6347 genotype and total ventricular expansion, independent of age, sex, and diagnosis.
Bold font indicates significant results (p<0.05), and regular font indicates results that did not reach statistical significance.
3.2 Post-hoc analyses
3.2.1 Ventricular expansion within each hemisphere
Carrying the minor C allele was associated with greater expansion in the left (p=0.013 and p=0.035 for the recessive and additive models, respectively) and right ventricle (p=0.035 and p=0.034), after controlling for age, sex, and diagnosis (Table 6). These results remained significant after controlling for ApoE genotype (Table 6).
3.2.2 Genotype by diagnosis interaction
We found no significant genotype by diagnosis interaction in the analyses of total (p=0.715 and p=0.320 for the recessive and additive models, respectively), left (p=0.784 and p=0.677), or right ventricular expansion (p=0.726 and p=0.110, Table 6).
3.2.3 Effects of genotype and diagnosis on participants’ ages
Table 7 indicates mean baseline ages in participants stratified by diagnosis, and substratified by rs6347 genotype groups. Diagnosis was not significantly related to age after controlling for sex, rs6347 genotype, and ApoE status (p=0.310, F-ratio=1.173). Likewise, genotype at rs6347 was not significantly related to age after controlling for sex, diagnosis, and ApoE status, though there was a trend for more C alleles to be associated with younger age (p=0.078, F-ratio=2.557).
Table 7.
Baseline age by diagnostic and genotype groups: Mean +/− SEM
| CON | MCI | AD | Total | |
|---|---|---|---|---|
| TT | 76.45 (± 0.421) | 75.59 (± 0.505) | 75.61 (± 0.857) | 75.86 (± 0.329) |
| TC | 76.15 (± 0.621) | 74.86 (± 0.629) | 75.74 (± 0.934) | 75.41 (± 0.420) |
| CC | 73.00 (± 1.701) | 72.80 (± 1.514) | 74.95 (± 1.391) | 73.60 (± 0.894) |
| Total | 76.15 (± 0.347) | 75.13 (± 0.382) | 75.58 (± 0.579) | 75.52 (± 0.250) |
3.2.4 Ethno-racial differences in allele frequency, linkage disequilibrium, and predicted target genes
As shown in Figure 2, the minor C allele at the rs6347 locus is more frequent in Europeans (CEU & TSI) than in Asians (CHD & CHB). In people of African ancestry (ASN & YRI), the minor allele at rs6347 is not the C but the T allele. Figure 3 illustrates the pattern of linkage disequilibrium (LD) across the DAT1 (SLC6A3) gene in the Caucasian-American (CEU) sample. It shows that rs6347 forms a haplotype block with two other variants: rs3776511 (D′=.70) and rs3776512 (D’=.71), both of which are intronic SNPs with no known effects on gene function.
Figure 2. Ethno-racial differences in genotype and minor allele frequencies at rs6347.
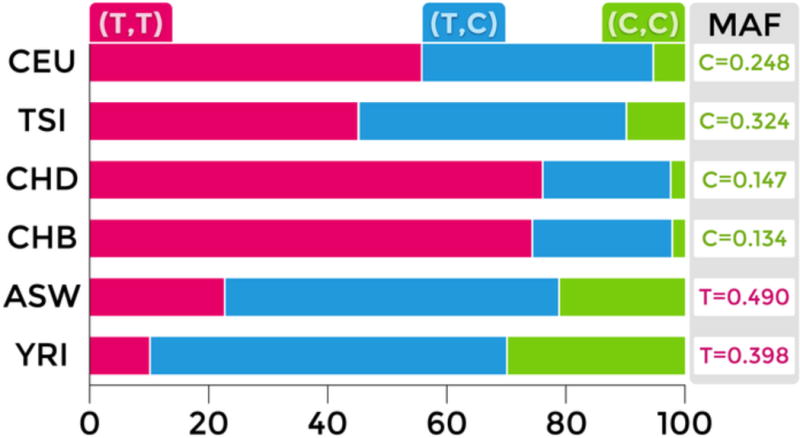
Bar graph adapted from SNPedia (http://www.snpedia.com/index.php/SNPedia); minor allele frequencies (MAFs) are obtained from dbSNP (http://www.ncbi.nlm.nih.gov/SNP/). CEU: Caucasian-Americans; TSI: Italians from Tuscany; CHD: Chinese-Americans; CHB; Han Chinese from Beijing; ASW: African-Americans; YRI: Yoruba Africans from Nigeria.
Figure 3. Patterns of linkage disequilibrium (LD) in the DAT1 (SLC6A3) gene genotyped in the HapMap Caucasian-American (CEU) sample.

