

Abstract
Although mankind has been suffering from osteoarthritis (OA) dating to the dawn of humankind, its pathogenesis remains poorly understood. OA is no longer considered a “wear and tear” condition but rather one driven by proteases where chronic low-grade inflammation may play a role in perpetuating proteolytic activity. While multiple factors are likely active in this process, recent evidence has implicated the importance of the innate immune system, the older or more primitive part of our body’s immune defense mechanisms. The role of some of the components of the innate immune system have been tested in OA models in vivo including the role of synovial macrophages and the complement system. This review is a selective overview of a large and evolving field. Insights into these mechanisms might inform our ability to phenotype patient subsets and give hope for the advent of novel OA therapies.
Key indexing terms: osteoarthritis, innate immunity, macrophages, complement
Introduction
Osteoarthritis (OA) is considered an “old” disease. Not only is it a disease of the elderly but evidence for OA exists in the archeological record of ancient man (1). OA involves the “whole joint”, including articular and meniscal cartilage degeneration and loss, sclerotic changes to the subchondral bone, bony osteophytosis and synovial inflammation (2). Although this disease is widely prevalent, the exact mechanisms involved in its pathogenesis are not well understood. However, OA is no longer thought to be a purely non-inflammatory or a biomechanical (“wear and tear”) process but rather one that has been increasingly recognized to include low grade inflammation, often subclinical (3), that is predictive of articular chondropathy. In one study (4), 422 patients (85% with moderate radiographic Kellgren-Lawrence grade 2–3 OA at baseline) underwent knee arthroscopy at the beginning of the study and 12 months later. Those noted to have inflammatory changes in the medial perimeniscal synovium at baseline were more likely to have progression of tibiofemoral cartilage damage observed upon follow-up arthroscopy. This study did not adjust for baseline severity of OA which itself is correlated with synovitis (5), so taken alone cannot directly prove an independent effect of inflammation on structural progression. However, at least two other studies more convincingly show a direct effect of inflammation on OA progression. In one recent study with the novel imaging agent 99mTc-Etarfolatide that detects activated macrophages (6), a soluble macrophage marker (CD163) in synovial fluid was strongly associated with 99mTc-Etarfolatide positivity of the knee and was also associated with OA progression based on osteophyte controlling for baseline osteophyte severity (7). Another study showed that effusion synovitis, assessed by MRI, was an independent predictor of cartilage loss in the tibiofemoral joint at 30 months follow-up in subjects with neither cartilage damage nor tibiofemoral radiographic OA of the knee at baseline (8). Based on histological and cytokine expression profiling, synovial membranes from patients with OA show increased cellular infiltrates (9) and a pannus similar but not as extensive as that observed in rheumatoid arthritis (RA) (10). A number of inflammatory cytokines, most notably IL-1β and TNFα, are increased in synovial fluid, and both are produced by synovial membranes and chondrocytes from OA patients (11, 12).
The latest theories of OA pathogenesis implicate the interplay between mechanical damage and chronic inflammation (13, 14). Activation of the innate immune system is intricately involved in initiation and perpetuation of this low-grade inflammation (15–17). Thus, OA pathology is the result of an imbalance between the anabolic and catabolic processes in the joint (11). It seems only fitting that the innate immune system, considered the older or more “primitive” branch of our body’s defense, plays a key role in this “oldest” known disease of humans (1). This article is a non-systematic review of in vitro and in vivo studies that examine the role of the innate immune system in OA pathogenesis. We provide a brief overview of innate immunity and the basic mechanisms by which it becomes activated; secondly, we review the literature that implicates the innate immune system, including the complement system and synovial macrophages, in the pathogenesis of OA. Although we will discuss the evidence implicating each, in actuality, this process involves a complex interaction between the various branches of the innate immune system.
Overview of Innate Immunity
How does innate immunity, which serves as our first line of defense, lead to inflammation and joint pathology? The answer lies in how the innate immune system reacts to changes that take place in the joint over time. Unlike the adaptive immune system, innate immunity relies on recognition of conserved motifs generated by pathogens or damage within the body (18). Damage to cellular and cartilage extracellular matrix products from trauma, microtrauma (from repetitive overuse) or normal aging generates damage-associated molecular patterns (DAMPs) that activate the innate immune system (15, 17). DAMPs can be fragments generated from proteins, proteoglycans or remnants of cellular breakdown, such a uric acid (16, 18, 19) (Table 1). DAMPs elicit a sterile inflammatory response through interaction with particle recognition receptors (PRR), such as toll-like receptors (TLR), on the surface of immune cells, or with PRRs in the cell cytoplasm, such as nod-like receptors (NLRs) (15, 17, 18).
Table 1.
Extracellular matrix breakdown products that can trigger innate immunity.
| COMP | Happonen et al. 201036 | Regulates complement |
|---|---|---|
| Collagen IX (NC4 domain) | Kalchishkova et al. 201138 | Direct/indirect inhibition of complement |
| Fibromodulin | Sjoberg A et al. 200537 | Activates classical complement pathway via C1q |
| Fibromodulin | Wang et al. 201135 | Upregulates C5b-9 (MAC) from human OA sera |
| Fibronectin (EC domain) | Okamura et al. 200129 | Triggers TLR-4 |
| Fibronectin (EC domain) | Gondokaryono et al. 200730 | Triggers TLR-4 mast cells |
| Hyaluronan | Yamasaki et al. 200965 | HA triggers inflammasome->IL-1β |
| Hyaluronan | Scheibner et al. 200631 | HA triggers TLR-2 |
| Hyaluronan | Taylor et al. 200732 | HA triggers TLR4/CD44/MD-2 |
| Tenascin-C | Midwood et al. 200923 | TLR-4 agonist leading to persistent synovial inflammation |
COMP-cartilage oligomeric matrix protein, MAC=membrane attack complex, OA=osteoarthritis, TLR=toll like receptor
TLR activation leads to increased expression of pro-inflammatory cytokines via a number of transcription factors, such as activator protein 1 (AP1), cyclic AMP responsive element binding (CREB) protein, interferon regulatory factors (IRF) and NF-kB (20); the latter has been found to play a role in OA (15). The PRRs, TLR-2 and TLR-4, have both been thought to play a role in OA. TLR-2 and TLR-4 are upregulated in the synovial tissue from patients with OA, although not to the same extent as those with RA (21). Histological studies have shown increased expression of both TLR-2 and TLR-4 in articular cartilage lesions in OA patient samples (22) as well as the synovial membranes of patients with OA (21). Human chondrocytes express TLRs and their activation in tissue culture by TLR agonists leads to upregulation of matrix metalloproteases (MMPs), nitric oxide, and prostaglandin E2 (PGE2) (22). Tenascin-C, a ECM glycoprotein, has been shown in experimental models to cause persistence of synovial inflammation via TLR-4 (23). The plasma proteins Gc-globulin (vitamin D-binding protein), a1-microglobulin, and a2-macroglobulin, found to be enriched in OA synovial fluid (24), can signal via TLR4 to induce macrophage production of inflammatory cytokines implicated in OA (25). Whereas knockout of TLR-4 resulted in a less severe phenotype in a mouse IL-1 driven model of arthritis, knockout of TLR-2 showed a more severe disease phenotype suggesting its activation may be a countermeasure to joint catabolism (26). Opposing actions of TLR-2 and TLR-4 have also been described in other tissues including presynaptic terminals in the spinal cord and astroglia (27) as well as hippocampal neurons (28). Cell culture studies revealed that the extracellular domain A of fibronectin can trigger TLR-4 to produce an inflammatory response (29, 30). Both in vitro cell culture studies as well as an animal model of inflammatory arthritis have suggested that low molecular weight hyaluronic acid (HA) can also trigger either TLR-2 or TLR-4 to produce an inflammatory response (31, 32).
