

Abstract
Single-electron-mediated alkyl transfer affords a novel mechanism for transmetalation, enabling cross-coupling under mild conditions. Here, general conditions are reported for cross-coupling of secondary alkyltrifluoroborates with an array of aryl bromides mediated by an Ir photoredox catalyst and a Ni cross-coupling catalyst.
Transition-metal-catalyzed cross-coupling reactions are an indispensable mainstay in the repertoire of modern synthetic chemists. Their tremendous impact is evidenced by ever-increasing prevalence in laboratories ranging from academic settings to large-scale industrial manufacture. As an illustrative example, a recent study revealed that >60% of all C−C bondforming reactions performed by medicinal chemists can be classified as transition-metal-catalyzed cross-couplings.1 Despite widespread adaptation of cross-coupling methods to construct C−C bonds, these methods remain largely limited to Csp2-hybridized centers owing to the challenges associated with their Csp3-hybridized counterparts.
Often, the complications associated with alkyl cross-coupling are inherent to the nature of the Csp3 organometallic reagents themselves (Figure 1). The instability of alkylboranes, alkylzincs, or alkyl Grignard reagents often requires their preparation and immediate use in superstoichiometric amounts (2−4 equiv) to achieve high yields.2 Further, for many alkylmetallic reagents, harsher reaction conditions can lead to proto-demetalation3 instead of the desired cross-coupling. Particularly for the more attractive organoboron nucleophiles, this decomposition pathway is difficult to suppress because the conditions that exacerbate these side reactions (high temperatures and strong basicity4) are typically required for efficient transmetalation.
Figure 1.

General catalytic cycle for secondary alkyl cross-coupling, depicting side reactions and off-cycle intermediates that may occur.
In those Pd-catalyzed alkyl cross-couplings where efficient transmetalation can be achieved, often an ensuing β-hydride elimination generates alkene byproducts and off-cycle catalyst intermediates. Successive β-hydride eliminations and reinsertions lead to a mixture of difficult-to-remove regioisomeric products from an isomerically pure alkylmetal.5 Careful ligand and transition metal selection can overcome these issues to some extent, but regioselective cross-coupling is often impossible.4b
The most challenging aspect of traditional alkylboron cross-coupling is the slow rate of transmetalation,6 a problem innate to the two-electron nature of the process. Because of the inherently low nucleophilicity of organoborons and the associated high energy of activation in the transmetalation, general approaches to secondary alkyl cross-coupling default to the more reactive alkylboranes,6a alkylzincs,7 alkyl Grignard reagents,8 and alkyllithiums.9 These reagents can normally be cross-coupled in high yields, but their poor shelf life and lack of functional group tolerability limit their widespread application, particularly in the context of parallel synthesis for medicinal chemistry.
Azastannatranes provide a viable solution to this challenge, as methods developed with these stable reagents are mild, if untested with highly hindered systems.10 However, their lack of both atom economy and commercial availability would appear to inhibit their widespread adoption, particularly given the perceived toxicity of organostannanes.
Among the more preferable organoboron reagents, a number of substrate-specific secondary alkyl cross-coupling reactions have been reported, but these always required electronic or functional group stabilization (β-carbonyl,11 α-boryl,12 α-alkoxy,13 benzylic,14 or allylic15) to allow effective reaction.16 A few isolated examples of cross-coupling with unactivated boronic acid derivatives exist, but these do not comprise a general strategy for alkyl cross-coupling.4a,17 The most universal transformations to date involve cross-coupling with alkyltrifluoroborates or boronic acids under rigorous reaction conditions (3 equiv K2CO3, 60−100 °C, 24−48 h).4b−d Most importantly, these methods are uniformly ineffective for more highly substituted systems such as 2-methylcycloalkylborons.
Recently, a novel concept for transmetalation based on a single-electron transfer (SET)-mediated pathway was reported.18 In this alternate mechanistic approach, the traditional four-coordinate transition structure19 was circumvented by a series of SET processes,20 enabling cross-coupling at room temperature. It was quickly recognized that this method had profound implications for Csp3−Csp2 cross-coupling because of the mild nature of the reaction conditions. Instead of a high activation energy transmetalation, alkyl transfer was effected through radical combination with a transition metal-based catalyst complex―a nearly barrierless energetic process.21 In such a transformation, exciting the photocatalyst provides virtually all of the energy necessary for the cross-coupling event, alleviating the need to heat the reactions. These conditions were anticipated to increase yields by limiting the side reactions that doomed previous alkyl Suzuki couplings.
