

Abstract
This study aimed to characterize the role of Pseudomonas aeruginosa low-molecular-mass penicillin-binding proteins (LMM PBPs), namely, PBP4 (DacB), PBP5 (DacC), and PBP7 (PbpG), in peptidoglycan composition, β-lactam resistance, and ampC regulation. For this purpose, we constructed all single and multiple mutants of dacB, dacC, pbpG, and ampC from the wild-type P. aeruginosa PAO1 strain. Peptidoglycan composition was determined by high-performance liquid chromatography (HPLC), ampC expression by reverse transcription-PCR (RT-PCR), PBP patterns by a Bocillin FL-binding test, and antimicrobial susceptibility by MIC testing for a panel of β-lactams. Microscopy and growth rate analyses revealed no apparent major morphological changes for any of the mutants compared to the wild-type PAO1 strain. Of the single mutants, only dacC mutation led to significantly increased pentapeptide levels, showing that PBP5 is the major dd-carboxypeptidase in P. aeruginosa. Moreover, our results indicate that PBP4 and PBP7 play a significant role as dd-carboxypeptidase only if PBP5 is absent, and their dd-endopeptidase activity is also inferred. As expected, the inactivation of PBP4 led to a significant increase in ampC expression (around 50-fold), but, remarkably, the sequential inactivation of the three LMM PBPs produced a much greater increase (1,000-fold), which correlated with peptidoglycan pentapeptide levels. Finally, the β-lactam susceptibility profiles of the LMM PBP mutants correlated well with the ampC expression data. However, the inactivation of ampC in these mutants also evidenced a role of LMM PBPs, especially PBP5, in intrinsic β-lactam resistance. In summary, in addition to assessing the effect of P. aeruginosa LMM PBPs on peptidoglycan structure for the first time, we obtained results that represent a step forward in understanding the impact of these PBPs on β-lactam resistance, apparently driven by the interplay between their roles in AmpC induction, β-lactam trapping, and dd-carboxypeptidase/β-lactamase activity.
INTRODUCTION
Pseudomonas aeruginosa is a frequent cause of nosocomial infections, especially affecting patients in intensive care units (ICUs) with mechanical ventilation-associated pneumonia or burn wound infections, both of which are associated with a high mortality rate (1). This pathogen is also the major cause of chronic respiratory infections in patients with cystic fibrosis and other underlying chronic respiratory diseases (2). One of the most striking features of P. aeruginosa is its extraordinary capacity for developing resistance to almost any available antibiotic by the selection of mutations in chromosomal genes (3). Among the mutation-mediated β-lactam resistance mechanisms, particularly noteworthy are those leading to the constitutive overexpression of the inducible chromosomal cephalosporinase AmpC, which confers resistance to penicillins, cephalosporins, and monobactams (4). Additionally, mutations that lead to the repression or inactivation of the porin OprD, acting synergistically with inducible or constitutively overexpressed AmpC, confer resistance to carbapenems (5, 6).
AmpC is a group I class C β-lactamase that hydrolyzes efficiently penicillins and cephalosporins but not carbapenems. An AmpC-produced phenotype can be plasmidic or chromosomal. In P. aeruginosa, ampC is chromosomally carried and can be induced by certain β-lactams, such as cefoxitin and carbapenems, which are designated AmpC inducers (7, 8). AmpC expression is tightly linked to peptidoglycan recycling and involves multiple enzymes, including the AmpG permease, AmpD amidase homologs (AmpD, AmpDh2, and AmpDh3), NagZ, and the LysR superfamily transcriptional regulator AmpR. Additionally, two competing AmpR-binding muropeptides, the UDP-MurNAc-pentapeptides (AmpC repressors) and the 1,6-anhydromuropeptides (AmpC inducers), play a major role in the regulation of AmpC expression (9, 10). In the absence of β-lactams, GlcNAc-N-acetylmuramic acid (MurNAc)–1,6-anhydromuropeptides are shed from the peptidoglycan and find their way via AmpG permease to the cytoplasm. In the cytoplasm, they are processed by the β-N-acetylglucosaminidase NagZ to generate MurNAc–1,6-anhydromuropeptides (11). These peptides replace the repressor UDP-MurNAc-pentapeptides from the AmpR-binding site, which in turn undergoes a conformational change that leads to AmpC induction (12). On the other hand, AmpD eliminates peptide stems (tri-, tetra-, and pentapeptides) from anhydromuropeptides. This reaction results in the repression of ampC expression, because it cleaves the inducer anhydromuropeptides and generates the peptidoglycan recycling components needed for the synthesis of the repressor UDP-MurNAc-pentapeptides. However, during exposure to AmpC-inducing β-lactams, MurNAc–1,6-anhydromuropeptides accumulate in the cytoplasm, leading to AmpC induction (13, 14).
The classical mechanisms of ampC overexpression include the mutational inactivation of AmpD or specific mutations in the ampR-ampC intergenic region or in AmpR itself (4, 15). More recently, mutations of the nonessential dacB gene encoding the dd-carboxypeptidase PBP4 were found to frequently determine AmpC overexpression and high-level β-lactam resistance in vitro and among P. aeruginosa clinical strains (16). Interestingly, mutations of P. aeruginosa PBP4 were also shown to lead to the activation of the CreBC-BlrAB two-component regulator that also plays a significant role in β-lactam resistance. Moreover, BlrAB is the regulator of several β-lactamases in Aeromonas spp., and recent studies suggest that the disruption of DacB triggers activation of the system through the elevation of the monomer-disaccharide-pentapeptide levels (17). Further recent studies show that several other P. aeruginosa enzymes involved in cell wall metabolism, including UDP-N-acetylmuramate:l-alanyl-γ-d-glutamyl-meso-diaminopimelate ligase (Mpl), NADH dehydrogenase I chain N (NuoN), and several lytic transglycosylases (SltB1 and MltB), may also have an effect on ampC expression (18, 19). Indeed, the targeting of β-lactamase expression pathways, particularly through the inhibition of NagZ or AmpG, has been proposed as a useful approach to combat β-lactam resistance in P. aeruginosa and other AmpC-producing Gram-negative rods (6, 20–22).
Penicillin-binding proteins (PBPs) are a group of periplasmic enzymes responsible for polymerization, cross-linking, and modification of the bacterial peptidoglycan (23, 24). Peptidoglycan is the sacculus envelope outside the cytoplasmic membrane. It maintains cell shape and strength against intracellular pressure (23, 25). According to their molecular structure, PBPs are classified into high-molecular-mass (HMM) and low-molecular-mass (LMM) PBPs (3). All of them have a penicillin-binding domain. HMM PBPs were further classified into class A (e.g., PBP1) and class B (e.g., PBP2 and PBP3). They are responsible for peptidoglycan polymerization, cross-linking, and insertion of the peptidoglycan precursors into the preexisting strands through transglycosylation and transpeptidation reactions (3). LMM PBPs were grouped as class C PBPs and subdivided into 4 subgroups (types 4, 5, 7 and AmpH) with reference to Escherichia coli (3). Type 4 class C PBPs (e.g., PBP4) have endopeptidase and carboxypeptidase activity. Type 5 class C PBPs (e.g., PBP5) are the main dd-carboxypeptidases. Type 7 class C PBPs (e.g., PBP7) are dd-endopeptidases. Type AmpH class C PBPs (e.g., AmpH) have been characterized in E. coli as bifunctional dd-carboxypeptidase and dd-endopeptidase enzymes with a structure similar to that of class C β-lactamases (3). PBP5 is the most abundant LMM PBP of E. coli, and it has an important role in the control of cell diameter and correct septum formation (3). Recent studies suggest that E. coli LMM PBPs, particularly PBP5, play a role in intrinsic β-lactam resistance (26, 27). Moreover, the recently crystalized P. aeruginosa PBP5 shows certain β-lactamase activities, adding further interest to the role of LMM PBPs in antibiotic resistance (28).
In the light of all these findings, the objective of the present work was to systematically investigate the potential roles of the main P. aeruginosa LMM PBPs (DacB, PBP4; DacC, PBP5; and PbpG, PBP7) in peptidoglycan structure, β-lactam resistance, and AmpC regulation. For this purpose, all possible combinations of single and multiple LMM PBPs and AmpC mutants were generated and analyzed.
MATERIALS AND METHODS
Construction of P. aeruginosa strain PAO1 knockout mutants.
