

Abstract
Microbial protease-mediated disruption of the intestinal epithelium is a potential mechanism whereby a dysbiotic enteric microbiota can lead to disease. This mechanism was investigated using the colitogenic, protease-secreting enteric microbe Enterococcus faecalis. Caco-2 and T-84 epithelial cell monolayers and the mouse colonic epithelium were exposed to concentrated conditioned media (CCM) from E. faecalis V583 and E. faecalis lacking the gelatinase gene (gelE). The flux of fluorescein isothiocyanate (FITC)-labeled dextran across monolayers or the mouse epithelium following exposure to CCM from parental or mutant E. faecalis strains indicated paracellular permeability. A protease-activated receptor 2 (PAR2) antagonist and PAR2-deficient (PAR2−/−) mice were used to investigate the role of this receptor in E. faecalis-induced permeability. Gelatinase (GelE) purified from E. faecalis V583 was used to confirm the ability of this protease to induce epithelial cell permeability and activate PAR2. The protease-mediated permeability of colonic epithelia from wild-type (WT) and PAR2−/− mice by fecal supernatants from ulcerative colitis patients was assessed. Secreted E. faecalis proteins induced permeability in epithelial cell monolayers, which was reduced in the absence of gelE or by blocking PAR2 activity. Secreted E. faecalis proteins induced permeability in the colonic epithelia of WT mice that was absent in tissues from PAR2−/− mice. Purified GelE confirmed the ability of this protease to induce epithelial cell permeability via PAR2 activation. Fecal supernatants from ulcerative colitis patients induced permeability in the colonic epithelia of WT mice that was reduced in tissues from PAR2−/− mice. Our investigations demonstrate that GelE from E. faecalis can regulate enteric epithelial permeability via PAR2.
INTRODUCTION
A potential mechanism for the pathogenesis of inflammatory bowel diseases (IBD) and irritable bowel syndrome (IBS) involves disruption of the intestinal epithelial barrier and exposure of a genetically defective immune system to enteric microbial antigens. Indeed, IBD and IBS patients exhibit a higher level of intestinal permeability than healthy controls (1–7). Additionally, first-degree relatives of IBD patients display elevated levels of enteric permeability (8–10), suggesting that this abnormality is independent of inflammation. Consistent with this hypothesis are animal models of colitis that use chemical disruption of the epithelial barrier with trinitrobenzene sulfonic acid, dextran sodium sulfate, or nonsteroidal anti-inflammatory drugs (NSAIDs) to elicit inflammation (11). Further, disruption of the intestinal epithelial barrier by exposure of susceptible patients to NSAIDs (blockers of prostaglandin synthesis) is a risk factor for intestinal inflammation (12).
Enteric proteases can act broadly as catalysts of protein degradation or specifically as selective agents that control physiological processes (13). Additionally, these enzymes can disrupt mucosal barriers and modulate the host immune response (14). Proteases can mediate their activity on mammalian cells through the activation of protease-activated receptors (PARs). The PAR family has four members, PARs 1 to 4 (15–19). These are transmembrane domain G-protein-coupled receptors that mediate multiple responses to external stimuli, such as hemostasis, thrombosis, and inflammation. PARs are activated through proteolytic cleavage of the extracellular N-terminal component of the receptor, unmasking a tethered peptide ligand residue that binds with another region of the receptor, causing a conformational change (20). Except for one study, reporting that PAR2 activation results in decreased colonic inflammation (21), the majority of investigations have found that activating PAR isoforms increases intestinal permeability and inflammation (22–25). Indeed, fecal proteases from ulcerative colitis (UC) patients have been shown to induce intestinal permeability in a PAR-dependent manner (24). However, it is unclear whether fecal protease-induced intestinal permeability is of mammalian or bacterial origin. Accumulating evidence shows that endogenous enteric microbes produce proteases that possess the ability to disrupt the epithelial barrier (26, 27). These commensal proteases may be involved in the pathogenesis of intestinal diseases in the context of a genetically predisposed host or an intestinal microbial dysbiosis. The interaction between the enteric microbiota and PARs may present a unique mechanism in which a microbial dysbiosis is associated with intestinal diseases via protease-mediated enteric permeability.
Enterococcus faecalis is a constituent of the normal intestinal microbiota that can induce intestinal inflammation in the interleukin 10 (IL-10)-deficient mouse model of colitis (28). E. faecalis secretes two dominant proteases, gelatinase (GelE) and serine protease (SprE) (29), making this species an ideal model with which to investigate bacterial protease-mediated enteric permeability and inflammation. Seminal work has demonstrated that E. faecalis-induced intestinal inflammation is associated with enteric permeability mediated by GelE degradation of an epithelial junctional protein (E-cadherin) (27). Here we suggest a novel mechanism by which E. faecalis can induce intestinal inflammation via enteric permeability mediated by GelE activation of PAR2.
MATERIALS AND METHODS
Bacterial strains, cell culture, and preparation of CCM.
E. faecalis V583 (originally isolated from infected human blood) (30) was used in our study. The ΔgelE isogenic mutant was generated as described previously (29). Bacteria were cultivated in brain heart infusion (BHI) medium (BD, Sparks, MD) at 37°C under aerobic conditions. For the preparation of concentrated conditioned media (CCM), bacterial culture supernatants from overnight cultures were concentrated using the Centriprep filter system (Millipore, Carrigtwohill, Cork, Ireland) with an exclusion size of 10 kDa. The abilities of parental E. faecalis V583 and mutant E. faecalis CCM to degrade casein were tested using BHI agar containing 2.5% skim milk with a methodology similar to that of previously described work (31). Briefly, parental or mutant E. faecalis cells were cultured overnight at 37°C; then 20 μl of this culture was spotted onto skim milk agar plates and was incubated overnight at 37°C. Additionally, 20 μl of parental or mutant CCM was pipetted directly onto skim milk agar plates and was incubated overnight at 37°C. A zone of clearing surrounding E. faecalis cells or CCM indicated caseinolytic activity. The Caco-2 and T-84 human colonic epithelial cell lines were cultured in Dulbecco's modified Eagle medium (DMEM; Gibco-Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich), 1% l-glutamine (Gibco-Invitrogen), and 1% penicillin-streptomycin (Gibco-Invitrogen) and were grown at 37°C in a 5% CO2 humidified incubator. For epithelial cell monolayer development, 2 × 105 Caco-2 or T-84 cells were seeded into transwell plates with a pore size of 0.4 or 3.0 μM, respectively, and were cultured under standard conditions. Monolayers were cultured until tight junctions (T-Js) formed between cells, as determined by transepithelial electrical resistance (TEER) measurements as described previously (32).
Exposure of epithelial cells to E. faecalis CCM and monolayer permeability assay.
Caco-2 and T-84 cell monolayers were exposed to CCM from either E. faecalis V583 or E. faecalis ΔgelE. Caco-2 cell monolayers were exposed to CCM apically, whereas T-84 cell monolayers were exposed to CCM basally, since PARs are expressed basolaterally in this cell line (33). After 20 h of CCM exposure, fluorescein isothiocyanate (FITC)-labeled dextran (50 ng/ml) was added to the upper chamber of the transwell, and monolayers were incubated for a further 4 h. Fluorescence was measured in the upper and lower chambers of the transwell plates using a fluorometer (SLM Aminco SPF 500; SLM Instruments, Urbana, IL) at an excitation wavelength of 485 nm and an emission wavelength of 530 nm to determine the percent flux of FITC-dextran across the monolayer. The PAR2 antagonist (FSLLRY-NH2; Tocris, Bristol, United Kingdom) was incubated with Caco-2 and T-84 cell monolayers for 24 h prior to exposure to experimental conditions at final concentrations of 5 and 20 μM, respectively.