Direct application of the previously reported SET strategy to secondary alkyltrifluoroborates was recognized as challenging because of their relatively high reduction potentials [+1.50 V vs SCE22] as compared to their benzylic counterparts [+1.10 V vs SCE22]. Exceeding the electrochemical potentials of highly oxidizing photocatalyst Ir(dFCF3ppy)2(bpy)PF6 (1) [+1.32 V vs SCE23], these substrates appeared to be destined for failure because of their high electrochemical demands. Nevertheless, encouragement was found in cases where thermodynamically unfavorable SETs occur, particularly within the context of an irreversible oxidation or reduction.22,24 Thus, only a small portion of the oxidation wave need overlap with the potential of the photocatalyst for the reaction to proceed, driven forward by irreversible C−B bond fragmentation.
Using this rationale, it was reasoned that visible light excitation of heteroleptic IrIII photocatalyst 1 would generate the long-lived photoexcited *IrIII complex 2, which could perform the thermodynamically challenging oxidation of secondary alkyltrifluoroborate 3 to the corresponding secondary alkyl radical 4 and the reduced ground-state photocatalyst 5 (Figure 2). Combining this radical with ligated Ni0 complex 625 would generate the alkyl-NiI complex 7, which is capable of engaging aryl halide 8 in oxidative addition to form the high-valent NiIII intermediate 9. Subsequent reductive elimination would afford the cross-coupled product 10, returning NiI-X species 11. Reduction of 11, with concurrent oxidation of 5, regenerates the Ni0 and Ir catalysts.26
Figure 2.

Comparison between traditional transmetalation and single-electron-mediated activation, and proposed catalytic cycle for photo-redox cross-coupling.
As a result of recent computational studies, the catalytic cycle proposed here differs from that initially hypothesized,18 where oxidative addition at Ni0 precedes radical combination. It is interesting, however, that both pathways converge on common intermediate 9 and, regardless of step order (oxidative addition before radical combination or vice versa), 9 is predicted to exist in equilibrium with ligated ArNiIIX species 12 and alkyl radical 4 through rapid and reversible homolytic cleavage of the Ni−C bond. Both pathways are energetically viable, but the low barrier for radical addition at Ni0 relative to oxidative addition at Ni0 would appear to favor the mechanism proposed in Figure 2 as being more representative of the predominant reaction pathway. Full computational details are forthcoming.27
Initially, attempts to apply previous conditions for the crosscoupling of benzylic and secondary α-alkoxyalkyltrifluoroborates [2.0 mol% 1, 3.0 mol% Ni(COD)2/dtbbpy, acetone/MeOH (10:1), 3.5 equiv lutidine] proved ineffective (<10% yield). The low yields achieved using that protocol appeared to be a consequence of the differing nature of the unstabilized, secondary alkyl radicals, where radical alkylation of arenes28 and H-atom abstraction from the solvent29 could outcompete combination with the Ni catalyst, which was present in low concentration. Using knowledge of established H-atom abstraction rates,30 these side reactions could be suppressed by the use of dioxane as solvent with careful monitoring of the reaction temperature. Next, various additives were determined to have a profound effect on the rate of the reaction and the amount of protodehalogenation side product observed. Of those additives screened, Cs2CO3 was the best for maximizing productive cross-coupling while limiting side reactions. The precise role of the base is not defined, but rate studies reveal that BF3 appears to inhibit the reaction. Its removal by added base allows reactions to proceed to completion in a more timely manner. Applying these conditions to the cross-coupling of methyl 4-bromobenzoate with potassium cyclopentyltrifluoroborate furnished product 14 in a satisfactory 92% yield (see Supporting Information for full optimization studies). Control experiments in the absence of Ni, Ir, and light all confirmed the dual catalytic nature of this cross-coupling.
