

Abstract
The atypical BH3-only protein Bcl-2/adenovirus E1B 19-kDa interacting protein 3 (BNIP3) is an important regulator of hypoxia-mediated cell death. Interestingly, the susceptibility to BNIP3-mediated cell death differs between cells. In this study we examined whether there are mechanistic differences in BNIP3-mediated cell death between neonatal and adult cardiac myocytes. We discovered that BNIP3 is a potent inducer of cell death in neonatal myocytes, whereas adult myocytes are remarkably resistant to BNIP3. When exploring the potential underlying basis for the resistance, we discovered that adult myocytes express significantly higher levels of the mitochondrial antioxidant manganese superoxide dismutase (MnSOD) than neonatal myocytes. Overexpression of MnSOD confers resistance to BNIP3-mediated cell death in neonatal myocytes. In contrast, the presence of a pharmacological MnSOD inhibitor, 2-methoxyestradiol, results in increased sensitivity to BNIP3-mediated cell death in adult myocytes. Cotreatment with the mitochondria-targeted antioxidant MitoTEMPO or the MnSOD mimetic manganese (III) tetrakis (4-benzoic acid) porphyrin chloride abrogates the increased cell death by 2-methoxyestradiol. Moreover, increased oxidative stress also restores the ability of BNIP3 to induce cell death in adult myocytes. Taken together, these data indicate that redox status determines cell susceptibility to BNIP3-mediated cell death. These findings are clinically relevant, given that pediatric hearts are known to be more vulnerable than the adult heart to ischemic injury. Our studies provide important insight into why pediatric hearts are more sensitive to ischemic injury and may help in the clinical management of childhood heart disease.
Keywords: BNIP3, cardiac myocytes, mitochondria, autophagy, apoptosis, oxidative stress
the atypical BH3-only protein Bcl-2/adenovirus E1B 19-kDa interacting protein 3 (BNIP3) is a proapoptotic member of the BCL-2 family. BNIP3 is localized primarily to the mitochondria, where it induces cell death via activation of BAX/BAK and opening of the mitochondrial permeability transition pore (22, 33, 38). BNIP3 is upregulated during hypoxia by the transcription factor hypoxia-inducible factor-1 and plays a role in hypoxia-mediated cell death in many different cell types, including neonatal myocytes (2, 10, 33). Paradoxically, some cells are resistant to death caused by overexpression of BNIP3 (1, 30, 37); in some other cell types, BNIP3 causes slower and more modest cell death than other BH3-only proteins (32, 39). It is unclear why some cells are more resistant to BNIP3-mediated cell death.
Recent studies have found that BNIP3 also regulates mitochondrial morphology and autophagy (mitophagy) in cells (6, 9). Specifically, BNIP3 promotes fission of mitochondria by its interaction with OPA1 (23). BNIP3 also functions as an autophagy receptor on mitochondria, where its microtubule-associated protein 1 light chain (LC) 3-interacting region tethers mitochondria to autophagosomes (11, 24). The proautophagy function of BNIP3 is independent of its prodeath activity (11), but how BNIP3 switches between these seemingly opposing functions is unclear.
There is increasing evidence that deregulation of BNIP3 plays a role in human disease. For instance, BNIP3 expression is downregulated in malignant glioma (4) and gastrointestinal malignancies (18, 29), which renders these cells more resistant to chemotherapy. In contrast, elevated BNIP3 expression in non-small-cell lung carcinoma and ductal carcinoma of the breast correlates with a more aggressive tumor (8, 36). In addition, BNIP3 contributes to loss of myocytes during myocardial ischemia (10, 19, 33) and to the remodeling process after a myocardial infarction (5). Because of its implication in human disease, BNIP3 signifies an important potential future therapeutic target. Therefore, it is critical to increase our understanding of how BNIP3 regulates survival and death in different cells.