The strains and plasmids used and constructed in this study are listed in Table S1 in the supplemental material. The conditions for knockout constructions were adapted from those described by Moya et al. (16) based on the cre-lox system for gene deletion and antibiotic resistance marker recycling in P. aeruginosa (29). Previously constructed plasmids (pEXTΔampC::Gm and pEXTΔdacB::Gm) were used for the generation of dacB and ampC mutants (16, 30). For the construction of plasmids for dacC or pbpG inactivation, the PCR products (using PAO1 DNA as the template) of the upstream and downstream sequences (see Table S2 in the supplemental material) were digested with either BamHI or EcoRI and HindIII and cloned by three-way ligation into pEX100Tlink with the HindIII site deleted and opened by EcoRI and BamHI. The resulting plasmids (pEXTΔdacC and pEXTΔpbpG) were transformed into the E. coli XL1-Blue strain. Transformants were selected in 30 μg/ml ampicillin LB agar plates. The lox-flanked gentamicin resistance cassette (aac1) obtained by HindIII restriction of plasmid pUCGmlox was cloned into the single site for this enzyme, formed by the ligation of the two flanking fragments. The resulting plasmids (pEXTΔdacC::Gm and pEXTΔpbpG::Gm) were again transformed into E. coli XL1-Blue. Transformants were selected in 30 μg/ml ampicillin-5 μg/ml gentamicin LB agar plates. The plasmids were then transformed into the E. coli S17-1 helper strain. Knockout mutants were generated by conjugation, followed by the selection of double recombinants using 5% sucrose-1 μg/ml cefotaxime-30 μg/ml gentamicin LB agar plates. Double recombinants were checked by first screening for carbenicillin (200 μg/ml) susceptibility and afterwards by PCR amplification and sequencing. For the recycling of the gentamicin resistance cassettes, plasmid pCM157 was electroporated into the different mutants. Transformants were selected in LB agar plates with 250 μg/ml tetracycline. One transformant for each mutant was grown overnight in 250 μg/ml tetracycline LB broth to allow the expression of the cre recombinase. Plasmid pCM157 was then cured from the strains by successive passages in LB broth. Selected colonies were then screened for their tetracycline (250 μg/ml) and gentamicin (30 μg/ml) susceptibilities and checked by PCR amplification and DNA sequencing. Double, triple, and quadruple mutants were then generated sequentially, using the same procedure.
ampC expression.
The expression of the gene encoding P. aeruginosa AmpC (ampC) was determined by real-time reverse transcription-PCR (RT-PCR) for the constructed mutants and PAO1 (as a control), according to previously described protocols (31). For the quantification of ampC induction, the strains were incubated in the presence of 50 μg/ml cefoxitin. Briefly, total RNA from logarithmic-phase-grown LB cultures was obtained with an RNeasy minikit (Qiagen, Hilden, Germany). Fifty nanograms of purified RNA was then used for one-step reverse transcription and real-time PCR using a QuantiTect SYBR green reverse transcription-PCR kit (Qiagen) in an Eco real-time PCR system (Illumina, Inc.). Previously described conditions and primers were used (31). The rpsL housekeeping gene was used to normalize the expression levels, and the results were always referenced against PAO1 basal expression. All RT-PCRs were performed in duplicate, and the mean values of mRNA expression resulting from three independent experiments were considered in all cases.
Antimicrobial susceptibility testing.
The MICs of ampicillin, piperacillin, aztreonam, cefotaxime, ceftazidime, cefepime, cefoxitin, imipenem, meropenem, and vancomycin were determined by microdilution in 100 μl of cation-adjusted Mueller-Hinton broth, according to Clinical and Laboratory Standards Institute (CLSI) guidelines (32). Vancomycin permeates the outer membranes of Gram-negative bacteria very slowly because of its large size, which demonstrates that these microorganisms show intrinsic clinical resistance to this antibiotic. However, vancomycin can still kill Gram-negative bacteria at a clinically unobtainable concentration through the same mechanism by which it kills Gram-positive bacteria: binding to the terminal d-alanine–d-alanine of muropentapeptides in peptidoglycan. Thus, vancomycin MICs can be used as markers of the peptidoglycan pentapeptide levels (17).
Preparation of peptidoglycan and analysis of muropeptides.
Well-established previously described procedures were used for peptidoglycan preparation (33, 34). The wild type and the different mutants of P. aeruginosa PAO1 were cultured in LB medium treated with and without 50 μg/ml cefoxitin (FOX) at 37°C and 180 rpm agitation until an optical density at 600 nm (OD600) of ∼0.75 to 0.8 was achieved. The cells were then collected by centrifugation at 5,000 rpm and 4°C and resuspended in 1× phosphate-buffered saline (PBS) buffer (pH 7.5). One fraction from this cell suspension was left at −20°C for membrane preparation (see below). The rest of cell suspension was added drop by drop to an equal volume of boiling 6% SDS solution with strong stirring. The final cell-SDS suspension was left under boiling conditions for 12 h with stirring. The cell-SDS suspensions were centrifuged at 60,000 rpm for 10 min to collect the sacculi from the pellet fraction, which was then washed with warm sterile Milli-Q water at least three times. Peptidoglycan was suspended in 10 ml of 10 mM Tris-HCl (pH 7.2) and digested with 100 μg/ml α-amylase (Sigma-Aldrich, St. Louis, MO) for 1 h at 37°C and then with 100 μg/ml preactivated pronase E (Merck, Darmstadt, Germany) at 60°C for 90 min. The enzymes were inactivated by boiling for 20 min in 1% (final concentration) SDS. Next, peptidoglycan was collected and washed as described above. After that, peptidoglycan was digested with 100 μg/ml Cellosyl muramidase (Hoechst AG, Frankfurt, Germany) in 50 mM phosphate buffer (pH 4.9) at 37°C overnight. Next, the enzyme was inactivated by boiling the sample for 10 min in a water bath and centrifuged at 14,000 rpm for 5 min to remove insoluble debris. The supernatant was mixed with 1/3 volume of 0.5 M sodium borate buffer (pH 9.0) and reduced with excess sodium borohydride (NaBH4) for 30 min at room temperature. The pH was tested with pH indicator strips (Acilit; Merck) and adjusted to pH 3 with orthophosphoric acid. All samples were filtered (Millex-GV filters, 0.22-μm pore size, 2.5-mm diameter; Millipore, Cork, Ireland) and injected into the HPLC. Separations were performed on a Breeze 2 HPLC system, consisting of a 1525 binary HPLC pump model code 5CH (Waters), a UV-visible detector 2489 (Waters), a manual injector model 7725i (Rheodyne), and an Aeris Peptide XB-C18, 3.6 μm, 250 by 4.6 mm reverse-phase column (Phenomenex). Separation of individual components (muropeptides) of peptidoglycan was performed in a linear gradient, the column was equilibrated at 45°C, and the eluted compounds were detected at a wavelength of 204 nm. The mobile-phase (A = 50 mM sodium phosphate [pH 4.35]; B = 75 mM sodium phosphate, 15% methanol [pH 4.95]) gradient consisted of elution at 1.0 ml/min with 100% A for 5 min, followed by a 60-min linear gradient to 0% A/100% B and then 100% B for 5 min.
The identification of individual muropeptides was carried out according to retention time, using a comparison analysis with the retention times of known muropeptides. When a difference was found in the retention time of a particular peak, this peak was purified, and the structure was confirmed or characterized by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry with the autoflex spectrometer (Bruker Daltonics). Finally, the relative abundances of muropeptides present in each sample were determined by integrating their respective areas of absorption (Breeze 2, Waters program) and expressed as the molar fraction (mol%) relative to the total content. The average values from three biological replicates, showing in all cases a variation of ≤5%, are shown.
Cell membrane preparation for Bocillin FL-binding test.
The frozen fractions of cell suspension (during peptidoglycan preparation) of the different PAO1 mutants were thawed, sonicated, and centrifuged at 265,000 × g for 40 min using the TL-100 ultracentrifuge at 4°C. The pellet was resuspended in 1× PBS (pH 7.5) and used for the Bocillin FL-binding test. In order to avoid possible Bocillin FL degradation by the presence of AmpC in the membrane fractions, an alternative protocol that included several washing steps was also performed. Briefly, 500-ml late-log-phase (OD600, 1) LB cultures were collected by centrifugation and then washed and suspended in 50 ml of 20 mM KH2PO4-140 mM NaCl (pH 7.5). The cells were then sonicated and centrifuged at 12,000 × g for 10 min. Membranes containing the PBPs were isolated from the supernatant through one step of ultracentrifugation at 150,000 × g and 4°C for 1 h, followed by two washing steps, using an Optima L-XP series preparative ultracentrifuge (Beckman Coulter, Inc., Palo Alto, CA).
Bocillin FL-binding test.
Previously described procedures (33) were used for the Bocillin FL-binding test, with some modifications. Briefly, 100 μg of membrane proteins was incubated with 10 μM Bocillin FL (Invitrogen, Carlsbad, CA) in 1× PBS (pH 7.5) at 37°C for 30 min. Next, a proper volume of loading sample buffer was added. The samples were left at 100°C for 10 min, centrifuged using an Eppendorf centrifuge at maximum speed for 5 min, and loaded to 8% acrylamide gels in an SDS-PAGE system and run at 90 V. After the run, the gels were left in fixing solution (10% methanol and 7% acetic acid) for 1 to 2 h and then visualized on a Typhoon 9410 variable-mode imager (General Electric) at 588 nm, with a 520 BP 40 emission filter. For the determination of cefoxitin 50% inhibitory concentrations (IC50) for the different PBPs, 100 μg of membrane proteins was incubated first with serial concentrations from 0 to 1,500 μg/ml cefoxitin at 37°C for 30 min, and then they were incubated with 20 μM Bocillin FL at 37°C for 30 min and processed as described above. The IC50 was calculated as the cefoxitin concentration producing a 50% reduction in Bocillin FL binding for each individual PBP.
