

Letter to the Editor
Pityriasis rubra pilaris (PRP) is a rare inflammatory papulo-squamous disorder manifesting with palmoplantar keratoderma and follicular hyperkeratotic papules which tend to coalesce into large, scaly, erythematous plaques often progressing to exfoliative erythroderma (Klein et al., 2010; Petrof et al., 2013). PRP is often misdiagnosed as psoriasis, a more common papulo-squamous inflammatory disorder. Nevertheless, the two conditions, in their classic presentations, are clearly distinct, and can be distinguished by clinical findings and histopathologic features (Magro and Crowson, 1997). Clinically, PRP manifests with characteristic “sparing islands” of apparently normal skin, palmoplantar keratoderma and follicular papules. The disease is frequently self-limiting within a few years’ timeframe. Histopathology of PRP is characterized by alternating ortho- and parakeratosis rete ridges oriented in vertical and horizontal arrays (“checkerboard pattern”), acanthosis with broadened bases, follicular plugging, perivascular lymphocytic infiltrate in the dermis, and lack of neutrophils in the epidermis. Currently, there is no specific or uniformly effective treatment for PRP. Most cases of PRP are sporadic and without family history, but a familial form with an autosomal dominant inheritance with partial penetrance represents <6% of all cases. We recently demonstrated that patients with the familial form of PRP harbor gain-of-function mutations in the CARD14 gene encoding the caspase recruitment domain family, member 14 (CARD14) (Fuchs-Telem et al., 2012). This protein is an activator of nuclear factor κB (NF-κB) (Blonska and Lin, 2011), and it has also been implicated in cases of familial psoriasis (Jordan et al., 2012a; Jordan et al., 2012b). This study investigates whether CARD14 mutations might also underlie cases of sporadic PRP.
Patients with PRP were solicited through a website (www.prp-support.org) which serves as a focus of PRP information exchange, frequently visited by patients. A total of 156 patients requesting enrollment were sent an IRB-approved informed consent, a questionnaire and a saliva collection kit for DNA isolation. This study was approved by the Institutional Review Board of Thomas Jefferson University. Of these, 48 patients returned a blood or saliva sample with study documents, including written, informed patient consent. Careful review of the available clinical, photographic and histopathologic information independently by two clinical dermatologists (HJC and MK), allowed us to establish a definitive diagnosis of PRP in 22 patients using predetermined criteria (Ross et al., manuscript in preparation). Another 7 patients had findings suggestive but not definitive for PRP. Seventeen patients had findings associated with PRP, but there were insufficient data to either confirm or rule out the diagnosis by our stringent, predetermined criteria. Finally, 2 patients were concluded not to have PRP. None of the patients reported family history of PRP.
Genomic DNA was isolated from saliva samples or in some cases from blood by standard techniques, and the CARD14 gene was examined by sequencing of the exons and the flanking intronic sequences by PCR utilizing specific previously published primers (Fuchs-Telem et al., 2012). Initial amplification of DNA from all 48 patients focused on exons 3 and 4, previously shown to harbor a cluster of mutations in the familial form of PRP and psoriasis. In addition, the remaining 18 exons and flanking intronic sequences were determined in a subset of 20 patients that had a definitive diagnosis of PRP.
Sequencing of CARD14 in PRP patients identified a total of 15 sequence variants, many of which were neutral and none of which resulted in premature termination codon for translation (Supplementary Table S1). A total of 8 missense mutations and 2 single nucleotide variants within the splice site junction were evaluated by computer programs predicting the consequences of the mutations at protein levels or on mRNA splicing, as well as by comparison with the SNP databases. By this approach, 6 sequence variants were considered to be inconsequential polymorphisms present in populations at large. The remaining four sequence variants (Table 1), all present in the SNP database in the minor allelic frequency of <1.5% were considered pathogenic (see Table 1), because (a) bioinformatics prediction programs suggested that the mutation was either damaging, or probably damaging, to the protein function (Variants 2, 3 and 4), (b) the mutated amino acid is conserved in CARD14 through evolution (Variants 2 from M. musculus and Variant 3 from D. rerio to H. sapiens), or (c) they have been previously reported to be present in patients with familial PRP and psoriasis (Variant 1) (Fuchs-Telem et al., 2012; Jordan et al., 2012a; Jordan et al., 2012b). Variant 4 is located in the C-terminus of CARD14 which is not involved in NF-κB activation (Bertin et al., 2001). Note that Variant 1 (c.599G7A; p.S200N) was present in three patients, and, therefore, a mutant CARD14 allele was present in a total of 6 out of 48 patients studied (12.5%). Among these variants, p.L228R and p.S802R are previously unpublished.