With suitable conditions [2.5 mol% 4, 5 mol% NiCl2·dme, 5 mol% dtbbpy, 1.5 equiv Cs2CO3, dioxane, 26 W compact fluorescent lightbulb (CFL), 24 °C, 24 h] in hand, the scope of the alkyltrifluoroborate component was explored (Table 1). Organoborons possessing various ring sizes were coupled in good to excellent yields. To ensure the scalability and efficiency of this secondary alkyl coupling, a gram-scale reaction generating 13 was performed with reduced loadings of Ir and Ni catalysts. Importantly, only 1.0 mol% of the photoredox catalyst and 2.0 mol% of the Ni catalyst were required, and under these conditions the cross-coupling of methyl 4-bromobenzoate with potassium cyclohexyltrifluoroborate was accomplished to afford 13 in 73% yield after 36 h.
Table 1.
Scope of Secondary Potassium Alkyltrifluoroborates in Ni/Photoredox Cross-Coupling
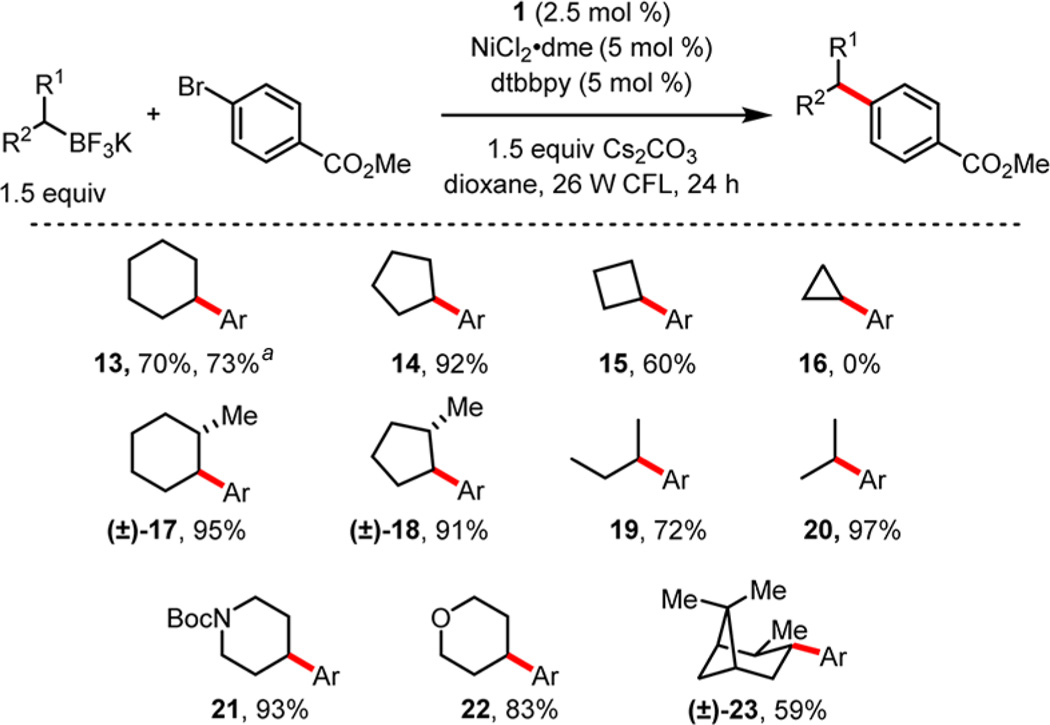 |
Reaction run on gram scale with 1.0 mol% Ir(dFCF3ppy)2bpy·PF6 and 2.0 mol% NiCl2·dme/dtbbpy.
When moving to smaller ring sizes, particularly cyclobutyl and cyclopropyl systems, where occupying an orbital with greater s-character destabilizes the resultant radical, yields suffered. In fact, for the cyclopropyl substrate, no reaction was observed. This poor reactivity serves to highlight the complementary nature of this method compared to established methods for Csp2- and Csp2-like cross-coupling, where the two-electron transmetalation regime has proven quite effective.31 Isopropyl- and sec-butyltrifluoroborates were also coupled in good yields. For these substrates, where isomerization would lead to different products, only the desired regioisomer was observed.
