

Abstract
Technological advances in computing, imaging and genomics have created new opportunities for exploring relationships between histology, molecular events and clinical outcomes using quantitative methods. Slide scanning devices are now capable of rapidly producing massive digital image archives that capture histological details in high-resolution. Commensurate advances in computing and image analysis algorithms enable mining of archives to extract descriptions of histology, ranging from basic human annotations to automatic and precisely quantitative morphometric characterization of hundreds of millions of cells. These imaging capabilities represent a new dimension in tissue-based studies, and when combined with genomic and clinical endpoints, can be used to explore biologic characteristics of the tumor microenvironment and to discover new morphologic biomarkers of genetic alterations and patient outcomes. In this paper we review developments in quantitative imaging technology and illustrate how image features can be integrated with clinical and genomic data to investigate fundamental problems in cancer. Using motivating examples from the study of glioblastomas (GBMs), we demonstrate how public data from The Cancer Genome Atlas (TCGA) can serve as an open platform to conduct in silico tissue based studies that integrate existing data resources. We show how these approaches can be used to explore the relation of the tumor microenvironment to genomic alterations and gene expression patterns and to define nuclear morphometric features that are predictive of genetic alterations and clinical outcomes. Challenges, limitations and emerging opportunities in the area of quantitative imaging and integrative analyses are also discussed.
Keywords: image analysis, morphometrics, tumor microenvironment, glioma, genomics
INTRODUCTION
Visual information embedded in histologic specimens carries prognostic value and reflects the underlying molecular traits of disease. Human evaluation of histology is a time-honored practice and serves as the basis of modern pathology, yet is highly subjective and known for its inter- and intra-observer variations (1). Human observers are also limited by scale and the need to reduce information into summary categorical descriptions. Diagnostic evaluation of histologic specimens is often performed over a prescribed number of high-power fields, and reasonable reporting cannot possibly capture detailed descriptions of the tissue heterogeneities observed in many diseases.
The digitization of pathologic specimens has advanced with improvements in charge-coupled device (CCD) sensors, storage and network performance. Early versions of slide scanning hardware suffered from slow image acquisition, and their practical use was limited by the expense of storage and network limitations that made file transfer and remote viewing difficult. Contemporary slide scanning devices are now capable of digitizing a single slide at 40X objective magnification in two minutes or less, and can produce hundreds of whole-slide images (WSIs) in a single day. With each image occupying hundreds of megabytes to several gigabytes, the recent precipitous decline in storage costs in the past decade has made generation and analysis of large WSI archives more practical. Faster networks and improved software have enabled users to fluidly view and interact with large WSI archives at their desktop by streaming imaging data directly from remote servers. Currently, no universal standards exists for file format or image compression within a WSI, despite some work by the DICOM Working-Group 26, creating significant challenges in the interoperability of various hardware and software platforms from different WSI vendors. Improvements in computing performance and image analysis algorithms enable large WSI archives to be mined to extract quantitative and objective imaging features that describe the visual characteristics of tissue architecture and microanatomy (2, 3). Advances in the theory of image analysis algorithms make it possible to reliably delineate objects across biological scales from cell nuclei and membranes (where stained) to complex multicellular structures and tissue interfaces (4–16). With these objects delineated, a set of descriptive features can be calculated to describe their appearance including shape, texture, and spatial relation to one another. New computing hardware like multi-core processors and graphics cards enable these techniques to be scaled to WSI archives that can contain billions of such objects.
A collection of algorithms has even emerged to mitigate technical effects introduced by the physical processing of tissues, allowing the automatic detection of artifacts, and the correction of color differences caused by variations in section thickness and staining (17–22). These procedures improve the robustness of image segmentation processes and result in uniform features that reflect biological properties, while reducing noise introduced by technical artifacts. The size in bytes of features extracted from an image can rival that of the image itself, and the management and standardization of image features and their provenance is not trivial.
Image analysis algorithms that precisely describe microscopic features within pathologic specimens provide tremendous opportunities for integration with genomic analyses and a new platform for advancing genotype-phenotype comparisons. Contemporary genomic platforms have generated a new view of the genetic, transcriptional and epigenetic events that are embedded within tissue samples. Deep molecular characterizations of biospecimens are increasingly available and gaining clinical relevance and the complementary nature of genomic and quantitative imaging descriptions creates new opportunities for their integrated analysis. Genomics provide extremely high molecular resolution but poor spatial resolution, and the genomic signature of a specimen therefore represents an aggregate measure of heterogeneous molecular profiles within distinct components of the tissue analyzed. Laser capture microdissection provides a way to increase the purity of genomic measurements, but is labor-intensive and difficult to carry out on large cohorts, although image analysis has been used to reduce this burden (23). An alternative approach is the integration of genomic and imaging features through computational means to deconvolve distinct profiles from the aggregate profile, with the goal of recovering information that is lost when tissue is homogenized for genomic analysis. Histology is also a manifestation of underlying molecular profiles within tissues, so quantitative imaging features can be expected to contain predictive power as biomarkers of genetic alterations and gene expression patterns. By integrating imaging and genomic features into risk models, prognostic variance may be reduced compared to genomics or histopathology alone.