NLR activation leads to inflammasome assembly and activation of the inflammasome mediated inflammatory pathways (33). In addition, in response to inflammatory cytokines, chondrocytes have the ability to produce complement (34), another component of the innate immune response. Various ECM components, such as Cartilage Oligomeric Matrix Protein (COMP) (35–37), and the NC4 domain of type 4 collagen (38), can also fix complement. Finally, activation of mechanoreceptors in the cartilage and the synovium can lead to upregulation of various inflammatory mediators (39)
Once initiated, this inflammatory response leads to upregulation of catabolic factors, such as pro-inflammatory cytokines, proteolytic enzymes and chemokines, and downregulation of anabolic factors, such as anti-inflammatory cytokines and growth factors (11). From a teleological prospective, the ability of DAMPs to trigger the innate immune system probably is meant to promote wound healing and tissue repair (18, 40). However, these events can lead to further tissue breakdown, which contributes to an on-going sterile wound healing cycle resulting in joint tissue pathology (see Figure 1). There are other mechanisms activated in joint tissues in response to injury and an altered mechanical environment including altered mechanoreceptor signaling (41) and release of growth factors such as fibroblast growth factor (42). The balance of these responses in conjunction with the level of activation of the innate immune response likely orchestrates the net rate and severity of joint tissue catabolism.
Figure 1. Osteoarthritis Pathogenesis.

This figure depicts the self-perpetuating cycle of joint degeneration that characterizes the pathogenesis of osteoarthritis. In this paradigm, an inciting injury to the joint tissue causes the breakdown of the extracellular matrix (ECM), which initiates activation of innate immunity and a cyclic cascade of inflammatory events leading to further and ongoing joint damage.
Overall, the pathologic response of the joint results from a combination of anabolic (growth factors and anti-inflammatory cytokines) and catabolic forces (proteolytic enzymes and pro-inflammatory cytokines) (43). The two major pro-inflammatory cytokines implicated in OA are IL-1β and TNFα (11); synovial membrane biopsies from patients with early OA (symptomatic but no radiographic changes) had greater immunostaining of these two cytokines compared with late OA (requiring hip arthroplasty) (44), implying that inflammation may play an important role early in the disease course. In these early OA samples they also observed upregulation of indicators of inflammation such as cellular infiltrates, ICAM-1, VEGF, NF-κB and COX-2 (44). Another group found increased concentrations of IL-15 in the synovial fluid from patients with early versus late-stage OA suggesting activation of an innate immune response in the synovial membrane (45). Analysis of synovial membranes from 54 patients requiring arthroplasty for hip or knee OA revealed that the majority (57%) had inflammatory infiltrates (46); the subgroup with inflammatory infiltrates had higher mean levels of plasma high sensitivity CRP, which was strongly correlated with IL-6 concentrations in the synovial fluid (46). In addition, various other inflammatory cytokines and chemokines have possible links to OA pathogenesis; these include IL-8, IL-17, IL-18, IL-21 and leukemia inhibitory factor (LIF) (11, 43).
While the pro-inflammatory cytokines and chemokines represent the “marching orders”, proteolytic enzymes are the actual mediators on “the frontline” responsible for actual degradation of the articular cartilage. The two main groups of enzymes that mediate this catabolic process are the MMPs and ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs) (11). Various MMPs and tissue inhibitor of metalloproteinases (TIMP) were found to be upregulated in the synovial fluid from patients with OA (47). Also, MMP-1, MMP-3, and MMP-13 were isolated from both OA pannus cells and chrondocytes--with MMP-3 being the most highly expressed from both (48). Both bovine and human chondrocytes have shown the ability to produce ADAMTS protein (49). Furthermore, RNA expression of ADAMTs from human OA synovial cells can be altered by exposure to IL-1β and TNFα and pharmacologic blockade of thes cytokines (50).
The Complement System
The complement system consists of over 30 proteins. It includes serine proteases that contribute to an enzymatic cascade that yields proteins involved with opsonization, chemotaxis, and cell lysis as well as naturally occurring inhibitors, such as CD59 (also known as protectin) and factor H, which serve to keep the complement system in check (51). There are three different pathways by which the complement system can become activated (Figure 2) but all converge into the membrane attack complex (MAC) formed from C5b to C9. The MAC forms a cytotoxic ring-structure that perforates its target (51). As shown by some recent studies, MAC forms in response to the presence of certain extracellular matrix (ECM) proteins, such as fibromodulin (35). Furthermore, MAC also has sublytic properties that can upregulate inflammatory mediators without causing direct cytotoxic effects (35).
Figure 2. The Complement System.
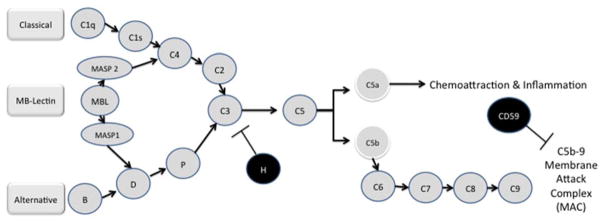
The complement cascade is a complex system that can become activated by one of three separate pathways: the classical, the mannose-binding (MB)-Lectin and alternative pathways. All three pathways converge on the C3 protein. C3 cleavage products participate in the activation of C5 whose cleaved components contribute to a local inflammatory response (C5a) or form part of the membrane attack complex that plays a role in cell lysis (C5b). Abbreviations: CD, cluster of differentiation; H, complement factor H; MAC, membrane attack complex; MASP, mannan-binding lectin serine protease; MB, mannose binding; MBL, mannose binding lectin. Complement effectors=blue, complement inhibitors=red. Adapted from Wang 2011 (35) and Sturfelt 2012 (51).