The SNPs are shown on a scale representing their position on chromosome 5. The rs6347 variant is located in haplotype block 2. Figure modified from the HapMap website (http://hapmap.ncbi.nlm.nih.gov/).
Regulatory elements can be located by mapping DNase I Hypersensitive Sites (DHSs), which indicate open or accessible chromatin where DNA is not tightly wrapped within a nucleosome [23]. The first extensive map of human DHSs was recently identified through genome-wide profiling in more than 100 human cell types [24]. The DNAse I data are publicly accessible through the UCSC Genome Browser. Sheffield and colleagues created a database and web interface to help visualize results from genome browsers [25]. The Regulatory Elements Database can be used to make predictions about the linkage between regulatory regions and genes, based on the statistical association of DHSs and gene expression across more than 100 samples consisting of over 70 diverse cell types [25]. We searched a region located ± 10 kb of rs6347 and a list of 4 DHSs found in this region was outputted. Two of these sites were predicted to regulate the expression of the adjacent LPCAT1 gene (p=0.012 and p=0.019), which encodes lysophosphatidylcholine acyltransferase. The other two were expected to regulate the neighboring CLPTM1L gene (p=0.006 and p=0.011), which codes for the cisplatin resistance-related protein 9. Figure 4 illustrates the relative positions of the DAT1 gene (SLC6A3, red arrow) and flanking LPCAT1 and CLPTM1L genes on chromosome 5.
Figure 4.
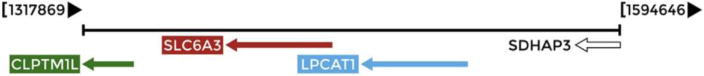
Relative positions of DAT1 (SLC6A3, red arrow) and the adjacent LPCAT1 CLPTM1L and genes on chromosome 5.
4. DISCUSSION
This study is the first to report that a genetic variant in the dopamine transporter gene is associated with several brain-related health factors, including diagnostic, cognitive, and anatomical indices of brain aging and neurodegeneration. Genotype at the rs6347 locus was related to the trajectory of ventricular volume expansion within each hemisphere, assuming both recessive and additive models of minor allele effects, and all results remained significant after controlling for ApoE status. Genotype and diagnosis were not significant predictors of participants’ ages.
We reported a statistically significant association of the minor allele at rs6347 with greater cognitive impairment, but the effect size was small, and we did not establish the clinical significance of this association [26]. These differences in MMSE scores between genotype groups remained significant after controlling for diagnosis, suggesting that the variant’s association with cognitive performance was not mediated by its association with AD. This SNP was more strongly associated with an objective index of atrophy (i.e., ventricular expansion) than with both cognitive impairment and dementia, suggesting a direct effect of this variant on the rate of brain aging, which may in turn mediate its weaker association with cognitive decline and diagnostic categories, as these are influenced by many other factors. Cognitive dysfunction in MCI does not always reflect neurodegenerative processes, and may be related to reversible conditions such as depression, delirium, vitamin B12 deficiency, hypothyroidism, and anticholinergic drug use [27]. This may be why allele frequency significantly differed across the 3 diagnostic groups, but pairwise comparisons (in a less well powered sample) revealed no significant differences between the smaller MCI and CON groups.
By showing that the minor C allele at rs6347 was more common AD than both in MCI and CON in the ADNI cohort, we replicated the association between this allele and severe dementia (initially reported in a Taiwanese sample [3]) in individuals of European descent. The minor C allele is more frequent in Caucasians than in Asians, and AD prevalence tends to be higher in individuals of European descent than in people of Asian ancestry. This is true both when comparing ethno-racial groups within the US [28] and in studies of AD prevalence across countries [29]. In this respect, our findings are consistent with the epidemiology of AD in different populations. In individuals of African descent, the minor allele is not the C but the T allele. In light of our results, people of African ancestry would be expected to have higher rates of AD than Caucasians and Asians, which is the case within the US [30,31]. Intriguingly, however, worldwide studies of AD prevalence show that Sub-Saharan African countries have lower AD rates than North American, European, and Asian countries [29]. Results from studies of AD prevalence across countries are complicated by a number of factors, most importantly life expectancy; therefore, a possible explanation for this discrepancy may be the much lower life expectancy in Sub-Saharan Africa [32]. This is only a conjecture, however, as cultural and educational differences, gene-environment interactions, admixture and other factors make it difficult to compare reports of AD prevalence in various ethno-racial groups with findings of allele frequency differences between diagnostic categories.