The availability of large de-identified data-sets from The Cancer Genome Atlas (TCGA) has greatly facilitated integrated analyses that use imaging, genomic and clinical data. This well-characterized and comprehensive data set would be difficult to duplicate at a single institution due to prohibitive cost, privacy concerns, and patient volumes. TCGA is a large public resource that provides comprehensive molecular characterizations of more than 22 cancers types. Although intended primarily as a genomic resource, TCGA contains over 22,000 whole-slide images from more than 10,000 tumors, in addition to detailed clinical descriptions, and serves as an open platform to perform studies that integrate quantitative histology with molecular and clinical data. The use of these existing resources to conduct in silico scientific investigations has enabled researchers in this area to focus effort on developing analysis methods rather than data production, and to scale studies to a number of samples that would be otherwise difficult to achieve (Fig 1). While TCGA is an exceptional resource at this point in time, such multifaceted descriptions of tissues will likely become more commonplace within academic research institutions with increasing clinical adoption of genomics and digital pathology, and as the information management systems that manage these data improve.
Figure 1.
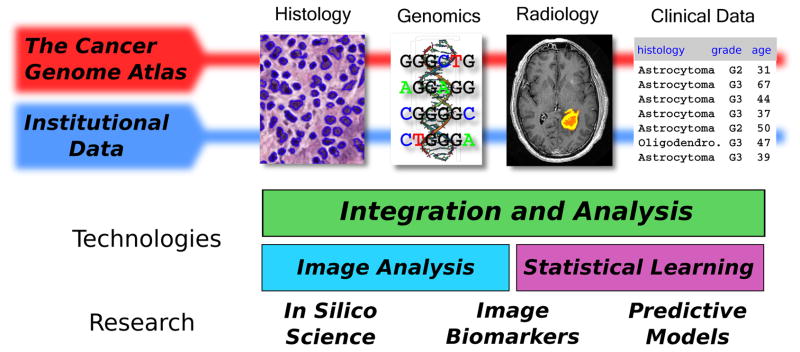
Integration of quantitative histology with multifaceted clinical and genomic data. Image analysis algorithms can extract features that describe the histology in digital whole-slide image datasets. This information can be combined with genomic, clinical and radiology data to identify image biomarkers of genetic alterations, to build predictive models of clinical outcomes, and to better understand tumor biology. Public data provided by The Cancer Genome Atlas (TCGA) makes it possible to explore these topics in large cohorts of more than 22 types of cancers. Discoveries made in analysis of public TCGA data can be validated in smaller institutional datasets.
In this paper we present a review of developments in the area of quantitative histology, using examples from glioblastoma to illustrate how imaging features can be integrated with genomic and clinical data to improve understanding. The first example explores issues of tumor microenvironment (TME), and how imaging features can illuminate the impact of the TME on the genomic signatures and molecular classifications. In the second example we present a pipeline for the morphometric characterization of nuclei that is capable of extracting quantitative descriptions of billions of cell nuclei in digital WSI archives. We show how this pipeline can be used along with statistical and statistical learning techniques to define imaging biomarkers of genetic alterations and epigenetic and transcriptional patterns, as well as clinical outcomes. We finish by describing near-term potential opportunities for quantitative imaging and integrated studies, and discuss the limitations and challenges associated with these approaches.
MAIN BODY
The Cancer Genome Atlas
The Cancer Genome Atlas (TCGA) was established in 2005 to improve understanding of the molecular basis of human cancers through large-scale genomic analysis. With a goal of accruing 500 tumors for each cancer selected for study, TCGA has expanded beyond initial pilot projects in glioblastoma, lung and ovarian carcinoma to now span more than 22 tumor types. This effort relies on a pipeline of participating institutions that submit frozen tissues and clinical data to a central repository, a set of de-centralized genomic analysis centers that produce messenger RNA, micro RNA, DNA exome sequencing, DNA copy number, DNA methylation, and protein expression profiles, and an electronic clearinghouse that then makes this data available to the public (https://tcga-data.nci.nih.gov).
An important, yet underappreciated aspect of acquiring clinical data from tissue source sites includes the collection of digitized whole-slide images of submitted tumors. Frozen sections are produced from the top and bottom of tissue samples that are submitted for genomic analysis, and are used for quality control to evaluate the percentage of tumor, the presence of necrosis and other factors that will influence the quality of genomic results. These images are a valuable resource since they are immediately adjacent to tissues used for genomics, and provide the most faithful representation of genomic-annotated tissues. Diagnostic permanent section slides are also solicited from participating institutions. The higher quality of these images and lack of freezing artifacts makes them more suitable for algorithmic analysis, particularly at high magnification. Expert pathology committees that are selected by disease area review these permanent sections to ensure correct diagnosis and to evaluate the presence of important pathologic criteria. Examples from the GBM project include the categorical scoring (0, 1+, 2+) of qualities like microvascular proliferation, pseudopalisading necrosis and lymphocytic infiltration. All permanent and frozen sections are digitized at 20X or 40X objective magnification and made publicly available for download.
The Tumor Microenvironment and Transcriptional Classification of Glioblastomas
One of the main outcomes of the TCGA analyses has been the development of genomic sub-classifications of many cancers. Using clustering analysis of gene expression and other molecular platforms, the goal of these analyses is to define cohesive sub-classes of tumors with distinct molecular signatures that may benefit from class-specific targeted therapies. In glioblastoma, two studies using TCGA data have identified tumor sub-classifications based on gene expression and DNA methylation (24, 25). The initial TCGA analysis of GBMs identified four gene expression classes (GESs): proneural, neural, classic and mesenchymal. These classes exhibit clear and distinct patterns of gene expression, and are highly correlated with genetic alterations in EGFR, IDH1, NF1, PDGFRA, and TP53. A subsequent analysis of DNA methylation data revealed that pro-neural GBMs are further subdivided into two groups - those with IDH mutations that have significant hypermethylation of CpG islands (GCIMP) and are typically secondary GBMs afflicting younger patients, and IDH wildtype tumors that do not exhibit DNA hypermethylation patterns.
