

Abstract
The constant emergence of new bacterial strains that resist the effectiveness of marketed antimicrobials has led to an urgent demand for and intensive research on new classes of compounds to combat bacterial infections. Antimicrobial peptoids comprise one group of potential candidates for antimicrobial drug development. The present study highlights a library of 22 cationic amphipathic peptoids designed to target bacteria. All the peptoids share an overall net charge of +4 and are 8 to 9 residues long; however, the hydrophobicity and charge distribution along the abiotic backbone varied, thus allowing an examination of the structure-activity relationship within the library. In addition, the toxicity profiles of all peptoids were assessed in human red blood cells (hRBCs) and HeLa cells, revealing the low toxicity exerted by the majority of the peptoids. The structural optimization also identified two peptoid candidates, 3 and 4, with high selectivity ratios of 4 to 32 and 8 to 64, respectively, and a concentration-dependent bactericidal mode of action against Gram-negative Escherichia coli.
INTRODUCTION
Antimicrobial peptides, also known as host defense peptides, are a class of antimicrobial agents that have long served all living organisms in combating infectious diseases by killing or inhibiting the growth of pathogens. As a class, they are relatively short (<100 amino acid residues), positively charged, amphipathic (have both hydrophobic and hydrophilic domains), and exhibit various biological activities based on their structural properties (1). Despite their high potency against a variety of clinical strains of bacteria, what limits the clinical drug development of antimicrobial peptides is their susceptibility to enzymatic degradation. Antimicrobial peptides have been subjected to various modifications in order to design novel classes of potent peptidomimetic antimicrobials with improved stability and activity profiles. Peptoids, which are oligomers of N-substituted glycines, are a new class of synthetic compounds that mimic peptide structures. The functional side chains of commercial availability in peptoids are attached to the nitrogen rather than the α-carbon in the peptide counterparts (Fig. 1) through a two-step process (see Fig. S1 in the supplemental material). Peptoids circumvent proteolytic susceptibility while retaining the beneficial features of antimicrobial peptides (2–4); hence, they are promising structures for antimicrobial drug development. For >15 years, research on peptoid synthesis and application has increased dramatically due to their potential biological applications as antimicrobials (5–7), molecular transporters (8, 9), and more recently as anticancer agents (10) and nanostructured materials (11, 12).
FIG 1.
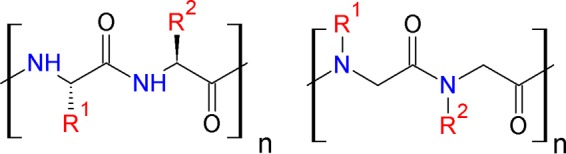
Chemical structures of peptides (left) and peptoids (right).
In the efforts to increase the effectiveness of antimicrobial peptides and their mimetics, two key structural elements that decide their overall antimicrobial activity are charge and hydrophobicity. These two elements contribute with different physiochemical properties to the interaction of peptides with bacterial and mammalian membranes in which hydrophobicity, in particular, directs the degree of peptide partitioning into the lipid bilayer (6, 13–16). With this in mind, the mechanism of action is greatly related to either the disruption of bacterial membranes through pore formation or passage through the bacterial membranes and targeting of cytoplasmic compartments (17). To date, few research studies have characterized the structural features of peptoids that mimic the structure of antimicrobial peptides with respect to their antimicrobial activities and toxicity profiles. Chongsiriwatana and coworkers (5) have described helical peptoids with broad-spectrum activity and have shown that net charge and moderate hydrophobicity are important for the observed antimicrobial activities, whereas high hydrophobicity and amphipathicity are associated with a high degree of hemolysis (5; A. M. Czyzewski, H. Jenssen, C. D. Fjell, M. Waldbrook, N. P. Chongsiriwatana, E. Yuen, R. E. W. Hancock, and A. E. Barron, unpublished data). Moreover, Bang and coworkers (3) provide evidence for the direct transformation of antimicrobial peptides into a peptoid analogues while retaining antimicrobial activity. Another study reported on a successful biomimicry of the peptide magainin with a minimal length of 12 residues, which has been suggested as a requirement for antimicrobial activity of peptoids (18). In the present study, we describe the direct transformation of known antimicrobial peptides into peptoid analogues. In addition, we designed a library of novel N-substituted glycines with a focus on dissecting the direct effect of hydrophobicity on peptoid antimicrobial activity and cytotoxicity. While fine-tuning the structure of the peptoids, we synthesized a systematic set of closely related analogs with different hydrophobic properties through single- or multiple monomer exchange. We correlated activity and toxicity with peptoid hydrophobicity using high-performance liquid chromatography (HPLC) retention times, as previously demonstrated for antimicrobial peptides (16). In order to identify the most promising peptoid, we first compared the MICs relative to those of the parent peptides GN-2, GN-4, and GN-6 (T. Godballe, B. Mojsoska, M. N. Hanne, and H. Jenssen, unpublished data). Next, we analyzed the effect of the structural changes along the peptoid backbone to the observed hemolytic and cytotoxic properties with respect to hydrophobicity. Furthermore, we investigated the mode of action of the most potent peptoids. We hypothesize that the novel peptoids will exhibit enhanced antimicrobial activities while retaining their already stable proteolytic stability with a mode of action similar to that of antimicrobial peptides.
MATERIALS AND METHODS
Bacterial strains.
The strains used in the present study were from our laboratory strain collection. Those obtained from the American Type Culture Collection (ATCC) (Rockville, MD) included Escherichia coli strain ATCC 25922 and Staphylococcus aureus strain ATCC 25923. Other strains (19) include Pseudomonas aeruginosa PAO1 H103 and Liverpool epidemic clinical H1027 strains, methicillin-resistant Staphylococcus aureus (MRSA) (strain C623 MRSA), and an E. coli clinical isolate expressing extended-spectrum β-lactamases (ESBL). All tested bacterial strains are categorized as biohazard level 2 pathogens.
Materials and reagents for peptoid synthesis.
The reagents used are as follows: tert-butoxycarbonyl (Boc), 9-fluoromethylmethoxylcarbonyl (Fmoc), trifluoroacetic acid (TFA), triisopropylsilane (TIPS), N,N-dimethylformamide (DMF), N,N-diisopropylcarbodiimide (DIC), N-methyl-2-pyrrolidone (NMP), and bromoacetic acid (BrAcOH). The solvents and reagents were purchased from commercial sources. Rink amide resin 4-methylbenzhydrylamine (MBHA) (100 to 200 mesh, 0.57 mmol g−1 loading capacity) was purchased from Novabiochem. The amine building blocks, 4-methylpiperidine, BrAcOH, DIC, and TFA were purchased from Sigma-Aldrich. DMF and NMP were purchased from VWR BDH Prolabo.