The minor C allele at rs6347 is associated with increased DAT1 expression in vitro [33]. Synonymous mutations can have direct functional effects, but rs6347 may also act as a surrogate marker for another regulatory polymorphism in high linkage disequilibrium (LD) with this variant. Rs6347 is in high LD with a variable number tandem repeat (VNTR) in intron 8 of DAT1 (D’ > 0.7) [33,34]. The 6-repeat variant is often co-inherited with the major rs6347 T allele, and the 5-repeat variant with the minor rs6347 C allele [8]. Consistent with the in vitro findings of Pinsonneault and colleagues [33], the 5-repeat is associated with a 32%–34% increase in DAT expression in cell cultures [34]. It is still unclear which polymorphism (the exon 9 rs6347 SNP or the intron 8 VNTR) is the actual regulatory variant. In addition, even if the pattern of LD in the DAT1 gene suggests that rs6347 forms a haplotype block with two intronic SNPs that have no known effects on gene function, we cannot rule out the possibility that rs6347 may be flagging a region that contains another functional variation.
Regardless of which SNP is the actual regulatory variant, individuals carrying these mutations in DAT1 are likely to express higher levels of DAT proteins, which pump dopamine into the cytoplasm, where DA is highly prone to spontaneous and enzymatic degradation. Free cytosolic dopamine has protons that dissociate from their corresponding hydroxyl groups, promoting the oxidation of DA to dopamine quinones (DAQ) [35] and other free radicals. DAQ are known to be involved in methamphetamine-induced neurotoxicity [36], and can lead to the stabilization of alpha-synuclein protofibrils [37], which may be involved in Lewy body formation and the etiology of Parkinson’s disease [38]. Furthermore, DAQ can cause microglial activation [39].
In AD and other dementias, it has been shown that reduced striatal uptake in vivo using single photon emission computed tomography (SPECT) results from nigral dopaminergic cell loss [40]. It is thus possible that increased DAT levels associated with these variants may result in elevated DAQ concentrations and degeneration of DA neurons. Interestingly, striatal dopamine transporter loss is much more characteristic of dementia with Lewy bodies (DLB) than of Alzheimer’s disease [41], and autopsy studies show that DLB is very commonly misdiagnosed as AD [42]. An important tool for differential diagnosis during a patient’s lifetime is the depiction of the dopaminergic system using SPECT [43]. In the ADNI cohort, SPECT was not used during the diagnostic process. It is unlikely that a large number of AD subjects in this cohort actually had “pure” DLB – as patients with DLB show ventricular expansion rates comparable to those of healthy controls [44] – but we cannot rule out the possibility that some participants had mixed AD/DLB pathology. The rs6347 C allele may confer increased risk for AD/DLB or other types of cognitive decline rather than AD. This would offer an alternative explanation for our finding that allele frequency did not significantly differ between the MCI and the control group, although all these comparisons are limited in statistical power.
While free cytosolic DA is associated with neurotoxic processes, several classes of compounds that increase dopaminergic transmission appear to have various neuroprotective effects. Monoamine oxidase B (MAO-B) inhibitors can prevent neuronal loss by inducing neuroprotective genes, anti-oxidant enzymes, and redox proteins [45]. In addition, some dopamine D2/D3 receptor agonists block neuronal cell death under oxidative stress [46], and protect against glutamate toxicity via inactivation of pro-apoptotic factors [47] and up-regulation of glutamate transporters [48]. Moreover, dopamine plays an important role in neuroplasticity [49–51], and activation of D1/D5 receptors can prevent the internalization of AMPA and NMDA receptors caused by oligomeric amyloid- beta (Aβ) peptides [52], suggesting that increased DA transmission may protect against Aβ-mediated impairments in synaptic plasticity and their neurotoxic repercussions [53–55]. Higher levels of DAT proteins likely result in reduced DA transmission, which may offer a possible explanation for the findings reported here. In carriers of the minor allele, the reduction in neuroprotective effects resulting from decreased DA transmission may drive the faster ventricular expansion, as ventricular expansion reflects increasing neuronal, myelin, and other cellular loss in neighboring tissues [12].