One of the first goals of our in silico research was to investigate the relationships between gene expression classifications and the tumor microenvironment in GBMs (26, 27). Most tissue-based transcriptional classification studies of tumors are subjective in that neoplasms are highly heterogeneous, and gene expression measurements can vary significantly among different samples from the same tumor. Glioblastomas are no exception, being spatially complex tumors that harbor a variety of non-neoplastic cell types and microenvironmental elements that can significantly impact gene expression measurements. Pseudopalisading necrosis and micro-vascular proliferation are perhaps the most notable elements, being part of the diagnostic criteria that distinguish glioblastomas from lower-grade gliomas, and indicators of poor prognosis (28). The development of necrosis and microvascular proliferation is can be focal at first, but then expands, and signals severe underlying hypoxia with resultant profound transcriptional changes.
Having access to both frozen sections and comprehensive molecular profiles from adjacent tissues from TCGA, we sought to measure the impact of necrosis and angiogenesis on gene expression patterns used to classify GBMs. We hypothesized the extent of necrosis and angiogenesis in a histologic section are tightly associated with the presence of hypoxia, which could play an important role in establishing GES signatures by activation of hypoxia-inducible transcription factors. With the degree of hypoxia varying spatially throughout a tumor, multiple GES classes could possibly co-exist within the same tumor, and so classification by gene expression could be subject to random effects in tissue sampling. Intra-tumoral variations in GES classification would have significant implications in using these classes as platforms for the development of targeted therapies.
Using a human-computer interface, we annotated 177 digitized frozen section images to define the boundaries of necrosis and angiogenesis for 99 tumors. The lumens within angiogenic regions were subtracted using an image analysis segmentation algorithm. The sections in these images are immediately adjacent to those used for genomic analysis, and these annotations therefore provide the most faithful representation of the microenvironmental conditions in genomically analyzed tissues (Fig 2A). The extent of necrosis and angiogenesis was calculated as a percentage of the total tissue area, and these quantitative features were linked to gene expression and other genomic measurements from the same tissue (Fig 2B).
Figure 2.

Tumor microenvironment study integrating histology and genomics from TCGA. (A) TCGA specimens are sections from the top and bottom to produce slides, and the middle portion is submitted for genomic analysis. (B) Digitized images from top/bottom sections were annotated to calculate the percentage of necrosis and angiogenesis for each tumor. (C) Tumors from the mesenchymal expression class are significantly enriched with necrosis. (D) As the amount of necrosis increases in non-mesenchymal GBMs, gene expressions patterns shift towards a mesenchymal expression signature.
We first examined the abundance of necrosis and angiogenesis in tumors organized by TCGA transcriptional class. Tumors with a mesenchymal GES were clearly enriched with higher amounts of necrosis (one-way ANOVA p = 8.7e-4, see Fig 2C), suggesting a strong association between the mesenchymal gene-expression signatures, necrosis, and hypoxia. All tumors with greater than 22% necrosis were members of the mesenchymal class. The relationship between angiogenesis and transcriptional class was less clear. The percentage of angiogenesis ranged from 0–4% for the large majority of tumors. There were only 4 outliers with much higher levels of angiogensis and 3 were from the mesenchymal GES and 1 was from the proneural GES. While we would expect the presence of angiogenesis to influence gene expression, the ability to detect this feature may in part be limited by the relatively small contribution these regions make to the total amount of DNA/RNA extracted for analysis.
Next we performed a genome-wide analysis of transcriptional data to discover the impact of necrosis on gene expression. A normalized linear regression coefficient was calculated for each transcript to measure the strength of relationship between extent of necrosis and gene expression for more than 22,000 transcripts measured with Affymetrix arrays. Significance Analysis of Microarrays (SAM) correction was applied to obtain multiple-test corrected p-values for each gene (29). This analysis identified 2422 genes that are significantly correlated with extent of necrosis at 5% false-discovery rate or below, suggesting that necrosis has tremendous influence on gene expression in GBMs. Among the genes most significantly correlated with necrosis were a set of transcription factors known as mesenchymal master regulators: CEBPB, FOSL2, CEBPD, STAT3, BHLHE40 (ranked 4th, 10th, 60th, 213th and 221st respectively). These transcription factors have been shown to form a small module regulating a much broader gene expression network that is responsible for mesenchymal tumor phenotype in glioblastomas (30). At the top of this regulatory module are the transcription factors CE-BPB/CEBPD and STAT3, whose coexpression is necessary and sufficient for activating the mesenchymal expression network. To explore the expression of these regulators in tissues, we performed immunohistochemistry on archived surgically resected glioblastomas from our own institution. We observed that CEBPB/CEBPD expression was strongly and specifically expressed in the hypoxic pseudopalisading cells surrounding areas of necrosis (Fig 1D). CEBPB was strongly expressed in nuclei of the first 2–5 cell layers immediately surrounding necrosis, and CEBPD expression was found in both nuclear and cytoplasmic regions of perinecrotic cells but extending slightly farther beyond CEBPD. In regions between foci of necrosis, only a small portion of cells expressed either CEBPB or CEBPD. STAT3 did not show a specific perinecrotic pattern of expression.