Complement proteins (see Table 2) have been found to be upregulated in both the synovial membranes as well as the synovial fluid of OA patients (24, 52, 53). The amount of MAC deposition in the synovial membrane is correlated with the level of synovial inflammation on histology (53). Chondrocytes are also capable of synthesizing complement components whose synthesis in OA can be upregulated by pro-inflammatory cytokines such as IL-1β and TNF-α (34). C5a receptors have been found to be upregulated on the surface of OA chondrocytes but not to the same extent as in RA (54). Other histological studies have found that complement deposition increases during an acute flare of the disease (52). Likewise, complement levels in the synovial fluid are elevated during the earlier acute phases of the disease (35). CD59, a natural occurring complement inhibitor, appears to be continuously upregulated in OA (52) implying that the complement system is chronically activated in OA. As described above, various ECM breakdown products, such as COMP, fibromodulin, and the NC4 domain of type 4 collagen, have all been shown to activate certain components of the complement pathway (see Table 1).
Table 2.
Components of Innate Immunity with a Putative Role in Osteoarthritis.
| C3c, C5, | Konttinen et al. 199652 | Increased in synovial membranes of OA patients -Further increased during acute flare |
| C3 | Gobezie et al. 200724 | Significantly increased from other SF proteins in proteomic assay. |
| C3a | Wang et al. 201135 | Increased in SF of OA patients |
| C4b | Gobezie et al. 200724 | Significantly increased from other SF proteins in proteomic assay. |
| C5b-9 (MAC) | Wang et al. 201135 | Increased in SF of OA patients |
| C5b-9 (MAC) | Corvetta et al. 199253 | Increased in synovial membrane of OA patients |
| C5, C6 | Wang et al. 201135 | Knockout mice for these complement proteins showed less OA damage |
| CD59 (inhibitor) | Konttinen et al. 199652 | Chronically upregulated in human OA synovium |
| CD59 (inhibitor) | Wang et al. 201135 | Knockout mice for this complement inhibitor showed more severe OA damage |
| Macrophages | Blom et al. 200767 | Depletion of synovial macrophages leads to MMP activity and less severe OA in mice |
| Macrophages | van Lent et al. 200469 | Macrophages secrete TGF-β that leads to osteophytes |
SF=synovial fluid, OA=osteoarthritis; TGF-tissue growth factor
While this evidence has implicated the complement system in the pathogenesis of OA, a series of recent studies in transgenic mouse models have more definitively demonstrated a pathological role of the complement system in OA. For instance, in a medial menisectomy mouse-model, knocking out components of the complement pathway (C5 and C6), attenuated joint damage (35). Conversely, knocking out CD59 (Protectin) increased degenerative changes compared to wild type mice (35). Pharmacologically blocking the complement system by CR2-fH, a fusion protein of a complement receptor and the naturally occurring inhibitor factor H, was associated with less severe joint damage (35). The same group showed that carboxypeptidase B (CPB) appeared to have a protective role in OA by inhibiting the complement system (55). Similar to their previous findings (35), in a medial menisectomy OA model, mice that were deficient for CPB showed more cartilage degeneration, osteophyte formation and synovitis than wild-type mice (55). In addition, they found that levels of CPB correlated to levels of MAC in the synovial fluid of patients with OA; suggesting that CPB has an anti-inflammatory role in the joint (55). Finally, in an in vitro model, CPB treated serum decreased MAC formation. Subsequently, they concluded that CPB has an anti-inflammatory effect in OA by inhibiting formation of MAC (55).
Synovial Macrophages
Similar to a war being fought in the air, land and sea, the overall innate immune response requires a concerted effort of multiple lines of defense. In addition to the complement system, innate immune cells, such as macrophages, serve vital functions to our body’s defense (56) and play a key role in innate immunity; they are involved in RA as well as OA (9) (see Table 2). Macrophages, as their name implies, are major phagocytic cells of the body, but they also carry out a number of other important functions, such as initiating inflammation, resolving inflammation, and restoring and repairing tissue damage (56, 57). Usually, macrophages exhibit a functional plasticity based on signals from their environment. However, their chronic activation can lead to deleterious effects (56, 57).
Macrophages can be activated in a variety of ways. As mentioned earlier, one of the primary ways is through activation of PRRs, which in turn activate a number of intracellular pathways, such as NF-κB (58). Another way macrophages can become activated is through inflammasome mediated pathways (59). Inflammasomes are large multimeric intracellular protein complexes that help process caspase-1 which is responsible for producing the mature forms of several pro-inflammatory cytokines such as IL-1β (60). NLRP3 is the most extensively studied of all the inflammasomes (59) and has been associated with crystal-induced inflammation triggered by uric acid and calcium pyrophosphate (61) as well as hydroxyapatite crystals (62, 63). One study of knee OA patients without gout suggested involvement of uric acid activated NLRP3 inflammasomes in the pathogenesis of OA (64). In this study, synovial fluid uric acid concentrations correlated with the concentrations of two cytokines, IL-18 and IL-1β, known to be produced by uric acid activated inflammasomes, and synovial fluid IL-18 was associated with OA progression. Hyaluronan also activates inflammasome pathways (65). Since there is a high degree of correlation of uric acid crystal deposition and cartilage lesions (66), and evidence for inflammasome activation in association with uric acid in OA (62, 64). it has been postulated that the chronic low-grade inflammasome activation helps drive OA progression (62, 64).
Experimental therapies aimed at macrophages have shown the ability to both decrease inflammation and progression of OA. Depletion of macrophages from a cell-culture suspension of human OA synovium decreases the inflammatory response, including both the cytokine response and the activity of proteolytic enzymes, such as matrix metalloproteases and aggrecanases, known to play a role in OA (50). Depletion of synovial macrophages via intra-articular injection of clodronate leads to less MMP activity and less cartilage damage in mouse model of OA (67). On the other hand, macrophages also secrete growth factors, such as TGFβ, that can enhance cartilage repair (68). However, intra-articular injections of TGFβ into the knees of mice can lead to fibrosis and extensive osteophyte formation; this response was abrogated by injecting clodronate beforehand which successfully depleted macrophages from the synovial lining (69). Thus, experimental therapies directed toward macrophages appear to be an attractive future target for OA.