Peptoid synthesis.
Peptoids were synthesized using standard submonomeric solid-phase synthesis (see Fig. S1 in the supplemental material), as described by Figliozzi et al. (20), with minor optimizations for the synthesis of peptoids containing tryptamine (Ntrp). The peptoid oligomers 7 to 18 were synthesized on an automated peptide synthesizer (ResPep SL; Intavis Bioanalytical Instruments AG) on a 15-μmol scale, and peptoids 1 to 6 and 19 to 22 were synthesized manually (100- to 200-mg scale) in polypropylene syringes fitted with a 2 × 20-μm polyethylene frit (Telos SPE columns; Mikrolab, Aarhus, Denmark). Briefly, rink amide MHBA resin was swelled in DMF for 20 min at room temperature, followed by Fmoc deprotection by 20% 4-methylpiperidine in DMF for 20 min. The resin was washed with DMF (5 × 1 ml). The acylation step was performed using 0.6 M BrAcOH in DMF and 50% DIC in DMF for 30 min. This step was optimized for tryptamine-rich peptoids. Bromine substitution was achieved through the addition of 1 M the amine of interest in NMP for 1 h (20 min for tryptamine) at room temperature. After completion of the peptoid chain, the peptoids were cleaved from the resin using TFA-water-TIPS (95:2.5:2.5) for 30 min to 1 h. Crude peptoids were purified by reverse-phase HPLC using a C18 column (10 μm, 250 × 10 mm; Higgins Analytical, Inc.) on an acetonitrile-water gradient. The correct masses and final purity (>95%) were determined using analytical Dionex UltiMate 3000 reverse-phase (RP)-ultrahigh performance liquid chromatography (UHPLC) (C18 Kinetex, 100 × 2.1 mm, 100 Å) electrospray ionization mass spectrometry (Finnigan LTQ) instrumentation.
MIC determination.
Antimicrobial susceptibility testing was performed using the microtiter broth dilution method described by Wiegand, Hilpert, and Hancock (21). Briefly, overnight cultures were prepared in Mueller-Hinton broth at 37°C in a heat shaker. The overnight culture was diluted 1:50 in fresh Mueller-Hinton broth and allowed to grow until an optical density at 600 nm (OD600) of 0.4 had been reached. A final concentration of bacteria in the range of 2 × 105 to 8 × 105 CFU/ml was challenged with serial dilutions of peptoid concentrations in a sterile 96-well polypropylene microtiter plates (catalog no. 3879; Costar) and incubated overnight at 37°C. The MIC was defined as the concentration of peptoid at which no visible bacterial growth was observed.
Determination of MICs with human blood plasma.
The MICs for peptides in the presence of human blood plasma were determined as described above. Briefly, peptoids 3 and 4 were premixed with 50% human blood plasma from different donors to a concentration of 1.28 μg/ml. A volume of 10 μl was then mixed with 90 μl of prepared bacterial suspension in a sterile 96-well polypropylene plate. The MIC was determined after 18 to 24 h.
Hemolysis assay.
The ability of peptoids to induce hemolysis in human erythrocytes was assessed using a hemolysis assay. Briefly, human red blood cells (hRBCs) were purified from freshly drawn human blood and centrifuged at 800 × g for 15 min between washes with 0.9% NaCl. A two-fold serial dilution of peptoids (1 to 128 μg/ml) in 0.9% NaCl was added to a sterile 96-well polypropylene microtiter plate for a total volume of 100 μl. The same volume of hRBCs was added to the wells to a final volume of 200 μl, followed by incubation for 18 to 24 h. The plate was centrifuged at 250 × g for 5 min, and 20 μl of the supernatant was added to 100 μl of 0.9% NaCl in a flat-bottomed polystyrene 96-well Greiner plate. As a control for 100% lysis and baseline correction, 1% Triton X-100 and sterile 0.9% NaCl were used, respectively. Absorbance at 546 nm was measured, and the percent hemolysis induced by the peptoids was calculated using equation 1. Hemolysis assays were performed with hRBCs from multiple donors.
| (1) |
In vitro cytotoxicity assay.
Cytotoxicity of the peptoids on HeLa cells was estimated using the CellTiter 96 aqueous nonradioactive cell proliferation assay (catalog no. G5421; Promega). This assay relies on the ability of viable cells to convert the novel tetrazolium salt MTS [(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] into a formazan product that is soluble in culture medium. The absorbance measured at 490 nm is directly proportional to the cell viability. Briefly, HeLa wild-type (WT) cells were cultured in Dulbecco's modified Eagle medium (DMEM) and medium containing 1% penicillin-streptomycin and 10% fetal bovine serum (FBS) for a minimum of 3 passages before use. Cells were seeded at a concentration of 5 × 105 per well in a sterile 96-well polystyrene plate. The next day, the cells were carefully washed with phosphate-buffered saline (PBS) without [w/o] Ca+2 and Mg+2, and peptoid in Dulbecco's modified Eagle medium (DMEM) at 4 different concentrations was added to each well in duplicate. The plate was incubated for 4 h in a humidified 5% CO2 atmosphere at 37°C, 20 μl of (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS)-phenazine methosulfate (PMS) solution was added, and the plate was further incubated for 1 h for color development. Absorbance at 490 nm was measured using the Synergy HT multidetection microplate reader. For viability controls and baseline correction, 1% Triton X-100 (0% viability), untreated cells (100% viability), and DMEM only were used. The percent viability was expressed as the concentration of peptoid that reduces cell viability by 50% and was calculated using equation 2 in the GraphPad Prism software. Y denotes percentage of cell viability and X denotes concentration.
| (2) |
Killing kinetics.
The kinetics of antimicrobial activity against E. coli (ATCC 25922) was assessed at peptoid concentrations corresponding to 1×, 2×, and 4× the MIC. Briefly, an overnight culture of bacteria was diluted 1:50 in fresh Mueller-Hinton broth and regrown to an OD600 of 0.4 before diluting with fresh Mueller-Hinton broth to a turbidity of 0.1. The bacterial suspension was added to a 96-well polystyrene flat-bottomed plate containing the peptoid of interest in addition to known antibiotics. The plate was incubated without shaking at 37°C for 180 min. Samples (100 μl) were taken at 20, 40, 80, 120, and 180 min and diluted in ice-cold 0.9% NaCl, from which 100 μl was plated on LB agar plates. The plates were incubated for 18 to 24 h at 30°C, and the CFU were counted. A sterility control was performed by plating 100 μl of Mueller-Hinton (MH) broth and 0.9% NaCl. Plates on which no detectable bacterial growth was observed were left for an additional 18 h of incubation. Three individual dilutions of each peptoid concentration at a given time were plated, and the data reported are from three individual experiments.
