

Abstract
OTS167 is a potent maternal embryonic leucine zipper kinase inhibitor undergoing clinical testing as antineoplastic agent. We aimed to identify the UDP-glucuronosyltransferases (UGTs) involved in OTS167 metabolism, study the relationship between UGT genetic polymorphisms and hepatic OTS167 glucuronidation, and investigate the inhibitory potential of OTS167 on UGTs. Formation of a single OTS167-glucuronide (OTS167-G) was observed in pooled human liver (HLM) (Km = 3.4 ± 0.2 µM), intestinal microsomes (HIM) (Km = 1.7 ± 0.1 µM), and UGTs. UGT1A1 (64 µl/min/mg) and UGT1A8 (72 µl/min/mg) exhibited the highest intrinsic clearances (CLint) for OTS167, followed by UGT1A3 (51 µl/min/mg) and UGT1A10 (47 µl/min/mg); UGT1A9 was a minor contributor. OTS167 glucuronidation in HLM was highly correlated with thyroxine glucuronidation (r = 0.91, P < 0.0001), SN-38 glucuronidation (r = 0.79, P < 0.0001), and UGT1A1 mRNA (r = 0.72, P < 0.0001). Nilotinib (UGT1A1 inhibitor) and emodin (UGT1A8 and UGT1A10 inhibitor) exhibited the highest inhibitory effects on OTS167-G formation in HLM (68%) and HIM (47%). We hypothesize that OTS167-G is an N-glucuronide according to mass spectrometry. A significant association was found between rs6706232 and reduced OTS167-G formation (P = 0.03). No or weak UGT inhibition (range: 0–21%) was observed using clinically relevant OTS167 concentrations (0.4–2 µM). We conclude that UGT1A1 and UGT1A3 are the main UGTs responsible for hepatic formation of OTS167-G. Intestinal UGT1A1, UGT1A8, and UGT1A10 may contribute to first-pass OTS167 metabolism after oral administration.
Introduction
OTS167 is a novel synthetic molecule undergoing clinical development as anticancer agent. It has been found to be a very potent inhibitor (IC50 of 0.41 nM) (Chung et al., 2012) of maternal embryonic leucine zipper kinase, a protein kinase aberrantly upregulated in many types of cancer and essential for survival and proliferation of undifferentiated cancer cells (Ganguly et al., 2014).
Glucuronidation is a common conjugation reaction catalyzed by UDP-glucuronosyltransferase (UGT) enzymes. The reaction involves the transfer of glucuronic acid from UDP-glucuronic acid (UDPGA) to an acceptor substrate containing hydroxyl, carboxyl, amino, or sulfhydryl groups to generate more polar and excretable compounds (Tukey and Strassburg, 2000). The human UGTs are classified into families UGT1 and UGT2 and subdivided into UGT1A, UGT2A, and UGT2B subfamilies based on gene structure and sequence homology (Guillemette, 2003). The UGT1A enzymes are generated by alternative splicing of unique exon 1 to the common exons 2–5, whereas the UGT2Bs are individually transcribed. There are nine UGT1As (UGT1A1, UGT1A3–UGT1A10), all of which are expressed in the gastrointestinal tract. UGT1A1, UGT1A3, UGT1A4, UGT1A5, UGT1A6, and UGT1A9 are expressed in the liver, the major site of glucuronidation, whereas UGT1A7, UGT1A8, and UGT1A10 are extrahepatic (Tukey and Strassburg, 2000, 2001; Finel et al., 2005). All seven UGT2B enzymes (UGT2B4, UGT2B7, UGT2B10, UGT2B11, UGT2B15, UGT2B17, and UGT2B28) are found in the liver (Tukey and Strassburg, 2000, 2001; Lévesque et al., 2001). Many polymorphisms exist in the UGT genes (http://www.pharmacogenomics.pha.ulaval.ca/cms/ugt_alleles/; accessed Nov. 26, 2014). UGT genetic variation could have a significant impact on metabolism of endogenous compounds such as bilirubin (Bosma, 2003), and on variability in response to drugs such as irinotecan among others (Ramírez et al., 2010).
The metabolism of OTS167 has not been previously described. The presence of phenolic and nitrogen-containing functional groups in its chemical structure (Fig. 1) suggested that the drug may undergo glucuronidation. In this study, we aimed to (1) evaluate whether OTS167 is glucuronidated in vitro by human liver microsomes (HLM), human intestinal microsomes (HIM), and UGTs; (2) investigate the relationship between UGT polymorphisms and OTS167 glucuronidation in human livers; and (3) evaluate the potential inhibitory effects of OTS167 on glucuronidation reactions.
Fig. 1.
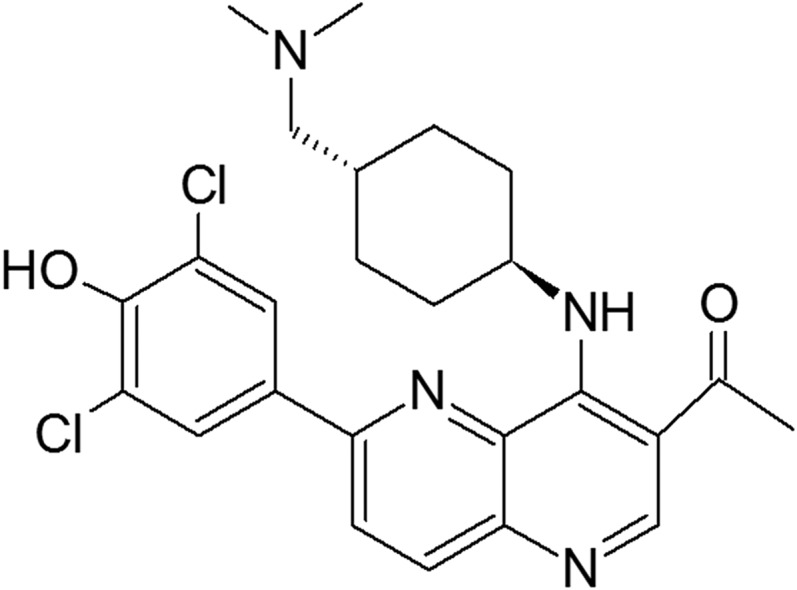
Chemical structure of OTS167.
Materials and Methods
Chemicals and Reagents
OTS167 was provided by OncoTherapy Science (Kawasaki City, Kanagawa, Japan). Coumarin, 4-methylumbelliferone (4-MU), 4-methylumbelliferyl-β-D-glucuronide hydrate (4-MU-G), diclofenac sodium salt, sulfinpyrazone, imipramine, hecogenin, niflumic acid, emodin, potassium phosphate monobasic, β-glucuronidase (type IX-A, from Escherichia coli), and dimethylsulfoxide (DMSO) were obtained from Sigma-Aldrich (St. Louis, MO). Imipramine N-β-D glucuronide (imipramine-G) and nilotinib were purchased from Toronto Research Chemicals (North York, Ontario, Canada). UGT Reaction Mix Solution A (25 mM UDPGA) and Solution B (250 mM Tris-HCl, 40 mM MgCl2, and 0.125 mg/ml alamethicin) were acquired from BD Biosciences (Bedford, MA). Acetonitrile, methanol, formic acid, glacial acetic acid, ammonium acetate, 7-hydroxycoumarin, and sodium phosphate monobasic and dibasic were purchased from Thermo Fisher Scientific (Hanover Park, IL).