Therapeutic Implications
Since OA has traditionally been thought to be a purely biomechanical disease, patients diagnosed with this condition are primarily treated to palliate symptoms. The growing body of evidence implicating the innate immune system in the pathogenesis of OA provides hope that insights into these mechanisms might inform our ability to phenotype patients who would stand to benefit the most from a particular therapy and treat these patient subsets more specifically than is currently possible. Although currently there are few effective pharmacologic treatment options for symptomatic OA, intra-articular glucocorticoids have shown some efficacy and are recommended by a number of international treatment guidelines (70, 71). Among their many effects, glucocorticoids lower expression of complement (72, 73), and induce macrophage polarization to an anti-inflammatory phenotype (74). However, their effects are broad and associated with numerous adverse effects including decreased bone formation, hyperglycemia and increased risk of infections (74). Development of more targeted therapies is critical for gaining clinical benefit without adverse effects.
Does the growing body of knowledge implicating the innate immune system in OA pathogenesis provide any hope for new treatments of OA in the future? Specifically, can slowing the inflammatory response lead to either symptomatic improvement or halt the progression in OA? Previous animal knockout models for COX-1 and COX-2 (75) and IL-1β and ICE have failed to show any chondroprotective effect (76) (and may have lead to increased disease). Knockout models are not always the most informative as it is difficult to ascertain any possible off-target effects (as illustrated by the previous study by Fukai et al (75)). Instead, are there other in vivo study designs that provide a more realistic but accelerated model for OA? As has been previously pointed out, one of the difficulties facing OA therapeutic studies is the long-natural history of the disease (77). As such, post-traumatic arthritis models might provide a way to evaluate a critical period of OA pathogenesis where inflammation may play a key role. Prior studies from our group have shown that IL-1β is upregulated in the synovial fluid of animals with post-traumatic arthritis (78, 79). A prior study found that recombinant IL-1RA used intra-articularly prevented OA development in an experimental animal model (80). More recent studies from our group have shown IL-1 inhibition to be effective in preventing progression of post-traumatic OA (81, 82). Several proof of concepts studies showed that a dual-variable domain immunoglobulin directed to both IL-1α and IL-1β prevented cartilage degradation in an animal model of OA (83, 84).
How close are some of these anti-inflammatory therapies that have been efficacious in preclinical OA, to going from “bench to bedside”? Prior human studies using current RA therapies to block cytokines in OA have met with mixed success. Intra-articular injections of adalimumab, an anti-TNF-α monoclonal antibody, showed some improvement in pain scores for knee OA (85) but showed no statistically significant improvement in pain for hand OA (86). Another small study showed improvement in pain but no changes in radiographic scores after 12 months for patients with hand OA that received intra-articular infliximab injections (87). Intra-articular injections of anakinra, an IL-1 receptor antagonist, have shown mixed results in improving pain in several small studies (88, 89). In a proof of concept from our group, the effects of intra-articular IL1RA injections were looked at following acute joint injury. Patients were randomized to either placebo intra-articular IL1RA. Those who received the intra-articular IL1RA were found to have less pain and improved function (90).
Also, targeting the cells or proteins of the innate immune system holds some promise for OA. There has been a growing body of literature on therapies targeting inflamed synovial tissue. Recently, a new recombinant protein (MT07), representing a fusion of an anti-C5 monoclonal antibody and a synovial-homing peptide, both prevented and successfully treated synovial inflammation in two different animal models of inflammatory arthritis (91). Another new strategy involved intra-articular injection of a DNA vector encoding an anti-C5 recombinant mini-antibody (MB12/22). This treatment lead to in situ production of this neutralizing antibody, which resulted in a statistically significant reduction in joint inflammation in a rat model of inflammatory arthritis (92). A human anti-DR5 antibody (TRA-8) was able to selectively induce apoptosis in a subset of inflammatory macrophages in a transgenic mouse model that led to less synovial hyperplasia and cellular infiltrates as well as improved clinical scores (93). As this therapy is directed toward a subset of inflammatory macrophages, theoretically it should have less off-target effects but further studies are needed. Tigatuzumab, a humanized monoclonal antibody to DR5, has been well tolerated in Phase I cancer studies (94). To the best of our knowledge, these therapies have not been studied in human for arthritis.
Conclusions
In addition to serving as our first line of defense, the innate immune system plays a key role in the pathogenesis of OA. Once activated, innate immunity “goes on the offensive”, leading to an inflammatory response that is a major driver of the disease process. The analogy of an innate immune system on the offensive is apt based on the failure of the innate immune response to resolve, which drives OA progression, if not development (43). A greater understanding of the basic mechanisms by which innate immunity becomes activated provides insights into OA pathogenesis. The advent of a much-improved understanding of the pathogenesis of OA is critical for effective phenotyping of OA patient subsets. Only through effective phenotyping will personalized medicine become a reality, the goals of which are to increase drug response rates, decrease adverse event rates, and improve the overall cost-effectiveness of medical therapy (95). It might be imagined that in addition to being able to identify inflammatory subsets of OA, the relative severity and profile of the innate immune response may reveal ‘subsets within subsets’ of OA. These advances could lead to potential new therapeutics for OA that would be expected to both modify symptoms and structural progression. While OA remains an “old” disease, our new understanding of it offers hope for more effective therapies in the future.
Acknowledgments
This work was supported, in whole or in part, by NIH 5T32AI007217-30 (EO), the NIH/NIA Grant 5P30 AG028716 (VBK) and NIH/NIAMS Grant P01 AR050245 (VBK).
We wish to thank Dr. David Pisetsky for editorial suggestions.
Glossary of Terms
- ADAMTS
a disintegrin and metalloproteinase with thrombospondin motifs
- CD
cluster of differentiation
- COMP
cartilage oligomeric matrix protein
- CPB
carboxypeptidase B
- CR
complement receptor
- ECM
extracellular matrix
- DAMP
damage associated molecular patterns
- DR
death receptor
- HA
hyaluronic acid
- ICAM
intracellular adhesion molecule 1
- ICE
IL-1β converting enzyme
- IL
interleukin
- IL-1RA
interleukin 1 receptor antagonist
- MAC
membrane attack complex
- MASP
mannan-binding lectin serine protease
- MB
mannose binding
- MBL
mannose binding lectin
- MMP
matrix metalloproteases
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NLR
NOD (nucleotide-binding domain)-like receptor
- OA
osteoarthritis
- PRR
particle recognition receptor
- RA
rheumatoid arthritis
- TGF
tissue growth factor
- TIMP
tissue inhibitor of metalloproteinases
- TLR
toll-like receptor
- TNF
tumor necrosis factor
- VEGF
vascular endothelial growth factor
Footnotes
Conflicts of Interest: No author has any conflicts related to commercial interests.
