

Key Points
A new approach to achieving immune tolerance and mixed chimerism with relevance for hematopoietic stem cell and organ transplantation.
Anti–third-party central memory T cells support engraftment with nonablative conditioning by sequestering and deleting anti-donor T cells.
Abstract
Transplantation of T cell–depleted BM (TDBM) under mild conditioning, associated with minimal toxicity and reduced risk of GVHD, offers an attractive therapeutic option for patients with nonmalignant hematologic disorders and can mediate immune tolerance to subsequent organ transplantation. However, overcoming TDBM rejection after reduced conditioning remains a challenge. Here, we address this barrier using donor-derived central memory CD8+ T cells (Tcms), directed against third-party antigens. Our results show that fully allogeneic or (hostXdonor)F1-Tcm, support donor chimerism (> 6 months) in sublethally irradiated (5.5Gy) mice, without GVHD symptoms. Chimerism under yet lower irradiation (4.5Gy) was achieved by combining Tcm with short-term administration of low-dose Rapamycin. Importantly, this chimerism resulted in successful donor skin acceptance, whereas third-party skin was rejected. Tracking of host anti-donor T cells (HADTCs), that mediate TDBMT rejection, in a novel bioluminescence-imaging model revealed that Tcms both induce accumulation and eradicate HADTCs in the LNs,concomitant with their elimination from other organs, including the BM. Further analysis with 2-photon microcopy revealed that Tcms form conjugates with HADTCs, resulting in decelerated and confined movement of HADTCs within the LNs in an antigen-specific manner. Thus, anti–third-party Tcms support TDBMT engraftment under reduced-conditioning through lymph-node sequestration and deletion of HADTCs, offering a novel and potentially safe approach for attaining stable hematopoietic chimerism.
Introduction
Hematopoietic stem cell transplantation (HSCT) can offer a cure for patients with a variety of nonmalignant hematologic disorders, such as sickle cell anemia and thalassemia. Furthermore, considering that chimerism induction is generally associated with immune tolerance to donor antigens1–4 it can serve as a platform for immune tolerance induction to be followed by either solid organ transplantation or by continuous cell therapy with donor cells in cancer patients. Therefore, attaining a well-tolerated HSCT protocol represents one of the most desirable goals in transplantation biology. However, the high risk for infections and GVHD, linked with procedures currently used in HSCT in leukemia patients, are not acceptable for patients with nonmalignant diseases associated with longer life expectancy. Thus, developing new, safer approaches for achieving hematopoietic chimerism under milder conditioning than that used in leukemia patients, and with reduced risk for GVHD, is of utmost importance.
The problem of GVHD could be adequately addressed, even in mismatched haploidentical transplants, by rigorous T-cell depletion or positive selection of CD34+ hematopoietic stem cells (HSCs), using megadoses of HSCs to overcome the residual host immunity remaining after myeloablative conditioning. This immune modulation is mediated, at least partially, by virtue of potent veto activity exhibited by CD34+ HSCs and their early myeloid derivatives5–7 which rapidly expand during the early posttransplant period. Veto activity, was defined as the capacity to specifically suppress CTL-precursor (CTLp) cells, directed against antigens (Ags) expressed by the veto cells. Therefore, use of donor-derived veto cells as specific immunosuppressants in transplantation settings, eliminating only host anti-donor T cells while sparing others, is highly attractive. However, the number of CD34+ HSC that can be harvested is limited, and insufficient for overcoming the robust host immunity surviving reduced-intensity conditioning (RIC; Gan et al, unpublished results). Therefore, combining megadoses of CD34+ HSC transplantation (HSCT) with other tolerizing veto cells could potentially support and promote successful engraftment of purified HSCs under a safer RIC protocol. One approach to address this challenge could be provided using donor CD8 T cells, shown to be endowed with the most potent veto activity.8 However, the utility of these cells for tolerance induction is limited because of their marked GVH reactivity. We previously described one approach to generate donor CD8 T cells with markedly reduced GVH reactivity by stimulation against third-party stimulators in the absence of exogenous cytokines.9 This approach was based on the observation that only activated anti–third-party T cells are capable of surviving a 6-day period of cytokine deprivation and that these anti–third-party clones can expand when further grown with exogenous IL-2.9 Subsequently, these anti–third-party CD8 T cells were shown to mediate potent veto activity via a mechanism involving both their CD8 molecule and the Fas-FasL pathway.10–12 More recently, we demonstrated that anti–third-party CD8 T cells can also actively respond by polarizing cytotoxic granules when recognized by the TCR of the host T cell (HTC), thereby eliminating HTCs through a perforin-dependent veto activity.13
Unfortunately, the attributes, exhibited ex vivo by anti–third-party CD8 T cells, did not translate to potent tolerizing activity in vivo. Considering the CD44+CD62L− effector phenotype of these cells, known to be associated with poor lymph node (LN) homing, we hypothesized that effector anti–third-party veto CD8 T cells fail to colocalize with rejecting HTCs at the LNS, which is the preferred site for priming and initiation of the cell-to-cell contact required for exerting their veto activity.14 To address this difficulty, we recently established new ex vivo culture conditions, using IL-15, that favor the induction of CD8 T cells expressing CD44+CD62L+ central memory (Tcm) phenotype.15,16 These anti–third-party Tcms generated in the presence of IL-15 were shown to home to LNs of BMT recipients,17 where they colocalize with the recipient's endogenous HTCs. Thereafter, the Tcms proliferate extensively and display long-term persistence in the recipients. Most importantly, these Tcms were shown to be endowed with potent in vivo veto activity, specifically inducing apoptosis on host anti-donor T cells in the secondary lymphoid organs without causing GVHD.17
Based on these findings, we now demonstrate the ability of anti–third-party Tcm cells to specifically delete anti-donor HTCs and thereby induce tolerance to fully mismatched TDBMT in a stringent mouse model for BM allografting, under a reduced conditioning protocol consisting of 5.5Gy sublethal total body irradiation (TBI). Moreover, by combining Tcm therapy with 5 days of a moderate rapamycin dose, we were able to further reduce the conditioning to 4.5Gy TBI.
In addition, using luciferase-expressing host anti-donor TCR transgenic T cells, we developed a novel in vivo BMT model that enables noninvasive monitoring of the rejection process. Using this model, as well as in vivo 2-photon microscopic imaging, we were able to interrogate the interaction between the Tcms and anti-donor HTCs that leads to HTC tolerization. Surprisingly, this analysis revealed a shift in the distribution of anti-donor HTCs, with preferential concentration at the LNs. Accordingly, the specific formation of conjugates at this site was shown to lead to reduced traffic velocity and confined movement of the anti-donor HTCs within the LNs. Subsequently, these host anti-donor cells were indeed deleted by the Tcms resulting in TDBMT acceptance.
Notably, the tolerance achieved by Tcm infusion under RIC protocols was strong enough to allow a successful acceptance of donor skin-graft, indicating that this Tcm-based protocol can offer a novel treatment for nonmalignant hematopoietic diseases, as well as a relatively safe platform for chimerism induction which can be followed by solid organ transplantation or by continuous cell therapy with donor cells in cancer patients.
Methods
Animals
Mice used were 6- to 13-week-old females. Balb/c(H-2d), CB6(H-2bd), FVB(H-2q), SJL(H-2s), C57BL/6(H-2b), and C3H(H-2k) were purchased from Harlan Israel. A breeding pair of transgenic (Tg) 2c-mice expressing a T-cell receptor (TCR) with H-2Ld specificity was kindly provided by Janko Nikolic-Zugic (Sloan-Kettering). Progeny of FVB luciferase expressing mice, kindly provided by Prof Negrin (Stanford), were backcrossed for 11 generations with C57BL/c mice, to generate H-2b luciferase expressing mice. Tg mice, Balb/c-nude, and C57BL/6-nude mice were bred at the Weizmann Institute Animal Breeding Center. Studies were approved by the Weizmann Institute of Science Institutional Animal Care and Use Committee.