Microsomal Preparations
Normal human livers were obtained with human subjects’ approval through the Liver Tissue Cell Distribution System (Pittsburgh, PA; National Institutes of Health Contract HHSN276201200017C) and the Cooperative Human Tissue Network. HLM were prepared using differential centrifugation methods (Ramírez et al., 2007). Pooled HIM and microsomes from cDNA-transfected baculovirus-insect cells (Supersomes) expressing UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B4, UGT2B7, UGT2B10, UGT2B15, and UGT2B17 as well as UGT control microsomes were obtained from BD Gentest (Woburn, MA).
OTS167 Glucuronidation Assay
Experiments were conducted to study the linearity of OTS167 glucuronide (OTS167-G) formation with respect to time and protein using pooled HLM and HIM. Initially, OTS167 (10 µM in methanol) was combined with UGT Reaction Mix Solution B (50 mM Tris-HCl, 8 mM MgCl2, and 0.025 mg/ml alamethicin) and 1 mg/ml pooled HLM or HIM on ice for 15 minutes. The concentration of methanol in the reaction was kept at 1% (v/v). After equilibration for 5 minutes in a 37°C water bath, reactions were initiated by addition of UGT Reaction Mix Solution A (2 mM UDPGA). The final reaction volume was 100 µl. Samples were incubated for various times (range: 10–120 minutes, 8 time points). Reactions were terminated with 100 µl cold acetonitrile, followed by addition of 10 μl internal standard (200 μM coumarin in acetonitrile). Samples were vortexed for 10 seconds and centrifuged at 20,817g for 15 minutes at 4°C. Aliquots (20–40 µl) of supernatant were injected into a high-performance liquid chromatography (HPLC) system (Hitachi High Technologies America, Schaumburg, IL). For determining optimal protein concentration, samples were incubated for 30 or 35 minutes (optimum time for HLM and HIM, respectively) with 10 protein concentrations (range: 0.1–1 mg/ml). Final incubation conditions obtained for subsequent experiments with HLM and HIM were 0.5 mg/ml protein (optimum for both HLM and HIM) and an incubation time of 30 or 35 minutes (for HLM and HIM, respectively). Control incubations were simultaneously performed in the absence of OTS167, UDPGA, HLM, and HIM.
Incubations with β-Glucuronidase
Enzymatic hydrolysis using β-glucuronidase was used to confirm OTS167-G formation by HLM and HIM. OTS167 incubations were dried down under nitrogen gas and reconstituted in 2000 U β-glucuronidase (dissolved in 200 µl 0.1 M sodium phosphate buffer). After 24-hour incubation at 37°C, 400 µl ice-cold methanol was added. Samples were dried under nitrogen gas and reconstituted with a 50/50 (v/v) mix of 2 mM ammonium acetate containing 0.1% formic acid (mobile phase A) and acetonitrile with 0.1% formic acid (mobile phase B) prior to injection into the HPLC. Control incubations were performed simultaneously with 0.1 M sodium phosphate buffer only (no β-glucuronidase added).
Measurement of OTS167 Glucuronidation
HPLC separation was achieved at room temperature using an Atlantis dC18 column (5 μM, 3.0 × 250 mm; Waters, Milford, MA) preceded by an Atlantis dC18 guard column (5 μM, 3.9 × 20 mm). The following mobile phase gradient was applied at a flow rate of 1 mL/min: 0–2 minutes, 90% A and 10% B; 2.1–17 minutes, switch from 90% A and 10% B to 65% A and 35% B; 17.1–20 minutes, hold at 65% A and 35% B; and 20.1–25 minutes, hold at 90% A and 10% B. The eluate was monitored for 25 minutes using UV detection at 305 nm. Retention times for OTS167, OTS167-G, and coumarin were 15.2, 12.7, and 17.7 minutes, respectively.
As OTS167-G was not available, its relative formation was estimated using a standard curve generated with OTS167. Standards were made by serial dilutions of OTS167 in methanol and further diluted 100× in deionized water. To be consistent with the microsomal incubations, standards (100 µl) were maintained at 37°C for 30 minutes (incubations with UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B4, UGT2B7, UGT2B10, UGT2B15, UGT2B17, and HLM) or 35 minutes (incubations with HIM, UGT1A7, UGT1A8, and UGT1A10), and processed, as described above. Calibration curves were linear over the concentration range of 0.31–10 µM (r2 > 0.99).
OTS167 Glucuronidation by Expressed Human UGT Enzymes
To screen for OTS167 glucuronidation activity, enzymes expressed in the liver (UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B4, UGT2B7, UGT2B10, UGT2B15, and UGT2B17) were incubated with 3.4 µM OTS167 (Km for HLM) for 30 minutes (optimum time for HLM). Extrahepatic UGTs (UGT1A7, UGT1A8, and UGT1A10) were incubated for 35 minutes (optimum time for HIM) with 1.7 µM OTS167 (Km for HIM). Protein concentration for all UGTs was 0.5 mg total protein/ml.
Enzyme Kinetics in HLM, HIM, and UGTs
Kinetic experiments with HLM, HIM, and UGTs were performed as described above using 11 concentrations of OTS167 (range: 0.5–100 µM).
Correlation Studies in HLM
Incubations with 46 individual HLM (34 male, 12 female) were performed, as described above, and contained 3.4 µM OTS167. Measurements of glucuronidation activities toward thyroxine (UGT1A1 and UGT1A3 substrate) (Tong et al., 2007; Yamanaka et al., 2007; Yoder Graber et al., 2007; Kato et al., 2008), SN-38 (UGT1A1 substrate) (Iyer et al., 1998; Hanioka et al., 2001; Gagné et al., 2002), mycophenolic acid (UGT1A9 substrate) (Bernard and Guillemette, 2004; Miles et al., 2005), and testosterone (UGT2B17 substrate) (Turgeon et al., 2001), and quantitation of mRNA levels (UGT1A1, UGT1A3, UGT1A9, and UGT2B17) in this set of HLM were previously described (Iyer et al., 1999; Ramírez et al., 2007; Yoder Graber et al., 2007; Kang et al., 2010; Liu et al., 2014).
Inhibition of OTS167 Glucuronidation
OTS167 glucuronidation by HLM, HIM, and UGTs (UGT1A1, UGT1A3, UGT1A8, UGT1A9, and UGT1A10) was investigated in the presence of UGT inhibitors. Incubations were performed, as described above, using OTS167 at the respective Km values (HLM, 3.4 µM; HIM, 1.7 µM; UGT1A1, 5.7 µM; UGT1A3, 2.2 µM; UGT1A8, 3.7 µM; UGT1A10, 0.9 µM) except for UGT1A9. Incubations with UGT1A9 were performed at 3.4 µM (Km for HLM) as its Km could not be estimated. Incubations included the following inhibitors: 2.5 μM nilotinib (dissolved in DMSO) for UGT1A1 (Ai et al., 2013), 500 μM imipramine (in deionized water) for UGT1A3 (Lu at al., 2009), 2.5 μM niflumic acid (in DMSO) for UGT1A9 (Miners et al., 2011), and 100 μM emodin (in DMSO) for UGT1A8 and UGT1A10 (Watanabe et al., 2002). Control incubations contained vehicle (no inhibitor added). Total concentration of organic solvents in incubations was 1% (v/v).