References
- 1.Dequeker J, Luyten FP. The history of osteoarthritis-osteoarthrosis. Ann Rheum Dis. 2008;67:5–10. doi: 10.1136/ard.2007.079764. [DOI] [PubMed] [Google Scholar]
- 2.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: A disease of the joint as an organ. Arthritis and Rheumatism. 2012;64:1697–707. doi: 10.1002/art.34453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fernandezmadrid F, Karvonen RL, Teitge RA, Miller PR, An T, Negendank WG. Synovial Thickening Detected by Mr-Imaging in Osteoarthritis of the Knee Confirmed by Biopsy as Synovitis. Magnetic Resonance Imaging. 1995;13:177–83. doi: 10.1016/0730-725x(94)00119-n. [DOI] [PubMed] [Google Scholar]
- 4.Ayral X, Pickering EH, Woodworth TG, Mackillop N, Dougados M. Synovitis: a potential predictive factor of structural progression of medial tibiofemoral knee osteoarthritis -- results of a 1 year longitudinal arthroscopic study in 422 patients. Osteoarthritis Cartilage. 2005;13:361–7. doi: 10.1016/j.joca.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 5.Guermazi A, Hayashi D, Roemer FW, Zhu Y, Niu J, Crema MD, et al. Synovitis in knee osteoarthritis assessed by contrast-enhanced magnetic resonance imaging (MRI) is associated with radiographic tibiofemoral osteoarthritis and MRI-detected widespread cartilage damage: the MOST study. J Rheumatol. 2014;41:501–8. doi: 10.3899/jrheum.130541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kraus V, McDaniel G, Huebner J, Stabler T, Pieper C, Coleman R, et al. Direct In vivo evidence of activated macrophages in human Osteoarthritis Osteo Cartilage. 2013;21:S42, 67. doi: 10.1016/j.joca.2016.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daghestani H, Pieper C, Kraus V. Soluble macrophage biomarkers indicate inflammatory phenotypes in patients with knee Osteoarthritis. Arthritis Rheum. 2014 doi: 10.1002/art.39006. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roemer FW, Guermazi A, Felson DT, Niu J, Nevitt MC, Crema MD, et al. Presence of MRI-detected joint effusion and synovitis increases the risk of cartilage loss in knees without osteoarthritis at 30-month follow-up: the MOST study. Ann Rheum Dis. 2011;70:1804–9. doi: 10.1136/ard.2011.150243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Revell PA, Mayston V, Lalor P, Mapp P. The synovial membrane in osteoarthritis: a histological study including the characterisation of the cellular infiltrate present in inflammatory osteoarthritis using monoclonal antibodies. Ann Rheum Dis. 1988;47:300–7. doi: 10.1136/ard.47.4.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furuzawa-Carballeda J, M-RP, Cabral AR. Osteoarthritis and rheumatoid arthritis pannus have similar qualitative metabolic characteristics and pro-inflammatory cytokine response. Clinical and Experimental Rheumatology. 2008;26:554–60. [PubMed] [Google Scholar]
- 11.Kapoor M, Martel-Pelletier J, Lajeunesse D, Pelletier JP, Fahmi H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat Rev Rheumatol. 2011;7:33–42. doi: 10.1038/nrrheum.2010.196. [DOI] [PubMed] [Google Scholar]
- 12.Pelletier J-P, Martel-Pelletier J, Abramson SB. Osteoarthritis, an inflammatory disease: Potential implication for the selection of new therapeutic targets. Arthritis & Rheumatism. 2001;44:1237–47. doi: 10.1002/1529-0131(200106)44:6<1237::AID-ART214>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 13.Kraus VB. Preclinical Osteoarthritis. In: Hochberg M, SA, Smolen J, Weinblatt M, Weisman M, editors. Rheumatology. 6. Philadelphia: Mosby Elsevier; 2014. [Google Scholar]
- 14.Vincent TL. Targeting mechanotransduction pathways in osteoarthritis: a focus on the pericellular matrix. Curr Opin Pharmacol. 2013;13:449–54. doi: 10.1016/j.coph.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 15.Scanzello CR, Plaas A, Crow MK. Innate immune system activation in osteoarthritis: is osteoarthritis a chronic wound? Curr Opin Rheumatol. 2008;20:565–72. doi: 10.1097/BOR.0b013e32830aba34. [DOI] [PubMed] [Google Scholar]
- 16.Scanzello CR, Goldring SR. The role of synovitis in osteoarthritis pathogenesis. Bone. 2012;51:249–57. doi: 10.1016/j.bone.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu-Bryan R. Synovium and the innate inflammatory network in osteoarthritis progression. Curr Rheumatol Rep. 2013;15:323. doi: 10.1007/s11926-013-0323-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nature Reviews Immunology. 2010;10:826–37. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sokolove J, Lepus CM. Role of inflammation in the pathogenesis of osteoarthritis: latest findings and interpretations. Ther Adv Musculoskelet Dis. 2013;5:77–94. doi: 10.1177/1759720X12467868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Neill LAJ, Golenbock D, Bowie AG. The history of Toll-like receptors [mdash] redefining innate immunity. Nature Reviews Immunology. 2013;13:453–60. doi: 10.1038/nri3446. [DOI] [PubMed] [Google Scholar]
- 21.Radstake TRDJ, Roelofs MF, Jenniskens YA, Oppers-Walgreen B, van Riel PLCA, Barrera P, et al. Expression of toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-gamma. Arthritis and Rheumatism. 2004;50:3856–65. doi: 10.1002/art.20678. [DOI] [PubMed] [Google Scholar]
- 22.Kim HA, Cho ML, Choi HY, Yoon CS, Jhun JY, Oh HJ, et al. The catabolic pathway mediated by Toll-like receptors in human osteoarthritic chondrocytes. Arthritis and Rheumatism. 2006;54:2152–63. doi: 10.1002/art.21951. [DOI] [PubMed] [Google Scholar]