RESULTS AND DISCUSSION
Peptoid design strategy.
To develop novel antimicrobial agents that mimic the structure and function of antimicrobial peptides, we targeted the design of peptoids with improved pharmacokinetic properties. The reference structures for the synthesis of the peptoids in this study were tryptophan-rich GN-2 (RWKRWWRWI-NH2) (Fig. 2A), GN-4 (RWKKWWRWL-NH2), and GN-6 (RKRWWWWFR-NH2) synthetic peptides with the highest selectivity profiles, derived from an in silico library of Bac2a analogues (Table 1) (22). First, we transformed these peptides into three peptoid analogs. In the structure of the new peptoids 1, 7, and 15, only N-(4-aminobutyl) glycine monomers were introduced to substitute for any charged residues found in the peptide structure. For the design of the related peptoid analogs, two central design parameters were considered, charge and hydrophobicity. Peptidomimetics with charged and hydrophobic groups segregate into amphiphilic structures that are important for interactions with bacterial membranes (5). The overall design of the current library of short linear peptoids provides fundamental knowledge about the effect of hydrophobicity while retaining a constant charge along the peptoid backbone for the observed biological activities.
FIG 2.
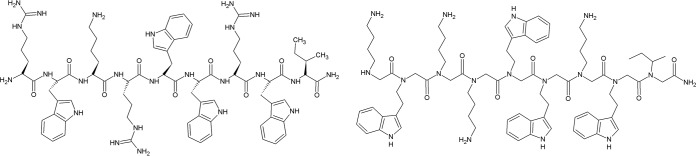
Chemical structures of GN-2 peptide (left) and GN-2 peptoid (right).
TABLE 1.
Retention times and in vitro antibacterial, hemolytic, and cytotoxic activities of compounds
| Peptoid no. | Peptoid nomenclature | Sequencef (N–C) | Rt (min)a | MIC (μg/ml) for strainb: |
Hemolytic concn (μg/ml)c | SRd | Cytotoxicity (μg/ml)e | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
S. aureus |
E. coli |
P. aeruginosa |
||||||||||
| ATCC 29213 | C623 MRSA | ATCC 25922 | 63103 ESBL | PAO1 | H1027 MDR | |||||||
| 1 | GN-2 | H-Nlys-Ntrp-Nlys-Nlys-Ntrp-Ntrp-Nlys-Ntrp-Nile-NH2 | 10.54 | 64 | 32 | 32 | 32 | 32 | 4 | >128 | >2–32 | 168 |
| 2 | GN-2-Ntrp1,2,8,9 Nile7Nlys3–6 | H-Ntrp-Ntrp-Nlys-Nlys-Nlys-Nlys-Nile-Ntrp-Ntrp-NH2 | 10.93 | 16 | 8 | 32 | 64 | 32 | 4 | >128 | >2–32 | 173 |
| 3 | GN-2-Nlys1–4Ntrp5–8 | H-Nlys-Nlys-Nlys-Nlys-Ntrp-Ntrp-Ntrp-Ntrp-Nile-NH2 | 11.48 | 8 | 8 | 32 | 8 | 32 | 4 | 128 | 4–32 | 110 |
| 4 | GN-2-Npm9 | H-Nlys-Ntrp-Nlys-Nlys-Ntrp-Ntrp-Nlys-Ntrp-Npm-NH2 | 10.79 | 8 | 8 | 16 | 16 | 16 | 2 | 128 | 8–64 | 104 |
| 5 | GN-2-Nae1,3,4,7 | H-Nae-Ntrp-Nae-Nae-Ntrp-Ntrp-Nae-Ntrp-Nile-NH2 | 10.81 | 32 | 16–32 | 16 | 64 | 16–32 | 8 | >128 | >4–16 | |
| 6 | GN-2-Ndpe6 | H-Nlys-Ntrp-Nlys-Nlys-Ntrp-Ndpe-Nlys-Ntrp-Nile-NH2 | 11.46 | 4 | 4 | 32 | 16 | 32 | 2 | 16 | 0.5-8 | 80 |
| 7 | GN-4 | H-Nlys-Ntrp-Nlys-Nlys-Ntrp-Ntrp-Nlys-Ntrp-Nleu-NH2 | 10.51 | 32 | ND | 64 | ND | 64 | ND | >128 | >2–4 | 166 |
| 8 | GN-4-Ndpe2,5,6,8 | H-Nlys-Ndpe-Nlys-Nlys-Ndpe-Ndpe-Nlys-Ndpe-Nleu-NH2 | 13.81 | 4 | 2 | 16 | 4 | 16 | 16 | 4 | 0.25–2 | 67 |
| 9 | GN-4-Nai2,5,6,8 | H-Nlys-Nai-Nlys-Nlys-Nai-Nai-Nlys-Nai-Nleu-NH2 | 12.31 | 4 | 4 | 16–32 | 32 | 16–32 | 4 | 32 | 1–8 | 172 |
| 10 | GN-4-Npe2,5,6,8 | H-Nlys-Npe-Nlys-Nlys-Npe-Npe-Nlys-Npe-Nleu-NH2 | ND | 64 | ND | 64–128 | ND | 64–128 | ND | >128 | >1–2 | |
| 11 | GN-4-Nspe2,5,6,8 | H-Nlys-Nspe-Nlys-Nlys-Nspe-Nspe-Nlys-Nspe-Nleu-NH2 | 10.69 | 128 | ND | 32 | ND | 32 | ND | >128 | >1–4 | 631 |
| 12 | GN-4-Ndpe2,5,6,8Nval9 | H-Nlys-Ndpe-Nlys-Nlys-Ndpe-Ndpe-Nlys-Ndpe-Nval-NH2 | 13.50 | 2 | ND | 16 | ND | 16 | ND | 4–8 | 0.5–4 | |
| 13 | GN-4-Ndpe2,5,6,8 Nma9 | H-Nlys-Ndpe-Nlys-Nlys-Ndpe-Ndpe-Nlys-Ndpe-Nma-NH2 | 14.07 | 2 | 4 | 32 | 4 | 32 | 16 | 4 | 0.125–2 | |
| 14 | GN-4-Nai2,5,6,8Nval9 | H-Nlys-Nai-Nlys-Nlys-Nai-Nai-Nlys-Nai-Nval-NH2 | 11.37 | 4 | 8 | 32 | 64 | 32 | 4 | >128 | >2–32 | |
| 15 | GN-6 | H-Nlys-Nlys-Nlys-Ntrp-Ntrp-Ntrp-Ntrp-Npm-Nlys-NH2 | 10.10 | 8–16 | ND | 64 | ND | 64 | ND | >128 | >2–16 | 203 |
| 16 | GN-6-Ndpe4–7 | H-Nlys-Nlys-Nlys-Ndpe-Ndpe-Ndpe-Ndpe-Npm-Nlys-NH2 | 15.43 | 16 | ND | 64 | ND | 64 | ND | 16 | 0.25–1 | |
| 17 | GN-6-Npe4–7 | H-Nlys-Nlys-Nlys-Npe-Npe-Npe-Npe-Npm-Nlys-NH2 | ND | 16 | ND | 32–64 | ND | 32–64 | ND | 32 | 0.5–2 | |
| 18 | GN-6-Ndpe4,5Npe6,7 | H-Nlys-Nlys-Nlys-Ndpe-Ndpe-Npe-Npe-Npm-Nlys-NH2 | 14.06 | 8 | ND | 32 | ND | 32 | ND | 32 | 1–4 | 118 |
| 19 | Nlys1,3,4,7 Ntrp2,5,6,8 | H-Nlys-Ntrp-Nlys-Nlys-Ntrp-Ntrp-Nlys-Ntrp-NH2 | 10.13 | 16 | ND | 32 | ND | ND | ND | >256 | >8–16 | 148 |
| 20 | Nae1–4 Ntrp5–8 | H-Nae-Nae-Nae-Nae-Ntrp-Ntrp-Ntrp-Ntrp-NH2 | 11.34 | 4 | ND | 16 | ND | ND | ND | 32 | 2–8 | ND |
| 21 | Nlys1–4 Ntrp5–8 | H-Nlys-Nlys-Nlys-Nlys-Ntrp-Ntrp-Ntrp-Ntrp-NH2 | 11.06 | 4 | ND | 16 | ND | ND | ND | 64 | 4–16 | ND |
| 22 | Nae1,2 Nlys3,4 Ntrp5–8 | H-Nae-Nae-Nlys-Nlys-Ntrp-Ntrp-Ntrp-Ntrp-NH2 | 11.11 | 16 | ND | 16 | ND | 8 | ND | 64 | 4–8 | ND |