Gene expression classifications are made by measuring the distances in gene expression space between a tumor’s expression profile and a set of points or centroids that represent each class. The tumor is assigned to the class with the “nearest” centroid, which can be measured using a variety of metrics including simple Euclidean distance. While a given tissue could potentially contain individual cells/regions with diverging gene expression profiles, these classifications force a selection of a single gene expression class that best defines the entire sample. To explore how the formation of necrosis influences the expression patterns of non-mesenchymal GBMs, we examined the relationship between extent of necrosis and distance to the mesenchymal expression centroid in this cohort. We observed a clear trend – the more necrosis that a sample contains, the more its expression profile resembles the mesenchymal centroid (Fig 1E). This finding further suggests that mesenchymal gene expression is strongly impacted by hypoxia and that expression signatures are strongly impacted by regionally varying elements of the microenvironment.
Molecular and Clinical Associations Revealed Through Quantitative Nuclear Morphometry
The morphologic characteristics of cell nuclei convey important clinical information in many types of neoplasms. Besides determining histologic classification and subtype, nuclear qualities including shape, texture and spatial arrangement can be indicative of more specific molecular alterations and patient prognosis. Gains, losses and rearrangements of DNA along with epigenetic modifications affecting chromatin structure can manifest in observable changes within nuclei of neoplastic cells. In the diffuse gliomas, nuclear features are of particular importance, as their classification of oligodendroglioma or astocytoma is based in large part on nuclear morphology. However, histopathologic classification based on human review is subjective and prone to substantial interobserver variation. Understanding the relationships between nuclear morphology, tumor genetics and clinical outcomes will provide a better understanding of tumor biology and further improve the precision of clinical predictions.
Our studies of tumor microenvironment used human markups and annotations to generate quantitative features from whole-slide images. The limitations of human annotations are apparent when dealing with nuclear morphology - nuclei can number in the hundreds of millions in even a modestly sized set of images, and qualities of interest like nuclear texture are difficult to accurately characterize objectively by human observers. To address these challenges we have developed a computational system for the study of nuclear morphometry in large archives of whole-slide images (Fig 3A). This system uses image analysis algorithms to delineate individual cell nuclei, and to calculate a set of objective nuclear features for each nucleus to describe its shape and texture. High performance and parallel computing approaches are used to scale this approach to hundreds of millions of cells. This system presents opportunities to define quantitative morphologic biomarkers of molecular and clinical endpoints by enabling the extraction of objective, repeatable measurements from WSI archives.
Figure 3.

Quantitative nuclear morphometry. (A) Image analysis algorithms are used to delineate nuclei in whole-slide images. A set of features is calculated to describe the appearance of each nucleus. This system is capable of processing thousands of slides and hundreds of millions of nuclei. (B) We developed a model-based system to score nuclei based on oligodendroglial differentiation. This model was validated by correlation of nuclear scores and gene expression data. (C) Model-free approaches were used to explore the clinical and genomic associations of nuclear features. Clustering of patient morphological signatures revealed three distinct patient clusters. Unsupervised analysis of features shows that proneural tumors are associated with more round, regular nuclei.
Our initial morphometric study focused on the quantitative characterization of oligodendroglial differentiation in glioblastomas (31). Although GBM is defined as a grade IV astrocytoma, an important subset exhibits varying degrees of oligodendroglial differentiation in addition to the dominant astrocytic component (28, 32–34). Neoplasms with pure oligodendroglial differentiation typically have slower growth and better survivals when compared with astrocytomas of the same grades. The morphologic characteristics of oligodendrogliomas distinguish them from astrocytomas: oligodendroglial nuclei tend to be smaller, round and hyperchromatic with a lack of detailed texture, in contrast with astrocytoma nuclei that are larger, irregularly shaped, typically elongated and unevenly textured. In most instances, GBMs contain a heterogeneous mixture of neoplastic cells with wide variations in nuclear characteristics, many of which are not clearly astrocytic or oligodendroglial. The volume and heterogeneity of cells present in GBMs combined with subtle differences in morphologic diversity make them an ideal candidate for computational morphometric approaches.
Using our computational pipeline, we analyzed 200 million nuclei from digitized images of diagnostic slides corresponding to 117 TCGA GBMs. Twenty-three quantitative features from four categories (shape, intensity, texture and gradient) were calculated to describe each nucleus. To represent the differentiation of each nucleus along the oligodendroglial/astrocytic spectrum, we built a regression model that uses the nuclear feature values to calculate a score for each nucleus representing its degree of oligodendroglial appearance (Fig 3B). Combining the 200 million scores obtained from our pipeline with gene expression, copy number, DNA sequence and methylation data from the same TCGA tumors, we were able to clearly separate a set of tumor enriched with oligodendroglial-like cells that had strong associations with PDGFRA amplification, proneural transcriptional class, and expression of the oligodendrocyte signature genes MBP, HOXD1, PLP1, MOBP and PDGFRA. These results provide molecular validation that the quantitative features extracted by our software pipeline can capture the morphologic variations of nuclei encountered in gliomas.