Infants with congenital heart defects often require corrective surgery to ensure survival. Several studies have found that the pediatric heart is more vulnerable than the adult heart to ischemic injury, which can occur during repair of congenital heart disease (12, 17). The underlying mechanism(s) for this difference in susceptibility to ischemic injury between pediatric and adult hearts is unknown. BNIP3 is a well-known contributor to hypoxia-mediated cell death, but no studies have examined whether there are mechanistic differences in BNIP3-mediated cell death between neonatal and adult myocytes. In this study we discovered that BNIP3 is a potent mediator of cell death in neonatal myocytes, whereas adult myocytes are resistant to BNIP3-mediated cell death as a result of increased antioxidant capacity.
MATERIALS AND METHODS
Cardiac myocyte isolation and cell culture.
All animal experiments were performed in accordance with institutional guidelines and approved by the Animal Care and Use Committee of the University of California, San Diego. Adult cardiac myocytes were isolated from 250- to 300-g male Sprague-Dawley rats, as previously described (21). Briefly, hearts were quickly excised, cannulated via the aorta, and perfused with heart medium [Joklik-modified MEM supplemented with 10 mM HEPES, 30 mM taurine, 2 mM carnitine, 2 mM creatine, and 0.1% collagenase II (Worthington Biochemicals, Lakewood, NJ) plus 0.1% BSA and 25 μM CaCl2]. After digestion, the ventricles were minced and myocytes were dispersed by gentle pipetting. Cells were filtered through a nylon mesh and washed twice at 100 g for 1 min. Calcium was reintroduced to select for calcium-tolerant cells. The cells were plated on laminin-coated dishes for experiments in plating medium (medium 199 supplemented with 10 mM HEPES, 5 mM taurine, 2 mM carnitine, 5 mM creatine, and 0.2% fatty acid-free BSA). After 1 h of plating, myocytes were infected with adenovirus.
Neonatal cardiac myocytes were isolated from 1- to 2-day-old Sprague-Dawley rat pups, digested with collagenase, and purified by a Percoll gradient (13). Myocytes were plated at a density of 3.5 × 104/cm2 and maintained overnight in 4:1 Dulbecco's modified Eagle's medium (DMEM)-medium 199, supplemented with 10% horse serum, 5% fetal calf serum, and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin). Cells were used for experiments on the following day.
Adenoviral infections.
Cultured neonatal and adult myocytes were infected with the adenovirus for 3 h in plating medium and DMEM + 2% heat-inactivated FBS, respectively. After infection, the cells were washed and then incubated in regular cell culture medium for 21 h.
Fluorescence microscopy analysis.
For measurement of mitochondrial membrane potential, myocytes overexpressing β-galactosidase (β-gal) or BNIP3 were stained with 100 nM tetramethylrhodamine methyl ester (TMRM; catalog no. T668, Life Technology) and Hoechst 33342 (Life Technology). Cell death was assessed by measurement of plasma membrane permeability to YO-PRO-1, as previously described (10). At 24 h postinfection, cells were stained with 100 nM TMRM or 1 μM YO-PRO-1 (catalog no. Y3603, Life Technology) and Hoechst 33342 for 20 min at 37°C and then examined by fluorescence microscopy. Autophagosomes were labeled by infection of cells with Ad-β-gal or Ad-BNIP3 + Ad-GFP-LC3. DMSO or 100 nM bafilomycin A1 was added for the last 3 h of the experiment to assess autophagic flux. The number of autophagosomes per cell was assessed at 20 h postinfection using a Carl Zeiss Axio Observer.Z1 equipped with a motorized z stage and ApoTome for optical sectioning at ×63 magnification. At least 50 cells per condition were analyzed.
Hypoxia experiments.
After 24 h of infection with β-gal, BNIP3ΔTM, or MnSOD, the medium was changed to DMEM high glucose (Invitrogen) saturated with 95% N2-5% CO2, and neonatal cardiac myocytes were placed in a hypoxic chamber (Billups-Rothenberg). The hypoxic chamber was flushed with 95% N2-5% CO2 for 10 min, sealed, and placed in the 37°C incubator for 30 h. Control cells were cultured at 37°C in 5% CO2. DNA fragmentation was detected using the cell death detection ELISAPLUS (Roche Applied Science), as previously described (35). Briefly, cells were washed with ice-cold PBS twice and harvested in cytosolic extraction buffer, nutated at 4°C for 10 min, and spun down at 20,000 g for 3 min, and supernatants were saved. Cell lysates were incubated with anti-histone-biotin antibody and anti-DNA-peroxidase antibody in a streptavidin-coated 96-well plate on a rocking shaker at room temperature for 2 h. Wells were washed with incubation buffer three times, and 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic) acid substrate was added. Absorbance was measured at 405 nm using an Infinite M200 PRO plate reader (Tecan).