Table 1.
Clinical features and CARD14 variants in patients with sporadic PRP1
| Variant2 | Age (Y)/Sex | Age at onset/diagnosis (Y) | Duration (Y) | Type3 | SNP rs | Variant | Minor allele frequency (%) | Functional consequences on the protein (Bioinformatics prediction programs)
|
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Polyphen-2 | SIFT | PMut | SNAP | PROVEAN | ||||||||
| 1a | 72/F | 47/57 | 20 | 2 | rs114688446 | c.599G>A, p.S200N | A: 0.8 | Benign | Tolerated | Neutral | Neutral | Neutral |
| b | 56/M | 51/51 | 2 | 1 | ||||||||
| c | 62/M | 57/57 | 3 | 1 | ||||||||
| 2 | 75/M | 70/70 | 2 | 1 | rs142246283 | c.683T>G, p.L228R | no data | Probably damaging | Tolerated | Pathological | Neutral | Neutral |
| 3 | 46/F | 21/21 | 2 | 1 | rs117918077 | c.2044C>T, p.R682W | T: 1.2 | Probably damaging | Damaging | Pathological | Non-neutral | Deleterious |
| 4 | 35/F | 32/32 | 1 | 1 | no data | c.2406C>A, p.S802R | no data | Benign | Damaging | Pathological | Neutral | Neutral |
A total of 15 genomic variants were identified in the CARD14 gene in 48 patients with sporadic PRP (Table S1). The above variants have minor allele frequency of less than 1.5%
Note that the Variant 1 was disclosed in three different families (a–c)
Type 1: Classic adult type; Type 2: Atypical presentation with prolonged manifestations
To examine the consequences of three variants (nos. 1–3) as putative pathogenic mutations on the activation of NF-κB, in vitro assays were performed in a HeLa cell line which constitutively expresses low level of luciferase reporter under a NF-κB responsive element (Signosis, Sunnyvale, CA) when transfected with a plasmid harboring CARD14 cDNA, either wild-type or mutant ones in which the corresponding sequence variants were introduced by QuikChange Site-Directed Mutagenesis Kit (Stratagene, LaJolla, CA). This approach was validated in a similar system of HEK293 cells by the analysis of two mutations (p.E138del and p.L156P) previously identified in patients with familial PRP (Fuchs-Telem et al., 2012). As indicated in Fig. 1a, both mutations resulted in NF-κB activation. The results with variants encountered in sporadic cases of PRP indicated that only one of the putative variants, Variant 2 (c.683T>G; p.L228R), present in a patient with definitive PRP, was capable of up-regulation of the NF-κB responsive element, as determined by the luciferase activity corrected for the transfection efficiency by β-galactosidase determination (Fig. 1b). Mutations p.R682W and p.S200N were not capable of upregulation of NF-κB, consistent with previous observations (Jordan et al., 2012a). This assay system was clearly functional and responsive to NF-κB activation, since incubation of the cells with recombinant human TNF-α (20 ng/ml), an activator of NF-κB, resulted in 10 to 20-fold upregulation of the NF-κB responsive element (Fig. 1c).
Figure 1. NF-κB activation by mutant CARD14 in cell culture systems in vitro.