Our differentiation study used a supervised approach to build a quantitative model of the oligodendroglial/astrocytic spectrum in gliomas. Model-based approaches are a powerful way to incorporate prior knowledge into morphologic analyses, and to use quantitative measures of recognized morphologic patterns to explore their molecular correlates. Because model-based approaches are built on prior knowledge, their ability to reveal previously unrecognized or unknown morphologic patterns is limited. To address this limitation, we have developed several unsupervised or model-free approaches that do not impose established constructs in the morphological analysis of WSI data. Instead, these approaches let data speak for itself, using clustering analysis and other statistical learning techniques to reveal natural structure within the feature data in a bottom-up fashion.
Our first study with unsupervised methods investigated patient clustering of GBMs into morphologically defined subtypes (35, 36). Using nuclear features, we sought to determine if there are clear and distinct groups of tumors that emerge from clustering analysis, similar to gene expression studies where transcriptional profiles are clustered to reveal molecular tumor subtypes. Taking the nuclear features from the TCGA cohort, a morphologic signature was calculated for each tumor to represent the morphologic properties of its average nuclei. These signatures were analyzed using a consensus-clustering algorithm to find natural groups within the data and to measure their robustness. Three clear clusters emerged from this analysis and we named them for themes observed in their molecular correlates: cell cycle (CC), protein biosynthesis (PB) and chromatin modification (CM). We observed that these clusters had significant differences in patient survival (logrank p=1.4e-3), with the PB cluster containing patients with relatively better outcomes and the CM cluster relatively worse. These clusters were also observed in an independent dataset of 84 GBMs where the relative differences in outcomes between the clusters were also confirmed. To explore the meaning of these clusters we used the various genomic platforms made available by TCGA including gene expression, DNA methylation, copy number and DNA sequencing. A pathway analysis found that the clusters varied in the extent of TP53 WNT, and NFKB signaling, and had variations in the extent of total DNA methylation. An analysis of the pathologic features using categorical human annotations (0,1+,2+) found that tumors in the CM cluster had enriched presence of lymphocytes, and that PB cluster tumors exhibited a conspicuous lack of inflammation.
To further explore model-free associations of nuclear morphometry in GBM and clinical and genomic endpoints, we took a more direct approach of correlating raw nuclear features with genomic and clinical endpoints (37). For each patient, we calculated the mean and standard deviation of each feature as metrics and correlating them directly with patient survival using Cox proportional hazards analysis using SAM. Notably, the mean circularity was significantly associated with longer patient survival, an observation consistent with prolonged clinical outcomes in gliomas with oligodendroglial differentiation. Other features that were significantly associated with outcome include major axis length, with longer nuclei associated with a shorter survival, and min nuclear pixel intensity, with higher values associated with longer survival. The fact that these features emerged from a more data-driven approach provides some level of confidence in our analysis workflow. To correlate these features with genomic measurements we performed a one-way ANOVA for each feature metric across transcriptional classifications, somatic mutations and DNA copy number alterations. Features distinguishing transcriptional classes include nuclear eccentricity (p = 3.81e-4), minor axis length (p = 8.87e-3) and nuclear extent (p = 3.2e-2). The greatest morphology differences were observed between the proneural and mesenchymal tumors. Those hypermethylated (GCIMP) tumors within the proneural group had greater variation of pixel intensities within their nuclei (nuclear energy, p = 2.28e-5), and greater variation in nuclear size. Genetic events having significant differences in nuclear morphometry included PTEN and TP53 mutations, and PDG-FRA amplification. PTEN and TP53 mutant tumors were both associated with less circular nuclei (p = 9.68e-3, 3.77e-2 respectively). PDGFRA amplified tumors were associated with greater circularity (p = 2.31e-2), consistent with PDGFRA amplifications being associated with oligodendroglial differentiation. Other genetic alterations with significant associations included EGFR amplification, which was associated with greater nuclear eccentricity and canny, and MDM2 amplifications, which were associated with greater minor axis length, area and circularity.
DISCUSSION
Emerging Challenges and Opportunities
Advances in whole-slide imaging and computing hardware have made it possible to approach increasingly difficult image analysis problems in pathology. At the same time, the increasing availability of rich genomic data have made pathology image analysis studies more interesting by allowing linkage of histologic features with comprehensive molecular measurements. Within the last decade, the goals of pathology image analysis have shifted from attempts to implement computer-aided diagnostic procedures, to more creative analyses that explore complex genotype-phenotype associations and define novel prognostication methods. The emerging goal is to go beyond computational replication of pathologists and to develop novel techniques to unmask latent content within image sets that has as yet unrecognized clinical and scientific value. This convergence of image analysis and bioinformatics has produced some exciting results in several different areas. In glioblastoma, morphometry-driven tumor subtypes were identified on the basis of nuclear morphometry and cellularity and found to be predictive of clinical outcomes and pathway activation (6, 38). In breast cancer, morphologic features describing stromal/tumor interface were found to be predictive of overall survival independent of other clinical, pathological and molecular features in two independent cohorts (39). Features of cellularity derived from images were found to improve the estimation of copy number variations, and a prognostic model that combines image measurements with gene expression features was developed and validated in independent cohorts (40). Focusing specifically on triple-negative breast cancer, a prognostic model based on morphologic features was also developed and validated in an independent dataset of triple negative breast cancers containing histology images (41). A gene-expression signature derived from this prognostic model was then developed and used to further validate the prognostic value of this model in gene expression datasets where histology images were not available.