Cell preparation for microscopic examination.
Overnight cultures of PAO1 wild-type and mutant strains were used to inoculate fresh LB medium and left to grow at 37°C and 180 rpm for about 8 h. The optical density at 600 nm was measured every 1 h with a U-2000 spectrophotometer (Hitachi). Also, at different time intervals, the cell morphology was tested in vivo using fluorescence resonance energy transfer (FRET) equipment comprising an Axiovert 200 inverted microscope (Zeiss) coupled to a monochrome CCD camera.
RESULTS
Construction of single and combined mutants in the three LMM PBPs and AmpC of P. aeruginosa.
In order to evaluate the role of the three P. aeruginosa LMM PBPs (DacB [PBP4], DacC [PBP5], and PbpG [PBP7]) and AmpC in β-lactam resistance, ampC expression, and peptidoglycan structure, the four single, six double, four triple, and two quadruple mutants were generated using the cre-lox system, as described in Materials and Methods. The PAO ΔdacB and PAO ΔampC single mutants were available from previous studies (16), while the PAO ΔpbpG and PAO ΔdacC mutants were constructed from wild-type PAO1 in this work. The PAO ΔdacB ΔdacC, PAO ΔdacB ΔpbpG, and PAO ΔdacB ΔampC double mutants were generated from PAO ΔdacB, while PAO ΔdacC ΔampC and PAO ΔpbpG ΔampC were constructed from PAO ΔampC, and PAO ΔdacC ΔpbpG was constructed from PAO ΔpbpG. The PAO ΔdacC ΔpbpG ΔampC triple mutant was constructed from PAO ΔdacC ΔpbpG and PAO ΔdacB ΔdacC ΔampC from PAO ΔdacB ΔdacC; both PAO ΔdacB ΔdacC ΔpbpG and PAO ΔdacB ΔpbpG ΔampC were generated from PAO ΔdacB ΔpbpG. Finally, the quadruple mutants were constructed in two ways, with PAO ΔdacB ΔpbpG ΔampC ΔdacC constructed from PAO ΔdacB ΔpbpG ΔampC and PAO ΔdacB ΔdacC ΔpbpG ΔampC constructed from PAO ΔdacB ΔdacC ΔpbpG. Although it was not a primary objective of this work, microscopy and growth rate analyses revealed no apparent major changes compared to wild-type PAO1, even for the quadruple mutants (not shown).
The Bocillin FL-binding patterns of PBPs (1a, 1b, 2, 3, 4 [DacB], 5 [DacC], and 7 [PbpG]) were checked through SDS-PAGE of membrane extracts from the different mutants (Fig. 1A). The observed patterns correlated well with the loss of the expected PBP for each mutant. However, as revealed in Fig. 1A, the band corresponding to DacC was almost absent in the PAO ΔdacB and PAO ΔdacB ΔpbpG mutants. On the other hand, the DacC band was present in the PAO ΔdacB ΔampC and PAO ΔdacB ΔpbpG ΔampC mutants. Therefore, these results suggested that the large amounts of AmpC produced by DacB mutants (see below) significantly compromised the Bocillin FL concentration required for DacC visualization. To confirm this hypothesis, the cell membrane preparation protocol was modified to include additional washing steps to avoid the contamination of the membrane fractions with AmpC. Indeed, as shown in Fig. 1B, the expression of DacC was not modified in the dacB, pbpG, or ampC mutants. Moreover, DNA sequencing and gene expression analysis (RT-PCR) revealed no modification of dacC in these mutants (not shown).
FIG 1.
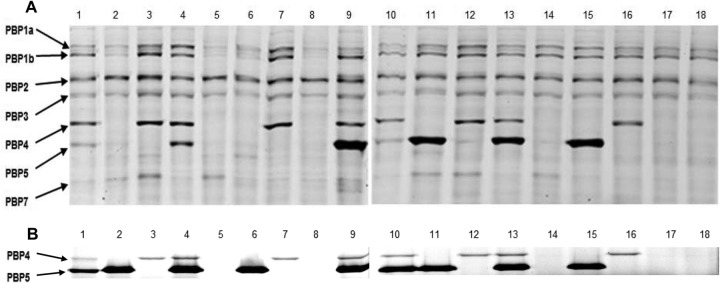
Bocillin FL binding test of PAO1 wild-type and derived mutants. (A) Conventional cell membrane preparation protocol. (B) Modified protocol to avoid AmpC contamination of cell membrane preparations leading to Bocillin FL hydrolysis. The PBP pattern (at left) of all the constructed P. aeruginosa mutants and the wild-type PAO1 (lanes 1 to 18) were visualized by fluorescence scanning using the Typhoon 9410 variable-mode imager at 588 nm, with a 520 BP 40 emission filter, after an SDS-PAGE run of the reaction samples in 8% acrylamide gels, in which each reaction involved an incubation of 100 μg of cell membrane protein with 10 μM Bocillin FL at 37°C for 30 min. Lanes 1 and 10, wild-type PAO1; lane 2, PAO ΔdacB; lane 3, PAO ΔdacC; lane 4, PAO ΔpbpG; lane 5, PAO ΔdacB ΔdacC; lane 6, PAO ΔdacB ΔpbpG; lane 7, PAO ΔdacC ΔpbpG; lane 8, PAO ΔdacB ΔdacC ΔpbpG; lane 9, PAO ΔampC; lane 11, PAO ΔdacB ΔampC; lane 12, PAO ΔdacC ΔampC; lane 13, PAO ΔpbpG ΔampC; lane 14, PAO ΔdacB ΔdacC ΔampC; lane 15, PAO ΔdacB ΔpbpG ΔampC; lane 16, PAO ΔdacC ΔpbpG ΔampC; lane 17, PAO ΔdacB ΔpbpG ΔampC ΔdacC; and lane 18, PAO ΔdacB ΔdacC ΔpbpG ΔampC.
Role of P. aeruginosa LMM PBPs in ampC expression.
The basal and cefoxitin-induced ampC expression levels for all the single and combined LMM PBP mutants of PAO1 are shown in Table 1. In agreement with previous data (16), the inactivation of DacB caused a marked (47-fold) increase in basal ampC expression. On the other hand, the inactivation of DacC or PbpG did not cause a significant modification of either basal or induced ampC expression levels. Likewise, the DacB-PbpG double mutant did not show modified ampC expression compared to that of the DacB mutant. In contrast, the inactivation of DacC in the DacB mutant caused a further major increase in basal ampC expression (478-fold compared to PAO1 and 10-fold compared to the DacB single mutant). Moreover, the basal and induced ampC expression levels were highest in the DacB-DacC-PbpG triple mutant, reaching levels >1,000-fold higher than those of PAO1.
TABLE 1.