| 23 | Bac8c | RIWVIWRR-NH2 | 8 (6.75) | ND | 4 (3.38) | ND | 4 (3.38) | ND | 128 | 16–32 | ND | |
| 24 | Indolicidin | ILPWKWPWWPWRR-NH2 | 16 (8.4) | ND | 32 (16.8) | ND | 32 (16.8) | ND | 64 | 2–4 | ND | |
| 25 | (Omiganan) MX-226 indolicidin variant | (25) | ND | 16 (10) | ND | 64 (40) | ND | 64 | ||||
| 26 | GN-2 peptide | RWKRWWRWI-NH2 (28) | 3.13 | 6.25 | 3.13 | 100 | 16–32 | 47* | ||||
| 27 | GN-4 peptide | RWKKWWRWL-NH2 (28) | 6.25 | 6.25 | 3.13 | 100 | 16–32 | 37* | ||||
| 28 | GN-6 peptide | RKRWWWWFR-NH2 (28) | 3.13 | 12.5 | 6.25 | 100 | 8–32 | 97* | ||||
Analytical retention times (Rt) estimated on a reverse-phase C18 Kinetex 100- by 2.1-mm 100-Å column at 60°C, run on a 15 to 65% acetonitrile gradient over 20 min.
Median MICs representative for 3 to 5 replicates, given in μg/ml. Due to different molecular weights, the MICs for Bac8c, indolicidin, and MX-226 are given in μM, which is in parenthses. ND, not determined.
The hemolysis results are from 3 individual experiments using different blood donors.
SR, selectivity ratio, or the quotient of 10% hemolysis and the lowest and highest MIC of the bacterial strains.
Cytotoxicity is reported as the IC50, which is defined as the concentration found to inhibit 50% of the metabolic activity of HeLa WT cells using the colorimetric tetrazolium salt-based MTS assay. *, IC50 for peptides as estimated by an MTT assay on HeLa WT cells. Antimicrobial peptides are taken as reference antimicrobial compounds.
Peptoids were synthesized using submonomeric solid-phase peptoid synthesis.
The positive charge was introduced by incorporating lysine-like monomers and was kept constant at +4 in total. The overall hydrophobicity was altered using different short and aromatic monomers. The chemical structures for the side chains used in creating this library were mimics of traditional amino acid side chains, in addition to a broader selection of bulky hydrophobic side chains, such as Npe, Ndpe, Nai, Ntrp, and Nspe (Fig. 3). The degree of hydrophobicity was measured by the retention time (Rt) on a reverse-phase C18 column, and the values are given in Table. 1. To achieve changes in the amphiphilicity of the peptoids, we also designed sequences in which we altered the position of certain monomers. Further, we investigated whether the first residue at the C-terminal end in the structure of peptoids 1 and 8 is necessary to retain antimicrobial activity by synthesizing several 8-mers (see Fig. S5 in the supplemental material). All 22 peptoids were screened for their antimicrobial activities against representative strains of S. aureus, E. coli, and P. aeruginosa, and their MICs are presented in Table 1.
FIG 3.

Chemical structures of peptoid monomers and their abbreviations.
Broad-spectrum antimicrobial activity.
All peptoids were initially screened for antimicrobial activity against various Gram-negative E. coli and P. aeruginosa and Gram-positive S. aureus bacterial strains. The MICs required to completely inhibit the growth of bacteria were within the range of 2 to 64 μg/ml and revealed high to moderate antimicrobial peptoid activity throughout the library compared to that of structurally similar antimicrobial peptides, such as Bac8c indolicidin and omiganan (Table 1.) (23). This is in agreement with other reported MICs of peptides and peptidomimetics that contain lysine and tryptophan side chains (3, 24, 25). Peptoids 1, 7, and 15 are mimics of GN-2, GN-4, and GN-6 peptides, respectively (22), and the broad-spectrum antimicrobial activities were not improved upon making these structural changes (Table 1; see also Fig. S3A and B in the supplemental material). Additionally, arginine residues present in the peptide structure were substituted with lysine in all positions, and this may be one of the reasons why the peptoid mimics exert lower antibacterial activity. In this context, it has been shown in the literature that peptides in which arginine residues have been replaced with lysine demonstrated reduced antibacterial killing, likely due to lower affinity to the membrane (26, 27). In general, peptoids with same or different structural features within the present library displayed distinctive selectivity toward killing specific bacterial strains, indicative of probable different mechanisms of antibacterial activity. In this context, peptoid 1 exhibited moderate activity (MIC, 32 μg/ml) against E. coli and S. aureus strains, in contrast to the high potency (MIC, 4 μg/ml) against the P. aeruginosa multidrug-resistant (MDR) strain. Moreover, peptoids 6, 9, 11, 12, and 14 showed high selectivity of inhibiting the growth of the two S. aureus strains and the P. aeruginosa MDR strain (MIC, 2 to 8 μg/ml) compared to that of E. coli strains and P. aeruginosa strain PAO1 (MIC, 16 to 64 μg/ml).