One barrier to progress in this area is the dissemination of algorithms and image features beyond image analysis experts to the broader research community. Making software and feature data publicly accessible will facilitate advances in this field by more fully engaging the pathology community and providing opportunities for comparative studies. Establishing the computational resources needed to execute image analysis algorithms on the primary images is difficult, and the sharing of derived feature data is limited by a lack of standardization. To begin to address these issues, we have developed web-based interfaces and data standards to support the visualization, federation and analysis of pathology image data. The Cancer Digital Slide Archive (CDSA, http://cancer.digitalslidearchive.net/) is a web-based resource that was originally developed to facilitate the visualization and analysis of pathology imaging and clinical data from TCGA (42). The CDSA currently hosts over 22,000 images and associated clinical data from over 22 different cancers represented in TCGA. This interface provides access to pathology and clinical data through a simple web-browser interface. Although currently focused on serving primary images for visualization, the CDSA and other similar resources could naturally serve as clearinghouses that allow a broader set of users to interact with image analysis algorithms and feature sets. Cloud-based services could be established that enable primary image data, derived features and computational tools to reside in a common computing environment, avoiding the need for costly transfer of massive amounts of image and feature data. Users could then perform end-to-end integrated analyses of pathology imaging online without the need to establish local computing resources or shepherding primary image or feature data between systems across the Internet. In cases where feature data and algorithms are exchanged, we have also developed the Pathology Analytic Imaging Standards (PAIS) to support the standardization of image analysis algorithms and image features (43, 44). PAIS provides data standards that enable users to capture software and algorithm parameter provenance, and a common file format that enables results to be stored in a database for search and exchange.
The validation of findings remains another significant problem in pathology imaging studies. This issue is particularly important in studies identifying image biomarkers of clinical outcomes or genomic features. One of the risks in using image features to predict outcome or genomic measurements is overfitting – are we are learning meaningful relationships that will generalize to new unseen datasets, or simply generating predictions that are specific to the noise and artifacts of the dataset that were analyzed? Validating findings in external datasets, when available, or by proper cross-validation of a single dataset is important for distinguishing true findings from artifacts. Sometimes the morphologic features that are predictive can be visualized, while other times their predictive power is clearly measurable but not apparent to the human eye. Some features are calculated (e.g. standard deviation of canny) are difficult to correlate with concrete histologic findings that can be visualized. In the cases where visualization is not possible or does not produce any obvious visible distinction, it is difficult to interpret the meaning of predictive features or to link them to existing knowledge about the histology of that disease. Greater availability of benchmark datasets containing whole-slide images, and genomic and clinical data could help to establish reproducibility and improve confidence in the relationships defined through computational analysis.
Another area for growth is the integration of radiology imaging with pathology, genomics and clinical data. Image analysis methods for radiology data are more mature than for pathology, and are able to extract meaningful features from MR, PET, CT and other medical imaging modalities. The global perspective of tumor provided by medical imaging is complementary to the tissue and molecular scale measurements provided by pathology and genomics, and a number of studies have already explored the relationships between quantitative radiology imaging features, genomic profiles and clinical outcomes (45–48). Integrating complementary features across biological scales into prognostic models is a promising avenue to improve the precision of clinical predictions and risk stratification of patients.
Supplementary Material
Acknowledgments
Support: This work was supported by US Public Health Service National Institutes of Health (NIH) grants K22LM011576 (LADC), R01CA176659 (DJB) and the Georgia Research Alliance (DJB).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
References
- 1.van den Bent MJ. Interobserver variation of the histopathological diagnosis in clinical trials on glioma: a clinician’s perspective. Acta neuropathologica. 2010;120(3):297–304. doi: 10.1007/s00401-010-0725-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gurcan MN, Boucheron LE, Can A, Madabhushi A, Rajpoot NM, Yener B. Histopathological image analysis: a review. IEEE reviews in biomedical engineering. 2009;2:147–71. doi: 10.1109/RBME.2009.2034865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cooper LAD, Carter AB, Farris AB, Fusheng W, Jun K, Gutman DA, et al. Digital Pathology: Data-Intensive Frontier in Medical Imaging. Proceedings of the IEEE. 2012;100(4):991–1003. doi: 10.1109/JPROC.2011.2182074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsu W, Markey MK, Wang MD. Biomedical imaging informatics in the era of precision medicine: progress, challenges, and opportunities. Journal of the American Medical Informatics Association: JAMIA. 2013;20(6):1010–3. doi: 10.1136/amiajnl-2013-002315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kothari S, Phan JH, Stokes TH, Wang MD. Pathology imaging informatics for quantitative analysis of whole-slide images. Journal of the American Medical Informatics Association: JAMIA. 