MICs and ampC expression under basal and cefoxitin induction conditions for all studied mutants
| Strain or mutant | MIC (μg/ml) fora: |
ampC expressionb |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AMP | PIP | ATM | CTX | CAZ | CEF | FOX | IMI | MER | VAN | Basal | Induced | |
| PAO1 | 1,024 | 2 | 4 | 12 | 1 | 1 | 1,024 | 0.5 | 0.5 | 512 | 347 ± 59 | |
| PAO ΔampC | 32 | 2 | 4 | 8 | 1 | 1 | 64 | 0.12 | 0.25 | 512 | ||
| PAO ΔdacB | 1,024 | 16 | 8 | 256 | 8 | 4 | 1,024 | 1 | 0.5 | 512 | 47 ± 29 | 569 ± 166 |
| PAO ΔdacB ΔampC | 32 | 2 | 2 | 8 | 1 | 0.5 | 96 | 0.12 | 0.25 | 512 | ||
| PAO ΔdacC | 1,536 | 4 | 2 | 8 | 0.75 | 0.5 | 1,024 | 0.5 | 0.5 | 1,024 | 1.3 ± 0.4 | 542 ± 380 |
| PAO ΔdacC ΔampC | 16 | 2 | 2 | 4 | 0.75 | 0.5 | 64 | 0.06 | 0.25 | 1,024 | ||
| PAO ΔpbpG | 512 | 4 | 4 | 16 | 1 | 1 | 1,024 | 1 | 0.5 | 512 | 0.6 ± 0.3 | 305 ± 152 |
| PAO ΔpbpG ΔampC | 32 | 4 | 3 | 8 | 1 | 0.5 | 96 | 0.12 | 0.25 | 512 | ||
| PAO ΔdacB ΔdacC | 1,024 | 128 | 16 | 512 | 16 | 4 | 1,024 | 0.5 | 0.5 | 2,048 | 478 ± 5.1 | 840 ± 245 |
| PAO ΔdacB ΔdacC ΔampC | 16 | 2 | 2 | 4 | 1 | 0.5 | 64 | 0.06 | 0.12 | 4,096 | ||
| PAO ΔdacB ΔpbpG | 2,048 | 16 | 8 | 256 | 8 | 4 | 1,024 | 0.5 | 0.25 | 512 | 45 ± 32 | 326 ± 106 |
| PAO ΔdacB ΔpbpG ΔampC | 32 | 2 | 2 | 6 | 0.75 | 0.5 | 64 | 0.12 | 0.5 | 4,096 | ||
| PAO ΔdacC ΔpbpG | 1,024 | 6 | 2 | 8 | 1 | 0.5 | 1,024 | 0.5 | 0.25 | 1,024 | 1.4 ± 0.7 | 162 ± 87 |
| PAO ΔdacC ΔpbpG ΔampC | 24 | 3 | 2 | 4 | 0.75 | 0.5 | 64 | 0.06 | 0.25 | 1,024 | ||
| PAO ΔdacB ΔdacC ΔpbpG | 1,024 | 128 | 16 | 512 | 16 | 4 | 1,024 | 0.5 | 0.5 | 4,096 | 1,207 ± 193 | 5,742 ± 1,975 |
| PAO ΔdacB ΔpbpG ΔampC ΔdacC | 16 | 2 | 2 | 4 | 0.5 | 0.5 | 64 | 0.06 | 0.12 | 4,096 | ||
| PAO ΔdacB ΔpbpG ΔdacC ΔampC | 16 | 2 | 1 | 4 | 0.5 | 0.5 | 64 | 0.06 | 0.12 | 4,096 | ||
AMP, ampicillin; PIP, piperacillin; ATM, aztreonam; CTX, cefotaxime; CAZ, ceftazidime; CEF, cefepime; FOX, cefoxitin; IMI, imipenem; MER, meropenem; VAN, vancomycin. The median values from 3 experiments are shown.
Relative ampC expression (with respect to wild-type PAO1) without induction (basal) and after induction with 50 μg/ml cefoxitin (induced). The mean values from three independent experiments ± standard deviation are shown.
Role of P. aeruginosa LMM PBPs in β-lactam resistance.
In agreement with previous data (16), the inactivation of DacB caused a marked increase in the MICs for the antipseudomonal penicillins (piperacillin), cephalosporins (cefotaxime, ceftazidime, and cefepime), and monobactams (aztreonam), which was consistent with the documented AmpC hyperproduction. As was also expected, the MICs of strong AmpC-inducing β-lactams, including the carbapenems (imipenem and meropenem), cefoxitin, and ampicillin, were barely modified. On the other hand, the inactivation of DacC or PbpG did not cause a significant modification of the MIC for any β-lactam, with the exception of a slight decrease in piperacillin susceptibility in the DacC mutant. Consistent with the ampC expression data, the MICs for antipseudomonal penicillins, cephalosporins, and monobactams were further increased in the DacB-DacC double mutant. Of all β-lactams tested, the highest MIC increase, compared to the DacB single mutant, was documented for piperacillin. On the other hand, unlike for ampC expression, β-lactam resistance was not further increased in the DacB-DacC-PbpG triple mutant (Table 1).
To determine the direct effect of the inactivation of the LMM PBPs on β-lactam resistance, susceptibility testing was also performed with all combinations of LMM PBPs and AmpC mutants (Table 1). As expected, the inactivation of AmpC in wild-type PAO1 produced a marked increase in the susceptibility of strong AmpC-inducing β-lactams, including the carbapenems, cefoxitin, and ampicillin, whereas the MICs of weak AmpC-inducing β-lactams (antipseudomonal penicillins, cephalosporins, and monobactams) were not significantly modified. Interestingly, the MICs for nearly all β-lactams were lower in the DacC-AmpC mutant than those in the AmpC single mutant, and this effect was further enhanced in the DacB-DacC-PbpG-AmpC mutant, indicating that LMM PBPs, particularly DacC, play a role in the intrinsic level of β-lactam resistance in P. aeruginosa.
Role of P. aeruginosa LMM PBPs in peptidoglycan composition.
The composition of the peptidoglycan of all PAO1 mutants was studied through muropeptide HPLC analysis, and the results are shown in Table 2; representative chromatograms of peptidoglycan muropeptides of the constructed mutants are also shown in Fig. 2 and Table S3 in the supplemental material. No major differences in DAP-DAP cross-linked and lipoprotein-binding muropeptides, both of them due to ld-transpeptidase activity, were found. Also, no major changes in peptidoglycan composition were observed for any of three LMM PBP single mutants, with the exception of a significant increase (4.4-fold) in the pentapeptide levels in the DacC mutant. Therefore, these results suggest that DacC is the primary dd-carboxypeptidase of P. aeruginosa. Moreover, pentapeptide levels were significantly increased (17.9-fold) in the DacB-DacC double mutant and still further enhanced (41.5-fold) in the DacB-DacC-PbpG triple mutant. Thus, these results indicate that DacB plays a significant role as a dd-carboxypeptidase when DacC is absent, and the dd-carboxypeptidase activity of PbpG is apparent only when both DacC and DacB are inactivated. Moreover, as expected, the pentapeptide levels correlated well with the MICs of vancomycin (Table 1), a glycopeptide that specifically binds to the d-Ala-d-Ala residues of pentapeptides.
TABLE 2.
HPLC analysis of muropeptides prepared from the peptidoglycan of the different mutants
| Strain or mutant | Relative abundance (mol%) of muropeptidea: |
Cross-linkb | D-D/Tc | Peptidoglycan lengthd | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mono | Di | Tri | D-D | Lpp | Anh | Penta | ||||
| PAO1 | 57.8 | 38.3 | 3.9 | 1.9 | 3.1 | 8.7 | 1.6 | 46.1 | 4.1 | 11.5 |
| PAO ΔdacB | 56.1 (1.0) | 39 (1.0) | 4.8 (1.2) | 1.5 (0.8) | 3.7 (1.2) | 9.6 (1.1) | 2.4 (1.5) | 48.9 (1.1) | 3.1 (0.8) | 10.5 (0.9) |
| PAO ΔdacC | 59.3 (1.0) | 37.1 (1.0) | 3.6 (0.9) | 1.1 (0.6) | 3.2 (1.0) | 9.1 (1.1) | 7.1 (4.4) | 44.4 (1.0) | 2.4 (0.6) | 11 (1.0) |
| PAO ΔpbpG | 58.7 (1.0) | 37.2 (1.0) | 4.1 (1.1) | 1.5 (0.8) | 2.8 (0.9) | 9.4 (1.1) | 1.4 (1.0) | 45.4 (1.0) | 3.2 (0.8) | 10.7 (0.9) |
| PAO ΔdacB ΔdacC | 58.4 (1.0) | 36.7 (1.0) | 4.8 (1.2) | 1.1 (0.6) | 3.4 (1.1) | 9.4 (1.1) | 28.6 (17.9) | 46.6 (1.0) | 2.3 (0.6) | 10.7 (0.9) |
| PAO ΔdacB ΔpbpG | 54.5 (0.9) | 39.5 (1.0) | 5.9 (1.5) | 1.5 (0.8) | 3.3 (1.1) | 14.2 (1.6) | 2.5 (1.6) | 51.6 (1.1) | 2.9 (0.7) | 7 (0.6) |
| PAO ΔdacC ΔpbpG | 55.9 (1.0) | 39.4 (1.0) | 4.6 (1.2) | 1.2 (0.6) | 3.3 (1.1) | 8.9 (1.0) | 9.8 (6.1) | 48.8 (1.1) | 2.6 (0.6) | 11.2 (1.0) |
| PAO ΔdacB ΔdacC ΔpbpG | 54.7 (1.0) | 40 (1.0) | 5.2 (1.3) | 1.3 (0.7) | 2.4 (0.8) | 7.6 (0.9) | 66.4 (41.5) | 50.6 (1.1) | 2.5 (0.6) | 13.2 (1.2) |
| PAO ΔampC | 59.9 (0.9) | 36.3 (1.0) | 3.8 (1.0) | 1.2 (0.6) | 3.3 (1.1) | 9 (1.0) | 2.5 (1.6) | 43.8 (1.0) | 2.8 (0.7) | 11.1 (1.0) |
| PAO ΔdacB ΔampC | 54.2 (1.0) | 40.3 (1.0) | 5.4 (1.4) | 2.1 (1.1) | 4.1 (1.3) | 10.3 (1.2) | 2.5 (1.6) | 51.2 (1.1) | 4.1 (1.0) | 9.8 (0.9) |
| PAO ΔdacC ΔampC | 59.7 (1.0) | 36.6 (1.0) | 3.7 (1.0) | 1.1 (0.6) | 3.2 (1.0) | 9.1 (1.0) | 8 (5.0) | 44.1 (1.0) | 2.4 (0.6) | 11 (1.0) |
| PAO ΔpbpG ΔampC | 56.4 (1.0) | 38.6 (1.0) | 4.9 (1.3) | 1.5 (0.8) | 3.7 (1.2) | 9.7 (1.1) | 2.5 (1.6) | 48.5 (1.1) | 3.1 (0.8) | 10.3 (0.9) |
| PAO ΔdacB ΔdacC ΔampC | 59.4 (1.0) | 36.1 (0.9) | 4.4 (1.1) | 1 (0.5) | 2.5 (0.8) | 7.9 (0.9) | 32 (20.0) | 45.2 (1.0) | 2 (0.5) | 12.6 (1.0) |
| PAO ΔdacB ΔpbpG ΔampC | 49.4 (0.9) | 43.5 (1.1) | 7 (1.8) | 1.7 (0.9) | 3.6 (1.2) | 11.3 (1.3) | 3.7 (2.3) | 57.8 (1.3) | 3 (0.7) | 8.8 (0.8) |
| PAO ΔdacC ΔpbpG ΔampC | 54.9 (1.0) | 39.9 (1.0) | 5.1 (1.3) | 1.8 (1.0) | 3.7 (119) | 9.4 (1.1) | 9.4 (5.9) | 50.4 (1.1) | 3.5 (0.9) | 10.7 (0.9) |
| PAO ΔdacB ΔdacC ΔpbpG ΔampC | 54.8 (1.0) | 40 (1.0) | 5.1 (1.3) | 0.6 (0.3) | 2.1 (68) | 7.2 (0.8) | 67.6 (42.2) | 50.4 (1.1) | 1.1 (0.3) | 13.9 (1.2) |
| PAO ΔdacB ΔpbpG ΔampC ΔdacC | 53 (0.9) | 40.4 (1.1) | 6.5 (1.7) | 1.5 (0.8) | 2.4 (77) | 8.8 (1.0) | 65.9 (41.2) | 53.6 (1.2) | 2.8 (0.7) | 11.4 (1.0) |
Mono, monomers; Di, dimers; Tri, trimers; D-D, muropeptides having Dap-Dap peptide bridges; Lpp, muropeptides bound to C-terminal Arg-Lys dipeptide of Braun's lipoprotein; Anh, muropeptides having anhydro-1,6-anhydromuramic acid; Penta, muropeptides having a pentapeptide stem. Values in parentheses represent the ratio of the values obtained for each mutant and wild-type PAO1.