Another possible mechanism underlying the observed association of rs6347 with brain aging and neurodegeneration may be related to the predicted target genes. CLPTM1L seems mainly involved in the etiology of lung cancer [56], but LPCAT1 is highly expressed in the brain [57]. Its gene product LPCAT is involved in the synthesis of phosphatidylcholine (PC) [58], a major component of biological membranes and a precursor of choline used for the synthesis of acetylcholine (ACh) [59], implicated in many cellular processes [60], including neurogenesis [61]. PC also plays a role in amyloid-β peptide aggregation [62], acts in conjunction with vitamin E to provide neuroprotection against oxidative damage, and can counteract the diminished oxidative buffering capacity of brains of ApoE-deficient mice [63]. The involvement of PC in ACh synthesis and neuroprotection against AD pathology suggests that rs6347 may affect brain atrophy rates through its predicted regulation of LPCAT1 expression. Moreover, as DLB, but not AD, is associated with early and profound cholinergic depletion [64], this hypothesis is also consistent with the minor C allele conferring increased risk for AD/DLB rather than AD. These two major mechanisms may well occur in parallel, with rs6347 affecting both DAT and LPCAT expression. In fact, several lines of evidence suggest numerous types of interactions between cholinergic and dopaminergic transmission. PC is involved in the activation and regulation of protein kinase C [65], and protein kinases play important roles in dopamine receptor signaling [66]. Moreover, the ACh and DA systems are related at the cortical level [67], and DA as well as DA receptor agonists increase cortical excitability and restore central cholinergic transmission in AD [67,68]. Our theoretical framework remains speculative, but our findings highlight possible links between DA, ACh, and AD neuropathology, suggesting potential novel strategies to prevent dementia by targeting more than one neurotransmitter system.
We found that a DAT1 SNP, which was a strong predictor of brain atrophy rates, also showed a significant association with cognitive decline, and may confer increased risk for dementia. Future studies should determine whether this polymorphism is indeed a regulatory variant (and if so, how it may affect protein expression) or whether it is simply flagging a region containing another functional variant. In the ADNI cohort, the minor C allele at rs6347 was more prevalent in AD than in both healthy control and MCI subjects, but did not significantly differ between MCI and control participants. The precise nature of this variant’s association with diagnostic categories remains to be determined, and future investigations should also clarify this. By modulating the expression of particular protein target(s), this variant may lead to disrupted transmission in specific brain circuits, which, over the years, results in a pattern of neuropathology associated with a mixed AD/DLB phenotype. Alternatively, this SNP may affect a range of neurodegenerative processes, which in turn mediate its association with AD, assuming that in some MCI subjects, the observed cognitive decline did not have a neurodegenerative etiology. In this case, this polymorphism may confer increased risk for various types of dementias, even possibly for a range of degenerative brain disorders, and each individual’s unique combination of genetic and environmental risk and protective factors ultimately results in the emergence of a specific disease phenotype.
This last possibility underscores the need to devise novel and personalized approaches for the prevention of degenerative brain disorders – as opposed to the symptomatic treatment of a specific syndrome. A therapeutic approach that interferes directly with neurodegenerative processes could override the detrimental effects of certain genetic variants, before the onset of neuropathology [69]. Our findings, combined with earlier reports describing the involvement of DA transmission in neuroprotective processes, suggest that dopaminergic agents may be a useful addition to cholinergic agents for the prevention of neurodegeneration and associated cognitive decline, particularly in carriers of this and other mutations thought to affect dopamine transporter expression.
Revised Research in Context.
Systematic review: Genetic variants in DAT1, the gene encoding the dopamine transporter protein (DAT), have been implicated in many brain disorders. Genetic factors play a role in most Alzheimer’s disease (AD) cases. We searched PubMed for published association studies of DAT1 variants and AD, and also reviewed studies about the role of dopaminergic transmission in neurodegenerative processes.
Interpretation: This study reveals that a common genetic variant in DAT1, previously linked with the clinical stages of dementia in humans, and with increased DAT expression in vitro, is associated with several brain-related health factors, including diagnostic, cognitive, and anatomical indices of neurodegeneration, providing new insight into the pathogenesis of AD.
Future directions: Findings from this study provide a strong basis to generate novel hypotheses and devise new experiments. In particular, it is important that future investigations: (a) clarify whether this polymorphism is indeed a regulatory variant (and if so, how it may affect protein expression) or is simply flagging a region that contains another functional variant; (b) elucidate the nature of this variant’s association with dementia phenotypes; (c) further characterize the role of dopamine, and the significance of its interactions with cholinergic transmission, in the etiology and pathophysiology of AD and other dementias; and (d) investigate the safety and efficacy of dopaminergic agents – alone and in combination in cholinergic agents – in the prevention of cognitive decline and neurodegeneration, particularly in carriers of this and other mutations affecting dopaminergic transmission.
Acknowledgments
F.F.R. was supported, in part, by a postdoctoral fellowship from the A. P. Giannini Foundation. This work was additionally supported by National Institute of Health grants (R01 MH097268, R01 AG040060) to P.M.T. Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; GE Healthcare; Innogenetics, N.V.; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129 and K01 AG030514.