Flow cytometric analysis
Fluorescence-activated cell sorting (FACS) analysis was performed using a modified Becton Dickinson FACScan. Cells were stained with labeled antibodies specific for: CD3-PE/FITC/APC, CD8α-PE/FITC/APC, CD11b-PE/FITC/APC, CD19-PE/FITC/APC, CD62L-PE/FITC/APC, CD44-PE/FITC/APC, H-2Kb-PE/FITC, H-2Dd-PE/FITC, H-2Kk-PE/FITC (BD Pharmigen); 1B2 biotinylated, and streptavidin-APC (Jackson Laboratories).
Preparation of host nonreactive donor anti–third-party cells
Anti–third-party Tcms were prepared as previously described.17 Briefly, splenocytes of donor mice were cultured against irradiated third-party splenocytes for 60 hours under cytokine deprivation. Subsequently, CD8 cells were positively selected using magnetic particles (BD Pharmingen) and cultured in an Ag-free environment. rhIL-15 (20 ng/mL; R&D Systems) was added every second day. At the end of the culture (day 16), Tcms were positively selected for CD62L expression (magnetic-activated cell sorting [MACS] cell separation, Miltenyi Biotec).
Reduced conditioning model: sublethal animal irradiation
Balb/c female mice (10-12 weeks of age) received sublethal TBI at doses indicated (4.5Gy or 5.5Gy), on day −1. On the following day (ie, day 0), animals were transplanted with a “megadose” (1 × 107 or 2 × 107) transplant of C57BL/6-nude BM cells with or without 5 × 106 anti–third-party Tcms. In some experiments, mice received subcutaneous injections (days −1 to +4) of 0.5 mg/kg-bw of rapamycin (Rapamune). Mice were evaluated twice weekly for overall appearance and weight. Chimerism analysis was conducted periodically. Skin grafting was performed to verify tolerance.
Skin grafting
Sections of skin (1 cm2) were cut from the backs of killed C57BL/6 donor mice and C3H third-party mice, and washed in saline. Patches of dorsal skin of 1 cm2 were removed from anesthetized recipient mice (ketamine 100 mg/kg and xylazine 20 mg/kg both intraperitoneally, Kepro) and replaced by the donor/third-party skin grafts. Histoacryl (Braun) was applied around the grafts, and 2 stitches using silk 5-0 suturing thread were applied to secure the graft. Animals were kept in individual cages for 15 days after skin transplantation. Skin graft status was monitored twice weekly.
Chimerism analysis
Chimerism was determined by cytofluorimetry. Peripheral blood was collected by retro-orbital bleeding, cells were fractionated on Ficoll-Paque Plus (Amersham Pharmacia Biotech), and the isolated mononuclear cells of each mouse were double-stained by direct immunofluorescence against donor and host surface markers. Chimerism within different cellular compartments was assessed by staining mononuclear-cell fraction against donor antigens and against either of the following: myeloid cells (CD11b), B cells (CD19), and T cells (CD4/CD8).
Graft-rejection imaging model
C57BL/6 female mice (10-12 weeks of age) were shaved on day −6. Mice were then supralethally irradiated with 4Gy TBI on day −5, and 10Gy TBI on day −2. On day −1, 700 CD8 2c-Luc+ purified host T cells (separated by MACS) together with 104 carrier cells (2Gy irradiated CD8-depleted 2c splenocytes), were injected to tail vein. On day 0, 3 × 106 allogeneic Balb/c-nude BM cells were transplanted, with or without 5 × 106 CB6-derived anti–third-party Tcms. Animals were monitored for 2c-Luc+ cell load by optical whole-body imaging (IVIS 100, Xenogen) at the indicated times after transplantation.
In vivo imaging
At the specified times after transplant, mice were monitored by optical whole-body imaging system (IVIS 100, Xenogen) coupled with a Pixelfly QE (PCO) charge-coupled device (CCD) camera. Mice were anesthetized with ketamine-xylazine as described and were then injected (intraperitoneally) with an aqueous solution of D-luciferin (150 mg/kg Cat#XR-1001, Xenogen; 30 mg/mL in PBS) 10 minutes before imaging. Image processing and data analysis were performed using Living Image 3.2 software.
Two-photon microscopy
C57BL/6 mice were lethally (10Gy) or sublethally (6.5Gy) irradiated on day −1; on day 0 stained cells were injected into the retro-orbital sinus, 4 or 16 hours before imaging. The injected cells, consisting of CD8 2c cells (2 × 106 per mouse) prepared from spleens and LNs of mice, and CD8 purified by MACS, were stained with carboxylic acid acetate, succinimidyl ester (SNARF-1, Invitrogen). Anti–third-party Tcm (107 per mouse) were stained with CellTracker Orange CMTMR (5-(and-6)-(4chloromethyl) benzoyl) amino) tetramethylrhodamine; Molecular Probes). Mice were anesthetized with ketamine-xylazine as described. Popliteal lymph node (PLN) was surgically exposed. Temperature was maintained at 37°C. Two-photon imaging was performed using an Ultima multiphoton microscope (Prairie Technologies) with a pulsed Mai Tai Ti-sapphire laser (Newport), using a water-immersed 20×/0.95 NA dipping objective (Olympus). Excitation was carried out at 890 nm. To create a typical time lapse sequence, a 65-μm thick section of the popliteal LN was scanned at 5-μm Z steps every 40 seconds. Videos were further processed using Volocity Version 4.3.2 software (Improvision) and ImageJ Version 1.4.3.67 software.
Statistical analysis
The analysis of survival data were performed using Kaplan-Meier curves (log-rank test). Comparison of means was conducted using the Student t test. Analysis of donor-skin graft acceptance using the χ2 test was preformed using SPSS Statistics 17.0 software. P values < .05 were considered statistically significant.
Results
Tcm support engraftment of TDBM allografts under RIC
To evaluate the ability of anti–third-party Tcms to overcome rejection of megadose TDBM after RIC, Balb/c (H-2d) mice were sublethally irradiated (5.5Gy TBI) on day −1, and on the next day, received a megadose of 20 × 106 C57BL/6-nude BM cells (H-2b), with or without 5 × 106 allogeneic donor C57BL/6 Tcm (H-2b). All 12 mice in the control group, receiving only TBI without BMT, survived for more than 6 months after the irradiation, demonstrating the nonmyeloablative nature of the conditioning. Likewise, all 14 recipients of megadose BMT survived, although exhibiting complete rejection of the BM by day 30 after transplantation, indicating that megadose transplant alone indeed fails to overcome the residual immunity remaining after the sublethal irradiation. In contrast, 9/9 of the mice that received allogeneic Tcms, in addition to the megadose TDBMT, displayed significant and stable donor chimerism (Figure 1A) comprising the myeloid, B cell, CD4 T cell, and CD8 T cell compartments (data not shown) that persisted for more than 6 months after transplant.
Figure 1.