Cell cytotoxicity (hemolysis and toxicity to HeLa cells).
Toxicity against human red blood cells was assessed using a hemolysis assay. The hemolytic concentrations (HC) corresponding to 10% hemolysis (HC10) are presented in Table 1. The most hemolytic peptoids that caused 10% lysis of human red blood cells at low concentrations were peptoids 13, 6, 8, 12, and 16 (Table 1). Cytotoxicity against HeLa cells was measured using a tetrazolium salt (MTS)-based colorimetric assay and is reported in Table 1 as the concentration that inhibits metabolic activity by 50% (IC50), calculated using equation 2. We succeeded in synthesizing peptoid analogs of GN2, GN-4, and GN-6 with lower toxicities to both red blood cells and HeLa cells. The most cytotoxic peptoids from those tested were peptoids 9 (HC10, 4 μg/ml; IC50, 67 μg/ml) and 7 (HC10, 16 μg/ml; IC50, 80 μg/ml). Cell selectivity was reported using the selectivity ratio (SR), which is an estimate of the tendency of the peptoid to kill bacteria over mammalian cells. The selectivity ratio is calculated by the ratio of the hemolytic concentration that causes 10% lysis of human red blood cells and the lowest and highest MICs. High selectivity ratios represent more selective peptoids, and in the present study, peptoids 3 and 4 exhibited the highest selectivity of killing bacterial over mammalian cells (Table 1).
Effects of increasing hydrophobicity by Nlys substitution.
Length variants of the amino acid lysine (e.g., ornithine) have been used in the synthesis of novel peptidomimetics with improved antibacterial and hemolytic activities (28, 29). In order to see whether the length of the charged residue in peptoid 1 affects activity, a shorter monomer, Nae, was substituted for Nlys in peptoid 1. This substitution resulted in an insignificant change in the retention time of the new peptoid 5, which resulted in a similar effect on the broad-spectrum activity and toxicity of these two peptoids. Comparable observations were deduced for the Nae-to-Nlys substitution in the 8-mer analogs (peptoid 20 and 21), in which the only minor influence on the shorter lysine variant (peptoid 20) could be observed in the low concentration needed to lyse 10% of human red blood cells (Table 1; see also Table S1 in the supplemental material). Furthermore, decreasing the content of this residue within the same sequence to 50% resulted in an optimized 8-mer (peptoid 22) with good antibacterial activity and lower toxicity (see Table S1).
Effects of increasing hydrophobicity by Ntrp substitution.
Structure-activity relationship studies on antimicrobial peptides have suggested a strong correlation between antimicrobial activity and overall hydrophobicity (14, 15, 30). This is partly because of the strong hydrophobic interactions between peptides and the target membranes, which is more pronounced if the membranes are composed of zwitterionic lipids, as is the case with mammalian membranes (30). Similar studies have been reported by the Barron group, in which an increase in hydrophobicity in peptoids that mimic antimicrobial peptides resulted in an increase in both antibacterial activity and hemolytic properties (5). To explore this fundamental correlation, residues of different hydrophobic character were substituted for tryptamine. These physiochemical changes lead to novel structures with different retention times, as measured by RP-HPLC on a C18 column (Table 1; see also Table S2 in the supplemental material). Hydrophobicity was increased (higher Rt) upon exchange of the Ntrp monomer in peptoid 7 with Ndpe and Nain, and this rise correlated well with an increase in antibacterial activity against all tested strains. Notably, despite the increase in retention time upon the exchange of Ntrp residues with Nspe, the antimicrobial activity of peptoid 11 against S. aureus decreased by 4-fold. No improvement in the antimicrobial activity was observed when Npe in peptoid 11 was substituted for Ntrp. In terms of the toxicity exerted by these peptoids, an overall linear correlation between hydrophobicity and toxicity was measured in both human red blood cells (Table 1; see also Table S2) and HeLa cells (see Fig. S2D in the supplemental material). This observation is consistent with findings in the literature for antimicrobial peptides with high hydrophobic profiles (30, 31). Similar changes in peptoid 15 led to the synthesis of two novel analog peptoids, 16 and 17, containing the Ndpe and Npe monomers, respectively. With regard to toxicity, there is an evident increase in the ability to lyse human red blood cells and kill HeLa cells when more bulky aromatic residues are introduced. While peptoid 7 analogs showed increased antimicrobial activity in parallel with an increase in hydrophobicity, the pronounced hydrophobicity shift upon this change in peptoid 15 analogs did not significantly affect antimicrobial activity toward E. coli, S. aureus, and P. aeruginosa strains. The difference in these two peptoid groups is the position of the individual monomers and the presence of Nleu at position 9 in peptoid 7 and its analogs and Npe in position 8 in peptoid 15 and its analogs. Therefore, the possibility that the position of the monomers along the peptoid backbone plays a role in the observed behavior should not be excluded. However, excessive hydrophobicity might explain the decreased antimicrobial activity observed for peptoid 16, in which a strong peptoid-self association will prevent the peptoid from passing through the cell wall in bacteria. This has been suggested as a plausible justification for the dramatic decrease in antibacterial activity of peptides upon increased hydrophobicity by Chen and coworkers (13). Our results lead to the conclusion that the overall hydrophobicity of the peptoids is not directly correlated with their antibacterial peptoid activity against E. coli and P. aeruginosa (Table 1; see also Table S2 and Fig. S2A and C in the supplemental material), while there is a direct relationship between hydrophobicity and antibacterial activity against S. aureus (see Table S2 and Fig. S2B in the supplemental material).
Effects of increasing hydrophobicity by single-monomer substitution.
Small hydrophobic amino acids, such as leucine, isoleucine, and valine, occur frequently in antimicrobial peptide structures found in bacteria and animals (32). To mimic the structure of the template peptides, initially, two analog monomers of leucine and isoleucine were retained in position 9 of peptoids 1 and 7. This single-monomer change did not show any significant shift in antibacterial activity, and the toxicity remained low at the highest tested concentrations of these peptoids. In contrary, when only one Ntrp residue at the center of peptoid 1 was replaced with a bulkier aromatic monomer, Ndpe, the increased hydrophobicity significantly enhanced its potency against S. aureus, human red blood cells, and HeLa cells (see Table S3 and Fig. S3A and B in the supplemental material). No significant change was observed for this peptoid 6 against E. coli (ATCC 25922) and P. aeruginosa (PAO1). Furthermore, to allow a further direct comparison of monomers with similar physiochemical characteristics but that differ in a single monomer at position 9, peptoids 8, 12, and 13, which a contain a more bulky residue, Ndpe, instead of Ntrp, were synthesized. The hydrophobicity order of the peptoids as a result of this change was greatest with 13, followed by 8, and then 12 (see Table S3). All three peptoids exhibited similar antimicrobial activities against killing S. aureus, E. coli, and P. aeruginosa strains, with high hemolytic properties, causing 10% hemolysis at very low concentrations. In regard to the two peptoids (peptoids 9 and 14) containing Nai instead of Ntrp along the backbone and that had variations in the Nleu and Nval residues, the more hydrophobic peptoid 9 exerts enhanced antibacterial activity against the methicillin-resistant S. aureus (MRSA) and the E. coli clinical isolate expressing extended spectrum β-lactamases (ESBL) by 2-fold. No significant difference against P. aeruginosa was observed. Regarding the toxicity profiles of these two peptoids, there is a clear correlation with toxicity, as the increase in hydrophobicity increases the red blood cell lysis of peptoid 9 by >4-fold.