Ex vivo epithelial permeability.
The ex vivo permeability of colonic tissue from wild-type (WT) C57BL/6 mice and PAR2−/− mice was determined using Ussing chambers as described previously (34). Briefly, distal colonic tissue sections were mounted between Lucite rings (aperture diameter, 2 mm; square area, 0.0314 cm2) in mini-Ussing chambers and were bathed on both sides with normal Ringer's solution. The flux was determined by adding FITC-dextran (50 ng/ml) to the bath exposed to the mucosal side of the tissue sample. An initial reading from the mucosal bath was taken, and the serosal bath was sampled at baseline and 2 h later. Samples were read for the presence of FITC-dextran using a fluorometer (SLM Aminco SPF 500; SLM Instruments, Urbana, IL) at an excitation wavelength of 485 nm and an emission wavelength of 530 nm. The flux as a percentage of the initial FITC-dextran concentration was calculated as (fluorescence in the serosal samples)/(fluorescence in the luminal bath) × 100.
Human fecal samples were collected from UC patients (n = 7) in the IBD Center, Department of Gastroenterology and Liver Diseases, Tel Aviv Medical Center, Tel Aviv, Israel. Upon collection, each sample was homogenized and frozen at −80°C. Fecal supernatants from all samples (n = 7) were generated as described previously (24, 35). Briefly, 500 mg of fecal material was thawed on ice. Four milliliters of a sterile 0.9% NaCl solution was added to each sample. All samples were homogenized using a sonicator (Sonic Dismembrator, set at level 10; Fisher Scientific) on ice. The sonicated mixture was then centrifuged for 10 min at 4,500 rpm at 4°C. Fecal supernatants were transferred to a syringe and were passed through a filter (0.45 μM), and pellets were discarded. The resulting fecal supernatants were stored at −80°C. Human experiments were approved by the local IRB at the Tel Aviv Medical Center.
Purification of gelatinase and Western blot analysis.
Gelatinase (GelE) secreted by E. faecalis V583 was partially purified using methods described by Hancock and Perego (36). The cell-free culture fluid of a 2-liter overnight culture of E. faecalis V583 grown in Todd-Hewitt broth was collected by centrifugation (at 5,000 × g, 4°C for 30 min). The conditioned medium was incubated overnight at 4°C with constant stirring, and ammonium sulfate was then added gradually to a final saturation of 60%. The resulting precipitate was collected by centrifugation (at 27,500 × g, 4°C for 30 min) and was resuspended in 200 ml of 200 mM Tris-HCl, 5 mM CaCl2 (pH 7.6). The resuspended protein was filter sterilized (0.2 μm) and was dialyzed (molecular weight cutoff [MWCO], 2,000) against dialysis buffer (50 mM Tris-HCl, 5 mM CaCl2 [pH 7.6]). Dialysis was performed at 4°C with constant stirring for 4 days with daily changes of buffer. The dialyzed sample was then concentrated to a volume of 20 ml by lyophilization using a VirTis Sentry Vacu Station 1SL system (SP Scientific, Gardiner, NY). Purified protein was run on a 4- to 20% gradient SDS gel (Bio-Rad Laboratories, Inc., Hercules, CA) to determine the sizes and purities of precipitated proteins.
The protein sample was digested using the filter-aided sample preparation protocol (37), and the resulting peptides were analyzed by liquid chromatography–tandem mass spectrometry (LC–MS-MS) as described previously (38). The spectra were searched using Peaks Studio software (version 7; Bioinformatics Solutions Inc.) with a downloaded UniProt/Swiss-Prot protein database.
Caco-2 cells were seeded onto 6-well plates and were grown to confluence. The cells were then exposed to 100 μl of purified GelE for 1 h. Total cellular protein was extracted with a lysis buffer (50 mmol/liter Tris HCl, 150 mmol/liter NaCl, 40 mmol/liter EDTA, 1% NP-40 [pH 7.5] plus Complete protease inhibitor cocktail [Roche, Indianapolis, IN]). Western blotting using an anti-PAR2 antibody (ab138479; Abcam, Cambridge, MA) and anti-β-actin (ICN601001; MP Biomedicals, Solon, OH) was carried out as described previously (39).
Statistical analysis.
Data are expressed as mean values ± standard errors (SE). Statistical examinations were performed using GraphPad Prism (version 5.01) software. Data comparing more than two groups were analyzed by one-way analysis of variance (ANOVA) with Tukey's multiple-comparison test. Data comparing two groups were analyzed with the nonparametric Mann-Whitney test. A P value of ≤0.05 was considered statistically significant. Data are presented as the fold change in the flux of FITC-dextran from that of the medium control or parental E. faecalis strain V583 in Caco-2 and T-84 cell experiments and as the fold change in the flux of FITC-dextran from that of the control (phosphate-buffered saline [PBS]) in Ussing chamber experiments.
RESULTS
E. faecalis V583 CCM exhibits caseinolytic activity that is eliminated in the absence of gelE.
Seminal work has demonstrated the ability of GelE secreted from E. faecalis to degrade casein (40). E. faecalis V583 cells and CCM exhibited the ability to degrade casein (Fig. 1A and B). Cells and CCM from E. faecalis lacking gelE (ΔgelE) failed to exhibit caseinolytic properties (Fig. 1A and B). Importantly, these data demonstrate that GelE is active in CCM from E. faecalis V583 and not in CCM from E. faecalis ΔgelE.
FIG 1.

Deletion of gelE from E. faecalis V583 results in the loss of casein degradation. E. faecalis V583, E. faecalis ΔgelE, and CCM from the growth of these strains were incubated overnight on agar plates containing 2.5% skim milk. (A) The parental E. faecalis strain V583 exhibits caseinolytic activity, while deletion of the gelE gene from E. faecalis V583 results in the loss of caseinolytic activity. (B) CCM from the parental E. faecalis strain V583 exhibits caseinolytic activity, while CCM from E. faecalis ΔgelE exhibits no caseinolytic activity. These observations were made consistently upon multiple incubations with parental and mutant E. faecalis cells and CCM on skim milk agar plates.