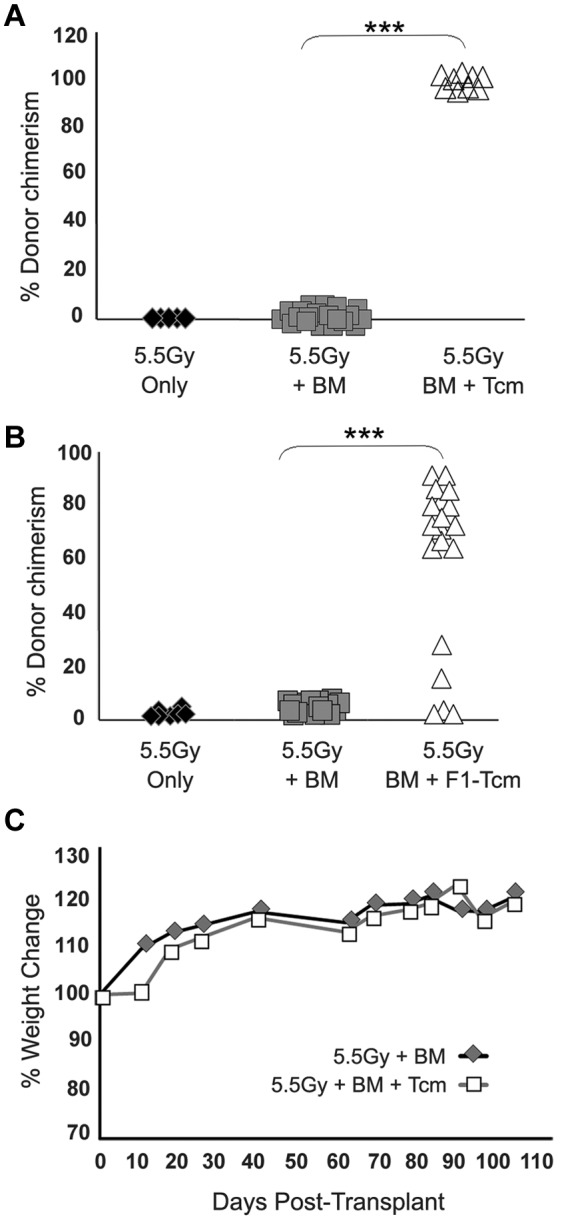
Anti–third-party Tcms without GVH reactivity support engraftment of TDBM allografts in sublethally irradiated mice. Sublethally irradiated (5.5Gy) Balb/c (H-2d) mice were transplanted with 20 × 106 C57BL/6-nude (H-2b) BM cells with or without: (A) Five × 106 allogeneic C57BL/6 Tcm (H-2b) or (B) 5 × 106 CB6 F1 Tcms (H-2bd). Percentage of donor cells in peripheral blood was analyzed 90 days after transplant by FACS using anti-donor (H-2Kb) antibodies. Data represent results of at least 3 independent experiments (***P < .001). (C) Average weight change during 100 days after transplant of nude-BM or nude-BM and allogeneic Tcms.
Importantly, and as previously demonstrated in lethally irradiated recipients,17 chimerism induced by the Tcms was not associated with manifestations of GVHD symptoms (eg, ruffled fur, hunched back, and reduced body weight). Thus, weight and overall appearance of mice receiving nude-BM and fully allogeneic anti–third-party Tcms were the same as that of control mice receiving a transplant of nude-BM alone (Figure 1C). This is in sharp contrast to severe GVHD induced by the same doses of unmanipulated naive CD8 T cells.17 To substantiate that the enhanced engraftment observed is not mediated by residual alloreactivity of the Tcm preparation, we also tested Tcms derived from (CB6)F1 (H-2bd) mice for their ability to support engraftment after RIC. As shown in Figure 1B, 18/23 transplanted mice that received F1 Tcms, exhibited significant and stable donor chimerism, which persisted for more than 6 months after transplantation. Again, this chimerism was of multilineage origin (data not shown). Importantly, enhancement of chimerism by Tcms was found to be MHC specific, in that no chimerism could be detected in mice that received BM megadose from C57BL/6 nude donors with host (Balb/c, H-2d)–derived Tcms (0/5) or C3H (H-2k)–derived Tcms (0/5), which do not express donor BM antigens.
Anti–third-party Tcms synergize with rapamycin
Considering that rapamycin was previously shown to synergize with veto anti–third-party CD8 T cells generated under conditions favoring induction of an effector phenotype,9 we attempted to test the possibility that rapamycin will also synergize with Tcms, to enable further reduction of the radiation dose. Initially, we evaluated the possibility that rapamycin might adversely affect the survival and proliferation of Tcms in vivo, by comparing rapamycin's effect on Tcms to its effect on host anti-donor naive T cells. To directly track host anti-donor T cells we used naive CD8 T cells derived from 2c T-cell receptor (TCR) transgenic mice. The CD8 T cells in these mice carry transgenic TCRs against H-2Ld and therefore represent a population of host cells with specificity against H-2Ld bearing donors. In addition, the 2c-derived CD8+ cells can be identified using the clonotypic 1B2 antibody directed against their transgenic TCR. Thus, lethally irradiated C57BL/6 (H-2b) hosts were transplanted with 3 × 106 Balb/c-nude (H-2d) BM cells, along with 0.5 × 106 host anti-donor naive 2c (H-2b) cells or 5 × 106 donor type Balb/c (H-2d) Tcms. In each group (“Tcm” or “2c”) a subgroup of mice received rapamycin (0.5 mg/kg bw) whereas another subgroup received vehicle (PBS) for 5 consecutive days after transplantation. Spleens were then harvested on day +5 and total numbers of Tcm or 2c cells were analyzed by FACS. Data showed that although rapamycin significantly inhibited (72% inhibition) the expansion of the naive host anti-donor 2c cells, it only slightly affected (13% inhibition) expansion of anti–third-party Tcms in vivo (supplemental Figure 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article).
Based on the demonstration that rapamycin efficiently inhibits host anti-donor cell expansion, but does not negatively affect the expansion of anti–third-party Tcms in vivo, we proceeded to evaluate whether Tcms and rapamycin synergize in overcoming TDBM graft rejection. Thus, on day −1, Balb/c mice were exposed to 4.5Gy sublethal TBI and then received on day 0, a megadose of 20 × 106 C57BL/6-nude BM, with or without 5 × 106 allogeneic C57BL/6 Tcm. In each group (BM and BM+Tcm), one subgroup of mice received rapamycin (0.5 mg/kg bw), whereas another subgroup received vehicle (PBS) for 5 consecutive days after transplantation (days −1 to +4). As can be seen in Figure 2A, stable donor chimerism for more than 6 months could be achieved in all 6 mice receiving transplantation of “megadose” BM supplemented with Tcms and short-term rapamycin administration. Here too, the chimerism spanned throughout the B cell, myeloid, and both CD4 and CD8 T-cell compartments (data not shown). In sharp contrast, all untreated mice, receiving BMT alone, rejected the graft, and only 3/6 mice receiving BM and Tcms, without rapamycin coadministration, displayed long-term donor chimerism (P < .05). The synergistic interaction between Tcms and Rapamycin was suggested by the observation that administration of rapamycin alone did not enable any level of chimerism, and all mice rejected the BM graft by day 30 after transplantation, whereas combined use of rapamycin and Tcms led to engraftment in the entire group of TDBMT recipients (Figure 2).
Figure 2.

Anti–third-party Tcms can be used with decreased irradiation dose when coadministered with low-dose rapamycin. Sublethally irradiated (4.5Gy TBI) Balb/c (H-2d) mice were transplanted with 20 × 106 C57BL/6-nude (H-2b) BM cells with or without: 5 × 106 allogeneic C57BL/6 Tcm (H-2b; ▵ and  ) or 5 × 106 CB6 F1 Tcm (H-2bd; ●). The indicated groups received subcutaneous injections of 0.5 mg/kg bw rapamycin during 5 days (days −1 to +4). Percentage of donor cells in peripheral blood, analyzed 90 days after transplant by FACS using anti-donor (H-2Kb) antibodies is presented. Data represents results of at least 2 independent experiments (*P < .05; ***P < .001).
) or 5 × 106 CB6 F1 Tcm (H-2bd; ●). The indicated groups received subcutaneous injections of 0.5 mg/kg bw rapamycin during 5 days (days −1 to +4). Percentage of donor cells in peripheral blood, analyzed 90 days after transplant by FACS using anti-donor (H-2Kb) antibodies is presented. Data represents results of at least 2 independent experiments (*P < .05; ***P < .001).
The ability of F1-derived Tcms to efficiently support the megadose BM engraftment under this conditioning protocol was also demonstrated. Here too, 8/9 of the mice that received BM+F1-Tcm+rapamycin were successfully engrafted (Figure 2) and displayed significant stable donor chimerism that spanned all compartments (data not shown) and persisted for more than 6 months after transplant.