Cross-link, degree of peptidoglycan cross-linking (percentage). Values in parentheses represent the ratio of the values obtained for each mutant and wild-type PAO1.
D-D/T, percent ratio of Dap-Dap cross-links to total peptidoglycan cross-links. Values in parentheses represent the ratio of the values obtained for each mutant and wild-type PAO1.
Values in parentheses represent the ratio of the values obtained for each mutant and wild-type PAO1.
FIG 2.

High-performance liquid chromatograms of peptidoglycan muropeptides of the wild-type and constructed PAO1 mutants. Each series displays peaks corresponding to the common muropeptides in peptidoglycan of the given PAO1 strain (indicated at right). Each peak corresponds to a muropeptide whose name (in bold) and retention time (RT) (in minutes) are indicated at the top. M3, disaccharide tripeptide; M4G, disaccharide tetrapeptide with Gly at position 4; M4, disaccharide tetrapeptide; M5, disaccharide pentapeptide in which l-Ala, d-Glu, Dap (meso-diaminopimelic acid), d-Ala, and d-Ala occupy positions 1, 2, 3, 4, and 5, respectively, and l-Ala is linked to N-acetylmuramic acid; M5G, disaccharide pentapeptide with Gly at position 5; D44, cross-linked dimer of disaccharide tetrapeptide-disaccharide tetrapeptide; D43, cross-linked dimer of disaccharide tetrapeptide-disaccharide tripeptide; D45, cross-linked dimer of disaccharide tetrapeptide-disaccharide pentapeptide; T444, cross-linked trimer of disaccharide tetrapeptide-disaccharide tetrapeptide-disaccharide tetrapeptide; T445, cross-linked trimer of disaccharide tetrapeptide-disaccharide tetrapeptide-disaccharide pentapeptide; anhydro-muropeptides D44N, D45N, T444N, and T445N have the same structures as muropeptides D44, D45, T444, and T445, respectively, but with anhydro-N-acetylmuramic acid instead of N-acetylmuramic acid. Each disaccharide is composed of N-acetylglucosamine and N-acetylmuramic acid.
Additionally, the peptidoglycan of the DacB-PbpG double mutant had more anhydromuropeptides and slightly higher cross-linking than those of the wild-type PAO1; these data should indicate the inhibition of dd-endopeptidase activity; this strongly suggests that both DacB and PbpG should also have this function and that they can complement each other (since only marginal effects are evidenced in the single mutants). No significant differences were found in the structure of the peptidoglycan of these mutants compared with those containing the ampC deletion (Table 2).
Effect of cefoxitin on peptidoglycan composition.
The effect of the exposure to the AmpC inducer cefoxitin on the composition of the peptidoglycan of all PAO1 mutants is shown in Table 3. Since cefoxitin induces AmpC expression and the β-lactamase efficiently degrades this antibiotic, information from the collection of AmpC mutants is of primary interest here (i.e., the absence of AmpC significantly enhances the effect of cefoxitin). Exposure to cefoxitin in the AmpC mutant (and, to a lower extent, also in wild-type PAO1) significantly increased pentapeptide levels (from 2.5 mol% to 14 mol%) but also resulted in fewer monomers (59.9 mol% versus 54.9 mol%), more dimers (36.3 mol% versus 40.3 mol%), and higher cross-linking (43.8% versus 50%). Therefore, these results strongly suggest that exposure to cefoxitin inhibits the dd-carboxypeptidase and dd-endopeptidase activities of LMM PBPs to some degree. To confirm and quantify the potency of such inhibition, cefoxitin IC50s were determined, and the results are shown in Table 4. As can be observed, of all the P. aeruginosa PBPs, the highest affinity (in the range of 1 to 2 μg/ml) was documented for DacB and PbpG. Although still <10 μg/ml, the affinity for DacC was significantly lower. As can be observed in Table 3, these results are consistent with the information obtained from the analysis of the peptidoglycan composition of the LMM PBP mutants exposed to cefoxitin. For instance, pentapeptide levels nearly reached the maximum values (>60 mol%) in the DacC-AmpC mutant, indicating that the dd-carboxypeptidase activity of DacB and PbpG is fully abolished upon cefoxitin exposure. On the other hand, the only significant change observed in the DacB mutant when exposed to cefoxitin is an increase in the anhydromuropeptide levels (from 9.6 mol% to 13.7 mol%), similar to what is observed for the DacB-PbpG double mutant in the absence of cefoxitin.
TABLE 3.