Genotyping
UGT polymorphisms significantly associated with UGT1A1 and UGT1A3 phenotypes (Yamamoto et al., 1998; Jinno et al., 2003; Liu et al., 2014) were genotyped, as previously described (Innocenti et al., 2004; Liu et al., 2014). These gene variants were the functional single nucleotide polymorphisms (SNPs) UGT1A1*28 (rs8175347, -53[TA]6 > 7, in the UGT1A1 promoter) and UGT1A1*6 (rs4148323, 211G>A, G71R, in UGT1A1 exon 1), and the tag SNPs (Liu et al., 2014), rs6706232 (UGT1A3 E27E), rs10203853 (in the UGT1A 3′-flanking region), and rs33979061 (in UGT1A1 intron 1).
Inhibition of UGTs by OTS167
4-MU Glucuronidation.
4-MU was used as nonspecific substrate to evaluate the inhibitory potential of OTS167 on the majority of the UGTs (UGT1A1, UGT1A3, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B4, UGT2B7, UGT2B15, and UGT2B17). Incubations contained 4-MU (concentrations from Dong et al., 2012), UGTs (concentrations reported by Liu et al., 2010, except that UGT1A10 was used at 0.25 mg/ml), 2.5 mM UDPGA, 50 mM Tris-HCl (pH 7.5), 8 mM MgCl2, 25 µg/ml alamethicin, and OTS167 (0.4 μM, 2 μM, and 10 μM). Incubation times for UGTs have been previously reported (Dong et al., 2012). Positive controls for inhibition were 500 μM diclofenac (in DMSO) for UGT1A1, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B4, UGT2B7, UGT2B15, and UGT2B17 (Liu et al., 2010), and 1 mM sulfinpyrazone (in DMSO) for UGT1A3 (Uchaipichat et al., 2006). Negative (vehicle) controls contained methanol (OTS167 incubations) or DMSO (incubations with diclofenac and sulfinpyrazone). Organic solvent concentrations were kept at 1% (v/v). Incubations were stopped with 100 μl ice-cold acetonitrile containing 100 μM 7-hydroxycoumarin (internal standard). Samples were centrifuged (20,817 relative centrifugal source, 15 minutes, 4°C) prior to analysis by HPLC (10 μl), as previously reported (Liu et al., 2010). 4-MU-G standards ranged from 1.25 μM to 100 μM.
Imipramine Glucuronidation.
Imipramine was used for investigation of the inhibitory effect of OTS167 on UGT1A4. Incubations were performed, as previously described (Nakajima et al., 2002), with minor modifications. Reactions contained 2.5 mM UDPGA, 50 mM Tris-HCl (pH 7.5), 8 mM MgCl2, 25 µg/ml alamethicin, 500 μM imipramine, 0.5 mg/ml protein, and OTS167 (0.4 μM, 2 μM, and 10 μM), or hecogenin (200 μM in methanol as positive control for inhibition). The negative (vehicle) control contained 1% methanol. Reactions were stopped after 60 minutes with ice-cold acetonitrile containing 100 μM 7-hydroxycoumarin as internal standard and centrifuged at 20,817 relative centrifugal force for 15 minutes (4°C). Supernatants (10 μl) were analyzed by HPLC. The mobile phase consisted of 28/72 (v/v) acetonitrile/10 mM potassium phosphate monobasic (pH 2.6) at a flow rate of 1 ml/min. Separation was achieved with a XTerra RP18 column (4.6 × 100 mm, 5 mm; Waters Corporation, Milford, MA) and a Nova-Pak C18 (4 μm) guard column (Waters Corporation). UV detection (254 nm) was used. Imipramine-G standards ranged from 1.9 μM to 30 μM.
Identification of OTS167-G
To determine the mass and structure for OTS167-G, microsomes (1 mg/ml HLM, 0.5 mg/ml UGT1A1, and 0.5 mg/ml UGT1A8) were incubated with 10 µM OTS167 for 90 minutes, as described above. The reaction was stopped with addition of 100 µL ice-cold acetonitrile, subjected to HPLC, and the metabolite peak (eluting around 13 minutes) was collected for mass spectrometry (MS). The latter was dried down under nitrogen gas, reconstituted in methanol, and subjected to MS using an API 2000 MS/MS triple-quadruple system (Applied Biosystems, Foster City, CA). Additionally, OTS167 was analyzed. OTS167 and its metabolite (from 3 different incubation mixtures: HLM, UGT1A1, and UGT1A8) were infused at 40 µl/min with a syringe pump. The electrospray ionization voltage was set at +5500 V with a temperature of 50°C. Positive ion Q1 monitoring was performed along with product ion monitoring (+MS2) of OTS167-G. The curtain, collision, nebulizing, and heater gases were set at 35, 5, 50, and 70 psi, respectively. The declustering potential, focusing potential, entrance potential, collision energy, and collision cell entrance potential voltages were 90, 400, 7, 50, and 18 V, respectively.
Data Analysis and Statistics.
Formation rates of OTS167-G, 4-MU-G, and imipramine-G were expressed as pmol/min/mg total protein. Results represent the mean of a single experiment performed in triplicate (assay optimization, UGT screening, kinetic studies, and inhibition of OTS167) or duplicates (correlation and inhibition studies by OTS167). Apparent kinetic parameters for HLM, HIM, and UGTs were estimated by fitting OTS167 glucuronidation rates versus OTS167 concentrations by nonlinear regression. The Enzyme Kinetics Module from SigmaPlot 12.3 (Systat Software, San Jose, CA) was used to analyze and plot kinetics data. The Kolmogorov-Smirnov (K-S) test was used to test whether glucuronidation rates and mRNA expression levels followed a normal distribution. The glucuronidation rates of thyroxine, mycophenolic acid, and testosterone were not normally distributed and were log transformed prior to statistical analyses, after which they passed the normality test (K-S distance = 0.09–0.11, P > 0.10). Formation rates of OTS167 and SN-38 glucuronides were apparently normal (K-S distance = 0.10, P > 0.01 for both). The mRNA levels of UGT1A1, UGT1A3, UGT1A9, and UGT2B17 were also log transformed to pass the normality test (K-S distance = 0.08–0.12, P > 0.10). Pearson correlation was used to test the association among glucuronidation activities measured with different substrates, and between activities and mRNA expression. Multivariate analysis to investigate the contribution of UGT mRNA levels to variability in OTS167 glucuronidation was done with Microsoft Excel 2010. In inhibition experiments, residual activity was calculated by dividing the amount of glucuronide formed in the presence of inhibitor by that formed in its absence. Correlations between UGT genotypes and OTS167 glucuronidation were tested using linear regression analysis. The P values of the linear regressions test the null hypothesis that the slope is equal to 0. Results were considered statistically significant when P < 0.05. Data were analyzed using GraphPad Prism 6.00 for Windows (GraphPad Software, La Jolla, CA, www.graphpad.com), unless specified otherwise.