Tcm-mediated chimerism specifically protects donor skin grafts from rejection
To test whether Tcm treatment in conjunction with donor TDBM cells induces durable immune tolerance, enabling subsequent acceptance of organ transplants, we tested the ability of Tcm-treated chimeric mice to tolerate donor-type skin transplants, known to be highly immunogenic.18–22 As can be seen in Figure 3B, 5/7 recipients of allogeneic C57BL/6-derived Tcms, and 8/10 recipients of (CB6)F1-derived Tcms, accepted the donor C57BL/6 (H-2b) skin (> 4 months follow-up), whereas the C3H thrid-party unrelated skin was promptly rejected. As expected, BM control nonchimeric animals, that received 5.5Gy TBI with megadose TDBMT alone, without Tcms, rapidly rejected both grafts.
Figure 3.
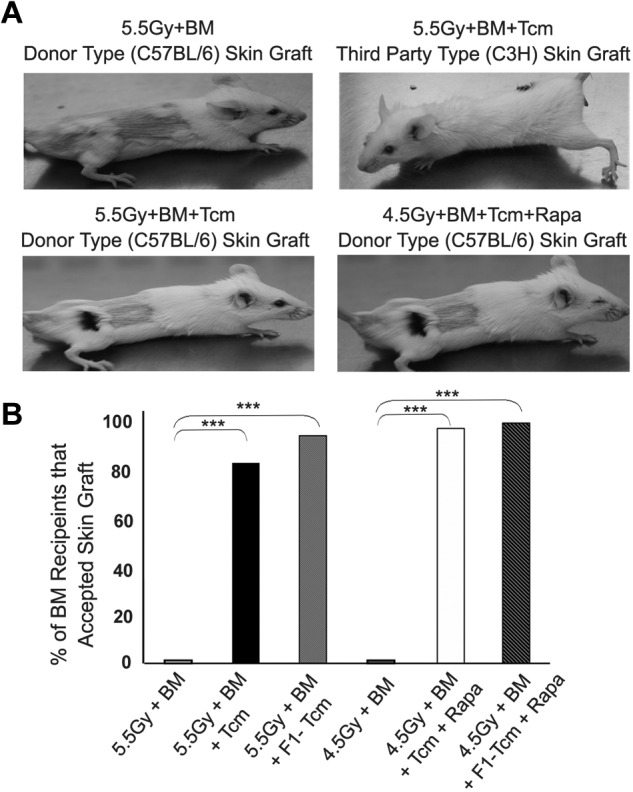
Anti–third-party Tcms mediated chimerism specifically protects donor skin graft from rejection. Sublethally irradiated (5.5Gy/4.5Gy TBI) Balb/c (H-2d) mice were transplanted with 20 × 106 C57BL/6-nude (H-2b) BM cells with or without administration of 5 × 106 C57BL/6 Tcm. The indicated groups received subcutaneous injections of 0.5 mg/kg rapamycin during 5 days (days −1 to +4). On day 30 after BM transplantation skin grafting of donor-type (C57BL/6, H-2b) as well as third-party (C3H, H-2k) was carried out (each recipient was transplanted with both grafts). Recipients of skin grafts were either BM control mice (“5.5Gy+BM” and “4.5+BM” Groups) which had rejected the donor-BM or those Tcm-treated mice which had achieved donor-chimerism. (A) Representative images of skin grafts across experimental groups taken 4 months after skin grafting. (B) Bars show percentage of chimeras that accepted donor skin grafts across experimental groups during a > 4-month follow-up period after skin grafting. Data were pooled from 6 independent experiments (***P < .001 by X2).
Notably, similar results were attained when skin grafting was carried out on Balb/c (H-2d) chimeric mice, where chimerism was achieved using a milder conditioning protocol including 4.5Gy TBI, megadose TDBMT, short-term rapamycin, and either allogeneic Tcms or F1 Tcms. Thus, 5/6 chimeras receiving allogeneic Tcms and 6/7 chimeras receiving F1 Tcms accepted the donor skin graft while promptly rejecting the third-party graft (Figure 3A-B). This successful skin engraftment suggests that the tolerance induced by the Tcms after RIC, can enable engraftment of donor-derived transplants, whereas HTCs that are not deleted by the Tcms retain their immune functionality toward nondonor antigens.
In vivo imaging reveals unique migration and expansion of anti-donor T cells in the presence of donor Tcms
To better understand the chain of events by which Tcms induce tolerance toward the transplanted bone marrow, we established a novel graft rejection imaging model. In this model, graft rejecting host immune cells can be monitored noninvasively over time in the same mouse. For this purpose, lethally irradiated C57BL/6 (H-2b) host mice received, on day −1, a fixed number of TCR transgenic 2c CD8+ T cells (H-2b) representing host cells with specificity against the Balb/c (H-2d) donor cells. These host anti-donor cells express the luciferase reporter gene (2c-Luc+) thereby enabling their noninvasive tracking, in vivo. On the following day (day 0), the mice were transplanted with 3 × 106 Balb/c-nude BM cells, with or without 5 × 106 CB6 derived anti–third-party Tcms. Both the migration and expansion of these TCR transgenic host anti-donor 2c-Luc+ cells were evaluated periodically after transplant using whole-body bioluminescence imaging. Notably, and in contrast to the sublethal irradiation used in the RIC model, in these experiments mice were supralethally irradiated. Here, the transferred luciferase-expressing host anti-donor T cells, reintroduced after irradiation, were expected to reject the transplanted bone marrow, consequently leading to death of mice of lethal graft rejection. In this way, we were able to follow the rejection process, mediated by the signal producing graft rejecting host cells.
As shown in Figure 4A, in the absence of Tcm treatment, 2c-Luc+ HTC expanded massively over time, leading to rejection of the BM transplant and consequently to the death of all mice in the group by day 27. However, this was not the case for mice treated with Tcm, where 2c-Luc+ HTC mostly expanded until day 10, after which their numbers gradually decreased until their luminescence signal became negligible by day 27 (Figure 4A). This diminished luminescence was consistent with the long-term survival of these Tcm-treated mice (> 5 months follow-up, Figure 4C). Moreover, the surviving recipients displayed stable donor chimerism (data not shown), validating the failure of HTCs to reject the BM graft when administered in conjunction with anti–third-party Tcms. Interestingly, Tcms treatment did not only result in the deletion of the rejecting HTCs but also affected their compartmentalization at the early posttransplant period. Thus, in mice that did not receive Tcms, 2c-Luc+ HTC localized mainly to the BM (femur, humerus, and sternum) and internal organs (abdominal area and lungs; Figure 5A), whereas in Tcm-treated mice, 2c-Luc+ HTC segregated mainly to peripheral LNs and internal organs (including the abdominal area, which may represent mesenteric LN localization, Figure 5C). Specifically, compartmentalization to the BM, the main site of graft rejection was notably different between Tcm-treated and untreated groups, such that in the Tcm untreated mice, the BM was heavily populated with 2c-Luc+ HTCs, whereas in the BM of Tcm-treated mice, these rejecting host cells could hardly be detected (Figure 5A-C).
Figure 4.