E. faecalis V583 CCM induces permeability in epithelial cell monolayers.
To investigate the ability of E. faecalis-secreted proteins to permeabilize the epithelial cell barrier, we exposed Caco-2 and T-84 cell monolayers to CCM from E. faecalis V583 and E. faecalis ΔgelE. Caco-2 cell monolayers exposed apically to E. faecalis V583 CCM for 24 h exhibited a significant (P < 0.0001) 3.2-fold increase in permeability over the medium control (Fig. 2A). The absence of gelE resulted in a significant decrease (P < 0.0001) in E. faecalis V583 CCM-mediated monolayer permeability (Fig. 2A). Additionally, T-84 cell monolayers exposed basally to E. faecalis V583 CCM for 24 h exhibited a significant (P < 0.0001) 2.3-fold increase in permeability over the medium control (Fig. 2B). CCM-mediated permeability was mirrored by a loss of barrier function, as determined by TEER (see Fig. S1 in the supplemental material). Conversely, apical exposure of T-84 cell monolayers to E. faecalis V583 CCM for 24 h did not impact permeability (see Fig. S2 in the supplemental material). Since PARs are expressed basolaterally and not apically on T-84 cell monolayers, this observation suggests the involvement of these receptors in CCM-mediated permeability. In agreement with the results for Caco-2 cell monolayers, the absence of gelE resulted in a significant decrease (P < 0.0001) in E. faecalis V583 CCM-mediated permeability of T-84 cell monolayers (Fig. 2B). These results suggest that secreted GelE is needed for E. faecalis V583 CCM-mediated monolayer permeability. Additionally, since gelatinase is one of many secreted proteases encoded in the E. faecalis V583 genome (41), and since the elimination of this single protease from this organism significantly reduces the ability of E. faecalis CCM to induce permeability in epithelial monolayers, we believe that not all proteases secreted by this microbe can carry out this process.
FIG 2.
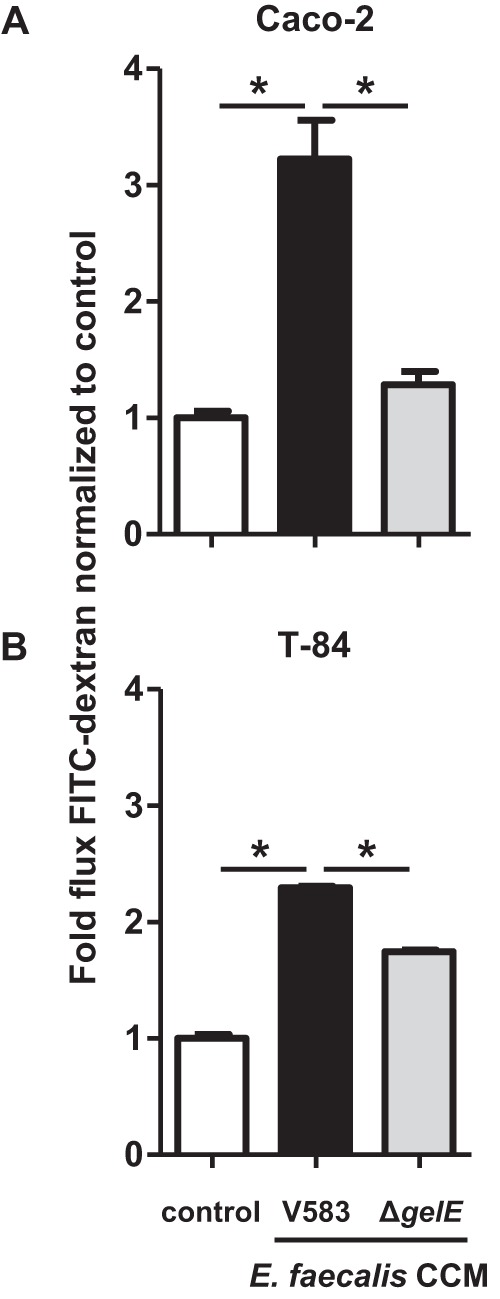
Deletion of gelE from E. faecalis V583 reduces the permeability of epithelial cell monolayers. (A) Caco-2 cell monolayers were incubated with CCM from E. faecalis V583 (filled bars) or from E. faecalis lacking the gelE gene (ΔgelE) (shaded bars). The flux of FITC-dextran across Caco-2 cell monolayers (indicative of paracellular permeability) was significantly greater when monolayers were apically cultured with E. faecalis V583 CCM for 24 h than with the medium control (open bars) (*, P < 0.0001). The flux of FITC-dextran across Caco-2 cell monolayers was significantly reduced to that of E. faecalis V583 CCM when cultured with CCM from E. faecalis ΔgelE for 24 h (*, P < 0.0001). (B) T-84 cell monolayers were incubated with CCM from E. faecalis V583 or E. faecalis ΔgelE. The flux of FITC-dextran across T-84 cell monolayers was significantly greater when monolayers were basally cultured with E. faecalis V583 CCM for 24 h than with the medium control (*, P < 0.0001). The flux of FITC-dextran across T-84 cell monolayers was significantly reduced to that of E. faecalis V583 CCM when cultured with CCM from E. faecalis ΔgelE for 24 h (*, P < 0.0001). Each experimental group represents at least 3 independent experiments with 2 technical replicates in each experiment. Data are expressed as mean values ± standard errors. Comparisons were made using one-way ANOVA.
Antagonism of PAR2 reduces the E. faecalis V583 CCM-induced permeability of epithelial cell monolayers.
To determine whether the induction of permeability in Caco-2 and T-84 cell monolayers by E. faecalis GelE acts via PAR2, we used an antagonist that has been reported to block PAR2 activation (42). A significant decrease (P < 0.0001) in the degree of permeability induced by E. faecalis V583 CCM was found in Caco-2 cell monolayers exposed to the PAR2 antagonist, while the PAR2 antagonist alone exhibited no significant effect on monolayer barrier permeability (Fig. 3). This reduction in E. faecalis V583 CCM-induced permeability by a PAR2 antagonist was observed at a range of concentrations (see Fig. S3A in the supplemental material). No decrease in the degree of permeability induced by E. faecalis V583 CCM was found in T-84 cell monolayers exposed to the PAR2 antagonist; however, barrier stability was impacted, as reflected by a change in TEER values (see Fig. S3B and C in the supplemental material).
FIG 3.
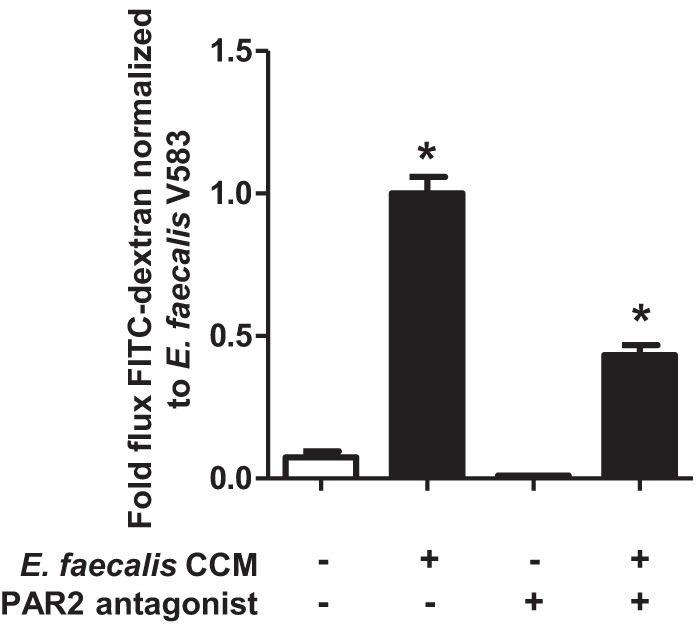
E. faecalis V583 mediates monolayer permeability via PAR2. Caco-2 cell monolayers were incubated with or without a PAR2 antagonist (FSLLRY-NH2) for 24 h. The monolayers were then incubated with E. faecalis V583 CCM (filled bars) or a medium control (open bars) for a further 24 h. The flux of FITC-dextran across Caco-2 cell monolayers was significantly greater when monolayers were cultured with E. faecalis V583 CCM than with the control (*, P < 0.0001). The E. faecalis V583 CCM-mediated permeability of Caco-2 cell monolayers was significantly reduced when PAR2 activation was blocked with an antagonist (*, P < 0.0001). Each experimental group represents at least 3 independent experiments with 2 technical replicates in each experiment. Data are expressed as mean values ± standard errors. Comparisons were made using one-way ANOVA.