2013;20(6):1099–108. doi: 10.1136/amiajnl-2012-001540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang H, Han J, Borowsky A, Loss L, Gray JW, Spellman PT, et al. Invariant delineation of nuclear architecture in glioblastoma multiforme for clinical and molecular association. IEEE transactions on medical imaging. 2013;32(4):670–82. doi: 10.1109/TMI.2012.2231420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang H, Nayak N, Spellman PT, Parvin B. Characterization of tissue histopathology via predictive sparse decomposition and spatial pyramid matching. Medical image computing and computer-assisted intervention: MICCAI International Conference on Medical Image Computing and Computer-Assisted Intervention. 2013;16(Pt 2):91–8. doi: 10.1007/978-3-642-40763-5_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hipp J, Smith SC, Cheng J, Tomlins SA, Monaco J, Madabhushi A, et al. Optimization of complex cancer morphology detection using the SIVQ pattern recognition algorithm. Analytical cellular pathology. 2012;35(1):41–50. doi: 10.3233/ACP-2011-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheng J, Hipp J, Monaco J, Lucas DR, Madabhushi A, Balis UJ. Automated vector selection of SIVQ and parallel computing integration MATLAB: Innovations supporting large-scale and high-throughput image analysis studies. Journal of pathology informatics. 2011;2:37. doi: 10.4103/2153-3539.83752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janowczyk A, Chandran S, Madabhushi A. Quantifying local heterogeneity via morphologic scale: Distinguishing tumoral from stromal regions. Journal of pathology informatics. 2013;4(Suppl):S8. doi: 10.4103/2153-3539.109865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janowczyk A, Chandran S, Singh R, Sasaroli D, Coukos G, Feldman MD, et al. High-throughput biomarker segmentation on ovarian cancer tissue microarrays via hierarchical normalized cuts. IEEE transactions on bio-medical engineering. 2012;59(5):1240–52. doi: 10.1109/TBME.2011.2179546. [DOI] [PubMed] [Google Scholar]
- 12.Song Y, Treanor D, Bulpitt AJ, Wijayathunga N, Roberts N, Wilcox R, et al. Unsupervised content classification based nonrigid registration of differently stained histology images. IEEE transactions on bio-medical engineering. 2014;61(1):96–108. doi: 10.1109/TBME.2013.2277777. [DOI] [PubMed] [Google Scholar]
- 13.Mosaliganti K, Janoos F, Irfanoglu O, Ridgway R, Machiraju R, Huang K, et al. Tensor classification of N-point correlation function features for histology tissue segmentation. Medical image analysis. 2009;13(1):156–66. doi: 10.1016/j.media.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooper L, Saltz J, Machiraju R, Huang K. Two-Point Correlation as a Feature for Histology Images: Feature Space Structure and Correlation Updating. Conference on Computer Vision and Pattern Recognition Workshops IEEE Computer Society Conference on Computer Vision and Pattern Recognition Workshops; 2010. pp. 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi X, Xing F, Foran DJ, Yang L. Robust segmentation of overlapping cells in histopathology specimens using parallel seed detection and repulsive level set. IEEE transactions on bio-medical engineering. 2012;59(3):754–65. doi: 10.1109/TBME.2011.2179298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samsi S, Krishnamurthy AK, Gurcan MN. An Efficient Computational Framework for the Analysis of Whole Slide Images: Application to Follicular Lymphoma Immunohistochemistry. Journal of computational science. 2012;3(5):269–79. doi: 10.1016/j.jocs.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan AM, Rajpoot N, Treanor D, Magee D. A Nonlinear Mapping Approach to Stain Normalization in Digital Histopathology Images Using Image-Specific Color Deconvolution. Biomedical Engineering, IEEE Transactions on. 2014;61(6):1729–38. doi: 10.1109/TBME.2014.2303294. [DOI] [PubMed] [Google Scholar]
- 18.Murakami Y, Abe T, Hashiguchi A, Yamaguchi M, Saito A, Sakamoto M. Color correction for automatic fibrosis quantification in liver biopsy specimens. Journal of pathology informatics. 2013;4:36. doi: 10.4103/2153-3539.124009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bautista PA, Hashimoto N, Yagi Y. Color standardization in whole slide imaging using a color calibration slide. Journal of pathology informatics. 2014;5:4. doi: 10.4103/2153-3539.126153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bautista PA, Yagi Y. Improving the visualization and detection of tissue folds in whole slide images through color enhancement. Journal of pathology informatics. 2010;1:25. doi: 10.4103/2153-3539.73320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chappelow J, Tomaszewski JE, Feldman M, Shih N, Madabhushi A. HistoStitcher((c)): an interactive program for accurate and rapid reconstruction of digitized whole histological sections from tissue fragments. Computerized medical imaging and graphics: the official journal of the Computerized Medical Imaging Society. 2011;35(7–8):557–67. doi: 10.1016/j.compmedimag.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kothari S, Phan JH, Wang MD. Eliminating tissue-fold artifacts in histopathological whole-slide images for improved image-based prediction of cancer grade. Journal of pathology informatics. 2013;4:22. doi: 10.4103/2153-3539.117448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roy Chowdhuri S, Hanson J, Cheng J, Rodriguez-Canales J, Fetsch P, Balis U, et al. Semiautomated laser capture microdissection of lung adenocarcinoma cytology samples. Acta cytologica. 2012;56(6):622–31. doi: 10.1159/000342984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer cell. 2010;17(5):510–22. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooper LA, Gutman DA, Chisolm C, Appin C, Kong J, Rong Y, et al. The tumor microenvironment strongly impacts master transcriptional regulators and gene expression class of glioblastoma. The American journal of pathology. 2012;180(5):2108–19. doi: 10.1016/j.ajpath.2012.