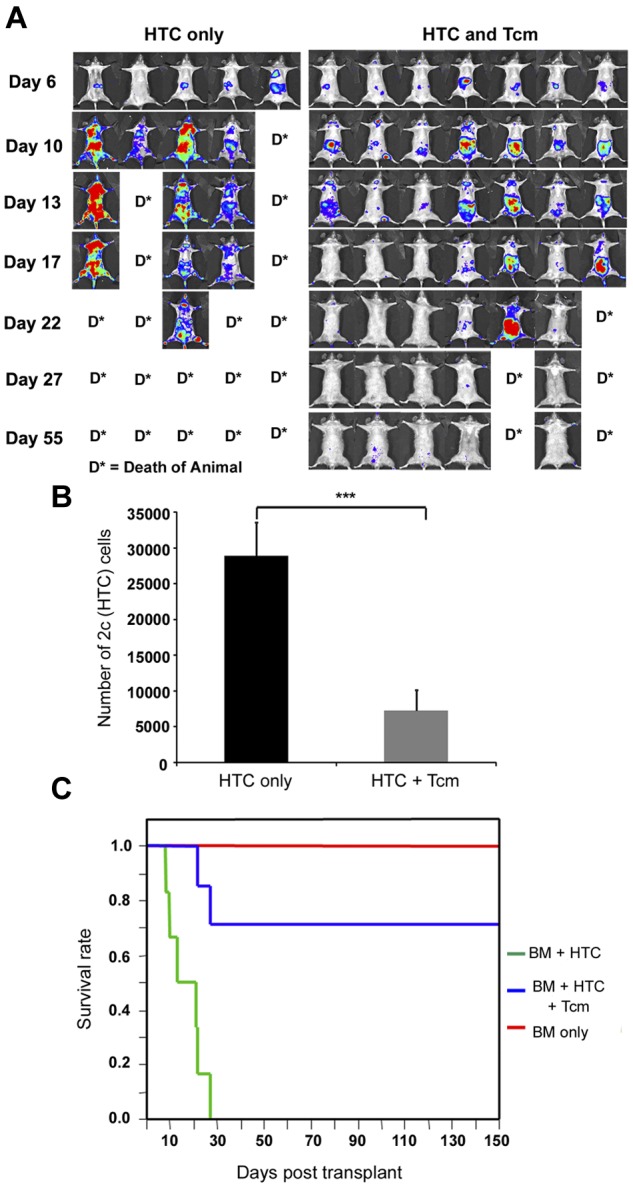
Tcms systemically eliminate host anti-donor T cells. Seven-hundred TCR transgenic 2c-Luc+ CD8+ cells were injected on day −1 into supralethally irradiated (4Gy on day −5 and 10Gy on day −2) C57BL/6 mice. One day after 2c administration, the mice received 3 × 106 Balb/c-nude BM cells (“HTC only”) or BM+5 × 106 (CB6)F1 Tcm (“HTC+Tcm”). (A) Images were taken at indicated days after transplantation in live anesthetized animals using IVIS. Color intensity is normalized for all images. (B) On day 9 after transplantation, organs (LN, spleen, BM, lung, and liver) were harvested from 5 animals from each group and numbers of live (7aad−) CD8+ 2c cells were assessed by FACS analysis. Total numbers of 2c cells harvested from all organs tested in the absence (black bar) or presence (gray bar) of Tcm treatment are presented. Data represent mean ± SD of total numbers of 2c cells from 5 animals in each group (***P < .001). (C) Survival curves of mice from the different treatment groups.
Figure 5.

Anti–third-party Tcm administration induces LN accumulation of host anti-donor HTCs. Seven-hundred TCR transgenic 2c-Luc+ CD8+ cells were injected on day −1 into supralethally irradiated (4Gy on day − 5, and 10Gy on day − 2) C57BL/6 mice. One day after 2c administration, the mice received 3 × 106 Balb/c-nude BM cells (“HTC only”) or BM+5 × 106 (CB6)F1 Tcm (“HTC+Tcm”). (A-C) Representative IVIS images depicting the difference in 2c-Luc+ cell distribution within the body of untreated (A, “HTC only”) versus treated (C, “HTC and Tcm”) mice on day 10 after transplantation. Color intensities were adjusted according to signal for each individual image. (B-D) On day 9 after transplantation, organs (LN, spleen, BM, lung, and liver) were harvested from 5 animals from each group and numbers of live (7aad negative, viability staining solution) CD8+ 2c cells were assessed by FACS analysis. (B) Distribution of 2c cells in the recipients' organs in the absence of Tcm treatment. (D) Distribution of 2c cells in the recipients' organs in the presence of Tcm treatment. Data represent mean ± SD of total number of 2c cells of 5 animals in each group (*P < .05; **P < .01; ***P < .001).
To better quantify the differences between Tcm recipients and Tcm untreated mice, organs were harvested from 5 animals in each group on day 9, and the number of 2c-Luc+ HTC in the organs, was analyzed by FACS. Figure 4B shows that the total number of 2c-Luc+ cells, harvested from all organs tested, is indeed significantly reduced in the Tcm-treated mice. Moreover, the different compartmentalization pattern of the 2c-Luc+ HTCs caused by the different treatments was verified by FACS, as well. In accordance with IVIS results, in the Tcm-treated group the 2c-Luc+ HTCs localized mainly to the LNs (Figure 5D), in contrast to mice not treated with Tcm, in which only a minority of the 2c-Luc+ HTCs were found in the LNs, and most of the cells localized to the BM, liver, and lungs (Figure 5B). Hence, the Tcms, not only significantly reduced the total number of host anti-donor T cells in the mice, but also affected their migration pattern, by causing accumulation of many of these rejecting HTCs in the LNs. This skewed host anti-donor T-cell distribution is most probably related to H-2Ld presentation by the Tcms at the LNs, as T cells are known to accumulate at sites presenting their cognate antigen.21,23–26
Anti-donor HTCs display decelerated and confined movement at the LNs after antigen-specific interactions with Tcms
To further understand the interactions of the Tcms in vivo, we used 2-photon microscopy, which permitted us to follow interactions between 2c anti-donor HTCs and Tcms in the LNs of live anesthetized animals. To that end, stained 2c CD8+ HTCs and CB6 Tcms (“specific” Tcms, recognized specifically by the 2c TCR transgene) were adoptively transferred at the indicated levels into lethally (10Gy TBI) and sublethally (6.5Gy TBI) irradiated C57BL/6 mice, 4 to 16 hours before imaging (as C57BL/6 mice are more resistant to TBI compared with Balb/c mice, we chose a radiation dose of 6.5Gy, which according to our unpublished results is equivalent to 5.5 Gy TBI in the Balb/c setting). As can be seen in Figure 6A and B, conjugates were formed between the 2c HTCs and the Tcms inside the LNs (see also supplemental Video 1 Lethal conjugates and supplemental Video 2 Sublethal conjugates).
Figure 6.

Tcm conjugation with antigen-specific HTC leads to decelerated and confined HTC movement in the LNs. Lethally (10Gy) and sublethally (6.5Gy) irradiated C57BL/6 mice received 0.5 to 2 × 106 2c CD8+ cells (dyed with SNARF; red for lethal irradiation and magenta for sublethal irradiation [RIC]), and 1.5 to 10 × 106 CB6 (“specific”) or C57BL/6 (“nonspecific”) derived Tcms (green, each dyed with CMTMR). Videos, using 2-photon microscopy were recorded at the indicated time points (see supplemental Methods). (A-B) 2c-CD8+-Tcm conjugates detected 4 to 16 hours after injection of 2 × 106 2c CD8+ and 10 × 106 CB6 cells (arrows mark conjugates). (Picture presented is a single frame from supplemental Videos 1 and 2). A video was recorded 16 hours after injection of 0.5 × 106 2c CD8+, and 1.5 × 106 CB6 or C57BL/6 cells. (C-D) Dot plot depicting velocity of individual cells at the LN (2c HTCs in the presence of “nonspecific” or “specific” Tcms). (E-F) Dot plot depicting displacement rate of individual 2c HTCs at the LN (in the presence of “nonspecific” or “specific” Tcms). Red horizontal bars indicate mean. (G-J) 3-D paths of tracked cells normalized to their starting coordinates in the presence of “nonspecific” (G-I), or “specific” (H-J) Tcms. Axes represent a distance of ± 100 μm for lethal irradiation and ± 30 μm for sublethal irradiation (RIC). Results presented represent 1 of 3 independent experiments (***P < .001).