Effects of amphiphilicity by sequence rearrangement.
Several structural analogs were synthesized to assess whether potency could be the influenced by the rearrangement of monomers along the peptoid chain that would result in altered amphiphilicity and charge distribution. One scrambled version of peptoid 15 resulted in peptoid 4. By disrupting the charge cluster at the N-terminal end of peptoid 15, the new peptoid, peptoid 4, has more pronounced hydrophobic character and, as such it, does not show significantly enhanced activity against S. aureus. Notably, it shows 4 times higher activity against Gram-negative strains of E. coli and P. aeruginosa. This shift does not contribute significantly to the observed hemolytic activity, although peptoid 4 appears to be more toxic to HeLa cells than peptoid 15 (see Table S4 in the supplemental material). The increase in potency against Gram-negative bacteria by the disrupted amphiphilicity in peptoid 4 is in agreement with the proposed model by Wimley, in which peptides with disrupted amphipathicity; i.e., peptides with imperfect segregation of charged and hydrophobic residues, have increased potency in disrupting bacterial membranes (33). Additionally, disrupting the hydrophobic region in melittin increased the ability of pore formation (34). Another sequence rearrangement is demonstrated for peptoid 1, in which the charged monomers were positioned either in the center or the at the N-terminal end (terminal amphiphile) of the peptoid chain. The resulting peptoids, 2 and 3, have three significantly different retention times, with peptoid 3 having a pronounced hydrophobic cluster at the C terminus exhibiting the highest hydrophobicity. In contrast to the observations of the disruption of amphiphilicity resulting in increased activity, the perfect amphiphilicity in peptoid 3 contributes to an enhanced activity of this peptoid against Gram-positive S. aureus. A significant change in activity against a Gram-negative strain is observed only for an E. coli clinical isolate expressing extended-spectrum β-lactamases (ESBL), with this peptoid showing 4- to 8-fold higher effectiveness. Interestingly, there is no difference in the antibacterial activity observed against P. aeruginosa strains. With regard to toxicity, all three peptoids show low hemolytic activity and differ slightly in their toxicity profiles against HeLa cells. Correspondingly, similar rearrangements and observations are associated with peptoids 19 and 21, with the only pronounced difference being the low hemolytic activity exerted by peptoid 19. The incorporation of a charged (lysine) residue in the hydrophobic region of peptide V13KL led to a significant reduction in the toxicity of the peptide, most likely due to disrupted dimerization of the peptide in aqueous solution, which allows easy access to the interface region of mammalian membranes while preventing membrane permeation (35). In summary, one plausible explanation for the different behavior observed upon the same structural changes in the above-mentioned peptoids is the exchange of Npm with Nile. Sequence rearrangement of peptoids 1 and 15 gave two peptoids, 3 and 4, with optimum hydrophobicity that resulted in the highest selectivity ratios among all peptoids in this study.
Effects of main chain length.
The maternal peptoids 1 and 7 share the exact same structural composition, except for the 9th residue. Thus, these sequences were truncated from the C-terminal end to understand the effect of chain length on their observed potency. The resulting peptoid 19 had decreased retention time and enhanced antimicrobial activity against S. aureus. No change in activity against E. coli was observed (see Table S5 in the supplemental material). In the case of peptoid 3, the deletion of the 9th residue gave similar results, indicating that higher antimicrobial activity is achieved by shortening the peptoid by one monomer. As a consequence, the deletion of the first monomer at the C terminus in peptoid 3 reflects a higher toxicity of this peptoid, which a factor by which peptoid 19 was not affected. Similarly, the deletion of the leucine residue (underlined) from the C-terminal end of peptide Ac-LKLLKKLL-KKLKKLLKKL-NH2 resulted in a peptide with increased antimicrobial activity and lower hemolytic property (16). Thus, the deletion of one small hydrophobic residue from the C-terminal end lowers the hydrophobicity of the resulting peptoids and enhances the antimicrobial activity of peptoids 7 and 3 against S. aureus and E. coli.
Antimicrobial activities of peptoids 3 and 4 in plasma.
Antimicrobial peptides are susceptible to enzymatic degradation, which limits their bioavailability in vivo. Bang and coworkers reported on a novel tryptamine-rich model of antimicrobial peptoids, compositionally similar to the peptoids in this study (1 to 5, 7, 15, and 19 to 22), with increased protease stability compared to that of the parental peptides (3). This is one of the major advantages of peptidomimetics, as antimicrobial activities are preserved but the pharmacokinetic properties are enhanced. The MICs of peptoids 3 and 4 were assayed by the broth microdilution method in the presence of 50% blood plasma from different donors. The MIC values remained unchanged: the MIC for peptoid 3 was 32 μg/ml, and the MIC for peptoid 4 was 16 μg/ml in the presence of blood plasma.
Killing kinetics.
Two peptoid candidates with optimized structural elements that exhibited good bacterial selectivity and high potency against E. coli (high SR values) were peptoids 3 and 4. The killing kinetics of these peptoids was evaluated using growth inhibition assays at different concentrations. When evaluating the killing kinetics assays, the bacterial load is 100 times higher than the one used in the determination of the MIC values. Therefore, the killing kinetics is in many respects conducted at sub-MIC values. The growth inhibition profiles for each peptoid at 1×, 2×, and 4× the MIC are presented in Fig. S4A and B in the supplemental material, in which concentration-dependent inhibition is observed for both peptoids. Furthermore, there is a distinct difference between the two peptoids at 1× the MIC, for which peptoid 3 shows noticeable growth inhibition (Fig. 4A). Ciprofloxacin is a fluoroquinolone antibiotic with high activity against Gram-negative bacteria. It blocks DNA replication by inhibiting DNA gyrase, an enzyme that catalyzes chromosomal DNA supercoiling (36). At 4× the MIC, both peptoids exhibit a bactericidal mode of action that resembles that of ciprofloxacin (Fig. 4B). The bactericidal rate against E. coli has been also reported for tryptamine-rich peptoid 11-mers in a comparative study between peptides and peptoid mimics (3). Here, a significant decrease in bacterial viability with 1× or 2× the MIC was established within the first 30 min of incubation. Compared to the results in this study, there is significant inhibition, which is more pronounced within the first 3 h with 4× the MIC for peptoids 3 and 4. If the compounds are not rapidly bactericidal, a mechanism other than membrane lysis can be attributed as a mode of action for these peptoids.