- 23.Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15:774–80. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- 24.Gobezie R, Kho A, Krastins B, Sarracino DA, Thornhill TS, Chase M, et al. High abundance synovial fluid proteome: distinct profiles in health and osteoarthritis. Arthritis Res Ther. 2007;9:R36. doi: 10.1186/ar2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sohn DH, Sokolove J, Sharpe O, Erhart JC, Chandra PE, Lahey LJ, et al. Plasma proteins present in osteoarthritic synovial fluid can stimulate cytokine production via Toll-like receptor 4. Arthritis Res Ther. 2012;14:R7. doi: 10.1186/ar3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abdollahi-Roodsaz S, Joosten LAB, Koenders MI, Devesa I, Roelofs MF, Radstake TRDJ, et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. The Journal of Clinical Investigation. 2008;118:205–16. doi: 10.1172/JCI32639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freria CM, Velloso LA, Oliveira AL. Opposing effects of Toll-like receptors 2 and 4 on synaptic stability in the spinal cord after peripheral nerve injury. Journal of neuroinflammation. 2012;9:240. doi: 10.1186/1742-2094-9-240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rolls A, Shechter R, London A, Ziv Y, Ronen A, Levy R, et al. Toll-like receptors modulate adult hippocampal neurogenesis. Nature cell biology. 2007;9:1081–8. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- 29.Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, et al. The extra domain A of fibronectin activates toll-like receptor 4. Journal of Biological Chemistry. 2001;276:10229–33. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 30.Gondokaryono SP, Ushio H, Niyonsaba F, Hara M, Takenaka H, Jayawardana STM, et al. The extra domain A of fibronectin stimulates murine mast cells via Toll-like receptor 4. Journal of Leukocyte Biology. 2007;82:657–65. doi: 10.1189/jlb.1206730. [DOI] [PubMed] [Google Scholar]
- 31.Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. Journal of Immunology. 2006;177:1272–81. doi: 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- 32.Taylor KR, Yamasaki K, Radek KA, Di Nardo A, Goodarzi H, Golenbock D, et al. Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on toll-like receptor 4, CD44, and MD-2. Journal of Biological Chemistry. 2007;282:18265–75. doi: 10.1074/jbc.M606352200. [DOI] [PubMed] [Google Scholar]
- 33.Frommer KW, Zimmermann B, Meier FM, Schroder D, Heil M, Schaffler A, et al. Adiponectin-mediated changes in effector cells involved in the pathophysiology of rheumatoid arthritis. Arthritis Rheum. 2010;62:2886–99. doi: 10.1002/art.27616. [DOI] [PubMed] [Google Scholar]
- 34.Bradley K, North J, Saunders D, Schwaeble W, Jeziorska M, Woolley DE, et al. Synthesis of classical pathway complement components by chondrocytes. Immunology. 1996;88:648–56. [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Q, Rozelle AL, Lepus CM, Scanzello CR, Song JJ, Larsen DM, et al. Identification of a central role for complement in osteoarthritis. Nat Med. 2011;17:1674–9. doi: 10.1038/nm.2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Happonen KE, Saxne T, Aspberg A, Morgelin M, Heinegard D, Blom AM. Regulation of complement by cartilage oligomeric matrix protein allows for a novel molecular diagnostic principle in rheumatoid arthritis. Arthritis Rheum. 2010;62:3574–83. doi: 10.1002/art.27720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sjoberg A, Onnerfjord P, Morgelin M, Heinegard D, Blom AM. The extracellular matrix and inflammation: fibromodulin activates the classical pathway of complement by directly binding C1q. J Biol Chem. 2005;280:32301–8. doi: 10.1074/jbc.M504828200. [DOI] [PubMed] [Google Scholar]
- 38.Kalchishkova N, Furst CM, Heinegard D, Blom AM. NC4 Domain of cartilage-specific collagen IX inhibits complement directly due to attenuation of membrane attack formation and indirectly through binding and enhancing activity of complement inhibitors C4B-binding protein and factor H. Journal of Biological Chemistry. 2011;286:27915–26. doi: 10.1074/jbc.M111.242834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loeser RF. Molecular mechanisms of cartilage destruction: Mechanics, inflammatory mediators, and aging collide. Arthritis & Rheumatism. 2006;54:1357–60. doi: 10.1002/art.21813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kraus VB. Osteoarthritis: The zinc link. Nature. 2014;507:441–2. doi: 10.1038/507441a. [DOI] [PubMed] [Google Scholar]
- 41.O’Conor CJ, Leddy HA, Benefield HC, Liedtke WB, Guilak F. TRPV4-mediated mechanotransduction regulates the metabolic response of chondrocytes to dynamic loading. Proceedings of the National Academy of Sciences. 2014;111:1316–21. doi: 10.1073/pnas.1319569111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vincent T, Hermansson M, Bolton M, Wait R, Saklatvala J. Basic FGF mediates an immediate response of articular cartilage to mechanical injury. Proc Natl Acad Sci U S A. 2002;99:8259–64. doi: 10.1073/pnas.122033199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sellam J, Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat Rev Rheumatol. 2010;6:625–35. doi: 10.1038/nrrheum.2010.159. [DOI] [PubMed] [Google Scholar]
- 44.Benito MJ, Veale DJ, FitzGerald O, van den Berg WB, Bresnihan B. Synovial tissue inflammation in early and late osteoarthritis. Ann Rheum Dis. 2005;64:1263–7. doi: 10.1136/ard.2004.025270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scanzello CR, Umoh E, Pessler F, Diaz-Torne C, Miles T, Dicarlo E, et al. Local cytokine profiles in knee osteoarthritis: elevated synovial fluid interleukin-15 differentiates early from end-stage disease. Osteoarthritis Cartilage. 2009;17:1040–8. doi: 10.1016/j.joca.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 46.Pearle AD, Scanzello CR, George S, Mandl LA, DiCarlo EF, Peterson M, et al. Elevated high-sensitivity C-reactive protein levels are associated with local inflammatory findings in patients with osteoarthritis. Osteoarthritis Cartilage. 2007;15:516–23. doi: 10.1016/j.joca.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 47.Ishiguro N, Ito T, Ito H, Iwata H, Jugessur H, Ionescu M, et al. Relationship of matrix metalloproteinases and their inhibitors to cartilage proteoglycan and collagen turnover: analyses of synovial fluid from patients with osteoarthritis. Arthritis Rheum. 1999;42:129–36. doi: 10.1002/1529-0131(199901)42:1<129::AID-ANR16>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 48.Yuan GH, Tanaka M, Masuko-Hongo K, Shibakawa A, Kato T, Nishioka K, et al. Characterization of cells from pannus-like tissue over articular cartilage of advanced osteoarthritis. Osteoarthritis Cartilage. 2004;12:38–45. doi: 10.1016/j.joca.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 49.Pratta MA, Scherle PA, Yang G, Liu R-Q, Newton RC. Induction of aggrecanase 1 (ADAM-TS4) by interleukin-1 occurs through activation of constitutively produced protein. Arthritis & Rheumatism. 2003;48:119–33. doi: 10.1002/art.10726. [DOI] [PubMed] [Google Scholar]