In contrast, C57BL/6 Tcm (“nonspecific” Tcms), which do not express the H-2Ld molecule, did not form conjugates with the 2c cells, under either lethal (supplemental Video 3) or sublethal (supplemental Video 4) conditioning. Furthermore, movement of 2c-HTC was different in the presence of their cognate antigen (ie, in the presence of “specific” CB6 Tcms, supplemental Videos 5 and 6 showing results in mice conditioned with lethal and sublethal TBI, respectively) compared with that found in the presence of “nonspecific” Tcms. Thus, in both lethally and sublethally irradiated mice, 2c-HTC migrating in the presence of their cognate antigens, presented by the “specific” Tcms, displayed considerably reduced velocity (Figure 6C-D), moved a significantly shorter average distance with respect to their point of origin (Figure 6E-F), and their movement was confined to a more compact area within the LN compared with their migration in LNs containing “nonspecific” Tcms (Figure 6G-J). Taken together, these results show that on antigen recognition, the 2c-HTC form conjugates with donor Tcms and display decelerated motion, and a confined migratory pattern. These findings are consistent with numerous studies that have demonstrated T-cell deceleration and restricted movement in response to antigen recognition in vivo.24,27–29 Moreover, similar deceleration and confined movement of the 2c-HTC in the presence of their cognate antigen (presented by the CB6 Tcm) was observed when 2-photon microscopy was carried out 9 days after transplant (data not shown). These results correlate with our in vivo imaging results, which demonstrate the accumulation of 2c-HTC in the LNs of Tcm-treated mice, on days 7 to 10 after transplant.
Discussion
The marked level of host T cells surviving RIC protocols and capable of mounting fierce rejection of donor cells, represents a major challenge preventing the application of “GVHD-free” purified CD34+ HSCT under safer nonmyeloablative protocols in patients with nonmalignant disorders. In this study, we demonstrate a new approach for overcoming this barrier using T cell–depleted BM supplemented with anti–third-party CD8+ T cells, generated ex vivo from naive cells to attain a central memory phenotype (Tcm). This donor CD8+ T-cell preparation was shown to be free of GVH reactivity by virtue of stimulation against a third-party under cytokine starvation, enabling selective survival of anti–third-party T-cell clones and subsequent dilution and death of anti-host clones not stimulated under these culture conditions.9,17
Our results clearly demonstrate that allogeneic Tcm support engraftment of TDBM allografts under a nonmyeloablative sublethal irradiation dose of 5.5Gy, without causing GVHD. Importantly and in line with the definition of veto activity, chimerism enhancement by Tcms was found to be MHC specific, in that no chimerism could be detected in mice that received BM megadose from C57BL/6 nude donors with host (Balb/c)–derived or C3H-derived Tcms which do not express donor BM antigens. These results, obtained as proof of concept under a rigorous fully mismatched allogeneic model, could probably be translated into partial and completely matched settings, more common in clinical BMT for nonmalignant disorders. Importantly, the TDBMT was also engrafted successfully when F1-derived Tcm were used, demonstrating that the enhanced engraftment was not solely mediated by residual alloreactivity of the allogeneic Tcms. However, it should be noted that allogeneic Tcms induced a higher degree of chimerism in a larger proportion of the treated mice. This alloreactive effect, which is not associated with any manifestations of GVHD, probably results from a minute population of residual alloreactive T cells, which are only able to mediate a form of subclinical alloreactivity, below the threshold for GVHD induction. Nonetheless, considering that human patients may be more prone to GVHD than inbred mice (which are maintained under sterile conditions from birth), clinical translation of this approach must be pursued with caution. Additional allo-depleting steps, such as photodepletion30 or selection of activated cells31 at the end of the anti–third-party allo-stimulation period, might be required to further reduce the risk of GVHD. Our results using F1 Tcms, confirm that such depletion of alloreactivity does not abolish the ability of allogeneic Tcms to induce tolerance.
To further reduce the toxic TBI level, we evaluated the use of rapamycin, which was previously shown to synergize with veto activity of anti–third-party CD8+ T cells, bearing an effector phenotype.9 Initially, we demonstrated that rapamycin abrogates expansion of anti-donor HTCs without affecting the expansion of ex vivo induced Tcms. These results are in line with those of previous studies that showed rapamycin's inability to control the expansion of “natural” memory cells.32,33 This well-known resistance of memory cells to various immunosuppressive drugs32,34 is attributed to their higher activation state, requiring lower threshold for activation. Thus, immunosuppressive drugs, such as rapamycin, which interfere with activation signals of immune cells, exert very mild suppression on previously activated memory cells.35–38 Subsequently, we were able to show that combined treatment using short-term low-dose rapamycin and Tcm therapy (both allogeneic and F1-derived) supports TDBMT engraftment under mild 4.5Gy TBI conditioning.
In vivo events, occurring during tolerance induction by anti–third-party Tcms, were followed in a model specifically designed to track relevant luciferase-expressing host anti-donor TCR transgenic T cells by bioluminescence imaging. The use of such transgenic cells allowed us to specifically monitor the population of host T cells, which mediates the rejection of the graft. Notably, the use of high affinity TCR transgenic cells in this model may limit the interpretation of our results by not accounting for the role of lower affinity clones in graft rejection. Nevertheless, this novel system allows us for the first time to follow BMT graft rejection noninvasively by specifically monitoring the cells that mediate BMT rejection. Importantly, in preliminary experiments, the number of 2c cells (700 cells) was adjusted so as to induce BM allograft rejection at around the same time frame found on infusion of a much higher number of WT CD8 T cells (3 × 104). Considering that alloreactive CD8 T cells probably represent around 1% to 5% of the total naive CD8 T cells, we expect that, in this model, rejection is mediated by 300 to 1500 cells, which is in the same range of the 2c cell number used. Interestingly, a similar range was suggested by Obar et al, who quantitated naive antigen-specific CD8+ T cells and the early response to infection, and found that among 6 different specificities measured, the number of naive antigen-specific precursors ranged from ∼ 80 to 1200 cells/mouse.39 Thus, the number of 2c TCR Tg cells used is probably within the physiologic range. Moreover, further dilution in this model of the 2c cells was associated with reduced BM allograft rejection (data not shown).
This system has enabled us to study the effects of Tcm-treatment on the kinetics and compartmentalization of anti-donor HTC dispersion throughout the body of the BMT recipient. Our results show that Tcms not only significantly reduce the number of anti-donor HTCs within the recipients, but also affect the migration pattern of these HTCs by inducing them to accumulate in the LNs; this effect further reduces the number of HTCs in all other organs, including the BM, critical for prevention of graft rejection. The probable explanation for this skewed distribution with preferential localization to the LNs is perhaps related to the known capacity of T cells to accumulate at sites presenting their cognate antigen.21,23–26,40 As Tcms home and proliferate in the LNs of the BMT recipient17,40–42 it is probable that the Tcm population within the LNs is large enough to cause accumulation of naive HTCs. Thus, we hypothesize that although the naive HTCs reach the LNs as part of their normal immuno-surveillance circulation,40–42 they accumulate there as a result of specific interactions formed with their cognate MHC antigen, presented by donor Tcms. Other studies also indicated that after T-cell entrance into their target tissue, they are retained and accumulate at the antigen site only on antigenic recognition.25,43,44 Notably, and in contrast to other target cells, Tcms are not rejected by anti-donor T cells, but rather induce apoptosis of the latter, as was previously demonstrated.17 Thus, Tcms can persist in the LNs where they can present their antigens, and “trap” HTCs, for extended periods of time. Importantly, we now show for the first time, using 2-photon microscopy, the ability of Tcm to form stable conjugates with host anti-donor naive HTCs, within the intact LNs of anesthetized animals. Furthermore, when cognate target Tcms are present in the LN, HTCs move in more confined areas and with diminished velocity compared with their movement in the presence of “nonspecific” Tcms, which do not display the cognate antigen. Such changes in T-cell migratory pattern, in response to antigen presentation in vivo, have been documented in several studies,24,27–29 and were shown to result from increased levels of adhesion molecules and cytoskeletal changes, induced by TCR signaling.45,46 Taken together, these findings suggest that the positive tolerizing effect of Tcms may occur in 2 stages; initially, the Tcms mediate accumulation of anti-donor HTCs in the LNs, thereby reducing HTC numbers in other organs including the most relevant site, the bone marrow. Subsequently, the Tcms eliminate these HTCs in the LNs through a specific veto-based mechanism, typical of activated CD8 T cells.11,13,17 In addition, the 2 steps involved in the suggested mechanism may partially overlap, because the induction of apoptosis by veto activity of the Tcms in the LNs may by itself, contribute to the prevention of LN egress of the anti-donor host cells. In conclusion, administration of anti–third-party Tcms enables TDBMT graft rejection to be overcome, in the absence of GVHD, by skewing the distribution of anti-donor HTCs and inducing their elimination in the LNs by induction apoptotic cell death, as previously described.