HPLC analysis of muropeptides prepared from the peptidoglycan of the different PAO1 mutants with FOX treatment
| Strain or mutant | Relative abundance (mol%) of muropeptidea: |
Cross-linkb | D-D/Tc | Peptidoglycan lengthd | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mono | Di | Tri | D-D | Lpp | Anh | Penta | ||||
| PAO1 | 55.5 (1.0) | 39.3 (1.0) | 5.1 (1.3) | 1.9 (1.0) | 3.7 (1.2) | 10.1 (1.2) | 4.4 (2.8) | 49.9 (1.1) | 3.7 (0.9) | 9.9 (0.9) |
| PAO ΔdacB | 57.1 (1.0) | 37.2 (1.0) | 5.6 (1.2) | 1.7 (1.1) | 3.4 (0.9) | 13.7 (1.4) | 3.7 (1.5) | 48.6 (1.0) | 3.5 (1.1) | 7.3 (0.7) |
| PAO ΔdacC | 55.2 (0.9) | 39.6 (1.1) | 5.1 (1.4) | 1.2 (1.1) | 2.5 (0.8) | 8.2 (0.9) | 38.8 (5.5) | 50.1 (1.1) | 2.3 (1.0) | 12.3 (1.1) |
| PAO ΔpbpG | 56.6 (1.0) | 37.9 (1.0) | 5.4 (1.3) | 1.9 (1.3) | 3.6 (1.3) | 8.9 (1.0) | 4.2 (3.0) | 49.0 (1.1) | 3.9 (1.2) | 11.3 (1.1) |
| PAO ΔdacB ΔdacC | 58.8 (1.0) | 36.7 (1.0) | 4.4 (0.9) | 1.1 (1.0) | 3.2 (0.9) | 8.1 (0.9) | 39.8 (1.4) | 45.8 (1.0) | 2.4 (1.0) | 12.4 (1.2) |
| PAO ΔdacB ΔpbpG | 51.0 (0.9) | 42.1 (1.1) | 6.8 (1.2) | 2.1 (1.4) | 4.9 (1.5) | 12.3 (0.9) | 4.0 (1.6) | 56.1 (1.1) | 3.7 (1.3) | 8.2 (1.2) |
| PAO ΔdacC ΔpbpG | 54.3 (1.0) | 40.3 (1.0) | 5.3 (1.2) | 1.0 (0.8) | 2.6 (0.8) | 7.5 (0.8) | 44.0 (4.5) | 51.3 (1.1) | 1.8 (0.7) | 13.4 (1.2) |
| PAO ΔdacB ΔdacC ΔpbpG | 54.8 (1.0) | 39.0 (1.0) | 6.0 (1.2) | 1.1 (0.9) | 3.1 (1.3) | 9.3 (1.2) | 66.0 (1.0) | 51.6 (1.0) | 2.1 (0.8) | 10.8 (0.8) |
| PAO ΔampC | 54.9 (1.0) | 40.3 (1.1) | 4.6 (1.2) | 1.3 (1.1) | 2.6 (0.8) | 7.2 (0.8) | 14.0 (5.6) | 50.0 (1.1) | 2.6 (0.9) | 14.0 (1.3) |
| PAO ΔdacB ΔampC | 55.8 (1.0) | 39.5 (1.0) | 4.5 (0.8) | 1.3 (0.6) | 3.4 (0.8) | 7.7 (0.8) | 16.5 (6.6) | 48.9 (1.0) | 2.7 (0.7) | 13.1 (1.3) |
| PAO ΔdacC ΔampC | 58.1 (1.0) | 37.0 (1.0) | 4.8 (1.3) | 1.4 (1.3) | 3.4 (1.1) | 7.5 (0.8) | 62.3 (7.8) | 46.9 (1.1) | 3.0 (1.3) | 13.3 (1.2) |
| PAO ΔpbpG ΔampC | 53.9 (1.0) | 40.6 (1.1) | 5.4 (1.1) | 1.8 (1.2) | 3.1 (0.8) | 8.2 (0.9) | 14.1 (5.6) | 51.7 (1.1) | 3.5 (1.2) | 12.3 (1.2) |
| PAO ΔdacB ΔdacC ΔampC | 56.9 (1.0) | 37.6 (1.0) | 5.3 (1.2) | 1.2 (1.2) | 2.6 (1.0) | 7.5 (1.0) | 63.1 (2.0) | 48.8 (1.1) | 2.5 (1.3) | 13.3 (1.1) |
| PAO ΔdacB ΔpbpG ΔampC | 53.4 (1.1) | 40.6 (0.9) | 5.8 (0.8) | 1.9 (1.1) | 3.6 (1.0) | 8.3 (0.7) | 14.7 (4.0) | 52.8 (0.9) | 3.6 (1.2) | 12.0 (1.4) |
| PAO ΔdacC ΔpbpG ΔampC | 55.8 (1.0) | 38.5 (1.0) | 5.5 (1.1) | 1.3 (0.7) | 2.8 (0.8) | 7.3 (0.8) | 64.6 (6.9) | 50.2 (1.0) | 2.5 (0.7) | 13.7 (1.3) |
| PAO ΔdacB ΔdacC ΔpbpG ΔampC | 57.0 (1.0) | 37.2 (0.9) | 5.6 (1.1) | 1.3 (2.2) | 2.7 (1.3) | 6.7 (0.9) | 63.1 (0.9) | 49.1 (1.0) | 2.7 (2.3) | 15.0 (1.1) |
| PAO ΔdacB ΔpbpG ΔampC ΔdacC | 57.2 (1.1) | 36.7 (0.9) | 5.9 (0.9) | 1.0 (0.7) | 2.0 (0.8) | 7.4 (0.8) | 65.5 (1.0) | 49.0 (0.9) | 1.9 (0.7) | 13.5 (1.2) |
Mono, monomers; Di, dimers; Tri, trimers; D-D, muropeptides having Dap-Dap peptide bridges; Lpp, muropeptides bound to C-terminal Arg-Lys dipeptide of Braun's lipoprotein; Anh, muropeptides having anhydro-1,6-anhydromuramic acid; Penta, muropeptides having a pentapeptide stem.
Cross-link, degree of peptidoglycan cross-linking (percentage). Values in parentheses represent the ratio of the values obtained for each strain with and without cefoxitin exposure.
D-D/T, percent ratio of Dap-Dap cross-links to total peptidoglycan cross-links. Values in parentheses represent the ratio of the values obtained for each strain with and without cefoxitin exposure.
Values in parentheses represent the ratio of the values obtained for each strain with and without cefoxitin exposure.
TABLE 4.
Estimated IC50s of cefoxitin for PAO1 and PAO ΔampC PBPs using Bocillin FL test
| Strain or mutant | IC50 of FOX (μg/ml) fora: |
||||||
|---|---|---|---|---|---|---|---|
| PBP1a | PBP1b | PBP2 | PBP3 | PBP4 (DacB) | PBP5 (DacC) | PBP7 (PbpG) | |
| PAO1 | 6.4 | 30.7 | 16.4 | 8 | 1.5 | 9.1 | <1.5 |
| PAO ΔampC | 4.8 | 27.2 | 15.4 | 7.7 | 1.3 | 6.5 | <1.5 |
Cefoxitin (FOX) concentration producing a 50% reduction in Bocillin FL binding for each individual PBP.
DISCUSSION
Role of P. aeruginosa LMM PBPs in cell wall physiology.
Previous analyses of the P. aeruginosa cell membrane identified eight proteins able to bind [3H]benzylpenicillin or 125I-ampicillin, (PBP1a, PBP1b, PBP2, PBP3, PBP3b, PBP4, PBP5, and PBP7) (35–37), and in silico analysis, using the Pseudomonas Genome Database (38), revealed the presence of eight open reading frames annotated as encoding potential penicillin-binding proteins. In this study, we used a fluorescence-labeled antibiotic (Bocillin FL) to identify PBPs of wild-type and mutant strains of PAO1. The PBP patterns of single- and multiple-deletion mutants correlated well with the loss of the expected PBP for each mutant (PBP4 [DacB], PBP5 [DacC], and PBP7 [PbpG]). These PBPs belong to the class C LMM PBP types 4, 5, and 7, respectively. All PBPs in these subclasses have dd-endopeptidase and/or dd-carboxypeptidase activity. The largest changes in peptidoglycan structure (increase in pentapeptide content) were observed for the DacB-DacC-PbpG triple mutant, with a structure similar to the nine-PBP deletion mutant of E. coli, in which all dd-endopeptidase and dd-carboxypeptidase activities were depleted, causing aberrant cellular morphology in E. coli (39). Therefore, these three PBPs must represent the major endolytic machinery of P. aeruginosa. The crystal structure of P. aeruginosa PBP5 (DacC) reveals a protein fold that is highly similar to the related E. coli PBP5 and PBP6 and also more closely resemble features seen previously only in the class A β-lactamases (28). Gram-negative bacteria most often have a major type 5 PBP, which is the most abundant PBP they produce (24). The most highly expressed PBP in P. aeruginosa membranes has been documented to be PBP5 (40), consistent with the results of our work. It was recently shown that PBP5 is a dd-carboxypeptidase that preferentially degrades low-molecular-weight substrates (28). In this work, we confirm that PBP5 is the major dd-carboxypeptidase in P. aeruginosa, as evidenced by the fact that of the three LMM PBP single mutants, only dacC mutation led to significantly increased pentapeptide levels. Moreover, our results indicate that DacB plays a significant role as dd-carboxypeptidase only when DacC is absent, and the dd-carboxypeptidase activity of PbpG is apparent only when both DacC and DacB are inactivated. On the other hand, the peptidoglycan structure of dacB and pbpG single and double mutants indicated that P. aeruginosa PBP4 and PBP7 have dd-endopeptidase activity, as previously suggested for E. coli (41). Moreover, our results are consistent with very recent data demonstrating that purified P. aeruginosa PBP4 shows both dd-carboxypeptidase and dd-endopeptidase activities (42).
No major effect on cell morphology or growth parameters was seen for any of the single, double, or triple mutants, suggesting that the major changes observed in the peptidoglycan structure do not affect significantly the morphology of the cell under laboratory conditions. In E. coli, it was reported that PBP5 inactivation was the only single mutation of LMM PBPs to produce an aberrant cellular shape; however, the further inactivation of PBP6 or PBP4 and PBP7 caused more deformation in cell morphology (43–45). In parallel with our findings within P. aeruginosa, it was found in E. coli that multiple mutants of all possible LMM PBPs did not affect their growth curves in LB medium at 37°C, and the cells were viable (39, 46). However, the in vivo role, and particularly the impact on virulence, of P. aeruginosa LMM PBPs still needs to be explored.