E. faecalis V583 CCM-induced permeability of the mouse colonic epithelium is reduced in the absence of PAR2.
The PAR2 antagonist used in this study is a peptide that binds to the PAR2 receptor and inhibits subsequent activation (42). Since this antagonist may exhibit nonspecific inhibition of PAR2 signaling, we used the intestinal epithelia from PAR2−/− mice to confirm the role of this receptor in the induction of permeability by E. faecalis V583 CCM. Indeed, a significant 2-fold increase (P < 0.04) in permeability was observed when the colonic epithelia from WT mice were exposed to E. faecalis V583 CCM (Fig. 4). Additionally, E. faecalis V583 CCM-induced permeability was not observed in the colonic epithelia of mice lacking the PAR2 receptor. Interestingly, the baseline permeabilities of the epithelia from WT and PAR2−/− mice did not differ when the absolute flux of FITC-dextran across these barriers was analyzed (see Fig. S4 in the supplemental material). These data confirm that secreted proteins from E. faecalis are capable of inducing permeability in the mouse colon via PAR2.
FIG 4.
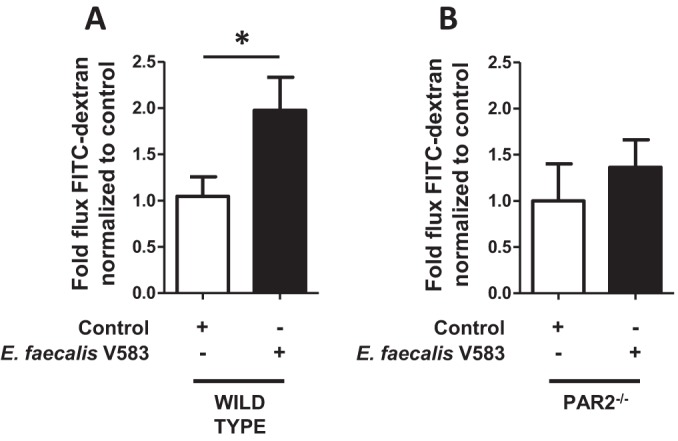
The E. faecalis V583 CCM-induced change in the permeability of the mouse colonic epithelium is absent in PAR2−/− mice. (A) The colonic epithelia of WT mice were exposed to CCM from E. faecalis V583 (filled bars), and the flux of FITC-dextran (paracellular permeability) was measured using Ussing chambers following 2 h of exposure. E. faecalis V583 CCM increased the flux of FITC-dextran across the epithelia from WT mice over that of the control (PBS) (open bars) (P < 0.04). (B) CCM from E. faecalis V583 could not induce a change in permeability in mice lacking the PAR2 receptor. Each bar represents experimental results for 10 WT or 5 PAR2−/− mice. Data are expressed as mean values ± standard errors. Comparisons were made using the nonparametric Mann-Whitney test.
GelE purified from E. faecalis V583 induces permeability in epithelial cell monolayers and activates PAR2.
Although deletion of the gelE gene from E. faecalis V583 resulted in decreased CCM-mediated permeability of epithelial cell monolayers, the removal of this gene may functionally impact the neighboring secreted protease, serine protease, which could potentially play a role in this process. We therefore sought to purify GelE from E. faecalis V583 in order to confirm the role of this protease in the induction of epithelial cell permeability via PAR2. Using previously described methods to isolate GelE (36), we purified an approximately 38 kDa protein from E. faecalis V583 that exhibited caseinolytic properties (Fig. 5A and B). The 38-kDa protein was confirmed to be GelE following trypsin digestion and LC–MS-MS (27 matching peptides with 48% coverage) (Fig. 5C). Purified GelE induced permeability in Caco-2 and T-84 cell monolayers (Fig. 6), demonstrating that this protease alone is capable of inducing these physiological changes.
FIG 5.

GelE purified from E. faecalis V583 exhibits caseinolytic activity. (A) Putatively isolated GelE was run on an SDS gel to confirm its size and purity. The isolated protein had a molecular mass of approximately 38 kDa. (B) The putatively purified GelE exhibited caseinolytic activity following overnight incubation on agar plates containing 2.5% skim milk. These data demonstrate that purified GelE retained its functional activity. (C) Alignment of the 27 digested fragments of the putative GelE protein generated by LC–MS-MS. All digested fragments aligned with GelE from E. faecalis V583, confirming the identity of this protein. The shaded sequence indicates the areas homologous to digested fragments. Blue lines represent digested fragments.
FIG 6.
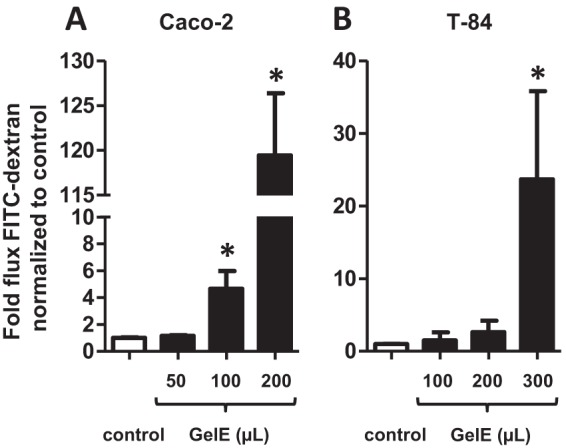
GelE from E. faecalis V583 induces permeability in epithelial cell monolayers. Caco-2 (A) and T-84 (B) cell monolayers were incubated with GelE purified from E. faecalis V583 (filled bars). The flux of FITC-dextran across monolayers (indicative of paracellular permeability) was significantly greater when monolayers were apically (Caco-2) or basally (T-84) cultured with GelE from E. faecalis V583 for 24 h than when monolayers were cultured with the medium control (open bars) (*, P < 0.0001). Each experimental group represents at least 3 independent experiments with 2 technical replicates in each experiment. Data are expressed as mean values ± standard errors. Comparisons were made using the nonparametric Mann-Whitney test.
Next, we wanted to determine whether purified GelE could activate PAR2. We incubated GelE with Caco-2 cells for 1 h. The duration of the exposure of Caco-2 cells to GelE was based on the influence of CCM from E. faecalis V583 on PAR2 detection over time (see Fig. S5 in the supplemental material). Exposure of this epithelial cell line to GelE resulted in the elimination of PAR2 detection by Western blotting (Fig. 7). Given that the anti-PAR2 antibody we used recognizes the N-terminal end of the receptor (amino acids 1 to 100) and that PAR2 is cleaved at the extracellular NH2 terminus (43), the observed reduction in antibody binding further suggests that, in addition to increasing permeability, E. faecalis V583 GelE activates PAR2.
FIG 7.