Footnotes
Conflict of interest disclosure: The authors declare no competing interests.
References
- 1.Nieoullon A. Dopamine and the regulation of cognition and attention. Prog Neurobiol. 2002;67:53–83. doi: 10.1016/s0301-0082(02)00011-4. [DOI] [PubMed] [Google Scholar]
- 2.Schuck NW, Frensch PA, Schjeide BM, Schroder J, Bertram L, et al. Effects of aging and dopamine genotypes on the emergence of explicit memory during sequence learning. Neuropsychologia. 2013 doi: 10.1016/j.neuropsychologia.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Lin WY, Wu BT, Lee CC, Sheu JJ, Liu SH, et al. Association analysis of dopaminergic gene variants (Comt, Drd4 And Dat1) with Alzheimer s disease. J Biol Regul Homeost Agents. 2012;26:401–410. [PubMed] [Google Scholar]
- 4.Shumay E, Fowler JS, Volkow ND. Genomic features of the human dopamine transporter gene and its potential epigenetic States: implications for phenotypic diversity. PLoS One. 2010;5:e11067. doi: 10.1371/journal.pone.0011067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vandenbergh DJ, Persico AM, Hawkins AL, Griffin CA, Li X, et al. Human dopamine transporter gene (DAT1) maps to chromosome 5p15.3 and displays a VNTR. Genomics. 1992;14:1104–1106. doi: 10.1016/s0888-7543(05)80138-7. [DOI] [PubMed] [Google Scholar]
- 6.Giros B, el Mestikawy S, Godinot N, Zheng K, Han H, et al. Cloning, pharmacological characterization, and chromosome assignment of the human dopamine transporter. Molecular pharmacology. 1992;42:383–390. [PubMed] [Google Scholar]
- 7.Ciliax BJ, Heilman C, Demchyshyn LL, Pristupa ZB, Ince E, et al. The dopamine transporter: immunochemical characterization and localization in brain. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1995;15:1714–1723. doi: 10.1523/JNEUROSCI.15-03-01714.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sullivan D, Pinsonneault JK, Papp AC, Zhu H, Lemeshow S, et al. Dopamine transporter DAT and receptor DRD2 variants affect risk of lethal cocaine abuse: a gene-gene-environment interaction. Transl Psychiatry. 2013;3:e222. doi: 10.1038/tp.2012.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haddley K, Vasiliou AS, Ali FR, Paredes UM, Bubb VJ, et al. Molecular genetics of monoamine transporters: relevance to brain disorders. Neurochem Res. 2008;33:652–667. doi: 10.1007/s11064-007-9521-8. [DOI] [PubMed] [Google Scholar]
- 10.Greenwood TA, Alexander M, Keck PE, McElroy S, Sadovnick AD, et al. Segmental linkage disequilibrium within the dopamine transporter gene. Mol Psychiatry. 2002;7:165–173. doi: 10.1038/sj.mp.4000958. [DOI] [PubMed] [Google Scholar]
- 11.Ono K, Takasaki J, Takahashi R, Ikeda T, Yamada M. Effects of antiparkinsonian agents on beta-amyloid and alpha-synuclein oligomer formation in vitro. J Neurosci Res. 2013;91:1371–1381. doi: 10.1002/jnr.23256. [DOI] [PubMed] [Google Scholar]
- 12.Hua X, Hibar DP, Ching CR, Boyle CP, Rajagopalan P, et al. Unbiased tensor-based morphometry: improved robustness and sample size estimates for Alzheimer’s disease clinical trials. Neuroimage. 2013;66:648–661. doi: 10.1016/j.neuroimage.2012.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stein JL, Hua X, Morra JH, Lee S, Hibar DP, et al. Genome-wide analysis reveals novel genes influencing temporal lobe structure with relevance to neurodegeneration in Alzheimer’s disease. Neuroimage. 2010;51:542–554. doi: 10.1016/j.neuroimage.2010.02.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 15.Leow AD, Klunder AD, Jack CR, Jr, Toga AW, Dale AM, et al. Longitudinal stability of MRI for mapping brain change using tensor-based morphometry. Neuroimage. 2006;31:627–640. doi: 10.1016/j.neuroimage.2005.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jack CR, Jr, Bernstein MA, Fox NC, Thompson P, Alexander G, et al. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27:685–691. doi: 10.1002/jmri.21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chou YY, Lepore N, de Zubicaray GI, Carmichael OT, Becker JT, et al. Automated ventricular mapping with multi-atlas fluid image alignment reveals genetic effects in Alzheimer’s disease. Neuroimage. 2008;40:615–630. doi: 10.1016/j.neuroimage.2007.