- 50.Bondeson J, Wainwright SD, Lauder S, Amos N, Hughes CE. The role of synovial macrophages and macrophage-produced cytokines in driving aggrecanases, matrix metalloproteinases, and other destructive and inflammatory responses in osteoarthritis. Arthritis Res Ther. 2006;8:R187. doi: 10.1186/ar2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sturfelt G, Truedsson L. Complement in the immunopathogenesis of rheumatic disease. Nature Reviews Rheumatology. 2012;8:458–68. doi: 10.1038/nrrheum.2012.75. [DOI] [PubMed] [Google Scholar]
- 52.Konttinen YT, Ceponis A, Meri S, Vuorikoski A, Kortekangas P, Sorsa T, et al. Complement in acute and chronic arthritides: assessment of C3c, C9, and protectin (CD59) in synovial membrane. Ann Rheum Dis. 1996;55:888–94. doi: 10.1136/ard.55.12.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corvetta A, Pomponio G, Rinaldi N, Luchetti MM, Diloreto C, Stramazzotti D. Terminal Complement Complex in Synovial Tissue from Patients Affected by Rheumatoid-Arthritis, Osteoarthritis and Acute Joint Trauma. Clinical and Experimental Rheumatology. 1992;10:433–8. [PubMed] [Google Scholar]
- 54.Onuma H, Masuko-Hongo K, Yuan GH, Sakata M, Nakamura H, Kato T, et al. Expression of the anaphylatoxin receptor C5aR (CD88) by human articular chondrocytes. Rheumatology International. 2002;22:52–5. doi: 10.1007/s00296-002-0199-6. [DOI] [PubMed] [Google Scholar]
- 55.Lepus CM, Song JJ, Wang Q, Wagner CA, Lindstrom TM, Chu CR, et al. Brief Report: Carboxypeptidase B Serves as a Protective Mediator in Osteoarthritis. Arthritis & Rheumatology. 2014;66:101–6. doi: 10.1002/art.38213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nature Reviews Immunology. 2011;11:723–37. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amos N, Lauder S, Evans A, Feldmann M, Bondeson J. Adenoviral gene transfer into osteoarthritis synovial cells using the endogenous inhibitor I kappa B alpha reveals that most, but not all, inflammatory and destructive mediators are NF kappa B dependent. Rheumatology. 2006;45:1201–9. doi: 10.1093/rheumatology/kel078. [DOI] [PubMed] [Google Scholar]
- 59.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 60.Dinarello CA. Anti-inflammatory Agents: Present and Future. Cell. 2010;140:935–50. doi: 10.1016/j.cell.2010.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 62.Jin C, Frayssinet P, Pelker R, Cwirka D, Hu B, Vignery A, et al. NLRP3 inflammasome plays a critical role in the pathogenesis of hydroxyapatite-associated arthropathy. Proceedings of the National Academy of Sciences. 2011;108:14867–72. doi: 10.1073/pnas.1111101108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCarthy G, Cheung H. Point: Hydroxyapatite crystal deposition is intimately involved in the pathogenesis and progression of human osteoarthritis. Current Rheumatology Reports. 2009;11:141–7. doi: 10.1007/s11926-009-0020-6. [DOI] [PubMed] [Google Scholar]
- 64.Denoble AE, Huffman KM, Stabler TV, Kelly SJ, Hershfield MS, McDaniel GE, et al. Uric acid is a danger signal of increasing risk for osteoarthritis through inflammasome activation. Proc Natl Acad Sci U S A. 2011;108:2088–93. doi: 10.1073/pnas.1012743108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamasaki K, Muto J, Taylor KR, Cogen AL, Audish D, Bertin J, et al. NLRP3/cryopyrin is necessary for interleukin-1beta (IL-1beta) release in response to hyaluronan, an endogenous trigger of inflammation in response to injury. J Biol Chem. 2009;284:12762–71. doi: 10.1074/jbc.M806084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Muehleman C, Li J, Aigner T, Rappoport L, Mattison E, Hirschmugl C, et al. Association Between Crystals and Cartilage Degeneration in the Ankle. The Journal of Rheumatology. 2008;35:1108–17. [PMC free article] [PubMed] [Google Scholar]
- 67.Blom AB, van Lent PL, Libregts S, Holthuysen AE, van der Kraan PM, van Rooijen N, et al. Crucial role of macrophages in matrix metalloproteinase-mediated cartilage destruction during experimental osteoarthritis: involvement of matrix metalloproteinase 3. Arthritis Rheum. 2007;56:147–57. doi: 10.1002/art.22337. [DOI] [PubMed] [Google Scholar]
- 68.Davidson ENB, van der Kraan PM, van den Berg WB. TGF-beta and osteoarthritis. Osteoarthr Cartilage. 2007;15:597–604. doi: 10.1016/j.joca.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 69.van Lent PLEM, Blom AB, van der Kraan P, Holthuysen AEM, Vitters E, van Rooijen N, et al. Crucial role of synovial lining macrophages in the promotion of transforming growth factor beta-mediated osteophyte formation. Arthritis and Rheumatism. 2004;50:103–11. doi: 10.1002/art.11422. [DOI] [PubMed] [Google Scholar]
- 70.Hochberg MC, Altman RD, April KT, Benkhalti M, Guyatt G, McGowan J, et al. American College of Rheumatology 2012 recommendations for the use of nonpharmacologic and pharmacologic therapies in osteoarthritis of the hand, hip, and knee. Arthritis Care Res (Hoboken) 2012;64:455–74. doi: 10.1002/acr.21596. [DOI] [PubMed] [Google Scholar]
- 71.McAlindon TE, Bannuru RR, Sullivan MC, Arden NK, Berenbaum F, Bierma-Zeinstra SM, et al. OARSI guidelines for the non-surgical management of knee osteoarthritis. Osteoarthritis Cartilage. 2014;22:363–88. doi: 10.1016/j.joca.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 72.Firestein G, Paine M, Littman B. Gene expression (collagenase, tissue inhibitor of metalloproteinases, complement, and HLA-DR) in rheumatoid arthritis and osteoarthritis synovium. Quantitative analysis and effect of intraarticular corticosteroids. Arthritis Rheum. 1991;34:1094–105. doi: 10.1002/art.1780340905. [DOI] [PubMed] [Google Scholar]
- 73.Bureeva S, Andia-Pravdivy J, Symon A, Bichucher A, Moskaleva V, Popenko V, et al. Selective inhibition of the interaction of C1q with immunoglobulins and the classical pathway of complement activation by steroids and triterpenoids sulfates. Bioorganic & Medicinal Chemistry. 2007;15:3489–98. doi: 10.1016/j.bmc.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 74.Baschant U, Lane NE, Tuckermann J. The multiple facets of glucocorticoid action in rheumatoid arthritis. Nature Reviews Rheumatology. 2012;8:645–55. doi: 10.1038/nrrheum.2012.166. [DOI] [PubMed] [Google Scholar]