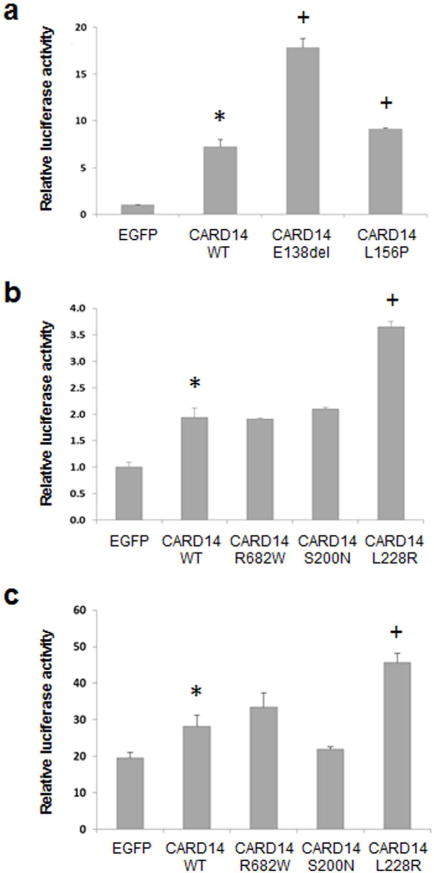
The cells were transfected with wild type (WT) or mutant CARD14 cDNA constructs using Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA), followed by assay of luciferase activity after 24 hours of incubation. The CARD14 cDNA (coding for 740 amino acids; GenBank BC018142) and EGFP cDNA as a control were cloned into pReceiver-M11 (Capital Biosciences, Rockville, MD) vector. (a) HEK293 cells were co-transfected with CARD14 constructs together with kB-Luc plasmid (kindly provided by Professor Yinor Ben-Neriah, Hebrew University, Jerusalem) and Renilla luciferase plasmid expression vector. Luciferase activity was measured using Dual-Luciferase® Reporter (DLT™) Assay System (Promega, Mullion, WI). (b) HeLa cells stably expressing NF-κB luciferase reporter (Signosis, Sunnyvale, CA) were co-transfected with the CARD14 constructs together with pRSV-galactosidase expression plasmid as a control of transfection efficiency. (c) The cultures as in (b) were supplemented after 24 hours of incubation with 20 ng/ml of recombinant human TNF-α (PeproTech, Rocky Hill, NJ). After an additional 24 hrs, luciferase activity was measured using Luciferase Assay System (Promega). The experiments were performed in triplicate cultures, and the values are expressed as mean + S.E. Statistical differences were evaluated by Student’s two-tailed t-test: *, p<0.05 as compared with EGFP as a control construct; +, p<0.05 as compared with CARD14 WT construct.
In conclusion, CARD14 mutations were only identified in a limited number (12.5%) of patients with sporadic PRP. This is consistent with a recent study wherein CARD14 mutations were undetectable in 8 cases of sporadic PRP (Hong et al., 2014). However, recent studies on sporadic PRP, similar to sporadic psoriasis, have suggested that NF-κB signaling is activated in the epidermis of patients with PRP, even in the absence of pathogenic CARD14 mutations (Eytan et al., 2014a). There could be several explanations for the lack of identifiable mutations in the CARD14 gene in most sporadic cases of PRP despite apparent activation of NF-κB. First, our mutation analysis is limited to exons and flanking intronic sequences, and does not detect possible mutations in the regulatory 5′-sequences or those embedded deeper in the introns. Secondly, it is possible that mutations in other components of the CARD14 signaling cascade, such as IKBKG/NEMO, can result in activation of NF-κB which is implicated in other genetic diseases (Conte et al., 2014). Finally, NF-κB activation could occur in a CARD14 independent, non-canonical signaling pathway (Wullaert et al., 2011). The importance of NF-κB signaling in the pathogenesis of PRP may have implications for development of specific therapies for the management of this therapeutically challenging disorder (Eytan et al., 2014b). In summary, while NF-κB activation may be a common mechanism in inflammatory skin diseases, such as familial PRP, CARD14 mutations may be rare in sporadic cases, and alternate mechanisms may be responsible for activation of the NF-κB signaling pathway.