To probe further the limits of the isomeric fidelity observed in this transformation, several more bulky secondary alkyltrifluoroborates were explored. Gratifyingly, products derived from 2-methylcyclopentyl and 2-methylcyclohexyl substrates were also isolated without rearrangement, and the products were exclusively the trans diastereomer. Remarkably, an even more sterically demanding pinane derivative, 23, was an able partner.
Aliphatic heterocyclic ring systems containing oxygen (22) and protected nitrogen (21), which have proven to be recalcitrant in previous cross-coupling methods,4b did not significantly reduce yields under the photoredox cross-coupling protocol. In particular, aryl piperidines structurally analogous to 21, which are highly desired for incorporation into pharmaceutical libraries,32 could be accessed in excellent yield.
Substituted aryl bromides were screened to explore the scope of functional group tolerance (Table 2). During this process, a variety of trifluoroborates were used, particularly those that were both unsymmetrical and sterically encumbered, in an effort to showcase the mild nature of these cross-couplings. In previous work under Pd catalysis, 2-methylcycloalkylborons were observed to isomerize during the reaction, leading to a complex mixture where cross-coupling at the less sterically crowded positions was preferred.4b,d In contrast, the ability to couple even more crowded trifluoroborates with a range of aryl bromides under the current paradigm serves to highlight the mechanistic differences between a traditional transmetalation and the radical-mediated alkyl transfer described here.
Table 2.
Aryl Bromide Scope with Both Symmetric and Nonsymmetric Secondary Potassium Alkyltrifluoroborates
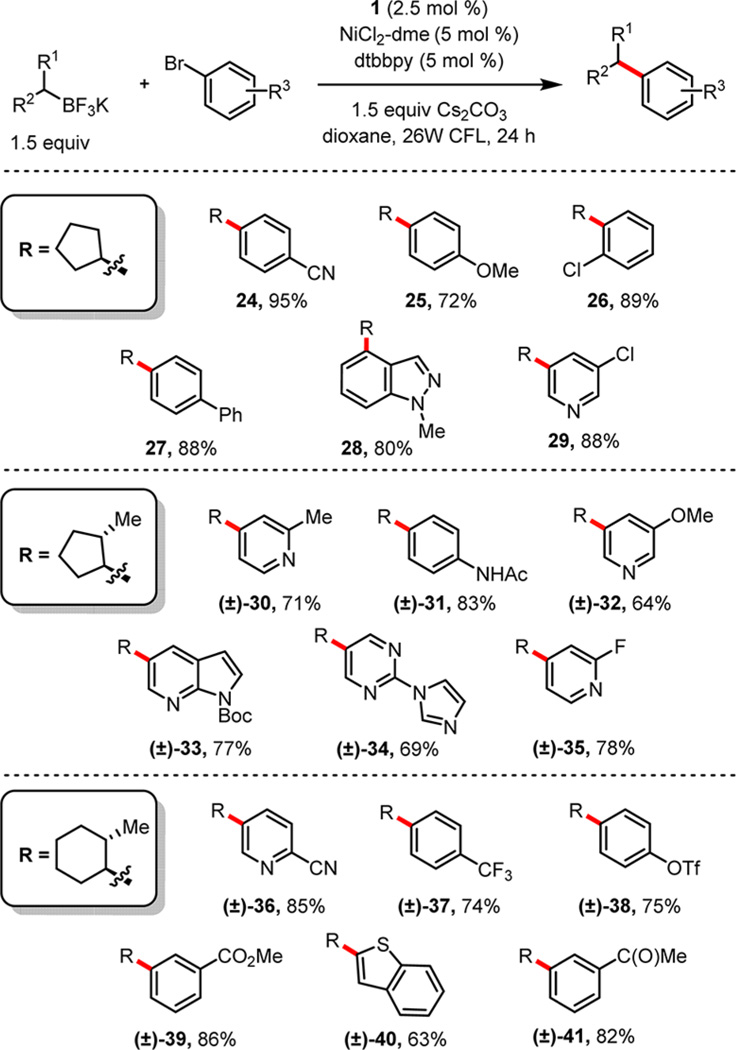 |
For the aryl bromide component, both electron-poor (24) and electron-rich systems (25) were coupled in good to excellent yields. Substitution ortho to the halide in 26 and 28 was tolerated without significant reduction in yield. Selective cross-coupling was observed in compounds with multiple electrophilic handles; both bifunctional aryl chlorides (leading to 26 and 29) and an aryl triflate, providing 38, coupled exclusively at the bromide. Ketones and esters (39 and 41), which can be intolerant of Kumada-type conditions, could be embedded within substrates to afford products in excellent yields. Further, nitrile-substituted bromides 24 and 36, which were unreactive under the previously developed Pd chemistry for alkyltrifluoroborates,4b served as competent reaction partners.