This tolerizing activity, probably translates into the powerful ability of the Tcms to induce tolerance to TDBMT under nonmyeloablative, reduced intensity conditioning regimens. Importantly, the immune tolerance induced by the Tcms was indeed successfully translated into acceptance of donor skin grafts, whereas third-party skin was promptly rejected. This selective acceptance of donor skin graft, further demonstrates the specific nature of the tolerance achieved, which is directed only toward the donor antigens, without inducing a general immunosuppressive effect.
Taken together, our results strongly suggest that anti–third-party Tcms, combined with “megadose” TDBMT, offer an attractive and safe modality for achieving hematopoietic chimerism under mild conditioning and without any risk for GVHD. This new approach could offer safe treatment for nonmalignant hematopoietic diseases, such as sickle cell anemia and thalassemia, and could serve as a platform for induction of durable immune tolerance in organ transplantation or for continuous cell therapy with donor cells in cancer patients.
Acknowledgments
This work was partially supported by the European Union 7th Framework Program as part of the project NanoII, grant agreement No. 229289, and by research grants from Mrs E. Drake and from Roberto and Renata Ruhman.
Footnotes
There is an Inside Blood commentary on this article in this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: E.O., N.O.-G., and Y.R. designed the research and wrote the paper; E.O., N.O.-G., I.G., O.T., Y.E., E.S., R.M., A.L., D.H., E.B.-L., and S.R.-Z. performed the research; E.O., N.O.-G., I.G., O.T., E.S., and Y.R. analyzed the data; G.S. provided expertise on 2-photon microscopy; and A.B. and R.N. contributed mice.
Conflict-of-interest disclosure: Y.R. serves as a consultant to Cell Source Ltd. The remaining authors declare no competing financial interests.
Correspondence: Yair Reisner, Dept of Immunology, Weizmann Institute of Science, 234 Herzl Street, Rehovot, Israel 7600; e-mail: yair.reisner@weizmann.ac.il.
References
- 1.Reisner Y, Hagin D, Martelli MF. Haploidentical hematopoietic transplantation: current status and future perspectives. Blood. 2011;118(23):6006–6017. doi: 10.1182/blood-2011-07-338822. [DOI] [PubMed] [Google Scholar]
- 2.Ophir E, Reisner Y. Induction of tolerance in organ recipients by hematopoietic stem cell transplantation. Int Immunopharmacol. 2009;9(6):694–700. doi: 10.1016/j.intimp.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 3.Sykes M, Sachs DH. Mixed allogeneic chimerism as an approach to transplantation tolerance. Immunol Today. 1988;9(1):23–27. doi: 10.1016/0167-5699(88)91352-7. [DOI] [PubMed] [Google Scholar]
- 4.Tomita Y, Khan A, Sykes M. Role of intrathymic clonal deletion and peripheral anergy in transplantation tolerance induced by bone marrow transplantation in mice conditioned with a nonmyeloablative regimen. J Immunol. 1994;153(3):1087–1098. [PubMed] [Google Scholar]
- 5.Gur H, Krauthgamer R, Berrebi A, et al. Tolerance induction by megadose hematopoietic progenitor cells: expansion of veto cells by short-term culture of purified human CD34(+) cells. Blood. 2002;99(11):4174–4181. doi: 10.1182/blood.v99.11.4174. [DOI] [PubMed] [Google Scholar]
- 6.Zangi L, Klionsky YZ, Yarimi L, et al. Deletion of cognate CD8 T cells by immature dendritic cells: a novel role for perforin, granzyme A, TREM-1, and TLR7. Blood. 2012;120(8):1647–1657. doi: 10.1182/blood-2012-02-410803. [DOI] [PubMed] [Google Scholar]
- 7.Rachamim N, Gan J, Segall H, et al. Tolerance induction by “megadose” hematopoietic transplants: donor-type human CD34 stem cells induce potent specific reduction of host anti-donor cytotoxic T lymphocyte precursors in mixed lymphocyte culture. Transplantation. 1998;65(10):1386–1393. doi: 10.1097/00007890-199805270-00017. [DOI] [PubMed] [Google Scholar]
- 8.Reich-Zeliger S, Bachar-Lustig E, Gan J, Reisner Y. Tolerance induction by veto CTLs in the TCR transgenic 2c mouse model I: relative reactivity of different veto cells. J Immunol. 2004;173(11):6654–6659. doi: 10.4049/jimmunol.173.11.6654. [DOI] [PubMed] [Google Scholar]
- 9.Bachar-Lustig E, Reich-Zeliger S, Reisner Y. Anti-third-party veto CTLs overcome rejection of hematopoietic allografts: synergism with rapamycin and BM cell dose. Blood. 2003;102(6):1943–1950. doi: 10.1182/blood-2003-03-0759. [DOI] [PubMed] [Google Scholar]
- 10.George JF, Thomas JM. The molecular mechanisms of veto mediated regulation of alloresponsiveness. J Mol Med. 1999;77(7):519–526. doi: 10.1007/s001099900027. [DOI] [PubMed] [Google Scholar]
- 11.Reich-Zeliger S, Zhao Y, Krauthgamer R, Bachar-Lustig E, Reisner Y. Anti-third-party CD8+ CTLs as potent veto cells: coexpression of CD8 and FasL is a prerequisite. Immunity. 2000;13(4):507–515. doi: 10.1016/s1074-7613(00)00050-9. [DOI] [PubMed] [Google Scholar]
- 12.Reich-Zeliger S, Gan J, Bachar-Lustig E, Reisner Y. Tolerance induction by veto CTLs in the TCR transgenic 2c mouse model II: deletion of effector cells by Fas-Fas ligand apoptosis. J Immunol. 2004;173(11):6660–6666. doi: 10.4049/jimmunol.173.11.6660. [DOI] [PubMed] [Google Scholar]
- 13.Milstein O, Hagin D, Lask A, et al. CTLs respond with activation and granule secretion when serving target for T cell recognition. Blood. 2011;117(3):1042–1052. doi: 10.1182/blood-2010-05-283770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anderson KM, Zimring JC. Rapamycin prolongs susceptibility of responding T cells to tolerance induction by CD8+ veto cells. Transplantation. 2006;81(1):88–94. doi: 10.1097/01.tp.0000185302.38890.6b. [DOI] [PubMed] [Google Scholar]
- 15.Carrio R, Bathe OF, Malek TR. Initial antigen encounter programs CD8+ T cells competent to develop into memory cells that are activated in an antigen-free, IL-7 and IL-15-rich environment. J Immunol. 2004;172(12):7315–7323. doi: 10.4049/jimmunol.172.12.7315. [DOI] [PubMed] [Google Scholar]
- 16.Manjunath N, Shankar P, Wan J, et al. Effector differentiation is not prerequisite for generation of memory cytotoxic T lymphocytes. J Clin Invest. 2001;108(6):871–878. doi: 10.1172/JCI13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ophir E, Eidelstein Y, Afik R, Bachar-Lustig E, Reisner Y. Induction of tolerance to bone marrow allografts by donor-derived host nonreactive ex vivo-induced central memory CD8 T cells. Blood. 2010;115(10):2095–2104. doi: 10.1182/blood-2009-10-248716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayumi H, Good RA. Long-lasting skin allograft tolerance in adult mice induced across fully allogeneic (multimajor H-2 plus multiminor histocompatibility) antigen barriers by a tolerance-inducing method using cyclophosphamide. J Exp Med. 1989;169(1):213–238. doi: 10.1084/jem.169.1.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ikehara S. New strategies for allogeneic bone marrow transplantation and organ allografts. Acta Haematol. 1999;101(2):68–77. doi: 10.1159/000040927. [DOI] [PubMed] [Google Scholar]