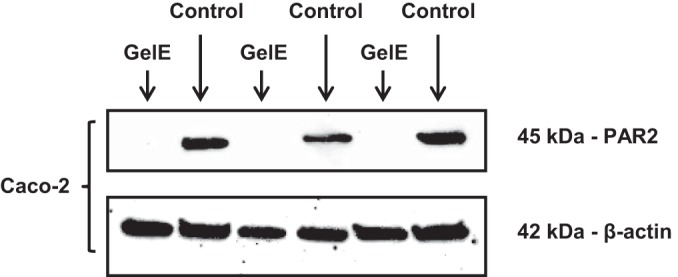
GelE from E. faecalis V583 activates PAR2 on epithelial cells. GelE was incubated with Caco-2 cells for 1 h. Antibodies could not detect PAR2 in epithelial cells exposed to GelE. Antibodies for β-actin demonstrated equal loading of proteins in each lane. This blot represents results from at least 3 independent experiments with 2 technical replicates in each experiment.
Human fecal proteases from UC patients induce permeability in the mouse colonic epithelium via PAR2.
We next investigated whether fecal supernatants generated from a human source (healthy individuals or IBD patients), which harbor a complex mixture of human and microbial proteases, can induce colonic epithelial permeability via PARs. A significant 2-fold increase in permeability was observed in the colonic epithelia from WT mice exposed to fecal supernatants from UC patients (n = 7), and that increase in permeability was reduced when PAR2 was absent from the epithelium (Fig. 8A and B). The permeability of mouse colonic epithelia induced by fecal supernatants from UC patients was reduced in the presence of protease inhibitors (Fig. 8C). A subset of supernatants (n = 4) was used, because 3 of the 7 supernatants from UC patients did not produce sufficient volume to be included in all 3 experiments. These data suggest that fecal proteases from UC patients can induce permeability in the colonic epithelium via PAR2.
FIG 8.
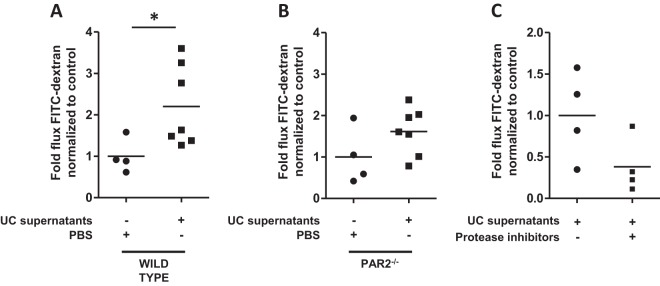
The permeability of the epithelium mediated by fecal supernatants from UC patients is lost in PAR2−/− mice. The colonic epithelia of mice were exposed to fecal supernatants generated from UC patients, and permeability was measured using Ussing chambers. (A) Exposure of the colonic epithelia of WT mice to supernatants from UC patients (n = 7) for 2 h induced a significant 2-fold increase in paracellular permeability over that of controls (P < 0.05). (B) Exposure of the colonic epithelia of PAR2−/− mice to supernatants from UC patients (n = 7) for 2 h did not exhibit a significant difference in paracellular permeability from controls. (C) Exposure of the colonic epithelia of WT mice to a subset of supernatants from UC patients (n = 4) for 2 h induced a reduced level of paracellular permeability when these supernatants were incubated with protease inhibitors. Comparisons were made using the nonparametric Mann-Whitney test.
DISCUSSION
The intestinal epithelial barrier is made up of a single layer of cells that provides a first line of defense against infection. Although the initiating events of intestinal diseases such as IBD and IBS have not been fully defined, one potential mechanism involves disruption of the intestinal epithelial barrier and exposure of a genetically defective mucosal immune system to enteric luminal antigens. Host factors, including proteases and cytokines, can regulate barrier function (44, 45); however, it is possible that host and microbial factors can synergistically modulate the intestinal barrier. The relationship between the intestinal microbiota and fecal protease activity has been reported previously (46), yet the functional impact of enteric microbial proteases on host enteric epithelial receptors is relatively unexplored. We therefore sought to demonstrate that an enteric protease-secreting microbe could activate specific intestinal epithelial receptors leading to permeability. The genus Enterococcus encompasses protease-secreting species and is reported to be enriched in patients with IBD (47). Additionally, the mucosal densities of E. faecalis, a species within this genus, have been shown to correlate positively with clinical activity indices and sigmoidoscopy scores in UC patients (48). The enteric microbe E. faecalis has been shown previously to circumvent the defense of the intestinal epithelial barrier directly through proteolytic degradation of an adherence junction protein (E-cadherin) and to contribute to intestinal inflammation in the IL-10-deficient mouse model of colitis (27). Thus, this organism was chosen as a representative enteric protease-secreting microbe to demonstrate a proof of principle that a microbial protease can impact the physiology of the host intestinal epithelium. Here we report a novel mechanism whereby this microbe, E. faecalis V583, can alter the intestinal epithelial barrier by the regulation of paracellular permeability via gelatinase secretion and PAR2 activation.
In the gastrointestinal (GI) tract, PARs can be activated by endogenous proteases secreted by the pancreas and mast cells, and potentially by the luminal enteric microbiota, resulting in permeability of the intestinal epithelium (49). Given that increased intestinal permeability is a hallmark of IBD and that PAR expression on the gut epithelium differs between IBD patients and healthy individuals (50, 51), it is possible that these receptors are involved in enteric microbial protease-mediated permeability of the intestine. Although the activation of PAR1, -2, and -4 isoforms has been linked to intestinal permeability (25, 52), increased intestinal permeability induced by PAR2 has been reported to occur via disruption of the tight junctions (T-Js) between epithelial cells (paracellular permeability). Additionally, blocking the activation of PAR2 was shown to be protective in an experimental model of colitis (53). Thus, we chose to focus our study on the role of PAR2 in enteric microbial protease-mediated paracellular permeability. Indeed, we found that a PAR2 antagonist inhibits the E. faecalis CCM-mediated change in the permeability of epithelial cell monolayers, suggesting a role for this receptor in this process. PARs are activated via cleavage of their N termini, resulting in intracellular signaling (54). Since the anti-PAR2 antibody used in this study recognizes the N terminus of the receptor, and since PAR2 detection was lost upon exposure of epithelial cell monolayers to E. faecalis V583 CCM over time, we conclude that secreted proteins from this enteric microbe result in the activation of PAR2. Using purified gelatinase and Western blotting, we further demonstrated the ability of this protease to activate PAR2. Taken together, these data highlight a role for secreted gelatinase from E. faecalis V583 in regulating the permeability of enteric epithelial cells via PAR2 activation. Given their association with intestinal permeability, it is possible that the PAR1 and -4 isoforms could also play a role in regulating the microbial protease-mediated permeability of enteric epithelial cells, a potential mechanism that warrants further investigation.
Our in vitro and ex vivo data highlight the ability of a colitogenic enteric microbe to induce intestinal epithelial permeability via PAR2 activation. In order to determine whether proteases secreted from a complex microbial community of human origin are able to induce the same physiological changes in the intestinal epithelium, we investigated fecal supernatants from a cohort of UC patients. It has been reported previously that fecal proteases from UC patients increase intestinal permeability via PAR4 (24); however, our findings suggest that proteases from UC patients can also contribute to increased permeability of the colonic epithelium via PAR2. Although the proteases in the fecal supernatants analyzed likely originate from both human and microbial sources, on the basis of our data, we speculate that there may be multiple enteric microbe-secreted proteases that can alter epithelial permeability via the activation of PARs or by alternative, yet undefined molecular mechanisms.