FIG 4.
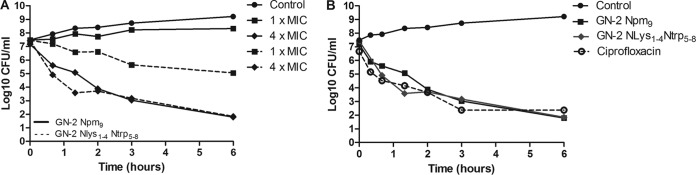
Growth inhibition curves for E. coli challenged with different antimicrobials. The data are log values of viable bacteria in log phase at 2 × 107 to 5 × 107 CFU/ml challenged with different antimicrobial agents, which were removed at various time points during 6 h incubation. Control growth is untreated bacteria. (A) Bacteria treated with 1× and 4× the MIC for peptoids 3 (GN-2-Nlys1–4 Ntrp5–8) and 4 (GN-2-Npm9). (B) Time-kill study of E. coli ATCC 25922 exposed to 4× the MIC of different antimicrobial agents: GN-2-Npm9 (64 μg/ml), GN-2- Nlys1–4 Ntrp5–8 (128 μg/ml), and ciprofloxacin (2 μg/ml). The data are representative of 3 independent experiments.
Conclusion.
Short peptidomimetics are promising candidates for the successful eradication of bacterial pathogens that show increasing resistance toward conventional or on-market antibiotics. In the present study, we employed a simple systematic design of short (9-mer) peptoid structures in order to generate novel antimicrobial peptoids. Changes, such as replacing Ntrp with monomers with specific aromatic side chains, rearranging the charge distribution along the peptoid backbone, and shortening the length, significantly impacted the overall hydrophobicity profiles of the peptoids. Our results conclude that peptoids with high hydrophobicity do not always appear as the most potent against E. coli and P. aeruginosa, while against S. aureus, there is a linear relationship between their hydrophobicity and potency. In addition, increased hydrophobicity caused by introducing highly aromatic residues, such as the N-(2,2-diphenylethyl)glycine (Ndpe) monomer, is strongly correlated with a loss of antibacterial specificity, resulting in high toxicity in mammalian cells. The monomer rearrangement within a peptoid sequence leads to increased antimicrobial activity against both E. coli and S. aureus. While fine-tuning the hydrophobicity, we have successfully optimized peptoid 1 by structural rearrangement and single-monomer exchange and identified two candidates with high selectivity ratios, 3 and 4, respectively. These peptoids show a bactericidal mode of action against E. coli similar to that of cationic antimicrobial peptides and peptidomimetics (37, 38). In summary, we report peptoids with an overall net charge of +4 and length of 8 or 9 residues that retain sufficient antimicrobial activity and toxicity profiles. Furthermore, while fine-tuning the hydrophobicity of peptoids to balance the structural requirements that allow selective bacterial killing, we propose N-(2-indolethyl)glycine (Ntrp) and N-(5-indene)glycine (Nai) to be used as preferred aromatic monomers.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by The Danish Council for Independent Research (grant 10-085287).
We also acknowledge the Molecular Foundry, whose work was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract DE-AC02-05CH11231.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.00237-15.
REFERENCES
- 1.Jenssen H, Hamill P, Hancock RE. 2006. Peptide antimicrobial agents. Clin Microbiol Rev 19:491–511. doi: 10.1128/CMR.00056-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tan NC, Yu P, Kwon YU, Kodadek T. 2008. High-throughput evaluation of relative cell permeability between peptoids and peptides. Bioorg Med Chem 16:5853–5861. doi: 10.1016/j.bmc.2008.04.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bang JK, Hai Y, Lee EK, Shin SY. 2010. A novel Trp-rich model antimicrobial peptide with increased protease stability. Bull Korean Chem Soc 31:2509–2513. doi: 10.5012/bkcs.2010.31.9.2509. [DOI] [Google Scholar]
- 4.Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. 1994. Proteolytic studies of homologous peptide and N-substituted glycine peptoid oligomers. Bioorg Med Chem Lett 4:2657–2662. doi: 10.1016/S0960-894X(01)80691-0. [DOI] [Google Scholar]
- 5.Chongsiriwatana NP, Patch JA, Czyzewski AM, Dohm MT, Ivankin A, Gidalevitz D, Zuckermann RN, Barron AE. 2008. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc Natl Acad Sci U S A 105:2794–2799. doi: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghosh C, Manjunath GB, Akkapeddi P, Yarlagadda V, Hoque J, Uppu DS, Konai MM, Haldar J. 2014. Small molecular antibacterial peptoid mimics: the simpler the better! J Med Chem 57:1428–1436. doi: 10.1021/jm401680a. [DOI] [PubMed] [Google Scholar]
- 7.Godballe T, Nilsson LL, Petersen PD, Jenssen H. 2011. Antimicrobial beta-peptides and alpha-peptoids. Chem Biol Drug Des 77:107–116. doi: 10.1111/j.1747-0285.2010.01067.x. [DOI] [PubMed] [Google Scholar]
- 8.Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. 2000. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci U S A 97:13003–13008. doi: 10.1073/pnas.97.24.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murphy JE, Uno T, Hamer JD, Cohen FE, Dwarki V, Zuckermann RN. 1998. A combinatorial approach to the discovery of efficient cationic peptoid reagents for gene delivery. Proc Natl Acad Sci U S A 95:1517–1522. doi: 10.1073/pnas.95.4.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang W, Seo J, Willingham SB, Czyzewski AM, Gonzalgo ML, Weissman IL, Barron AE. 2014. Learning from host-defense peptides: cationic, amphipathic peptoids with potent anticancer activity. PLoS One 9:e90397. doi: 10.1371/journal.pone.0090397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olivier GK, Cho A, Sanii B, Connolly MD, Tran H, Zuckermann RN. 2013. Antibody-mimetic peptoid nanosheets for molecular recognition. ACS Nano 7:9276–9286. doi: 10.1021/nn403899y. [DOI] [PubMed] [Google Scholar]
- 12.Olivier G, Proulx C, Zuckermann R. 2013. Functionalized peptoid nanosheets. Abstracts of papers of the American Chemical Society, vol 246 American Chemical Society, Washington, DC. [Google Scholar]