P. aeruginosa LMM PBPs and AmpC induction.
It is well known that exposure to certain β-lactams, such as cefoxitin or the carbapenems, leads to the induction of AmpC expression. Current models consider that the specific effects on the cell wall produced by subinhibitory concentrations of these drugs determine the accumulation of MurNAc-1,6-anhydromuropeptides in the cytoplasm, which replace UDP-MurNAc-pentapeptides from AmpR, leading to AmpC induction (9). Early studies found a correlation between the AmpC-inducing potency of β-lactams and their affinity for certain E. coli LMM PBPs, in particular PBP4 (47). Moreover, years later, we demonstrated that the inactivation of dacB, which encodes PBP4, is a major mechanism of AmpC overexpression in P. aeruginosa (16). These data suggested a major role of LMM PBPs in AmpC induction; however, the specific effects on the cell wall triggering the AmpC induction response are mostly unknown. A recent work using Aeromonas hydrophila as a model organism showed that PBP4 inactivation also led to β-lactamase overexpression, and this correlated with a 2-fold increase in peptidoglycan pentapeptide levels, presumably caused by reduced dd-carboxypeptidase activity (17). Our P. aeruginosa results show, however, that PBP5 is the major dd-carboxypeptidase, and the PBP5 mutant is the only LMM PBP single mutant producing a significant increase in pentapeptide levels (up to 4.4-fold higher than those of wild-type PAO1). Thus, increased peptidoglycan pentapeptide levels, or apparently any other effect on peptidoglycan structure (Table 2), does not explain, at least for P. aeruginosa, the major role of PBP4 in AmpC induction. Whether the PBP4 effect is driven by significantly increasing periplasmic soluble anhydromuropeptides levels leading to the activation of classical AmpC induction pathway needs still to be explored. This possibility is indeed supported by the fact that the phenotype requires a functional AmpG (6) and AmpR (16), and by very recent PBP4 catalytic data (42). In any case, our results suggest that increased peptidoglycan pentapeptide levels explain the major role of PBP5 in ampC expression when PBP4 is absent. Indeed, except for the specific effect of PBP4, a correlation between peptidoglycan pentapeptide levels and ampC expression was documented.
P. aeruginosa LMM PBPs and β-lactam resistance.
As could be anticipated, the MICs for the antipseudomonal penicillins (piperacillin), cephalosporins (cefotaxime, ceftazidime, and cefepime), and monobactams (aztreonam) correlated well with the ampC expression data (Table 1); they were significantly increased in the DacB mutant and further increased in the DacB-DacC double mutant. On the other hand, unlike for ampC expression, β-lactam resistance was not further increased in the DacB-DacC-PbpG triple mutant. Besides the obvious effect on resistance driven by the impact on ampC expression, we asked whether P. aeruginosa LMM PBPs had a direct effect on β-lactam susceptibility. For this purpose, we analyzed the β-lactam MICs for all combinations of LMM PBPs and AmpC mutants. As expected (5, 6), the inactivation of AmpC in wild-type PAO1 produced a marked increase in the susceptibility of strong AmpC-inducing β-lactams, including the carbapenems, cefoxitin, and ampicillin, whereas the MICs of weak AmpC-inducing β-lactams (antipseudomonal penicillins, cephalosporins, and monobactams) were not significantly modified. Remarkably, the MICs of nearly all β-lactams were lower for the DacC-AmpC mutant than those for the AmpC single mutant, and this effect was further enhanced in the DacB-DacC-PbpG-AmpC mutant, indicating that LMM PBPs, particularly DacC, play a role in the intrinsic level of β-lactam resistance in P. aeruginosa. Our results are therefore in agreement with recent studies suggesting that E. coli LMM PBPs, particularly PBP5, play a role in intrinsic β-lactam resistance (26, 27). Purified E. coli PBP5 failed to show significant β-lactamase activity, and therefore it was concluded that the role of this PBP in intrinsic β-resistance might be a consequence of β-lactam trapping. However, interestingly, the recently crystalized P. aeruginosa PBP5 does show certain broad-spectrum (including to penicillins, cephalosporins, and carbapenems) β-lactamase activity (28). Therefore, the observed effect of PBP5 in P. aeruginosa intrinsic resistance is expected to result from both trapping and hydrolysis of β-lactams.
In summary, we have assessed for the first time the effect of P. aeruginosa LMM PBPs in peptidoglycan structure, defining PBP5 as the major dd-carboxypeptidase, compensated for, if absent, by PBP4 and PBP7, which additionally show dd-endopeptidase activity. Moreover, our results represent a step forward in understanding the impact of LMM PBPs in β-lactam resistance, apparently driven by the interplay between their effects on AmpC induction, β-lactam trapping, and dd-carboxypeptidase/β-lactamase activity.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Ministerio de Economía y Competitividad of Spain and the Instituto de Salud Carlos III through the Spanish Network for the Research in Infectious Diseases (grants RD06/0008 and RD12/0015) and grants PS09/00033 and PI12/00103. The work at CBMSO was supported by grants BFU2009-09200 from the Ministerio de Economía y Competitividad of Spain and 223431 DIVINOCELL from the European Union. We acknowledge the predoctoral grant JAE/Predoc from the Consejo Superior de Investigaciones Cientificas to A.R. The study is also supported by the Direcció General d'Universitats, Recerca i Transferència del Coneixement del Govern de les Illes Balears, Spain.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.05150-14.
REFERENCES
- 1.Vincent JL. 2003. Nosocomial infections in adult intensive-care units. Lancet 361:2068–2077. doi: 10.1016/S0140-6736(03)13644-6. [DOI] [PubMed] [Google Scholar]
- 2.Lyczak JB, Cannon CL, Pier GB. 2002. Lung infections associated with cystic fibrosis. Clin Microbiol Rev 15:194–222. doi: 10.1128/CMR.15.2.194-222.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lister PD, Wolter DJ, Hanson ND. 2009. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev 22:582–610. doi: 10.1128/CMR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Juan C, Macia MD, Gutierrez O, Vidal C, Perez JL, Oliver A. 2005. Molecular mechanisms of beta-lactam resistance mediated by AmpC hyperproduction in Pseudomonas aeruginosa clinical strains. Antimicrob Agents Chemother 49:4733–4738. doi: 10.1128/AAC.49.11.4733-4738.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Livermore DM. 1992. Interplay of impermeability and chromosomal beta-lactamase activity in imipenem-resistant Pseudomonas aeruginosa. Antimicrob Agents Chemother 36:2046–2048. doi: 10.1128/AAC.36.9.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zamorano L, Reeve TM, Juan C, Moya B, Cabot G, Vocadlo DJ, Mark BL, Oliver A. 2011. AmpG inactivation restores susceptibility of pan-beta-lactam-resistant Pseudomonas aeruginosa clinical strains. Antimicrob Agents Chemother 55:1990–1996. doi: 10.1128/AAC.01688-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacoby GA. 2009. AmpC beta-lactamases. Clin Microbiol Rev 22:161–182. doi: 10.1128/CMR.00036-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bush K, Jacoby GA. 2010. Updated functional classification of beta-lactamases. Antimicrob Agents Chemother 54:969–976. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson JW, Fisher JF, Mobashery S. 2013. Bacterial cell wall recycling. Ann N Y Acad Sci 1277:54–75. doi: 10.1111/j.1749-6632.2012.06813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeng X, Lin J. 2013. Beta-lactamase induction and cell wall metabolism in Gram-negative bacteria. Front Microbiol 4:128. doi: 10.3389/fmicb.2013.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng Q, Li H, Merdek K, Park JT. 2000. Molecular characterization of the beta-N-acetylglucosaminidase of Escherichia coli and its role in cell wall recycling. J Bacteriol 182:4836–4840. doi: 10.1128/JB.182.17.4836-4840.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balcewich MD, Reeve TM, Orlikow EA, Donald LJ, Vocadlo DJ, Mark BL. 2010. Crystal structure of the AmpR effector binding domain provides insight into the molecular regulation of inducible ampc beta-lactamase. J Mol Biol 400:998–1010. doi: 10.1016/j.jmb.2010.05.040. [DOI] [PubMed] [Google Scholar]