Although it has been reported that different mammalian and prokaryotic proteases can activate PARs (e.g., human trypsin [55, 56] or Porphyromonas gingivalis gingipains [57]), different forms of these receptors exhibit specificities for various activating proteases (e.g., PAR2 can be activated by trypsin but not by thrombin). Additionally, most studies investigating how microbes disrupt the intestinal epithelium via PAR activation have focused on pathogens. Our study demonstrates that gelatinase secreted into the gut by a commensal microbe is also capable of activating PAR2. Gelatinase from E. faecalis has been reported as the principal mediator of pathogenesis in endocarditis (58) and a significant contributor to biofilm development (59); thus, we report a novel mechanism for this microbial protease that may extend to other enteric microbial proteases and may be a relevant risk factor for intestinal diseases.
In summary, we report a novel mechanism by which bacterial proteases from a commensal inhabitant of the intestinal microbiota can disrupt the enteric epithelial barrier. Given that elevated protease activity has been found in UC patients and dysbiosis has been established in IBD, our data suggest a possible mechanism whereby proteases from a dysbiotic intestinal microbiota may increase the permeability of the enteric epithelium and lead to inflammation. Since increased intestinal permeability is prevalent in IBD patients and their apparently healthy first-degree family members (1, 8), our study highlights gelatinase from E. faecalis, and by extension gelatinase from other protease-secreting enteric microbes, as a potential target for therapeutic agents used for the prevention of intestinal inflammation or the maintenance of remission. PAR activation is known to regulate the T-Js of epithelial cells (49), and gelatinase from E. faecalis has been shown previously to disrupt the epithelial barrier directly through proteolytic degradation of E-cadherin (27). Since we have shown that gelatinase can activate PAR2, and since E-cadherin lies below epithelial T-Js, it is speculated that gelatinase may disrupt epithelial T-Js via PAR2 activation, which, in turn, exposes E-cadherin to gelatinase and further contributes to epithelial barrier disruption. This model will need to be delineated in future mechanistic studies investigating this interesting phenomenon.
Supplementary Material
ACKNOWLEDGMENTS
We thank the UNC-CH National Gnotobiotic Rodent Resource Center (funded by USPHS P40 OD010995) for its contribution to this study. This research is based in part on work conducted using the UNC Michael Hooker Proteomics Center, which is supported in part by NIH-NCI grant CA016086 to the Lineberger Comprehensive Cancer Center. This study was supported by grant K01 DK 092330 and a CGIBD pilot feasibility grant (P30DK03498), awarded to I.M.C., and by grant RO1 HL096679, awarded to R.P. This study was partially supported by a generous grant from the Leona M. and Harry B. Helmsley Charitable Trust (ID).
We thank Susan J. Henning and Richard Von Furstenberg for helpful input. We further thank the Translational Pathology Laboratory for valuable help. PAR2−/− mice were originally provided by Patricia Andrade-Gordon (Johnson & Johnson Pharmaceutical).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00425-15.
REFERENCES
- 1.Arnott ID, Kingstone K, Ghosh S. 2000. Abnormal intestinal permeability predicts relapse in inactive Crohn disease. Scand J Gastroenterol 35:1163–1169. doi: 10.1080/003655200750056637. [DOI] [PubMed] [Google Scholar]
- 2.D'Incà R, Di Leo V, Corrao G, Martines D, D'Odorico A, Mestriner C, Venturi C, Longo G, Sturniolo GC. 1999. Intestinal permeability test as a predictor of clinical course in Crohn's disease. Am J Gastroenterol 94:2956–2960. doi: 10.1111/j.1572-0241.1999.01444.x. [DOI] [PubMed] [Google Scholar]
- 3.Dunlop SP, Hebden J, Campbell E, Naesdal J, Olbe L, Perkins AC, Spiller RC. 2006. Abnormal intestinal permeability in subgroups of diarrhea-predominant irritable bowel syndromes. Am J Gastroenterol 101:1288–1294. doi: 10.1111/j.1572-0241.2006.00672.x. [DOI] [PubMed] [Google Scholar]
- 4.Lee JW, Park JH, Park DI, Park JH, Kim HJ, Cho YK, Sohn CI, Jeon WK, Kim BI. 2010. Subjects with diarrhea-predominant IBS have increased rectal permeability responsive to tryptase. Dig Dis Sci 55:2922–2928. doi: 10.1007/s10620-009-1094-8. [DOI] [PubMed] [Google Scholar]
- 5.McOmber ME, Ou CN, Shulman RJ. 2010. Effects of timing, sex, and age on site-specific gastrointestinal permeability testing in children and adults. J Pediatr Gastroenterol Nutr 50:269–275. doi: 10.1097/MPG.0b013e3181aa3aa9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meddings JB, Sutherland LR, May GR. 1994. Intestinal permeability in patients with Crohn's disease. Gut 35:1675–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wyatt J, Vogelsang H, Hubl W, Waldhoer T, Lochs H. 1993. Intestinal permeability and the prediction of relapse in Crohn's disease. Lancet 341:1437–1439. doi: 10.1016/0140-6736(93)90882-H. [DOI] [PubMed] [Google Scholar]
- 8.Hollander D, Vadheim CM, Brettholz E, Petersen GM, Delahunty T, Rotter JI. 1986. Increased intestinal permeability in patients with Crohn's disease and their relatives. A possible etiologic factor. Ann Intern Med 105:883–885. [DOI] [PubMed] [Google Scholar]
- 9.Katz KD, Hollander D, Vadheim CM, McElree C, Delahunty T, Dadufalza VD, Krugliak P, Rotter JI. 1989. Intestinal permeability in patients with Crohn's disease and their healthy relatives. Gastroenterology 97:927–931. [DOI] [PubMed] [Google Scholar]
- 10.Munkholm P, Langholz E, Hollander D, Thornberg K, Orholm M, Katz KD, Binder V. 1994. Intestinal permeability in patients with Crohn's disease and ulcerative colitis and their first degree relatives. Gut 35:68–72. doi: 10.1136/gut.35.1.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reuter BK, Asfaha S, Buret A, Sharkey KA, Wallace JL. 1996. Exacerbation of inflammation-associated colonic injury in rat through inhibition of cyclooxygenase-2. J Clin Invest 98:2076–2085. doi: 10.1172/JCI119013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dubeau MF, Iacucci M, Beck PL, Moran GW, Kaplan GG, Ghosh S, Panaccione R. 2013. Drug-induced inflammatory bowel disease and IBD-like conditions. Inflamm Bowel Dis 19:445–456. doi: 10.1002/ibd.22990. [DOI] [PubMed] [Google Scholar]
- 13.Neurath H. 1984. Evolution of proteolytic enzymes. Science 224:350–357. doi: 10.1126/science.6369538. [DOI] [PubMed] [Google Scholar]
- 14.Biancheri P, Di Sabatino A, Corazza GR, MacDonald TT. 2013. Proteases and the gut barrier. Cell Tissue Res 351:269–280. doi: 10.1007/s00441-012-1390-z. [DOI] [PubMed] [Google Scholar]