- 13.Chen YX, Guarnieri MT, Vasil AI, Vasil ML, Mant CT, Hodges RS. 2007. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob Agents Chemother 51:1398–1406. doi: 10.1128/AAC.00925-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thaker HD, Cankaya A, Scott RW, Tew GN. 2013. Role of amphiphilicity in the design of synthetic mimics of antimicrobial peptides with Gram-negative activity. ACS Med Chem Lett 4:481–485. doi: 10.1021/ml300307b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park Y, Kim HN, Park SN, Jang SH, Choi CH, Lim HT, Hahm KS. 2004. Design of novel analogues with potent antibiotic activity based on the antimicrobial peptide, HP(2-9)-ME(1-12). Biotechnol Lett 26:493–498. doi: 10.1023/B:BILE.0000019556.79703.4b. [DOI] [PubMed] [Google Scholar]
- 16.Blondelle SE, Houghten RA. 1992. Design of model amphipathic peptides having potent antimicrobial activities. Biochemistry 31:12688–12694. doi: 10.1021/bi00165a020. [DOI] [PubMed] [Google Scholar]
- 17.Brogden KA. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 3:238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 18.Patch JA, Barron AE. 2003. Helical peptoid mimics of magainin-2 amide. J Am Chem Soc 125:12092–12093. doi: 10.1021/ja037320d. [DOI] [PubMed] [Google Scholar]
- 19.Fjell CD, Jenssen H, Hilpert K, Cheung WA, Pante N, Hancock REW, Cherkasov A. 2009. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J Med Chem 52:2006–2015. doi: 10.1021/jm8015365. [DOI] [PubMed] [Google Scholar]
- 20.Figliozzi GM, Goldsmith R, Ng SC, Banville SC, Zuckermann RN. 1996. Synthesis of N-substituted glycine peptoid libraries. Methods Enzymol 267:437–447. doi: 10.1016/S0076-6879(96)67027-X. [DOI] [PubMed] [Google Scholar]
- 21.Wiegand I, Hilpert K, Hancock RE. 2008. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc 3:163–175. doi: 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- 22.Fjell CD, Jenssen H, Cheung WA, Hancock RE, Cherkasov A. 2011. Optimization of antibacterial peptides by genetic algorithms and cheminformatics. Chem Biol Drug Des 77:48–56. doi: 10.1111/j.1747-0285.2010.01044.x. [DOI] [PubMed] [Google Scholar]
- 23.Cherkasov A, Hilpert K, Jenssen H, Fjell CD, Waldbrook M, Mullaly SC, Volkmer R, Hancock REW. 2009. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem Biol 4:65–74. doi: 10.1021/cb800240j. [DOI] [PubMed] [Google Scholar]
- 24.Ryge TS, Hansen PR. 2005. Novel lysine-peptoid hybrids with antibacterial properties. J Pept Sci 11:727–734. doi: 10.1002/psc.705. [DOI] [PubMed] [Google Scholar]
- 25.Gopal R, Seo CH, Song PI, Park Y. 2013. Effect of repetitive lysine-tryptophan motifs on the bactericidal activity of antimicrobial peptides. Amino Acids 44:645–660. doi: 10.1007/s00726-012-1388-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Leeuw E, Rajabi M, Zou G, Pazgier M, Lu W. 2009. Selective arginines are important for the antibacterial activity and host cell interaction of human alpha-defensin 5. FEBS Lett 583:2507–2512. doi: 10.1016/j.febslet.2009.06.051. [DOI] [PubMed] [Google Scholar]
- 27.Strøm MB, Haug BE, Skar ML, Stensen W, Stiberg T, Svendsen JS. 2003. The pharmacophore of short cationic antibacterial peptides. J Med Chem 46:1567–1570. doi: 10.1021/jm0340039. [DOI] [PubMed] [Google Scholar]
- 28.McPhee JB, Scott MG, Hancock RE. 2005. Design of host defence peptides for antimicrobial and immunity enhancing activities. Comb Chem High Throughput Screen 8:257–272. doi: 10.2174/1386207053764558. [DOI] [PubMed] [Google Scholar]
- 29.Zelezetsky I, Tossi A. 2006. Alpha-helical antimicrobial peptides–using a sequence template to guide structure-activity relationship studies. Biochim Biophys Acta 1758:1436–1449. doi: 10.1016/j.bbamem.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 30.Wieprecht T, Dathe M, Krause E, Beyermann M, Maloy WL, MacDonald DL, Bienert M. 1997. Modulation of membrane activity of amphipathic, antibacterial peptides by slight modifications of the hydrophobic moment. FEBS Lett 417:135–140. doi: 10.1016/S0014-5793(97)01266-0. [DOI] [PubMed] [Google Scholar]
- 31.Fernández-Vidal M, Jayasinghe S, Ladokhin AS, White SH. 2007. Folding amphipathic helices into membranes: amphiphilicity trumps hydrophobicity. J Mol Biol 370:459–470. doi: 10.1016/j.jmb.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tyagi A, Kapoor P, Kumar R, Chaudhary K, Gautam A, Raghava GP. 2013. In silico models for designing and discovering novel anticancer peptides. Sci Rep 3:2984. doi: 10.1038/srep02984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wimley WC. 2010. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem Biol 5:905–917. doi: 10.1021/cb1001558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mihajlovic M, Lazaridis T. 2012. Charge distribution and imperfect amphipathicity affect pore formation by antimicrobial peptides. Biochim Biophys Acta 1818:1274–1283. doi: 10.1016/j.bbamem.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Y, Mant CT, Farmer SW, Hancock RE, Vasil ML, Hodges RS. 2005. Rational design of alpha-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J Biol Chem 280:12316–12329. doi: 10.1074/jbc.M413406200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fisher LM, Lawrence JM, Josty IC, Hopewell R, Margerrison EEC, Cullen ME. 1989. Ciprofloxacin and the fluoroquinolones. New concepts on the mechanism of action and resistance. Am J Med 87:S2–S8. doi: 10.1016/0002-9343(89)90010-7. [DOI] [PubMed] [Google Scholar]
- 37.Chan DI, Prenner EJ, Vogel HJ. 2006. Tryptophan- and arginine-rich antimicrobial peptides: structures and mechanisms of action. Biochim Biophys Acta 1758:1184–1202. doi: 10.1016/j.bbamem.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 38.Jahnsen RD, Sandberg-Schaal A, Vissing KJ, Nielsen HM, Frimodt-Møller N, Franzyk H. 2014. Tailoring cytotoxicity of antimicrobial peptidomimetics with high activity against multidrug-resistant Escherichia coli. J Med Chem 57:2864–2873. doi: 10.1021/jm401335p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.