Oxidative stress experiments.
Adult cardiac myocytes overexpressing β-gal or BNIP3 were treated with 100 μM H2O2 for 3 h. For study of mitochondrial membrane potential and cell death, cells were stained with TMRM and YO-PRO-1, respectively. To inhibit MnSOD, adult cardiac myocytes were infected with Ad-β-gal or Ad-BNIP3 in the presence of 100 μM 2-methoxyestradiol (2-ME; catalog no. M6383, Sigma Aldrich), 5 μM MitoTEMPO, or 10 μM manganese (III) tetrakis (4-benzoic acid) porphyrin chloride (MnTBAP) for 24 h.
Western blotting.
Cells were lysed in ice-cold buffer containing 50 mM Tris·HCl, pH 7.4, 150 mM NaCl, 12 mM Na-deoxycholate, and 1% Triton X-100 plus complete protease inhibitor cocktail (Roche Applied Science) and then cleared by centrifugation at 20,000 g for 20 min. Proteins were separated by SDS-PAGE under reducing conditions, transferred to nitrocellulose, and incubated with antibodies [BNIP3 (Sigma-Aldrich; 1:1,000 dilution), LC3 (Cell Signaling; 1:1,000 dilution), MnSOD (Thermo Scientific; 1:1,000 dilution), and actin (Sigma-Aldrich; 1:1,000 dilution)]. Blots were quantified and analyzed using Quantity One software (Bio-Rad).
Statistical analysis.
Values are means ± SE. Statistical analyses were performed using one-way ANOVA followed by Tukey's post hoc test for multiple comparisons to identify statistical significance between groups. P < 0.05 was considered significant.
RESULTS
Adult cardiac myocytes are resistant to BNIP3-mediated cell death.
Since the prodeath activity of BNIP3 has been found to differ in various cells, we investigated whether susceptibility to BNIP3-mediated cell death differs between neonatal and adult cardiac myocytes. Neonatal and adult myocytes were infected with an adenovirus encoding β-gal or BNIP3 to assess the effect on cell viability. Interestingly, we found that BNIP3 caused loss of mitochondrial membrane potential in a significant number of neonatal myocytes (Fig. 1, A and B). In contrast, BNIP3 had no effect on mitochondrial membrane potential in adult myocytes (Fig. 1, A and B). BNIP3 also compromised plasma membrane integrity, as measured by YO-PRO-1 uptake, in neonatal, but not adult, myocytes (Fig. 1, C and D). A dose-response experiment confirmed that adult cardiac myocytes were resistant to BNIP3-mediated cell death, even at much higher levels of BNIP3 infection (Fig. 1E).
Fig. 1.

BNIP3 overexpression induces cell death in neonatal cardiac myocytes (NCM), but not adult cardiac myocytes (ACM). Cells were infected with 50 multiplicity of infection (MOI) of β-galactosidase adenovirus (Ad-β-gal) or Ad-BNIP3 and, after 24 h, stained with tetramethylrhodamine methyl ester (TMRM, red) to assess mitochondrial membrane potential (Δψm) or with YO-PRO-1 (green) to determine plasma membrane permeability. Nuclei were counterstained with Hoechst 33342 (blue). Scale bars = 50 μm. A: representative images of neonatal and adult cardiac myocytes. B: quantitation of Δψm. Values are means ± SE; n = 3. *P < 0.05. C: representative images of neonatal and adult cardiac myocytes stained with YO-PRO-1. Scale bars = 100 μm. D: quantitation of cell death. BNIP3 overexpression causes significant cell death in neonatal, but not adult, cardiac myocytes. Values are means ± SE; n = 3. *P < 0.05. E: cells were infected with 50, 100, or 200 MOI of Ad-β-gal or Ad-BNIP3 for 24 h and then stained with TMRM or YO-PRO-1. Values are means ± SE; n = 3. *P < 0.05; **P < 0.01; ***P < 0.001.