- 15.Ishihara H, Connolly AJ, Zeng D, Kahn ML, Zheng YW, Timmons C, Tram T, Coughlin SR. 1997. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature 386:502–506. doi: 10.1038/386502a0. [DOI] [PubMed] [Google Scholar]
- 16.Kahn ML, Hammes SR, Botka C, Coughlin SR. 1998. Gene and locus structure and chromosomal localization of the protease-activated receptor gene family. J Biol Chem 273:23290–23296. doi: 10.1074/jbc.273.36.23290. [DOI] [PubMed] [Google Scholar]
- 17.Nystedt S, Emilsson K, Larsson AK, Strombeck B, Sundelin J. 1995. Molecular cloning and functional expression of the gene encoding the human proteinase-activated receptor 2. Eur J Biochem 232:84–89. doi: 10.1111/j.1432-1033.1995.tb20784.x. [DOI] [PubMed] [Google Scholar]
- 18.Vu TK, Hung DT, Wheaton VI, Coughlin SR. 1991. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64:1057–1068. doi: 10.1016/0092-8674(91)90261-V. [DOI] [PubMed] [Google Scholar]
- 19.Xu WF, Andersen H, Whitmore TE, Presnell SR, Yee DP, Ching A, Gilbert T, Davie EW, Foster DC. 1998. Cloning and characterization of human protease-activated receptor 4. Proc Natl Acad Sci U S A 95:6642–6646. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hollenberg MD, Compton SJ. 2002. International Union of Pharmacology. XXVIII. Proteinase-activated receptors. Pharmacol Rev 54:203–217. [DOI] [PubMed] [Google Scholar]
- 21.Fiorucci S, Mencarelli A, Palazzetti B, Distrutti E, Vergnolle N, Hollenberg MD, Wallace JL, Morelli A, Cirino G. 2001. Proteinase-activated receptor 2 is an anti-inflammatory signal for colonic lamina propria lymphocytes in a mouse model of colitis. Proc Natl Acad Sci U S A 98:13936–13941. doi: 10.1073/pnas.241377298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cenac N, Cellars L, Steinhoff M, Andrade-Gordon P, Hollenberg MD, Wallace JL, Fiorucci S, Vergnolle N. 2005. Proteinase-activated receptor-1 is an anti-inflammatory signal for colitis mediated by a type 2 immune response. Inflamm Bowel Dis 11:792–798. doi: 10.1097/01.mib.0000177506.71784.bd. [DOI] [PubMed] [Google Scholar]
- 23.Cenac N, Coelho AM, Nguyen C, Compton S, Andrade-Gordon P, MacNaughton WK, Wallace JL, Hollenberg MD, Bunnett NW, Garcia-Villar R, Bueno L, Vergnolle N. 2002. Induction of intestinal inflammation in mouse by activation of proteinase-activated receptor-2. Am J Pathol 161:1903–1915. doi: 10.1016/S0002-9440(10)64466-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dabek M, Ferrier L, Annahazi A, Bezirard V, Polizzi A, Cartier C, Leveque M, Roka R, Wittmann T, Theodorou V, Bueno L. 2011. Intracolonic infusion of fecal supernatants from ulcerative colitis patients triggers altered permeability and inflammation in mice: role of cathepsin G and protease-activated receptor-4. Inflamm Bowel Dis 17:1409–1414. doi: 10.1002/ibd.21454. [DOI] [PubMed] [Google Scholar]
- 25.Vergnolle N. 2005. Clinical relevance of proteinase activated receptors (PARs) in the gut. Gut 54:867–874. doi: 10.1136/gut.2004.048876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pruteanu M, Hyland NP, Clarke DJ, Kiely B, Shanahan F. 2011. Degradation of the extracellular matrix components by bacterial-derived metalloproteases: implications for inflammatory bowel diseases. Inflamm Bowel Dis 17:1189–1200. doi: 10.1002/ibd.21475. [DOI] [PubMed] [Google Scholar]
- 27.Steck N, Hoffmann M, Sava IG, Kim SC, Hahne H, Tonkonogy SL, Mair K, Krueger D, Pruteanu M, Shanahan F, Vogelmann R, Schemann M, Kuster B, Sartor RB, Haller D. 2011. Enterococcus faecalis metalloprotease compromises epithelial barrier and contributes to intestinal inflammation. Gastroenterology 141:959–971. doi: 10.1053/j.gastro.2011.05.035. [DOI] [PubMed] [Google Scholar]
- 28.Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, Braun J, Huycke MM, Sartor RB. 2005. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 128:891–906. doi: 10.1053/j.gastro.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 29.Thomas VC, Thurlow LR, Boyle D, Hancock LE. 2008. Regulation of autolysis-dependent extracellular DNA release by Enterococcus faecalis extracellular proteases influences biofilm development. J Bacteriol 190:5690–5698. doi: 10.1128/JB.00314-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aakra A, Vebo H, Snipen L, Hirt H, Aastveit A, Kapur V, Dunny G, Murray BE, Nes IF. 2005. Transcriptional response of Enterococcus faecalis V583 to erythromycin. Antimicrob Agents Chemother 49:2246–2259. doi: 10.1128/AAC.49.6.2246-2259.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shankar J, Walker RG, Ward D, Horsburgh MJ. 2012. The Enterococcus faecalis exoproteome: identification and temporal regulation by Fsr. PLoS One 7:e33450. doi: 10.1371/journal.pone.0033450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rao R, Baker RD, Baker SS. 1999. Inhibition of oxidant-induced barrier disruption and protein tyrosine phosphorylation in Caco-2 cell monolayers by epidermal growth factor. Biochem Pharmacol 57:685–695. doi: 10.1016/S0006-2952(98)00333-5. [DOI] [PubMed] [Google Scholar]
- 33.Chin AC, Lee WY, Nusrat A, Vergnolle N, Parkos CA. 2008. Neutrophil-mediated activation of epithelial protease-activated receptors-1 and -2 regulates barrier function and transepithelial migration. J Immunol 181:5702–5710. doi: 10.4049/jimmunol.181.8.5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jovov B, Shaheen NJ, Orlando GS, Djukic Z, Orlando RC. 2013. Defective barrier function in neosquamous epithelium. Am J Gastroenterol 108:386–391. doi: 10.1038/ajg.2012.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Róka R, Rosztoczy A, Leveque M, Izbeki F, Nagy F, Molnar T, Lonovics J, Garcia-Villar R, Fioramonti J, Wittmann T, Bueno L. 2007. A pilot study of fecal serine-protease activity: a pathophysiologic factor in diarrhea-predominant irritable bowel syndrome. Clin Gastroenterol Hepatol 5:550–555. doi: 10.1016/j.cgh.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 36.Hancock LE, Perego M. 2004. The Enterococcus faecalis fsr two-component system controls biofilm development through production of gelatinase. J Bacteriol 186:5629–5639. doi: 10.1128/JB.186.17.5629-5639.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wiśniewski JR, Zougman A, Nagaraj N, Mann M. 2009. Universal sample preparation method for proteome analysis. Nat Methods 6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 38.Turowec JP, Zukowski SA, Knight JD, Smalley DM, Graves LM, Johnson GL, Li SS, Lajoie GA, Litchfield DW. 2014. An unbiased proteomic screen reveals caspase cleavage is positively and negatively regulated by substrate phosphorylation. Mol Cell Proteomics 13:1184–1197. doi: 10.1074/mcp.M113.037374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cleynen I, Vazeille E, Artieda M, Verspaget HW, Szczypiorska M, Bringer MA, Lakatos PL, Seibold F, Parnell K, Weersma RK, Mahachie John JM, Morgan-Walsh R, Staelens D, Arijs I, De Hertogh G, Muller S, Tordai A, Hommes DW, Ahmad T, Wijmenga C, Pender S, Rutgeerts P, Van Steen K, Lottaz D, Vermeire S, Darfeuille-Michaud A. 2014. Genetic and microbial factors modulating the ubiquitin proteasome system in inflammatory bowel disease. Gut 63:1265–1274. doi: 10.1136/gutjnl-2012-303205. [DOI] [PubMed] [Google Scholar]