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chou YY, Lepore N, Avedissian C, Madsen SK, Parikshak N, et al. Mapping correlations between ventricular expansion and CSF amyloid and tau biomarkers in 240 subjects with Alzheimer’s disease, mild cognitive impairment and elderly controls. Neuroimage. 2009;46:394–410. doi: 10.1016/j.neuroimage.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gutman BA, Hua X, Rajagopalan P, Chou YY, Wang Y, et al. Maximizing power to track Alzheimer’s disease and MCI progression by LDA-based weighting of longitudinal ventricular surface features. Neuroimage. 2013;70:386–401. doi: 10.1016/j.neuroimage.2012.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leow AD, Yanovsky I, Chiang MC, Lee AD, Klunder AD, et al. Statistical properties of Jacobian maps and the realization of unbiased large-deformation nonlinear image registration. IEEE Trans Med Imaging. 2007;26:822–832. doi: 10.1109/TMI.2007.892646. [DOI] [PubMed] [Google Scholar]
- 21.Gutman BA, Wang Y, Thompson PM, Rajagopalan P, Toga AW. Shape matching with medial curves and 1-D group-wise registration. Biomedical Imaging (ISBI), 9th IEEE International Symposium 2012 [Google Scholar]
- 22.Yushkevich PA, Wang H, Pluta J, Das SR, Craige C, et al. Nearly automatic segmentation of hippocampal subfields in in vivo focal T2-weighted MRI. Neuroimage. 2010;53:1208–1224. doi: 10.1016/j.neuroimage.2010.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu C. The 5′ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature. 1980;286:854–860. doi: 10.1038/286854a0. [DOI] [PubMed] [Google Scholar]
- 24.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheffield NC, Thurman RE, Song L, Safi A, Stamatoyannopoulos JA, et al. Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions. Genome Res. 2013;23:777–788. doi: 10.1101/gr.152140.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skelly AC. Probability, proof, and clinical significance. Evid Based Spine Care J. 2011;2:9–11. doi: 10.1055/s-0031-1274751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel BB, Holland NW. Mild cognitive impairment: hope for stability, plan for progression. Cleveland Clinic journal of medicine. 2012;79:857–864. doi: 10.3949/ccjm.79a.11126. [DOI] [PubMed] [Google Scholar]
- 28.Chen H, Foo SH, Ury W. Recognizing dementia. The Western journal of medicine. 2002;176:267–270. [PMC free article] [PubMed] [Google Scholar]
- 29.Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, et al. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–2117. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang MX, Cross P, Andrews H, Jacobs DM, Small S, et al. Incidence of AD in African-Americans, Caribbean Hispanics, and Caucasians in northern Manhattan. Neurology. 2001;56:49–56. doi: 10.1212/wnl.56.1.49. [DOI] [PubMed] [Google Scholar]
- 31.Demirovic J, Prineas R, Loewenstein D, Bean J, Duara R, et al. Prevalence of dementia in three ethnic groups: the South Florida program on aging and health. Annals of epidemiology. 2003;13:472–478. doi: 10.1016/s1047-2797(02)00437-4. [DOI] [PubMed] [Google Scholar]
- 32.Murray CJ, Lopez AD. Regional patterns of disability-free life expectancy and disability-adjusted life expectancy: global Burden of Disease Study. Lancet. 1997;349:1347–1352. doi: 10.1016/S0140-6736(96)07494-6. [DOI] [PubMed] [Google Scholar]
- 33.Pinsonneault JK, Han DD, Burdick KE, Kataki M, Bertolino A, et al. Dopamine transporter gene variant affecting expression in human brain is associated with bipolar disorder. Neuropsychopharmacology. 2011;36:1644–1655. doi: 10.1038/npp.2011.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guindalini C, Howard M, Haddley K, Laranjeira R, Collier D, et al. A dopamine transporter gene functional variant associated with cocaine abuse in a Brazilian sample. Proc Natl Acad Sci U S A. 2006;103:4552–4557. doi: 10.1073/pnas.0504789103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munoz P, Huenchuguala S, Paris I, Segura-Aguilar J. Dopamine oxidation and autophagy. Parkinson’s disease. 2012;2012:920953. doi: 10.1155/2012/920953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Conway KA, Rochet JC, Bieganski RM, Lansbury PT., Jr Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science. 2001;294:1346–1349. doi: 10.1126/science.1063522. [DOI] [PubMed] [Google Scholar]