Results
Identification of OTS167-G in Microsomal Incubations.
Incubations containing OTS167, UDPGA, and HLM showed formation of a single product with a shorter retention time (12.7 minutes) than OTS167 (15.2 minutes) (Fig. 2A), suggesting formation of a metabolite that is more polar than its parent compound. The conjugated compound was absent from incubations without OTS167, UDPGA, or microsomes (data not shown). Incubations with β-glucuronidase showed complete disappearance of the new peak (Fig. 2B), proving that the metabolite formed is OTS167-G. Same results were obtained in incubations with HIM (data not shown).
Fig. 2.

Chromatograms of HLM incubations without (A) and with (B) β-glucuronidase. Following OTS167 incubations, samples were treated with 2000 U β-glucuronidase (dissolved in 0.1 M sodium phosphate buffer) for 24 hours at 37°C. Control incubations (no β-glucuronidase added) contained 0.1 M sodium phosphate buffer only. Coumarin was used as internal standard (IS).
Enzyme Kinetics in HLM and HIM.
HLM and HIM exhibited substrate inhibition kinetics (Fig. 3, A and B). Kinetic parameters are summarized in Table 1. Both microsomal systems showed high affinity for the substrate (Km = 3.4 µM for HLM and 1.7 µM for HIM). The difference in intrinsic metabolic clearances (CLint) for OTS167 by HLM and HIM was less than twofold. Glucuronidation activity was inhibited with OTS167 concentrations higher than 25 µM (Ki = 168 and 276 µM for HLM and HIM, respectively).
Fig. 3.

Michaelis-Menten and Eadie-Hofstee plots for formation of OTS167-G by HLM (A), HIM (B), UGT1A1 (C), and UGT1A8 (D). Incubations were performed over a substrate concentration range of 0.5–100 µM. Data are shown as the mean ± S.E.M. of triple determinations in a single experiment.
TABLE 1.
Enzyme kinetic parameters of UGTs toward OTS167
Incubations were performed in triplicate with OTS167 concentrations ranging from 0.5 µM to 100 µM. Data are shown as means ± S.E.M. Data were fitted using the substrate inhibition model Y = Vmax × X/[Km + X × (1 + X/Ki)], where Y is the reaction rate, X is substrate concentration, Vmax is apparent maximal velocity, Km is substrate concentration at which half-maximum velocity is achieved (Michaelis constant), and Ki is the inhibition constant. CLint = Vmax/Km.
| UGTs | r2 | Km (µM) | Vmax (pmol/min/mg) | Ki (µM) | CLint (μl/min/mg) |
|---|---|---|---|---|---|
| HLM | 0.991 | 3.4 ± 0.2 | 164 ± 4 | 168 ± 15 | 48 |
| HIM | 0.985 | 1.7 ± 0.1 | 151 ± 3 | 276 ± 33 | 89 |
| UGT1A1 | 0.991 | 5.7 ± 0.4 | 365 ± 13 | 179 ± 24 | 64 |
| UGT1A3 | 0.992 | 2.2 ± 0.1 | 112 ± 2 | 382 ± 41 | 51 |
| UGT1A8 | 0.997 | 3.7 ± 0.1 | 265 ± 4 | 382 ± 42 | 72 |
| UGT1A10 | 0.888 | 0.9 ± 0.1 | 42 ± 0.7 | 428 ± 57 | 47 |
OTS167 Glucuronidation by Expressed Human UGT Enzymes.
Incubations with UGT1A1, UGT1A3, UGT1A8, UGT1A9, and UGT1A10 showed formation of a single product with the same retention time as that observed with HLM and HIM. OTS167-G formation rates were 163 ± 4 pmol/min/mg, 95 ± 1 pmol/min/mg, 87 ± 1 pmol/min/mg, 33 ± 1 pmol/min/mg, and 27 ± 2 pmol/min/mg for UGT1A1, UGT1A8, UGT1A3, UGT1A10, and UGT1A9, respectively (Fig. 4). UGT1A4, UGT1A6, UGT1A7, UGT2B4, UGT2B7, UGT2B10, UGT2B15, and UGT2B17 microsomes did not metabolize OTS167.
Fig. 4.

OTS167-G formation by recombinant UGT microsomes. Bars show mean ± S.D. of triplicate measurements.
Kinetic Analyses in Recombinant Microsomes Expressing Human UGTs.
Kinetic experiments were conducted with the UGTs that metabolized OTS167 in the screening experiments. However, kinetic parameters could not be estimated in experiments with UGT1A9 due to the low rate of metabolite formation. The substrate inhibition model best fit the kinetic data generated with UGT1A1, UGT1A3, UGT1A8, and UGT1A10. Representative kinetic profiles are shown for UGT1A1 (Fig. 3C) and UGT1A8 (Fig. 3D). UGT1A8 (72 µl/min/mg) and UGT1A1 (64 µl/min/mg) had the highest CLint for OTS167, followed by UGT1A3 (51 µl/min/mg) and UGT1A10 (47 µl/min/mg) (Table 1). These UGTs all showed high affinity for the substrate (Km = 0.9–5.7 µM). OTS167-G formation was inhibited with substrate concentrations higher than 25 µM (UGT1A1, UGT1A3, and UGT1A8) or 10 µM (UGT1A10). Ki values were high (range: 179–428 µM).
Correlation of OTS167 Glucuronidation with UGT Activities and mRNA Levels.
OTS167-G formation was measured in 46 Caucasian HLM. The average rate of OTS167 glucuronidation (mean ± S.D.) was 100.7 ± 35.8 pmol/min/mg (range: 35.6–195.2 pmol/min/mg). The interindividual variation in OTS167-G formation was 36%. Rates of OTS167 glucuronidation correlated strongly with thyroxine (r = 0.91, P < 0.0001) (Fig. 5A) and SN-38 (r = 0.79, P < 0.0001) (Fig. 5B) glucuronidation, and with UGT1A1 mRNA levels (r = 0.72, P < 0.0001) (Fig. 5C). Moderate correlations were observed with mycophenolic acid glucuronidation (r = 0.50, P = 0.001) and mRNA levels of UGT1A3 (r = 0.36, P = 0.01) (Fig. 5D) and UGT1A9 (r = 0.42, P = 0.004). Multivariate analysis performed to investigate the contribution of UGT1A1, UGT1A3, and UGT1A9 mRNA levels to variability in OTS167 glucuronidation identified mRNA levels of UGT1A1 (P < 0.0001) as more important predictors than those of UGT1A3 (P = 0.30) and UGT1A9 (P = 0.11). Testosterone glucuronide formation and UGT2B17 mRNA levels were used as negative controls for correlations. OTS167 and testosterone glucuronidation rates were not correlated (r = −0.05, P = 0.77), and the correlation between OTS167-G and UGT2B17 mRNA levels was weak and insignificant (r = 0.25, P = 0.10).
Fig. 5.

Correlation analysis between OTS167-G formation and glucuronidation of thyroxine (A), glucuronidation of SN-38 (B), mRNA levels of UGT1A1 (C), and mRNA levels of UGT1A3 (D). Microsomes (0.5 mg/ml) were incubated with 3.4 µM OTS167 for 30 minutes at 37°C in duplicate.