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rutledge WC, Kong J, Gao J, Gutman DA, Cooper LA, Appin C, et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19(18):4951–60. doi: 10.1158/1078-0432.CCR-13-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta neuropathologica. 2007;114(2):97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(9):5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463(7279):318–25. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kong J, Cooper LA, Wang F, Gao J, Teodoro G, Scarpace L, et al. Machine-based morphologic analysis of glioblastoma using whole-slide pathology images uncovers clinically relevant molecular correlates. PloS one. 2013;8(11):e81049. doi: 10.1371/journal.pone.0081049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hegi ME, Janzer RC, Lambiv WL, Gorlia T, Kouwenhoven MC, Hartmann C, et al. Presence of an oligodendroglioma-like component in newly diagnosed glioblastoma identifies a pathogenetically heterogeneous subgroup and lacks prognostic value: central pathology review of the EORTC_26981/NCIC_CE.3 trial. Acta neuropathologica. 2012;123(6):841–52. doi: 10.1007/s00401-011-0938-4. [DOI] [PubMed] [Google Scholar]
- 33.Appin CL, Gao J, Chisolm C, Torian M, Alexis D, Vincentelli C, et al. Glioblastoma with oligodendroglioma component (GBM-O): molecular genetic and clinical characteristics. Brain pathology. 2013;23(4):454–61. doi: 10.1111/bpa.12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gupta M, Djalilvand A, Brat DJ. Clarifying the diffuse gliomas: an update on the morphologic features and markers that discriminate oligodendroglioma from astrocytoma. American journal of clinical pathology. 2005;124(5):755–68. doi: 10.1309/6JNX-4PA6-0TQ5-U5VG. [DOI] [PubMed] [Google Scholar]
- 35.Cooper LA, Kong J, Gutman DA, Wang F, Gao J, Appin C, et al. Integrated morphologic analysis for the identification and characterization of disease subtypes. Journal of the American Medical Informatics Association: JAMIA. 2012;19(2):317–23. doi: 10.1136/amiajnl-2011-000700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cooper LA, Kong J, Wang F, Kurc T, Moreno CS, Brat DJ, et al. Morphological Signatures and Genomic Correlates in Glioblastoma. Proceedings/IEEE International Symposium on Biomedical Imaging: from nano to macro IEEE International Symposium on Biomedical Imaging; 2011. pp. 1624–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jun K, Fusheng W, Teodoro G, Cooper L, Moreno CS, Kurc T, et al., editors. High-performance computational analysis of glioblastoma pathology images with database support identifies molecular and survival correlates; Bioinformatics and Biomedicine (BIBM), 2013 IEEE International Conference on; 2013 18–21 Dec; 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ju H, Hang C, Fontenay GV, Spellman PT, Borowsky A, Parvin B, editors. Molecular bases of morphometric composition in Glioblastoma multiforme; Biomedical Imaging (ISBI), 2012 9th IEEE International Symposium on; 2012 2–5 May; 2012. [Google Scholar]
- 39.Beck AH, Sangoi AR, Leung S, Marinelli RJ, Nielsen TO, van de Vijver MJ, et al. Systematic analysis of breast cancer morphology uncovers stromal features associated with survival. Science translational medicine. 2011;3(108):108ra13. doi: 10.1126/scitranslmed.3002564. [DOI] [PubMed] [Google Scholar]
- 40.Yuan Y, Failmezger H, Rueda OM, Ali HR, Graf S, Chin SF, et al. Quantitative image analysis of cellular heterogeneity in breast tumors complements genomic profiling. Science translational medicine. 2012;4(157):157ra43. doi: 10.1126/scitranslmed.3004330. [DOI] [PubMed] [Google Scholar]
- 41.Wang C, Pecot T, Zynger DL, Machiraju R, Shapiro CL, Huang K. Identifying survival associated morphological features of triple negative breast cancer using multiple datasets. Journal of the American Medical Informatics Association: JAMIA. 2013;20(4):680–7. doi: 10.1136/amiajnl-2012-001538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gutman DA, Cobb J, Somanna D, Park Y, Wang F, Kurc T, et al. Cancer Digital Slide Archive: an informatics resource to support integrated in silico analysis of TCGA pathology data. Journal of the American Medical Informatics Association: JAMIA. 2013;20(6):1091–8. doi: 10.1136/amiajnl-2012-001469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang F, Kong J, Cooper L, Pan T, Kurc T, Chen W, et al. A data model and database for high-resolution pathology analytical image informatics. Journal of pathology informatics. 2011;2:32. doi: 10.4103/2153-3539.83192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang F, Kong J, Gao J, Cooper LA, Kurc T, Zhou Z, et al. A high-performance spatial database based approach for pathology imaging algorithm evaluation. Journal of pathology informatics. 2013;4:5. doi: 10.4103/2153-3539.108543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gutman DA, Cooper LA, Hwang SN, Holder CA, Gao J, Aurora TD, et al. MR imaging predictors of molecular profile and survival: multi-institutional study of the TCGA glioblastoma data set. Radiology. 2013;267(2):560–9. doi: 10.1148/radiol.13120118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zinn PO, Mahajan B, Sathyan P, Singh SK, Majumder S, Jolesz FA, et al. Radiogenomic mapping of edema/cellular invasion MRI-phenotypes in glioblastoma multiforme. PloS one. 2011;6(10):e25451. doi: 10.1371/journal.pone.0025451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jain R, Poisson L, Narang J, Gutman D, Scarpace L, Hwang SN, et al. Genomic mapping and survival prediction in glioblastoma: molecular subclassification strengthened by hemodynamic imaging biomarkers. Radiology. 2013;267(1):212–20. doi: 10.1148/radiol.12120846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gevaert O, Xu J, Hoang CD, Leung AN, Xu Y, Quon A, et al. Non-small cell lung cancer: identifying prognostic imaging biomarkers by leveraging public gene expression microarray data--methods and preliminary results. Radiology. 2012;264(2):387–96. doi: 10.1148/radiol.12111607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.