- 20.Pree I, Wekerle T. Inducing mixed chimerism and transplantation tolerance through allogeneic bone marrow transplantation with costimulation blockade. Methods Mol Biol. 2007;380:391–403. doi: 10.1007/978-1-59745-395-0_25. [DOI] [PubMed] [Google Scholar]
- 21.Sirinek LP, O'Dorisio MS, Dunaway DJ. Accumulation of donor-specific cytotoxic T cells in intestinal lymphoid tissues following intestinal transplantation. J Clin Immunol. 1995;15(5):258–265. doi: 10.1007/BF01540883. [DOI] [PubMed] [Google Scholar]
- 22.Wekerle T, Sykes M. Mixed chimerism and transplantation tolerance. Annu Rev Med. 2001;52:353–370. doi: 10.1146/annurev.med.52.1.353. [DOI] [PubMed] [Google Scholar]
- 23.Honjo K, Xu XY, Kapp JA, Bucy RP. Activation and migration of allo-peptide specific TCR transgenic T cells in cardiac allograft rejection. Cell Immunol. 2004;230(1):44–55. doi: 10.1016/j.cellimm.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 24.Stoll S, Delon J, Brotz TM, Germain RN. Dynamic imaging of T cell-dendritic cell interactions in lymph nodes. Science. 2002;296(5574):1873–1876. doi: 10.1126/science.1071065. [DOI] [PubMed] [Google Scholar]
- 25.Reinhardt RL, Bullard DC, Weaver CT, Jenkins MK. Preferential accumulation of antigen-specific effectorCD4 t cells at an antigen injection site involves CD62E-dependent migration but not local proliferation. J Exp Med. 2003;197(6):751–762. doi: 10.1084/jem.20021690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhan, Reinisch CL, Levey RH, McCluskey RT. T-cell migration into allografts. J Exp Med. 1975;141(5):1210–1215. doi: 10.1084/jem.141.5.1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang Q, Adams JY, Tooley AJ, et al. Visualizing regulatory T-cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol. 2006;7(1):83–92. doi: 10.1038/ni1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skokos D, Shakhar G, Varma R, et al. Peptide-MHC potency governs dynamic interactions between T cells and dendritic cells in lymph nodes. Nat Immunol. 2007;8(8):835–844. doi: 10.1038/ni1490. [DOI] [PubMed] [Google Scholar]
- 29.Kawakami N, Nägerl UV, Odoardi F, Bonhoeffer T, Wekerle H, Flügel A. Live imaging of effector cell trafficking and autoantigen recognition within the unfolding autoimmune encephalomyelitis lesion. J Exp Med. 2005;201(11):1805–1814. doi: 10.1084/jem.20050011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perruccio K, Topini F, Tosti A, et al. Photodynamic purging of alloreactive T cells for adoptive immunotherapy after haploidentical stem cell transplantation. Blood Cells Mol Dis. 2008;40(1):76–83. doi: 10.1016/j.bcmd.2007.06.022. [DOI] [PubMed] [Google Scholar]
- 31.Barrett AJ, Rezvani K, Solomon S, et al. New developments in allotransplant immunology. Hematology Am Soc Hematol Educ Program. 2003:350–371. doi: 10.1182/asheducation-2003.1.350. [DOI] [PubMed] [Google Scholar]
- 32.Kim JS, Lee JI, Shin JY, et al. Bortezomib can suppress activation of rapamycin-resistant memory T cells without affecting regulatory T-cell viability in non-human primates. Transplantation. 2009;88(12):1349–1359. doi: 10.1097/TP.0b013e3181bd7b3a. [DOI] [PubMed] [Google Scholar]
- 33.Adams AB, Williams MA, Jones TR, et al. Heterologous immunity provides a potent barrier to transplantation tolerance. J Clin Invest. 2003;111(12):1887–1895. doi: 10.1172/JCI17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heeger PS, Greenspan NS, Kuhlenschmidt S, et al. Pretransplant frequency of donor-specific, IFN-gamma-producing lymphocytes is a manifestation of immunologic memory and correlates with the risk of posttransplant rejection episodes. J Immunol. 1999;163(4):2267–2275. [PubMed] [Google Scholar]
- 35.Lanzavecchia A, Sallusto F. Dynamics of T lymphocyte responses: intermediates, effectors, and memory cells. Science. 2000;290(5489):92–97. doi: 10.1126/science.290.5489.92. [DOI] [PubMed] [Google Scholar]
- 36.Zeng H, Chen Y, Yu M, et al. T Cell receptor-mediated activation of CD4 CD44hi T cells bypasses Bcl10: An Implication of differential NF-κB dependence of naive and memory T cells during T cell receptor-mediated responses. J Biol Chem. 2008;283(36):24392–24399. doi: 10.1074/jbc.M802344200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fontenot AP, Gharavi L, Bennett SR, Canavera SJ, Newman LS, Kotzin BL. CD28 costimulation independence of target organ versus circulating memory antigen-specific CD4 T cells. J Clin Invest. 2003;112(5):776–784. doi: 10.1172/JCI18317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.London CA, Lodge MP, Abbas AK. Functional responses and costimulator dependence of memory CD4+ T cells. J Immunol. 2000;164(1):265–272. doi: 10.4049/jimmunol.164.1.265. [DOI] [PubMed] [Google Scholar]
- 39.Obar JJ, Khanna KM, Lefrancois L. Endogenous naive CD8+ T cell precursor frequency regulates primary and memory responses to infection. Immunity. 2008;28(6):859–869. doi: 10.1016/j.immuni.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weninger W, Manjunath N, von Andrian UH. Migration and differentiation of CD8+ T cells. Immunol Rev. 2002;186:221–233. doi: 10.1034/j.1600-065x.2002.18618.x. [DOI] [PubMed] [Google Scholar]
- 41.Weninger W, Crowley MA, Manjunath N, von Andrian UH. Migratory Properties of naive, effector, and memory CD8 T Cells. J Exp Med. 2001;194(7):953–966. doi: 10.1084/jem.194.7.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mora JR, von Andrian UH. T-cell homing specificity and plasticity: new concepts and future challenges. Trends Immunol. 2006;27(5):235–243. doi: 10.1016/j.it.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 43.Zeine R, Owens T. Direct demonstration of the infiltration of murine central nervous system by Pgp-1/CD44high CD45RB (low) CD4 T cells that induce experimental allergic encephalomyelitis. J Neuroimmunol. 1992;40(1):57–69. doi: 10.1016/0165-5728(92)90213-5. [DOI] [PubMed] [Google Scholar]
- 44.Hogan RJ, Usherwood EJ, Zhong W, et al. Activated antigen-specific CD8+ T cells persist in the lungs following recovery from respiratory virus infections. J Immunol. 2001;166(3):1813–1822. doi: 10.4049/jimmunol.166.3.1813. [DOI] [PubMed] [Google Scholar]
- 45.Dustin ML, Bromley SK, Kan Z, Peterson DA, Unanue ER. Antigen receptor engagement delivers a stop signal to migrating T lymphocytes. Proc Natl Acad Sci U S A. 1997;94(8):3909–3913. doi: 10.1073/pnas.94.8.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cernuda-Morollón E, Millán J, Shipman M, Marelli-Berg FM, Ridley AJ. Rac activation by the T-cell receptor inhibits T-cell migration. PLoS One. 2010;5(8):e12393. doi: 10.1371/journal.pone.0012393. [DOI] [PMC free article] [PubMed] [Google Scholar]