Inhibition of OTS167 Glucuronidation.
The effect of chemical inhibitors was studied to confirm the UGTs involved in OTS167 glucuronidation. Results are shown in Fig. 6. Nilotinib showed strong inhibitory effects on OTS167-G formation by UGT1A1 (83%) (Fig. 6A) and HLM (68%) (Fig. 6B), whereas inhibition of metabolism by HIM (25%) (Fig. 6B) was modest. Imipramine was found to inhibit OTS167-G production by 45%, 34%, and 19% in incubations with UGT1A3 (Fig. 6A), HLM, and HIM (Fig. 6B), respectively. In incubations with emodin, OTS167 glucuronidation was inhibited by 76%, 100%, 65%, and 47% in incubations with UGT1A8 (Fig. 6A), UGT1A10 (Fig. 6A), HLM (Fig. 6B), and HIM (Fig. 6B), respectively. Niflumic acid completely inhibited OTS167 glucuronidation in the presence of UGT1A9 (Fig. 6A). Very low inhibition was found when coincubated with HLM (10%) and HIM (5%) (Fig. 6B).
Fig. 6.

Inhibition of OTS167 glucuronidation in UGTs (A), HLM (B), and HIM (B). HLM, HIM, UGT1A1, UGT1A3, UGT1A8, UGT1A9, and UGT1A10 were incubated with 3.4 µM, 1.7 µM, 5.7 µM, 2.2 µM, 3.7 µM, 3.4 µM, and 0.9 µM OTS167, respectively, in the presence of 2.5 µM nilotinib, 500 µM imipramine, 100 µM emodin, and 2.5 µM niflumic acid. Control incubations contained vehicle only. Data represent means ± S.D. of triple determinations in a single experiment.
Association of OTS167 Glucuronidation with Genotypes.
Reduced OTS167 glucuronidation rates were observed across UGT1A1*28, rs6706232, and rs33979061 genotypes; the opposite effect was observed with rs10203853 (Table 2). A modest and significant association was observed between rs6706232 genotypes and OTS167-G formation rates with G/G > G/A > A/A (P = 0.03). A borderline association (P = 0.05) was found with UGT1A1*28 (*1/*1 > *1/*28 > *28/*28). The differences in OTS167 glucuronidation rates among samples with rs33979061 and rs10203853 genotypes did not reach statistical significance (P > 0.05).
TABLE 2.
Association between UGT polymorphisms and OTS167-G formation
| Polymorphism | Genotype | N | OTS167-G Formation (pmol/min/mg) (mean ± S.D.) | r2 | P |
|---|---|---|---|---|---|
| UGT1A1*28 | *1/*1 | 18 | 111.7 ± 42.6 | 0.09 | 0.05 |
| *1/*28 | 21 | 98.9 ± 29.1 | |||
| *28/*28 | 6 | 78.9 ± 28.2 | |||
| rs6706232 | G/G | 11 | 115.4 ± 45.0 | 0.11 | 0.03 |
| G/A | 26 | 102.5 ± 32.2 | |||
| A/A | 8 | 77.9 ± 24.0 | |||
| rs10203853 | A/A | 17 | 89.5 ± 34.2 | 0.06 | 0.10 |
| A/T | 21 | 103.9 ± 36.7 | |||
| T/T | 7 | 113.9 ± 34.6 | |||
| rs33979061 | A/A | 21 | 105.5 ± 35.9 | 0.09 | 0.07 |
| A/C | 12 | 92.4 ± 23.2 | |||
| C/C | 5 | 78.6 ± 31.5 |
To assess the effect of the UGT1A1*6 polymorphism on OTS167 glucuronidation, incubations were performed using HLM from a donor heterozygous for UGT1A1*6. Additional samples with UGT1A1*6 were not available. The OTS167-G formation rate was low (57.8 pmol/min/mg) compared with the average velocity (100.7 pmol/min/mg) in 46 Caucasian HLM (all G/G). This UGT1A1*6 carrier did not have the UGT1A1*28 mutation. The other genotypes (rs6706232, rs33979061, and rs10203853) were not available.
Inhibition of UGTs by OTS167.
To investigate whether OTS167 is a UGT inhibitor, we performed coincubations of OTS167 with 4-MU (nonspecific substrate for all UGTs except for UGT1A4 and UGT2B10) and imipramine (specific UGT1A4 substrate). OTS167 showed no to marginal inhibition (<15%) of UGT1A3, UGT1A4, UGT1A6, UGT1A8, UGT1A9, UGT2B4, UGT2B7, UGT2B15, and UGT2B17 at all concentrations tested (range: 0.4–10 μM) (Fig. 7). Similar results were observed for UGT1A1 (0–7% inhibition with 0.4 and 2 μM OTS167), UGT1A7 (0–11% with 0.4 and 2 μM OTS167), and UGT1A10 (7% with 0.4 μM OTS167) (Fig. 7A). At higher concentrations, relatively weak inhibition of UGT1A1 (36% with 10 μM OTS167), UGT1A7 (22% with 10 μM), and UGT1A10 (21% and 33% with 2 and 10 μM OTS167) was obtained (Fig. 7A). Residual activity (%) left after treatment with diclofenac, hecogenin, and sulfinpyrazone (positive controls for inhibition) is shown in Table 3.
Fig. 7.

Inhibition of glucuronidation by OTS167 in recombinant UGT1As (A) and UGT2Bs (B). UGTs were incubated with 4-MU (substrate for UGT1A1, UGT1A3, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2B4, UGT2B7, UGT2B15, and UGT2B17) or imipramine (UGT1A4 substrate) and OTS-167. Control incubations contained vehicle only. Data represent means ± S.D. of duplicate determinations in a single experiment.
TABLE 3.
Residual glucuronidation activity after treatment with inhibitors
Control incubations contained vehicle only (DMSO for diclofenac and sulfynpirazone, and methanol for hecogenin). Data represent means ± S.D. of duplicate determinations in a single experiment.
| UGTs | Residual Activity (% Control) |
||
|---|---|---|---|
| 1 mM Sulfinpyrazone | 200 µM Hecogenin | 500 µM Diclofenac | |
| UGT1A1 | N.A. | N.A. | 14.5 ± 0.9 |
| UGT1A3 | 65.9 ± 0.4 | N.A. | N.A. |
| UGT1A4 | N.A. | 0 | N.A. |
| UGT1A6 | N.A. | N.A. | 11.7 ± 0.9 |
| UGT1A7 | N.A. | N.A. | 0 |
| UGT1A8 | N.A. | N.A. | 0 |
| UGT1A9 | N.A. | N.A. | 20.3 ± 0.1 |
| UGT1A10 | N.A. | N.A. | 0 |
| UGT2B4 | N.A. | N.A. | 0 |
| UGT2B7 | N.A. | N.A. | 4.5 ± 0.0 |
| UGT2B15 | N.A. | N.A. | 8.5 ± 0.1 |
| UGT2B17 | N.A. | N.A. | 0 |
N.A., not applicable.