Protic groups did not diminish reactivity for the acetanilide substrate leading to 31. A number of N-heterocyclic bromides, providing privileged substructures of use in medicinal chemistry,32 readily participated in the cross-coupling. Pyridines (29, 30, 32, 35, 36), pyrimidine (34), and protected heterocycles (indazole 28, azaindole 33) were all accessed in acceptable yields. In addition to the more desirable N-heterocycles, a bromobenzothiophene was also reactive under the conditions, returning coupled product 40 in reasonable yield. Overall, the functional group tolerance for bromides shown here rivals or exceeds that of any previously developed method for secondary alkyl Suzuki cross-coupling, particularly within the context of heteroaryl substrates.
In summary, extremely mild conditions (rt, 1.5 equiv base, 26 W CFL irradiation) for the cross-coupling of secondary alkyltrifluoroborates with aryl bromides have been developed. The best previously reported Pd-catalyzed conditions for secondary alkylboron coupling4b,d required a significant excess of base and elevated temperatures to achieve less general overall results. Moreover, extensive Pd source, ligand, and reaction condition screenings have so far failed to identify satisfactory results for systems such as 2-methyl-substituted cycloalkylborons. Only slight modification of the initially reported conditions for benzylic coupling was required to extend both the nucleophilic and electrophilic scope of secondary alkyl cross-couplings with the photoredox cross-coupling dual catalytic system. The ease of this transformation across a broad array of radical precursors (α-amino,33 α-alkoxy,18,33 benzylic,18 and now secondary alkyl) speaks to the power of the SET-mediated approach. Further investigations into Ni precatalysts and ligands (entities that yielded vast improvements in analogous Pd chemistry34) can arguably afford similar gains when applied to this system. With proper screening and catalyst design, extensions to extremely hindered electrophiles and aryl chlorides appear feasible.
Finally, it should be noted that several Pd-catalyzed processes allow the conversion of enantiomerically enriched organoborons to cross-coupled products with high stereochemical fidelity.4d,11,13−15 Although such a process is clearly not feasible under the current paradigm, where radical intermediates are generated from the organoboron precursors, the prochiral radicals generated can be engaged in stereoconvergent transformations by employing enantioenriched ligands around the Ni catalyst.18,35 The fine-tuning of stereoconvergent protocols by ligand development and screening has the potential to provide a unique tactic for asymmetric synthesis by eliminating the need to synthesize optically active organometallic reagents, approaches to which are often long, tedious, and intolerant of sensitive functional groups.36
The most attractive feature of the reported photoredox cross-coupling may be its operational simplicity. By eliminating the need to prepare sensitive organometallics, titrate reagents, or use glovebox techniques, these methods greatly enhance the approachability of secondary alkyl cross-couplings. Consequently, the methods described here provide a significant advancement to Csp3−Csp2 cross-coupling.
Supplementary Material
ACKNOWLEDGMENTS
Alexandra Nilova (University of Pennsylvania) is acknowledged for some substrate preparation and reaction screening. Dr. Simon Berritt and Seth Martin (University of Pennsylvania) developed a photoredox screening platform that greatly facilitated the research. Frontier Scientific is thanked for the donation of boron compounds used in this investigation. Johnson-Matthey is acknowledged for the donation of IrCl3 used for the synthesis of the photocatalyst. We thank Pfizer, NIGMS (R01 GM081376), and TÜBİTAK for financial support of this research.
Footnotes
ASSOCIATED CONTENT
Experimental details and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
NOTE ADDED AFTER ASAP PUBLICATION
Reference 16 was replaced February 9, 2015.