- 38.Sulzer D. alpha-synuclein and cytosolic dopamine: stabilizing a bad situation. Nature medicine. 2001;7:1280–1282. doi: 10.1038/nm1201-1280. [DOI] [PubMed] [Google Scholar]
- 39.Kuhn DM, Francescutti-Verbeem DM, Thomas DM. Dopamine quinones activate microglia and induce a neurotoxic gene expression profile: relationship to methamphetamine-induced nerve ending damage. Annals of the New York Academy of Sciences. 2006;1074:31–41. doi: 10.1196/annals.1369.003. [DOI] [PubMed] [Google Scholar]
- 40.Colloby SJ, McParland S, O’Brien JT, Attems J. Neuropathological correlates of dopaminergic imaging in Alzheimer’s disease and Lewy body dementias. Brain: a journal of neurology. 2012;135:2798–2808. doi: 10.1093/brain/aws211. [DOI] [PubMed] [Google Scholar]
- 41.Piggott MA, Marshall EF, Thomas N, Lloyd S, Court JA, et al. Striatal dopaminergic markers in dementia with Lewy bodies, Alzheimer’s and Parkinson’s diseases: rostrocaudal distribution. Brain: a journal of neurology. 1999;122(Pt 8):1449–1468. doi: 10.1093/brain/122.8.1449. [DOI] [PubMed] [Google Scholar]
- 42.Lopez OL, Becker JT, Kaufer DI, Hamilton RL, Sweet RA, et al. Research evaluation and prospective diagnosis of dementia with Lewy bodies. Archives of neurology. 2002;59:43–46. doi: 10.1001/archneur.59.1.43. [DOI] [PubMed] [Google Scholar]
- 43.Bittner V, Ullrich G, Thormann M, Muller NG, Friederichs C, et al. Positive FP-CIT SPECT (DaTSCAN) in Clinical Alzheimer’s Disease – An Unexpected Finding? Dementia and geriatric cognitive disorders extra. 2011;1:283–291. doi: 10.1159/000330470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whitwell JL, Jack CR, Jr, Parisi JE, Knopman DS, Boeve BF, et al. Rates of cerebral atrophy differ in different degenerative pathologies. Brain: a journal of neurology. 2007;130:1148–1158. doi: 10.1093/brain/awm021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maruyama W, Naoi M. “70th Birthday Professor Riederer” induction of glial cell line-derived and brain-derived neurotrophic factors by rasagiline and (−)deprenyl: a way to a disease-modifying therapy? J Neural Transm. 2013;120:83–89. doi: 10.1007/s00702-012-0876-x. [DOI] [PubMed] [Google Scholar]
- 46.Shah M, Rajagopalan S, Xu L, Voshavar C, Shurubor Y, et al. The high-affinity D2/D3 agonist D512 protects PC12 cells from 6-OHDA-induced apoptotic cell death and rescues dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Journal of neurochemistry. 2014 doi: 10.1111/jnc.12767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oster S, Radad K, Scheller D, Hesse M, Balanzew W, et al. Rotigotine protects against glutamate toxicity in primary dopaminergic cell culture. European journal of pharmacology. 2014;724:31–42. doi: 10.1016/j.ejphar.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 48.Odaka H, Numakawa T, Adachi N, Ooshima Y, Nakajima S, et al. Cabergoline, Dopamine D2 Receptor Agonist, Prevents Neuronal Cell Death under Oxidative Stress via Reducing Excitotoxicity. PloS one. 2014;9:e99271. doi: 10.1371/journal.pone.0099271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao C, Sun X, Wolf ME. Activation of D1 dopamine receptors increases surface expression of AMPA receptors and facilitates their synaptic incorporation in cultured hippocampal neurons. J Neurochem. 2006;98:1664–1677. doi: 10.1111/j.1471-4159.2006.03999.x. [DOI] [PubMed] [Google Scholar]
- 50.Mockett BG, Guevremont D, Williams JM, Abraham WC. Dopamine D1/D5 receptor activation reverses NMDA receptor-dependent long-term depression in rat hippocampus. J Neurosci. 2007;27:2918–2926. doi: 10.1523/JNEUROSCI.0838-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Navakkode S, Sajikumar S, Frey JU. Synergistic requirements for the induction of dopaminergic D1/D5-receptor-mediated LTP in hippocampal slices of rat CA1 in vitro. Neuropharmacology. 2007;52:1547–1554. doi: 10.1016/j.neuropharm.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 52.Jurgensen S, Antonio LL, Mussi GE, Brito-Moreira J, Bomfim TR, et al. Activation of D1/D5 dopamine receptors protects neurons from synapse dysfunction induced by amyloid-beta oligomers. J Biol Chem. 2011;286:3270–3276. doi: 10.1074/jbc.M110.177790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, et al. Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci. 1999;2:271–276. doi: 10.1038/6374. [DOI] [PubMed] [Google Scholar]