Identification of OTS167-G by MS/MS.
Based on the structure of OTS167, the metabolite could be either an O-glucuronide or an N-glucuronide. We hypothesize that the metabolite is an N-glucuronide due to the fragmentation patterns of the molecule in Q1 (Supplemental Fig. 1). OTS167 (486.9 m/z) fragmented ions at 140.1, 244.3, and 347.8 (Supplemental Figs. 1, B and C, and 2). For all three experiments (HLM, UGT1A1, and UGT1A8), OTS167-G (663 m/z) similarly fragmented ions at 347.9, 365, and 487 (Supplemental Figs. 1, A–D, 3, 4, and 5). The masses at 347.8 m/z (Supplemental Fig. 1C), 486.9 m/z (Supplemental Fig. 1B), and 663 m/z (Supplemental Fig. 1A) were characteristic of having two chlorine atoms in the molecule due to peaks of M+, M+2, and M+4 in a 9:6:1 ratio, respectively, whereas the fragment at 365 m/z (Supplemental Fig. 1D) lacked this characteristic and is consistent with a glucuronide attached to one of two aromatic N-heterocycles. A product ion scan of 663 m/z was not conclusive, as it produced fragments of 95.1, 139.8, 347.6, and 486.7 m/z (consistent with the fragments found in Q1, but missing the identifying fragment of 365 m/z; data not shown).
Discussion
This is the first in vitro study to report glucuronidation as a metabolic pathway involved in OTS167 clearance. We demonstrated that UGT1A1 and UGT1A3 are the major hepatic UGTs involved in OTS167 metabolism. Among these, UGT1A1 appears to play the main role based on our experimental data and its high level of hepatic and intestinal expression (Nakamura et al., 2008; Izukawa et al., 2009; Ohno and Nakajin, 2009; Wu et al., 2011; Court et al., 2012; Harbourt et al., 2012; Rowland et al., 2013). Supporting data include high CLint for OTS167, significant correlations of OTS167 glucuronidation with UGT1A1 activity and mRNA levels in HLM, and high level of HLM inhibition with nilotinib, a highly selective UGT1A1 inhibitor at low concentrations (Ai et al., 2013). The decreased glucuronidation observed in HIM after treatment with nilotinib reflects inhibition of intestinal UGT1A1 and unaffected OTS167 glucuronidation activity by intestinal UGT1A8 and UGT1A10. We hypothesize that UGT1A3 is involved in OTS167-G formation to a lesser extent than UGT1A1, as the correlation between OTS167-G formation and UGT1A3 mRNA (r = 0.36) was not as strong as with UGT1A1 mRNA (r = 0.72), and UGT1A3 is expressed in the liver at lower levels than UGT1A1 (Nakamura et al., 2008; Izukawa et al., 2009; Ohno and Nakajin, 2009; Wu et al., 2011; Court et al., 2012; Harbourt et al., 2012; Rowland et al., 2013). The high correlation observed between OTS167 and thyroxine glucuronidation (r = 0.91) reflects contribution from both UGT1A1 and UGT1A3. Use of imipramine as a UGT1A3 inhibitor (Lu et al., 2009) confirmed its involvement in the glucuronidation reaction. However, we cannot draw conclusions regarding the extent of UGT1A3 involvement compared with UGT1A1 based on inhibition results, as even though imipramine reduced glucuronidation in HLM (34%) to a lesser extent than nilotinib (68%), the latter was a more efficient inhibitor of OTS167 glucuronidation (in UGT1A1 microsomes) than imipramine (for UGT1A3). The low inhibition of OTS167-G formation observed in HIM with imipramine is most likely due to the lower expression of UGT1A3 in the intestines compared with UGT1A1, UGT1A8, and UGT1A10 (Nakamura et al., 2008; Ohno and Nakajin, 2009; Wu et al., 2011). UGT1A9 seems to have a marginal role in OTS167 metabolism. This enzyme has high hepatic expression (Nakamura et al., 2008; Izukawa et al., 2009; Ohno and Nakajin, 2009; Wu et al., 2011; Court et al., 2012; Harbourt et al., 2012; Rowland et al., 2013), but it did not glucuronidate OTS167 at a significant extent, the correlations with UGT1A9-marker activity and mRNA levels were weak compared with the results with UGT1A1, and glucuronide production in HLM and HIM was weakly inhibited (5–10%) by a very strong UGT1A9 inhibitor.
Because UGT1A8 and UGT1A10 are expressed in the intestine but not in the liver (Ohno and Nakajin, 2009; Harbourt et al., 2012), we hypothesize that these enzymes could contribute to first-pass drug metabolism after oral administration of OTS167. UGT1A8 had higher CLint for OTS167 than UGT1A10. Correlation studies could not be performed, as individual HIM were not available. The inhibition of OTS167-G formation observed in HIM very likely reflects complete inhibition of UGT1A10, high inhibition of UGT1A8, and inhibition of UGT1A1, as this enzyme is highly expressed in the intestine (Ohno and Nakajin, 2009; Wu et al., 2011; Court et al., 2012; Harbourt et al., 2012) and emodin inhibited glucuronidation in HLM. Emodin is a good UGT1A8 and UGT1A10 inhibitor, but is not highly selective (Yamanaka et al., 2007).
We hypothesize that OTS167 undergoes N-glucuronidation according to MS data. Various nitrogen-containing compounds are converted to N-glucuronides, a reaction catalyzed by multiple enzymes, including UGT1A1, UGT1A3, UGT1A4, UGT1A8, UGT1A9, UGT2B4, UGT2B7, and UGT2B10 (Kaivosaari et al., 2011). This reflects the fact that aromatic heterocyclic substrates are structurally very diverse and UGTs involved in their glucuronidation are substrate-dependent (Kaivosaari et al., 2011). Our hypothesized structure for OTS167-G includes three possible sites where the glucuronide molecule might attach to the amine. Further NMR studies will be required to confirm our results.
We also identified potential genotypic predictors of OTS167 glucuronidation. The trends observed, although not always statistically significant, are in agreement with those previously observed with the UGT1A1 substrate SN-38: decreased glucuronidation activities with UGT1A1*28, rs6706232, and rs33979061, and increased glucuronide formation with rs10203853 (Liu et al., 2014). However, as the associations of rs6706232 and UGT1A1*28 with OTS167-G formation rates were modest, the effect of these SNPs on OTS167 clearance may not be significant. UGT1A1*6, an allele present in Asians with a 13–23% frequency (Guillemette, 2003), reduces the catalytic efficiency of UGT1A1 (Premawardhena et al., 2003) and predicts irinotecan-related toxicity in that population (Ramírez et al., 2010). We observed reduced OTS167 glucuronidation observed in a liver with UGT1A1*6. Additional studies are needed to evaluate its impact on OTS167-G formation in individuals of Asian ancestry.
It seems unlikely that OTS167 will have clinically significant effects on the glucuronidation of other drugs. The degree of relevance of the in vitro inhibition of UGT1A1 (36%) and UGT1A10 (33%) by 10 μM OTS167 is of no concern, and it is unlikely that significantly higher plasma concentrations will be maintained in patients.