REFERENCES
- 1.Roughley SD, Jordan AM. J. Med. Chem. 2011;54:3451. doi: 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]
- 2.Jana R, Pathak TP, Sigman MS. Chem. Rev. 2011;111:1417. doi: 10.1021/cr100327p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lozada J, Liu Z, Perrin DM. J. Org. Chem. 2014;79:5365. doi: 10.1021/jo500734z. [DOI] [PubMed] [Google Scholar]
- 4.(a) Littke AF, Dai C, Fu GC. J. Am. Chem. Soc. 2000;122:4020. [Google Scholar]; (b) Dreher SD, Dormer PG, Sandrock DL, Molander GA. J. Am. Chem. Soc. 2008;130:9257. doi: 10.1021/ja8031423. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) van den Hoogenband A, Lange JHM, Terpstra JW, Koch M, Visser GM, Visser M, Korstanje TJ, Jastrzebski JTBH. Tetrahedron Lett. 2008;49:4122. [Google Scholar]; (d) Li L, Zhao S, Joshi-Pangu A, Diane M, Biscoe MR. J. Am. Chem. Soc. 2014;136:14027. doi: 10.1021/ja508815w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartwig JF. Organotransition Metal Chemistry: From Bonding to Catalysis. Sausalito, CA: University Science Books; 2009. [Google Scholar]
- 6. Miyaura N, Ishiyama T, Sasaki H, Ishikawa M, Satoh M, Suzuki A. J. Am. Chem. Soc. 1989;111:314. Organoboron transmetalation rates mirror the trends established for the corresponding organostannanes: Labadie JW, Stille JK. J. Am. Chem. Soc. 1983;105:6129.
- 7.(a) Han C, Buchwald SL. J. Am. Chem. Soc. 2009:7532. doi: 10.1021/ja902046m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pompeo M, Froese RDJ, Hadei N, Organ MG. Angew. Chem. Int. Ed. 2012;51:11354. doi: 10.1002/anie.201205747. [DOI] [PubMed] [Google Scholar]
- 8.Hayashi T, Konishi M, Kobori Y, Makoto K, Taiichi H, Hirotsu K. J. Am. Chem. Soc. 1984;106:158. [Google Scholar]
- 9.Vila C, Giannerini M, Hornillos V, Fañanás-Mastral M, Feringa BL. Chem. Sci. 2014;5:1361. doi: 10.1039/c4sc03117b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li L, Wang C-Y, Huang R, Biscoe MR. Nat. Chem. 2013;5:607. doi: 10.1038/nchem.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sandrock DL, Jean-Gerard L, Chen C, Dreher SD, Molander GA. J. Am. Chem. Soc. 2010;132:17108. doi: 10.1021/ja108949w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Endo K, Ohkubo T, Hirokami M, Shibata T. J. Am. Chem. Soc. 2010;132:11033. doi: 10.1021/ja105176v. [DOI] [PubMed] [Google Scholar]
- 13.Molander GA, Wisniewski SR. J. Am. Chem. Soc. 2012;132:16856. doi: 10.1021/ja307861n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Imao D, Glasspoole BW, Laberge S, Crudden CM. J. Am. Chem. Soc. 2009:5024. doi: 10.1021/ja8094075. [DOI] [PubMed] [Google Scholar]; (b) Ohmura T, Awano T, Suginome M. J. Am. Chem. Soc. 2010;132:13191. doi: 10.1021/ja106632j. [DOI] [PubMed] [Google Scholar]
- 15.Glasspoole BW, Ghozati K, Moir JW, Crudden CM. Chem. Commun. 2012;48:1230. doi: 10.1039/c2cc16076e. [DOI] [PubMed] [Google Scholar]
- 16.Bonet A, Odachowski M, Leonori D, Essafi S, Aggarwal VK. Nature Chem. 2014;6:584. doi: 10.1038/nchem.1971. [DOI] [PubMed] [Google Scholar]
- 17.Kataoka N, Shelby Q, Stambuli JP, Hartwig JF. J. Org. Chem. 2002;67:5553. doi: 10.1021/jo025732j. [DOI] [PubMed] [Google Scholar]
- 18.Tellis JC, Primer DN, Molander GA. Science. 2014;345:433. doi: 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lennox AJJ, Lloyd-Jones GC. Angew. Chem. Int. Ed. 2013;52:7362. doi: 10.1002/anie.201301737. [DOI] [PubMed] [Google Scholar]
- 20.Recent reviews on photoredox catalysis: Schultz DM, Yoon TP. Science. 2014;343:985. doi: 10.1126/science.1239176. Prier CK, Rankic DA, Macmillan DWC. Chem. Rev. 2013;113:5322. doi: 10.1021/cr300503r. Tucker JW, Stephenson CRJ. J. Org. Chem. 2012;77:1617. doi: 10.1021/jo202538x. Review on photoredox and transition metal catalysis: Hopkinson MN, Sahoo B, Li J-L, Glorius F. Chem.- Eur. J. 2014;20:3874. doi: 10.1002/chem.201304823.