- 13.Dietz H, Pfeifle D, Wiedemann B. 1997. The signal molecule for beta-lactamase induction in Enterobacter cloacae is the anhydromuramyl-pentapeptide. Antimicrob Agents Chemother 41:2113–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fisher JF, Mobashery S. 2014. The sentinel role of peptidoglycan recycling in the beta-lactam resistance of the Gram-negative Enterobacteriaceae and Pseudomonas aeruginosa. Bioorg Chem 56C:41–48. doi: 10.1016/j.bioorg.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cabot G, Ocampo-Sosa AA, Dominguez MA, Gago JF, Juan C, Tubau F, Rodriguez C, Moya B, Peña C, Martinez-Martinez L, Oliver A, Spanish Network for Research in Infectious Diseases (REIPI). 2012. Genetic markers of widespread extensively drug-resistant Pseudomonas aeruginosa high-risk clones. Antimicrob Agents Chemother 56:6349–6357. doi: 10.1128/AAC.01388-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moya B, Dotsch A, Juan C, Blazquez J, Zamorano L, Haussler S, Oliver A. 2009. Beta-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog 5:e1000353. doi: 10.1371/journal.ppat.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tayler AE, Ayala JA, Niumsup P, Westphal K, Baker JA, Zhang L, Walsh TR, Wiedemann B, Bennett PM, Avison MB. 2010. Induction of beta-lactamase production in Aeromonas hydrophila is responsive to beta-lactam-mediated changes in peptidoglycan composition. Microbiology 156:2327–2335. doi: 10.1099/mic.0.035220-0. [DOI] [PubMed] [Google Scholar]
- 18.Tsutsumi Y, Tomita H, Tanimoto K. 2013. Identification of novel genes responsible for overexpression of ampC in Pseudomonas aeruginosa PAO1. Antimicrob Agents Chemother 57:5987–5993. doi: 10.1128/AAC.01291-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cavallari JF, Lamers RP, Scheurwater EM, Matos AL, Burrows LL. 2013. Changes to its peptidoglycan-remodeling enzyme repertoire modulate β-lactam resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother 57:3078–3084. doi: 10.1128/AAC.00268-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zamorano L, Reeve TM, Deng L, Juan C, Moya B, Cabot G, Vocadlo DJ, Mark BL, Oliver A. 2010. NagZ inactivation prevents and reverts beta-lactam resistance, driven by AmpD and PBP 4 mutations, in Pseudomonas aeruginosa. Antimicrob Agents Chemother 54:3557–3563. doi: 10.1128/AAC.00385-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asgarali A, Stubbs KA, Oliver A, Vocadlo DJ, Mark BL. 2009. Inactivation of the glycoside hydrolase NagZ attenuates antipseudomonal beta-lactam resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother 53:2274–2282. doi: 10.1128/AAC.01617-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mark BL, Vocadlo DJ, Oliver A. 2011. Providing β-lactams a helping hand: targeting the AmpC β-lactamase induction pathway. Future Microbiol 6:1415–1427. doi: 10.2217/fmb.11.128. [DOI] [PubMed] [Google Scholar]
- 23.Typas A, Banzhaf M, Gross CA, Vollmer W. 2012. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol 10:123–136. doi: 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sauvage E, Kerff F, Terrak M, Ayala JA, Charlier P. 2008. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32:234–258. doi: 10.1111/j.1574-6976.2008.00105.x. [DOI] [PubMed] [Google Scholar]
- 25.Vollmer W, Bertsche U. 2008. Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim Biophys Acta 1778:1714–1734. doi: 10.1016/j.bbamem.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 26.Sarkar SK, Chowdhury C, Ghosh AS. 2010. Deletion of penicillin-binding protein 5 (PBP5) sensitises Escherichia coli cells to beta-lactam agents. Int J Antimicrob Agents 35:244–249. doi: 10.1016/j.ijantimicag.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 27.Sarkar SK, Dutta M, Chowdhury C, Kumar A, Ghosh AS. 2011. PBP5, PBP6 and DacD play different roles in intrinsic β-lactam resistance of Escherichia coli. Microbiology 157:2702–2707. doi: 10.1099/mic.0.046227-0. [DOI] [PubMed] [Google Scholar]
- 28.Smith JD, Kumarasiri M, Zhang W, Hesek D, Lee M, Toth M, Vakulenko S, Fisher JF, Mobashery S, Chen Y. 2013. Structural analysis of the role of Pseudomonas aeruginosa penicillin-binding protein 5 in β-lactam resistance. Antimicrob Agents Chemother 57:3137–3146. doi: 10.1128/AAC.00505-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Quénée L, Lamotte D, Polack B. 2005. Combined sacB-based negative selection and cre-lox antibiotic marker recycling for efficient gene deletion in Pseudomonas aeruginosa. Biotechniques 38:63–67. doi: 10.2144/05381ST01. [DOI] [PubMed] [Google Scholar]
- 30.Moya B, Juan C, Alberti S, Perez JL, Oliver A. 2008. Benefit of having multiple ampD genes for acquiring beta-lactam resistance without losing fitness and virulence in Pseudomonas aeruginosa. Antimicrob Agents Chemother 52:3694–3700. doi: 10.1128/AAC.00172-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Juan C, Moya B, Perez JL, Oliver A. 2006. Stepwise upregulation of the Pseudomonas aeruginosa chromosomal cephalosporinase conferring high-level beta-lactam resistance involves three AmpD homologues. Antimicrob Agents Chemother 50:1780–1787. doi: 10.1128/AAC.50.5.1780-1787.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.CLSI. 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard—9th ed, vol 32 CLSI document M07-A09. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 33.González-Leiza SM, de Pedro MA, Ayala JA. 2011. AmpH, a bifunctional dd-endopeptidase and dd-carboxypeptidase of Escherichia coli. J Bacteriol 193:6887–6894. doi: 10.1128/JB.05764-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Evans KL, Kannan S, Li G, de Pedro MA, Young KD. 2013. Eliminating a set of four penicillin binding proteins triggers the Rcs phosphorelay and Cpx stress responses in Escherichia coli. J Bacteriol 195:4415–4424. doi: 10.1128/JB.00596-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liao X, Hancock RE. 1997. Identification of a penicillin-binding protein 3 homolog, PBP3x, in Pseudomonas aeruginosa: gene cloning and growth phase-dependent expression. J Bacteriol 179:1490–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song J, Xie G, Elf PK, Young KD, Jensen RA. 1998. Comparative analysis of Pseudomonas aeruginosa penicillin-binding protein 7 in the context of its membership in the family of low-molecular-mass PBPs. Microbiology 144:975–983. doi: 10.1099/00221287-144-4-975. [DOI] [PubMed] [Google Scholar]
- 37.Weigel LM, Belisle JT, Radolf JD, Norgard MV. 1994. Digoxigenin-ampicillin conjugate for detection of penicillin-binding proteins by chemiluminescence. Antimicrob Agents Chemother 38:330–336. doi: 10.1128/AAC.38.2.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winsor GL, Lam DK, Fleming L, Lo R, Whiteside MD, Yu NY, Hancock RE, Brinkman FS. 2011. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res 39:D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vega D, Ayala JA. 2006. The dd-carboxypeptidase activity encoded by pbp4B is not essential for the cell growth of Escherichia coli. Arch Microbiol 185:23–27. doi: 10.1007/s00203-005-0057-5. [DOI] [PubMed] [Google Scholar]
- 40.Noguchi H, Matsuhashi M, Mitsuhashi S. 1979. Comparative studies of penicillin-binding proteins in Pseudomonas aeruginosa and Escherichia coli. Eur J Biochem 100:41–49. doi: 10.1111/j.1432-1033.1979.tb02031.x. [DOI] [PubMed] [Google Scholar]
- 41.Korat B, Mottl H, Keck W. 1991. Penicillin-binding protein 4 of Escherichia coli: molecular cloning of the dacB gene, controlled overexpression, and alterations in murein composition. Mol Microbiol 5:675–684. doi: 10.1111/j.1365-2958.1991.tb00739.x. [DOI] [PubMed] [Google Scholar]
- 42.Lee M, Hesek D, Blazquez B, Lastochkin E, Boggess B, Fisher JF, Mobashery S. 2014. Catalytic spectrum of the penicillin-binding protein 4 of Pseudomonas aeruginosa, a nexus for the induction of beta-lactam antibiotic resistance. J Am Chem Soc 137:190–200. doi: 10.1021/ja5111706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meberg BM, Paulson AL, Priyadarshini R, Young KD. 2004. Endopeptidase penicillin-binding proteins 4 and 7 play auxiliary roles in determining uniform morphology of Escherichia coli. J Bacteriol 186:8326–8336. doi: 10.1128/JB.186.24.8326-8336.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nelson DE, Young KD. 2000. Penicillin binding protein 5 affects cell diameter, contour, and morphology of Escherichia coli. J Bacteriol 182:1714–1721. doi: 10.1128/JB.182.6.1714-1721.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nelson DE, Young KD. 2001. Contributions of PBP 5 and dd-carboxypeptidase penicillin binding proteins to maintenance of cell shape in Escherichia coli. J Bacteriol 183:3055–3064. doi: 10.1128/JB.183.10.3055-3064.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Denome SA, Elf PK, Henderson TA, Nelson DE, Young KD. 1999. Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: viability, characteristics, and implications for peptidoglycan synthesis. J Bacteriol 181:3981–3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanders CC, Bradford PA, Ehrhardt AF, Bush K, Young KD, Henderson TA, Sanders WE Jr. 1997. Penicillin-binding proteins and induction of AmpC beta-lactamase. Antimicrob Agents Chemother 41:2013–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.