Supplementary Material
Acknowledgments
Human liver microsomal samples for correlation studies were provided by the Pharmacogenomics of Anticancer Agents Research Group.
Abbreviations
- CLint
intrinsic clearance
- DMSO
dimethylsulfoxide
- HIM
human intestinal microsomes
- HLM
human liver microsomes
- HPLC
high-performance liquid chromatography
- imipramine-G
imipramine N-β-D glucuronide
- K-S
Kolmogorov-Smirnov
- MS
mass spectrometry
- 4-MU
4-methylumbelliferone
- 4-MU-G
4-methylumbelliferyl-β-D-glucuronide hydrate
- OTS167-G
OTS167-glucuronide; single nucleotide polymorphisms (SNPs)
- UDPGA
UDP-glucuronic acid
- UGT
UDP-glucuronosyltransferase
Authorship Contributions
Participated in research design: Ramírez, Mirkov, House, Ratain.
Conducted experiments: Mirkov, House.
Performed data analysis: Ramírez, Mirkov, House.
Wrote or contributed to the writing of the manuscript: Ramírez, Mirkov, House, Ratain.
Footnotes
This work was supported by OncoTherapy Science (Kawasaki City, Kanagawa, Japan) and the National Institutes of Health National Institute of General Medical Sciences [Grant U01GM061393 to Pharmacogenomics of Anticancer Agents Research Group].
This work was previously presented as preliminary data in a poster session at the following meeting: 115th Annual Meeting of American Society for Clinical Pharmacology and Therapeutics; March 18-22, 2014; Atlanta, GA [Ramirez J, Mirkov S, Matsuo Y, Ratain MJ. In vitro glucuronidation of OTS167. Clin Pharmacol Ther, 95:S73, abstract no. PII-037].
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Ai L, Zhu L, Yang L, Ge G, Cao Y, Liu Y, Fang Z, Zhang Y. (2014) Selectivity for inhibition of nilotinib on the catalytic activity of human UDP-glucuronosyltransferases. Xenobiotica 44:320–325. [DOI] [PubMed] [Google Scholar]
- Bernard O, Guillemette C. (2004) The main role of UGT1A9 in the hepatic metabolism of mycophenolic acid and the effects of naturally occurring variants. Drug Metab Dispos 32:775–778. [DOI] [PubMed] [Google Scholar]
- Bosma PJ. (2003) Inherited disorders of bilirubin metabolism. J Hepatol 38:107–117. [DOI] [PubMed] [Google Scholar]
- Chung S, Suzuki H, Miyamoto T, Takamatsu N, Tatsuguchi A, Ueda K, Kijima K, Nakamura Y, Matsuo Y. (2012) Development of an orally-administrative MELK-targeting inhibitor that suppresses the growth of various types of human cancer. Oncotarget 3:1629–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court MH, Zhang X, Ding X, Yee KK, Hesse LM, Finel M. (2012) Quantitative distribution of mRNAs encoding the 19 human UDP-glucuronosyltransferase enzymes in 26 adult and 3 fetal tissues. Xenobiotica 42:266–277. [DOI] [PubMed] [Google Scholar]
- Dong RH, Fang ZZ, Zhu LL, Liang SC, Ge GB, Yang L, Liu ZY. (2012) Investigation of UDP-glucuronosyltransferases (UGTs) inhibitory properties of carvacrol. Phytother Res 26:86–90. [DOI] [PubMed] [Google Scholar]
- Finel M, Li X, Gardner-Stephen D, Bratton S, Mackenzie PI, Radominska-Pandya A. (2005) Human UDP-glucuronosyltransferase 1A5: identification, expression, and activity. J Pharmacol Exp Ther 315:1143–1149. [DOI] [PubMed] [Google Scholar]
- Gagné JF, Montminy V, Belanger P, Journault K, Gaucher G, Guillemette C. (2002) Common human UGT1A polymorphisms and the altered metabolism of irinotecan active metabolite 7-ethyl-10-hydroxycamptothecin (SN-38). Mol Pharmacol 62:608–617. [DOI] [PubMed] [Google Scholar]
- Ganguly R, Hong CS, Smith LG, Kornblum HI, Nakano I. (2014) Maternal embryonic leucine zipper kinase: key kinase for stem cell phenotype in glioma and other cancers. Mol Cancer Ther 13:1393–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemette C. (2003) Pharmacogenomics of human UDP-glucuronosyltransferase enzymes. Pharmacogenomics J 3:136–158. [DOI] [PubMed] [Google Scholar]
- Hanioka N, Ozawa S, Jinno H, Ando M, Saito Y, Sawada J. (2001) Human liver UDP-glucuronosyltransferase isoforms involved in the glucuronidation of 7-ethyl-10-hydroxycamptothecin. Xenobiotica 31:687–699. [DOI] [PubMed] [Google Scholar]
- Harbourt DE, Fallon JK, Ito S, Baba T, Ritter JK, Glish GL, Smith PC. (2012) Quantification of human uridine-diphosphate glucuronosyl transferase 1A isoforms in liver, intestine, and kidney using nanobore liquid chromatography-tandem mass spectrometry. Anal Chem 84:98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innocenti F, Undevia SD, Iyer L, Chen PX, Das S, Kocherginsky M, Karrison T, Janisch L, Ramírez J, Rudin CM, et al. (2004) Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol 22:1382–1388. [DOI] [PubMed] [Google Scholar]
- Iyer L, Hall D, Das S, Mortell MA, Ramírez J, Kim S, Di Rienzo A, Ratain MJ. (1999) Phenotype-genotype correlation of in vitro SN-38 (active metabolite of irinotecan) and bilirubin glucuronidation in human liver tissue with UGT1A1 promoter polymorphism. Clin Pharmacol Ther 65:576–582. [DOI] [PubMed] [Google Scholar]
- Iyer L, King CD, Whitington PF, Green MD, Roy SK, Tephly TR, Coffman BL, Ratain MJ. (1998) Genetic predisposition to the metabolism of irinotecan (CPT-11): role of uridine diphosphate glucuronosyltransferase isoform 1A1 in the glucuronidation of its active metabolite (SN-38) in human liver microsomes. J Clin Invest 101:847–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izukawa T, Nakajima M, Fujiwara R, Yamanaka H, Fukami T, Takamiya M, Aoki Y, Ikushiro S, Sakaki T, Yokoi T. (2009) Quantitative analysis of UDP-glucuronosyltransferase (UGT) 1A and UGT2B expression levels in human livers. Drug Metab Dispos 37:1759–1768. [DOI] [PubMed] [Google Scholar]
- Jinno H, Tanaka-Kagawa T, Hanioka N, Saeki M, Ishida S, Nishimura T, Ando M, Saito Y, Ozawa S, Sawada J. (2003) Glucuronidation of 7-ethyl-10-hydroxycamptothecin (SN-38), an active metabolite of irinotecan (CPT-11), by human UGT1A1 variants, G71R, P229Q, and Y486D. Drug Metab Dispos 31:108–113. [DOI] [PubMed] [Google Scholar]
- Kaivosaari S, Finel M, Koskinen M. (2011) N-glucuronidation of drugs and other xenobiotics by human and animal UDP-glucuronosyltransferases. Xenobiotica 41:652–669. [DOI] [PubMed] [Google Scholar]