- 21.Breitenfeld J, Ruiz J, Wodrich MD, Hu X. J. Am. Chem. Soc. 2013;135:12004. doi: 10.1021/ja4051923. [DOI] [PubMed] [Google Scholar]
- 22.Yasu Y, Koike T, Akita M. Adv. Synth. Catal. 2012;354:3414. [Google Scholar]
- 23.Koike T, Akita M. Inorg. Chem. Front. 2014;1:562. [Google Scholar]
- 24.Shih H-W, Vander Wal MN, Grange RL, MacMillan DWC. J. Am. Chem. Soc. 2010;132:13600. doi: 10.1021/ja106593m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.In this catalytic cycle, initial reduction of the NiIICl2 is required to access an active Ni0 catalyst. This is likely mediated by the photocatalyst through sacrificial oxidation of trifluoroborate and subsequent reduction of NiIICl2 to generate the active Ni0 species.
- 26.We also recognize the possibility of an alternative quenching cycle, where reduction of NiI occurs from *IrIII to generate an IrIV species, which can then oxidize the secondary potassium alkyltrifluoroborate and generate IrIII in the ground state. However, the high concentration of RBF3Krelative to Ni in the reaction mixture at the outset leads us to favor the mechanism outlined in the text and Figure 2.
- 27.Gutierrez O, Tellis JC, Primer DN, Molander GA, Kozlowski MC. manuscript submitted. [Google Scholar]
- 28.Giese B. Radicals in Organic Synthesis: Formation of Carbon- Carbon Bonds. New York: Pergamon Press; 1986. [Google Scholar]
- 29.For example, in THF as solvent coupling at the carbon alpha to oxygen was observed alongside the desired alkyl cross-coupling product.
- 30.Hallen RT, Gleicher GJ, Mahiou B, Clapp GE. J. Phys. Org. Chem. 1989;2:367. [Google Scholar]
- 31.Molander GA, Gormisky PE. J. Org. Chem. 2008;73:7481. doi: 10.1021/jo801269m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Welsch ME, Snyder SA, Stockwell BR. Curr. Opin. Chem. Biol. 2010;14:347. doi: 10.1016/j.cbpa.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zuo Z, Ahneman D, Chu L, Terrett J, Doyle AG, MacMillan DWC. Science. 2014;345:437. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.(a) Bruno NC, Niljianskul N, Buchwald SL. J. Org. Chem. 2014;79:4161. doi: 10.1021/jo500355k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Martin R, Buchwald SL. Acc. Chem. Res. 2008;41:1461. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.For list of asymmetric secondary bromide cross-coupling ligands and substrates see: Tasker SZ, Standley EA, Jamison TF. Nature. 2014;509:299. doi: 10.1038/nature13274. For an example of asymmetric cross-coupling of a secondary bromide see: Binder JT, Cordier CJ, Fu GC. J. Am. Chem. Soc. 2012;134:17003. doi: 10.1021/ja308460z.
- 36.(a) Matteson DS. Tetrahedron. 1998;54:10555. [Google Scholar]; (b) Brown HC, Singaram B. Acc. Chem. Res. 1988;21:287. [Google Scholar]; (c) Larouche-Gauthier R, Fletcher CJ, Couto I, Aggarwal VK. Chem. Commun. 2011;47:12592. doi: 10.1039/c1cc14469c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.