Supplementary Material
Acknowledgments
This study was supported by an NIH/NIAMS grant K01AR064766 and a Research Grant from Dermatology Foundation (QL), by a grant from the Cove Charitable Trust of Boston (JU), and donations of the Ram Family (ES). The authors thank Carol Kelly for assistance with manuscript preparations, and Drs. Emad Alnemri, Ulrich Rodeck and Joel Rosenbloom for helpful advice.
Abbreviations
- PRP
pityriasis rubra pilaris
- CARD14
caspase recruitment domain family, member 14
- NF-κB
nuclear factor κB
Footnotes
Conflict of Interest
The authors state no conflict of interest.
References
- Bertin J, Wang L, Guo Y, et al. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-kappa B. J Biol Chem. 2001;276:11877–82. doi: 10.1074/jbc.M010512200. [DOI] [PubMed] [Google Scholar]
- Blonska M, Lin X. NF-kappaB signaling pathways regulated by CARMA family of scaffold proteins. Cell Res. 2011;21:55–70. doi: 10.1038/cr.2010.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte MI, Pescatore A, Paciolla M, et al. Insight into IKBKG/NEMO locus: report of new mutations and complex genomic rearrangements leading to incontinentia pigmenti disease. Hum Mutat. 2014;35:165–77. doi: 10.1002/humu.22483. [DOI] [PubMed] [Google Scholar]
- Eytan O, Qiaoli L, Nousbeck J, et al. Increased epidermal expression and absence of mutations in CARD14 in a series of patients with sporadic pityriasis rubra pilaris. Br J Dermatol. 2014a;170:1196–8. doi: 10.1111/bjd.12799. [DOI] [PubMed] [Google Scholar]
- Eytan O, Sarig O, Sprecher E, et al. Clinical response to ustekinumab in familial pityriasis rubra pilaris caused by a novel mutation in CARD14. Br J Dermatol. 2014b;171:420–2. doi: 10.1111/bjd.12952. [DOI] [PubMed] [Google Scholar]
- Fuchs-Telem D, Sarig O, van Steensel MA, et al. Familial pityriasis rubra pilaris is caused by mutations in CARD14. Am J Hum Genet. 2012;91:163–70. doi: 10.1016/j.ajhg.2012.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JB, Chen PL, Chen YT, et al. Genetic analysis of CARD14 in non-familial pityriasis rubra pilaris: a case series. Acta Derm Venereol. 2014;94:587–8. doi: 10.2340/00015555-1814. [DOI] [PubMed] [Google Scholar]
- Jordan CT, Cao L, Roberson ED, et al. Rare and common variants in CARD14, encoding an epidermal regulator of NF-kappaB, in psoriasis. Am J Hum Genet. 2012a;90:796–808. doi: 10.1016/j.ajhg.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan CT, Cao L, Roberson ED, et al. PSORS2 is due to mutations in CARD14. Am J Hum Genet. 2012b;90:784–95. doi: 10.1016/j.ajhg.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A, Landthaler M, Karrer S. Pityriasis rubra pilaris: a review of diagnosis and treatment. Am J Clin Dermatol. 2010;11:157–70. doi: 10.2165/11530070-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Magro CM, Crowson AN. The clinical and histomorphological features of pityriasis rubra pilaris. A comparative analysis with psoriasis. J Cutan Pathol. 1997;24:416–24. doi: 10.1111/j.1600-0560.1997.tb00816.x. [DOI] [PubMed] [Google Scholar]
- Petrof G, Almaani N, Archer CB, et al. A systematic review of the literature on the treatment of pityriasis rubra pilaris type 1 with TNF-antagonists. J Eur Acad Dermatol Venereol. 2013;27:e131–5. doi: 10.1111/j.1468-3083.2012.04456.x. [DOI] [PubMed] [Google Scholar]
- Wullaert A, Bonnet MC, Pasparakis M. NF-kappaB in the regulation of epithelial homeostasis and inflammation. Cell Res. 2011;21:146–58. doi: 10.1038/cr.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.