In addition, BNIP3 is a potent inducer of autophagy (10). We confirmed that induction of autophagy by BNIP3 was independent of loss of mitochondrial membrane potential and activation of cell death. Overexpression of BNIP3 led to a significant increase in autophagy as measured by an increase in the ratio of LC3-II to LC3-I in neonatal and adult cardiac myocytes (Fig. 2, A and B). We also confirmed that BNIP3 caused an increase in the number of GFP-LC3-positive autophagosomes. The presence of bafilomycin A1 led to an additional increase in the number of autophagosomes in cells overexpressing BNIP3 (Fig. 2, C–F), confirming that the increased number of autophagosomes reflected an increase in autophagic activity, rather than reduced flux.
Fig. 2.

BNIP3 overexpression activates autophagy in neonatal and adult cardiac myocytes. A: representative Western blots for the autophagy marker microtubule-associated protein 1 light chain 3 (LC3-I and LC3-II). B: quantitation of the ratio of LC3-II to LC3-I. Values are means ± SE; n = 4. *P < 0.05. C: fluorescence microscopy analysis of GFP-LC3-positive puncta in neonatal cardiac myocytes. Baf A1, bafilomycin A1. Scale bars = 10 μm. D: quantitation of GFP-LC3-positive autophagosomes per neonatal cardiac myocyte. Bafilomycin A1 (100 nM) increases the number of autophagosomes. Values are means ± SE; n = 3. *P < 0.05. E: fluorescence microscopy analysis of GFP-LC3-positive puncta in adult cardiac myocytes. Scale bars = 20 μm. F: quantitation of GFP-LC3-positive autophagosomes per adult cardiac myocyte. Bafilomycin A1 (100 nM) increases the number of autophagosomes. Values are means ± SE; n = 4. *P < 0.05.
Autophagy has been reported to protect against cell death by clearing damaged organelles and cytotoxic protein aggregates (27). Therefore, we investigated whether the enhanced autophagy contributed to the resistance to BNIP3-mediated cell death in adult myocytes. Autophagy-related protein 5 (Atg5) is involved in elongation of the autophagosomes and is a critical component of autophagy (28). We found that overexpression of Atg5-K130R, a dominant-negative of Atg5 (31), did not render the adult myocytes susceptible to BNIP3-mediated cell death (Fig. 3A). Similarly, the presence of the pharmacological inhibitor of autophagy 3-methyladenine (3-MA) had no effect on cell viability, even in cells overexpressing BNIP3 (Fig. 3B). This suggests that BNIP3 induces autophagy in neonatal and adult myocytes but that autophagy does not contribute to resistance to the prodeath activity of BNIP3 in adult cardiac myocytes.
Fig. 3.
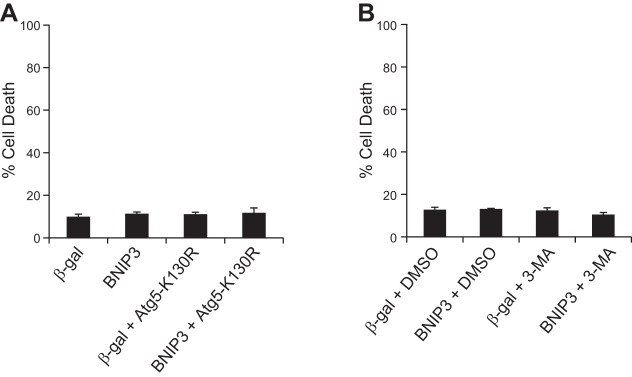
Inhibition of autophagy does not increase sensitivity to BNIP3-mediated cell death in adult cardiac myocytes. Cells were infected with 100 MOI of Ad-β-gal, Ad-BNIP3, Ad-β-gal + Ad-Atg5-K130R, or Ad-BNIP3 + Ad-Atg5-K130R (A) or Ad-β-gal or Ad-BNIP3 in the presence of vehicle (DMSO) or 10 mM 3-methyladenine (3-MA) (B), and cell death was assessed 24 h later by YO-PRO-1 staining. Values are means ± SE; n = 3–4.