- 40.Kawalec M, Potempa J, Moon JL, Travis J, Murray BE. 2005. Molecular diversity of a putative virulence factor: purification and characterization of isoforms of an extracellular serine glutamyl endopeptidase of Enterococcus faecalis with different enzymatic activities. J Bacteriol 187:266–275. doi: 10.1128/JB.187.1.266-275.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paulsen IT, Banerjei L, Myers GS, Nelson KE, Seshadri R, Read TD, Fouts DE, Eisen JA, Gill SR, Heidelberg JF, Tettelin H, Dodson RJ, Umayam L, Brinkac L, Beanan M, Daugherty S, DeBoy RT, Durkin S, Kolonay J, Madupu R, Nelson W, Vamathevan J, Tran B, Upton J, Hansen T, Shetty J, Khouri H, Utterback T, Radune D, Ketchum KA, Dougherty BA, Fraser CM. 2003. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299:2071–2074. doi: 10.1126/science.1080613. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y, Yang C, Wang ZJ. 2011. Proteinase-activated receptor 2 sensitizes transient receptor potential vanilloid 1, transient receptor potential vanilloid 4, and transient receptor potential ankyrin 1 in paclitaxel-induced neuropathic pain. Neuroscience 193:440–451. doi: 10.1016/j.neuroscience.2011.06.085. [DOI] [PubMed] [Google Scholar]
- 43.Adams MN, Ramachandran R, Yau MK, Suen JY, Fairlie DP, Hollenberg MD, Hooper JD. 2011. Structure, function and pathophysiology of protease activated receptors. Pharmacol Ther 130:248–282. doi: 10.1016/j.pharmthera.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 44.Fish SM, Proujansky R, Reenstra WW. 1999. Synergistic effects of interferon gamma and tumour necrosis factor alpha on T84 cell function. Gut 45:191–198. doi: 10.1136/gut.45.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patrick DM, Leone AK, Shellenberger JJ, Dudowicz KA, King JM. 2006. Proinflammatory cytokines tumor necrosis factor-alpha and interferon-gamma modulate epithelial barrier function in Madin-Darby canine kidney cells through mitogen activated protein kinase signaling. BMC Physiol 6:2. doi: 10.1186/1472-6793-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carroll IM, Ringel-Kulka T, Ferrier L, Wu MC, Siddle JP, Bueno L, Ringel Y. 2013. Fecal protease activity is associated with compositional alterations in the intestinal microbiota. PLoS One 8:e78017. doi: 10.1371/journal.pone.0078017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang S, Denman SE, Morrison M, Yu Z, Dore J, Leclerc M, McSweeney CS. 2010. Dysbiosis of fecal microbiota in Crohn's disease patients as revealed by a custom phylogenetic microarray. Inflamm Bowel Dis 16:2034–2042. doi: 10.1002/ibd.21319. [DOI] [PubMed] [Google Scholar]
- 48.Fite A, Macfarlane S, Furrie E, Bahrami B, Cummings JH, Steinke DT, Macfarlane GT. 2013. Longitudinal analyses of gut mucosal microbiotas in ulcerative colitis in relation to patient age and disease severity and duration. J Clin Microbiol 51:849–856. doi: 10.1128/JCM.02574-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carroll IM, Maharshak N. 2013. Enteric bacterial proteases in inflammatory bowel disease—pathophysiology and clinical implications. World J Gastroenterol 19:7531–7543. doi: 10.3748/wjg.v19.i43.7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim JA, Choi SC, Yun KJ, Kim DK, Han MK, Seo GS, Yeom JJ, Kim TH, Nah YH, Lee YM. 2003. Expression of protease-activated receptor 2 in ulcerative colitis. Inflamm Bowel Dis 9:224–229. doi: 10.1097/00054725-200307000-00002. [DOI] [PubMed] [Google Scholar]
- 51.Vergnolle N, Cellars L, Mencarelli A, Rizzo G, Swaminathan S, Beck P, Steinhoff M, Andrade-Gordon P, Bunnett NW, Hollenberg MD, Wallace JL, Cirino G, Fiorucci S. 2004. A role for proteinase-activated receptor-1 in inflammatory bowel diseases. J Clin Invest 114:1444–1456. doi: 10.1172/JCI21689. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Annaházi A, Gecse K, Dabek M, Ait-Belgnaoui A, Rosztoczy A, Roka R, Molnar T, Theodorou V, Wittmann T, Bueno L, Eutamene H. 2009. Fecal proteases from diarrheic-IBS and ulcerative colitis patients exert opposite effect on visceral sensitivity in mice. Pain 144:209–217. doi: 10.1016/j.pain.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 53.Lohman RJ, Cotterell AJ, Barry GD, Liu L, Suen JY, Vesey DA, Fairlie DP. 2012. An antagonist of human protease activated receptor-2 attenuates PAR2 signaling, macrophage activation, mast cell degranulation, and collagen-induced arthritis in rats. FASEB J 26:2877–2887. doi: 10.1096/fj.11-201004. [DOI] [PubMed] [Google Scholar]
- 54.Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. 1994. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci U S A 91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Molino M, Woolkalis MJ, Reavey-Cantwell J, Pratico D, Andrade-Gordon P, Barnathan ES, Brass LF. 1997. Endothelial cell thrombin receptors and PAR-2. Two protease-activated receptors located in a single cellular environment. J Biol Chem 272:11133–11141. [DOI] [PubMed] [Google Scholar]
- 56.Déry O, Corvera CU, Steinhoff M, Bunnett NW. 1998. Proteinase-activated receptors: novel mechanisms of signaling by serine proteases. Am J Physiol 274:C1429–C1452. [DOI] [PubMed] [Google Scholar]
- 57.Giacaman RA, Asrani AC, Ross KF, Herzberg MC. 2009. Cleavage of protease-activated receptors on an immortalized oral epithelial cell line by Porphyromonas gingivalis gingipains. Microbiology 155:3238–3246. doi: 10.1099/mic.0.029132-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thurlow LR, Thomas VC, Narayanan S, Olson S, Fleming SD, Hancock LE. 2010. Gelatinase contributes to the pathogenesis of endocarditis caused by Enterococcus faecalis. Infect Immun 78:4936–4943. doi: 10.1128/IAI.01118-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thomas VC, Hiromasa Y, Harms N, Thurlow L, Tomich J, Hancock LE. 2009. A fratricidal mechanism is responsible for eDNA release and contributes to biofilm development of Enterococcus faecalis. Mol Microbiol 72:1022–1036. doi: 10.1111/j.1365-2958.2009.06703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.