- 75.Fukai A, Kamekura S, Chikazu D, Nakagawa T, Hirata M, Saito T, et al. Lack of a chondroprotective effect of cyclooxygenase 2 inhibition in a surgically induced model of osteoarthritis in mice. Arthritis & Rheumatism. 2012;64:198–203. doi: 10.1002/art.33324. [DOI] [PubMed] [Google Scholar]
- 76.Clements KM, Price JS, Chambers MG, Visco DM, Poole AR, Mason RM. Gene deletion of either interleukin-1beta, interleukin-1beta-converting enzyme, inducible nitric oxide synthase, or stromelysin 1 accelerates the development of knee osteoarthritis in mice after surgical transection of the medial collateral ligament and partial medial meniscectomy [see comment] Arthritis & Rheumatism. 2003;48:3452–63. doi: 10.1002/art.11355. [DOI] [PubMed] [Google Scholar]
- 77.Hunter DJ. Pharmacologic therapy for osteoarthritis--the era of disease modification. Nat Rev Rheumatol. 2011;7:13–22. doi: 10.1038/nrrheum.2010.178. [DOI] [PubMed] [Google Scholar]
- 78.Huebner J, Furman B, Seifer D, Kraus V, Guilak F, Olson S. Assessment of serum and synovial fluid levels of IL-1α AND IL-1β in a mouse model of post-traumatic arthritis. Osteo Cartilage. 2009;17:S73, 122. [Google Scholar]
- 79.Lewis JS, Jr, Furman BD, Zeitler E, Huebner JL, Kraus VB, Guilak F, et al. Genetic and cellular evidence of decreased inflammation associated with reduced incidence of posttraumatic arthritis in MRL/MpJ mice. Arthritis Rheum. 2013;65:660–70. doi: 10.1002/art.37796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Caron JP, Fernandes JC, Martel-Pelletier J, Tardif G, Mineau F, Geng C, et al. Chondroprotective effect of intraarticular injections of interleukin-1 receptor antagonist in experimental osteoarthritis. Suppression of collagenase-1 expression. Arthritis & Rheumatism. 1996;39:1535–44. doi: 10.1002/art.1780390914. [DOI] [PubMed] [Google Scholar]
- 81.Kimmerling K, Furman B, Mangiapani D, Moverman M, Sinclair S, Huebner J, et al. Prolonged intra-articular delivery of IL-1Ra from a thermally-responsive elastin-like polypeptide depot prevents post-traumatic arthritis in mice. 2014 doi: 10.22203/ecm.v029a10. in review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Furman B, Mangiapani D, Zeitler E, Bailey K, Horne P, Huebner J, et al. Targeting pro-inflammatory cytokines following joint injury: Acute intra-articular inhibition of IL-1 following knee injury prevents post-traumatic arthritis. Arthritis Research & Therapy. 2014;16:R134. doi: 10.1186/ar4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kamath R, Hart M, Conlon D, Ghayur T. Simultaneous targeting of IL-1A and IL-1B by a dual-variable domain immunoglobulin (DVD-Ig™) prevents cartilage degradation in preclinical models of osteoarthritis. Osteoarthritis and Cartilage. 2011;19:S64, 126. [Google Scholar]
- 84.Kamath RV, Simler G, Zhou C, Hart M, Joshi S, Ghayur T, et al. Blockade of both IL-1A and IL-1B by a combination of monoclonal antibodies prevents the development and reverses established pain in a preclinical model of osteoarthritis. Osteoarthritis and Cartilage. 2012;20:S62, 109. [Google Scholar]
- 85.Grunke M, Schulze-Koops H. Successful treatment of inflammatory knee osteoarthritis with tumour necrosis factor blockade. Annals of the Rheumatic Diseases. 2006;65:555–6. doi: 10.1136/ard.2006.053272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fioravanti A, Fabbroni M, Cerase A, Galeazzi M. Treatment of erosive osteoarthritis of the hands by intra-articular infliximab injections: a pilot study. Rheumatol Int. 2009;29:961–5. doi: 10.1007/s00296-009-0872-0. [DOI] [PubMed] [Google Scholar]
- 87.Magnano MD, Chakravarty EF, Broudy C, Chung L, Kelman A, Hillygus J, et al. A pilot study of tumor necrosis factor inhibition in erosive/inflammatory osteoarthritis of the hands. The Journal of Rheumatology. 2007;34:1323–7. [PubMed] [Google Scholar]
- 88.Chevalier X, Goupille P, Beaulieu AD, Burch FX, Bensen WG, Conrozier T, et al. Intraarticular injection of anakinra in osteoarthritis of the knee: a multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2009;61:344–52. doi: 10.1002/art.24096. [DOI] [PubMed] [Google Scholar]
- 89.Bacconnier L, Jorgensen C, Fabre S. Erosive osteoarthritis of the hand: clinical experience with anakinra. Ann Rheum Dis. 2009;68:1078–9. doi: 10.1136/ard.2008.094284. [DOI] [PubMed] [Google Scholar]
- 90.Kraus VB, Birmingham J, Stabler TV, Feng S, Taylor DC, Moorman CT, 3rd, et al. Effects of intraarticular IL1-Ra for acute anterior cruciate ligament knee injury: a randomized controlled pilot trial ( NCT00332254) Osteoarthritis Cartilage. 2012;20:271–8. doi: 10.1016/j.joca.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 91.Macor P, Durigutto P, De Maso L, Garrovo C, Biffi S, Cortini A, et al. Treatment of experimental arthritis by targeting synovial endothelium with a neutralizing recombinant antibody to C5. Arthritis and Rheumatism. 2012;64:2559–67. doi: 10.1002/art.34430. [DOI] [PubMed] [Google Scholar]
- 92.Durigutto P, Macor P, Ziller F, De Maso L, Fischetti F, Marzari R, et al. Prevention of arthritis by locally synthesized recombinant antibody neutralizing complement component C5. Plos One. 2013;8:e58696. doi: 10.1371/journal.pone.0058696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li J, Hsu H-C, Yang P, Wu Q, Li H, Edgington LE, et al. Treatment of arthritis by macrophage depletion and immunomodulation: Testing an apoptosis-mediated therapy in a humanized death receptor mouse model. Arthritis & Rheumatism. 2012;64:1098–109. doi: 10.1002/art.33423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Forero-Torres ASJ, Wood T, Posey J, Carlisle R, Copigneaux C, Luo FR, Wojtowicz-Praga S, Percent I, Saleh M. Phase I trial of weekly tigatuzumab, an agonistic humanized monoclonal antibody targeting death receptor 5 (DR5) Cancer Biotherapy & Radiopharmaceuticals. 2010;25:13–9. doi: 10.1089/cbr.2009.0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Karsdal MA, Christiansen C, Ladel C, Henriksen K, Kraus VB, Bay-Jensen AC. Osteoarthritis – a case for personalized health care? Osteoarthritis and Cartilage. 2014;22:7–16. doi: 10.1016/j.joca.2013.10.018. [DOI] [PubMed] [Google Scholar]