- 54.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, et al. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ. Amyloid-beta oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans. 2002;30:552–557. doi: 10.1042/bst0300552. [DOI] [PubMed] [Google Scholar]
- 56.Yang IA, Holloway JW, Fong KM. Genetic susceptibility to lung cancer and co-morbidities. J Thorac Dis. 2013;5:S454–S462. doi: 10.3978/j.issn.2072-1439.2013.08.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cheng L, Han X, Shi Y. A regulatory role of LPCAT1 in the synthesis of inflammatory lipids, PAF and LPC, in the retina of diabetic mice. Am J Physiol Endocrinol Metab. 2009;297:E1276–1282. doi: 10.1152/ajpendo.00475.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nakanishi H, Shindou H, Hishikawa D, Harayama T, Ogasawara R, et al. Cloning and characterization of mouse lung-type acyl-CoA:lysophosphatidylcholine acyltransferase 1 (LPCAT1). Expression in alveolar type II cells and possible involvement in surfactant production. J Biol Chem. 2006;281:20140–20147. doi: 10.1074/jbc.M600225200. [DOI] [PubMed] [Google Scholar]
- 59.Blusztajn JK, Liscovitch M, Mauron C, Richardson UI, Wurtman RJ. Phosphatidylcholine as a precursor of choline for acetylcholine synthesis. J Neural Transm Suppl. 1987;24:247–259. [PubMed] [Google Scholar]
- 60.Nakamura S. Phosphatidylcholine hydrolysis and protein kinase C activation for intracellular signaling network. J Lipid Mediat Cell Signal. 1996;14:197–202. doi: 10.1016/0929-7855(96)00525-1. [DOI] [PubMed] [Google Scholar]
- 61.Zeisel SH. The supply of choline is important for fetal progenitor cells. Semin Cell Dev Biol. 2011;22:624–628. doi: 10.1016/j.semcdb.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gaudin M, Panchal M, Auzeil N, Duyckaerts C, Brunelle A, et al. Choline-containing phospholipids in microdissected human Alzheimer’s disease brain senile plaque versus neuropil. Bioanalysis. 2012;4:2153–5159. doi: 10.4155/bio.12.189. [DOI] [PubMed] [Google Scholar]
- 63.Shea TB, Ekinci FJ, Ortiz D, Dawn-Linsley M, Wilson TO, et al. Efficacy of vitamin E, phosphatidyl choline, and pyruvate on buffering neuronal degeneration and oxidative stress in cultured cortical neurons and in central nervous tissue of apolipoprotein E-deficient mice. Free Radic Biol Med. 2002;33:276–282. doi: 10.1016/s0891-5849(02)00872-9. [DOI] [PubMed] [Google Scholar]
- 64.Tiraboschi P, Hansen LA, Alford M, Merdes A, Masliah E, et al. Early and widespread cholinergic losses differentiate dementia with Lewy bodies from Alzheimer disease. Archives of general psychiatry. 2002;59:946–951. doi: 10.1001/archpsyc.59.10.946. [DOI] [PubMed] [Google Scholar]
- 65.Chen SG, Kulju D, Halt S, Murakami K. Phosphatidylcholine-dependent protein kinase C activation. Effects of cis-fatty acid and diacylglycerol on synergism, autophosphorylation and Ca(2+)-dependency. Biochem J. 1992;284(Pt 1):221–226. doi: 10.1042/bj2840221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- 67.Martorana A, Mori F, Esposito Z, Kusayanagi H, Monteleone F, et al. Dopamine modulates cholinergic cortical excitability in Alzheimer’s disease patients. Neuropsychopharmacology. 2009;34:2323–2328. doi: 10.1038/npp.2009.60. [DOI] [PubMed] [Google Scholar]
- 68.Martorana A, Di Lorenzo F, Esposito Z, Lo Giudice T, Bernardi G, et al. Dopamine D(2)-agonist rotigotine effects on cortical excitability and central cholinergic transmission in Alzheimer’s disease patients. Neuropharmacology. 2013;64:108–113. doi: 10.1016/j.neuropharm.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 69.Cummings JL. Integrating symptomatic- and disease-modifying treatments. CNS Spectr. 2008;13:28–30. doi: 10.1017/s1092852900027024. [DOI] [PubMed] [Google Scholar]