- Kang SP, Ramírez J, House L, Zhang W, Mirkov S, Liu W, Haverfield E, Ratain MJ. (2010) A pharmacogenetic study of vorinostat glucuronidation. Pharmacogenet Genomics 20:638–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Ikushiro S, Emi Y, Tamaki S, Suzuki H, Sakaki T, Yamada S, Degawa M. (2008) Hepatic UDP-glucuronosyltransferases responsible for glucuronidation of thyroxine in humans. Drug Metab Dispos 36:51–55. [DOI] [PubMed] [Google Scholar]
- Lévesque E, Turgeon D, Carrier JS, Montminy V, Beaulieu M, Bélanger A. (2001) Isolation and characterization of the UGT2B28 cDNA encoding a novel human steroid conjugating UDP-glucuronosyltransferase. Biochemistry 40:3869–3881. [DOI] [PubMed] [Google Scholar]
- Liu W, Ramírez J, Gamazon ER, Mirkov S, Chen P, Wu K, Sun C, Cox NJ, Cook E, Jr, Das S, et al. (2014) Genetic factors affecting gene transcription and catalytic activity of UDP-glucuronosyltransferases in human liver. Hum Mol Genet 23:5558–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Ramírez J, House L, Ratain MJ. (2010) Comparison of the drug-drug interactions potential of erlotinib and gefitinib via inhibition of UDP-glucuronosyltransferases. Drug Metab Dispos 38:32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhu J, Chen X, Li N, Fu F, He J, Wang G, Zhang L, Zheng Y, Qiu Z, et al. (2009) Identification of human UDP-glucuronosyltransferase isoforms responsible for the glucuronidation of glycyrrhetinic acid. Drug Metab Pharmacokinet 24:523–528. [DOI] [PubMed] [Google Scholar]
- Miles KK, Stern ST, Smith PC, Kessler FK, Ali S, Ritter JK. (2005) An investigation of human and rat liver microsomal mycophenolic acid glucuronidation: evidence for a principal role of UGT1A enzymes and species differences in UGT1A specificity. Drug Metab Dispos 33:1513–1520. [DOI] [PubMed] [Google Scholar]
- Miners JO, Bowalgaha K, Elliot DJ, Baranczewski P, Knights KM. (2011) Characterization of niflumic acid as a selective inhibitor of human liver microsomal UDP-glucuronosyltransferase 1A9: application to the reaction phenotyping of acetaminophen glucuronidation. Drug Metab Dispos 39:644–652. [DOI] [PubMed] [Google Scholar]
- Nakajima M, Tanaka E, Kobayashi T, Ohashi N, Kume T, Yokoi T. (2002) Imipramine N-glucuronidation in human liver microsomes: biphasic kinetics and characterization of UDP-glucuronosyltransferase isoforms. Drug Metab Dispos 30:636–642. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Nakajima M, Yamanaka H, Fujiwara R, Yokoi T. (2008) Expression of UGT1A and UGT2B mRNA in human normal tissues and various cell lines. Drug Metab Dispos 36:1461–1464. [DOI] [PubMed] [Google Scholar]
- Ohno S, Nakajin S. (2009) Determination of mRNA expression of human UDP-glucuronosyltransferases and application for localization in various human tissues by real-time reverse transcriptase-polymerase chain reaction. Drug Metab Dispos 37:32–40. [DOI] [PubMed] [Google Scholar]
- Premawardhena A, Fisher CA, Liu YT, Verma IC, de Silva S, Arambepola M, Clegg JB, Weatherall DJ. (2003) The global distribution of length polymorphisms of the promoters of the glucuronosyltransferase 1 gene (UGT1A1): hematologic and evolutionary implications. Blood Cells Mol Dis 31:98–101. [DOI] [PubMed] [Google Scholar]
- Ramírez J, Liu W, Mirkov S, Desai AA, Chen P, Das S, Innocenti F, Ratain MJ. (2007) Lack of association between common polymorphisms in UGT1A9 and gene expression and activity. Drug Metab Dispos 35:2149–2153. [DOI] [PubMed] [Google Scholar]
- Ramírez J, Ratain MJ, Innocenti F. (2010) Uridine 5′-diphospho-glucuronosyltransferase genetic polymorphisms and response to cancer chemotherapy. Future Oncol 6:563–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland A, Miners JO, Mackenzie PI. (2013) The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification. Int J Biochem Cell Biol 45:1121–1132 Review. [DOI] [PubMed] [Google Scholar]
- Tong Z, Li H, Goljer I, McConnell O, Chandrasekaran A. (2007) In vitro glucuronidation of thyroxine and triiodothyronine by liver microsomes and recombinant human UDP-glucuronosyltransferases. Drug Metab Dispos 35:2203–2210. [DOI] [PubMed] [Google Scholar]
- Tukey RH, Strassburg CP. (2000) Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu Rev Pharmacol Toxicol 40:581–616. [DOI] [PubMed] [Google Scholar]
- Tukey RH, Strassburg CP. (2001) Genetic multiplicity of the human UDP-glucuronosyltransferases and regulation in the gastrointestinal tract. Mol Pharmacol 59:405–414. [DOI] [PubMed] [Google Scholar]
- Turgeon D, Carrier JS, Lévesque E, Hum DW, Bélanger A. (2001) Relative enzymatic activity, protein stability, and tissue distribution of human steroid-metabolizing UGT2B subfamily members. Endocrinology 142:778–787. [DOI] [PubMed] [Google Scholar]
- Uchaipichat V, Mackenzie PI, Elliot DJ, Miners JO. (2006) Selectivity of substrate (trifluoperazine) and inhibitor (amitriptyline, androsterone, canrenoic acid, hecogenin, phenylbutazone, quinidine, quinine, and sulfinpyrazone) “probes” for human udp-glucuronosyltransferases. Drug Metab Dispos 34:449–456. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Nakajima M, Yokoi T. (2002) Troglitazone glucuronidation in human liver and intestine microsomes: high catalytic activity of UGT1A8 and UGT1A10. Drug Metab Dispos 30:1462–1469. [DOI] [PubMed] [Google Scholar]
- Wu B, Kulkarni K, Basu S, Zhang S, Hu M. (2011) First-pass metabolism via UDP-glucuronosyltransferase: a barrier to oral bioavailability of phenolics. J Pharm Sci 100:3655–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka H, Nakajima M, Katoh M, Yokoi T. (2007) Glucuronidation of thyroxine in human liver, jejunum, and kidney microsomes. Drug Metab Dispos 35:1642–1648. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Sato H, Fujiyama Y, Doida Y, Bamba T. (1998) Contribution of two missense mutations (G71R and Y486D) of the bilirubin UDP glycosyltransferase (UGT1A1) gene to phenotypes of Gilbert’s syndrome and Crigler-Najjar syndrome type II. Biochim Biophys Acta 1406:267–273. [DOI] [PubMed] [Google Scholar]
- Yoder Graber AL, Ramírez J, Innocenti F, Ratain MJ. (2007) UGT1A1*28 genotype affects the in-vitro glucuronidation of thyroxine in human livers. Pharmacogenet Genomics 17:619–627. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.