Reduced antioxidant capacity contributes to increased sensitivity to BNIP3-mediated cell death.
BNIP3 induces production of reactive oxygen species (ROS) (38), which contributes to opening of the mitochondrial permeability transition pore and cell death. Thus we postulated that neonatal and adult myocytes possess different levels of antioxidant capacity that might influence their susceptibility to BNIP3. To investigate this possibility, we compared the levels of the antioxidant enzyme MnSOD in neonatal and adult myocytes. MnSOD is localized in the mitochondria and converts superoxide, a by-product of oxidative phosphorylation, to H2O2 and O2 (14). Interestingly, we found significantly higher levels of MnSOD expression in adult than neonatal cells (Fig. 4A). To investigate if elevated MnSOD contributed to the resistance to BNIP3, we overexpressed MnSOD and BNIP3 in neonatal myocytes. We found that overexpression of MnSOD significantly suppressed BNIP3-induced cell death (Fig. 4, B and C), confirming that increased levels of MnSOD antagonized the prodeath activity of BNIP3.
Fig. 4.

Overexpression of MnSOD protects neonatal cardiac myocytes against BNIP3-mediated cell death. A: Western blot analysis of MnSOD expression in neonatal and adult cardiac myocytes. Significantly higher levels of MnSOD are expressed in adult cardiac myocytes. Values are means ± SE; n = 4. *P < 0.05. B: representative images of neonatal myocytes stained with YO-PRO-1. Scale bars = 100 μm. C: quantitation of cell death. Overexpression of MnSOD suppresses BNIP3-mediated cell death in neonatal myocytes. Values are means ± SE; n = 4. *P < 0.05.
Next, we investigated whether MnSOD could protect against cell death mediated by endogenous BNIP3. BNIP3 is rapidly induced on exposure to hypoxia in neonatal myocytes (Fig. 5, A and B), which correlated with increased cell death (Fig. 5C). Overexpression of BNIP3ΔTM, a dominant-negative of BNIP3 (33), or MnSOD significantly reduced hypoxia-mediated cell death (Fig. 5C). This confirms that endogenous BNIP3 contributes to hypoxia-mediated cell death and that enhancing MnSOD levels is protective.
Fig. 5.

Overexpression of MnSOD protects neonatal myocytes against hypoxia-mediated cell death. A: representative Western blot of BNIP3 expression in neonatal cardiac myocytes under normoxia and after exposure to hypoxia. B: BNIP3 is significantly increased after 30 h of hypoxia. Values are means ± SE; n = 4. *P < 0.05. C: overexpression of BNIP3ΔTM (30 MOI) or MnSOD (30 MOI) significantly reduces hypoxia-mediated cell death in neonatal myocytes. Cell death was assessed using cell death detection ELISA. Values are means ± SE; n = 4. *P < 0.05.
Inhibition of MnSOD restores susceptibility of adult cardiac myocytes to BNIP3-induced cell death.
To further investigate if the elevated expression of MnSOD in adult myocytes provides resistance to BNIP3-mediated cell death, we assessed whether inhibition of MnSOD would increase sensitivity to BNIP3-mediated cell death. We found that the presence of 2-ME, a pharmacological inhibitor of MnSOD (16), resulted in cell death in adult myocytes overexpressing BNIP3 (Fig. 6, A and B). The presence of the mitochondria-targeted ROS scavenger MitoTEMPO or MnTBAP, a MnSOD mimetic (25), significantly reduced cell death in cells treated with 2-ME (Fig. 6C). These results confirm that the enhanced antioxidant capacity of adult myocytes protects against BNIP3-induced cell death.
Fig. 6.

Inhibition of MnSOD restores sensitivity of adult myocytes to BNIP3-mediated cell death. A and B: cells were infected with 50 MOI of Ad-β-gal or Ad-BNIP3 in the presence of DMSO or 100 μM 2-methoxyestradiol (2-ME). A: representative images of adult myocytes stained with YO-PRO-1. Scale bars = 100 μm. B: quantitation of cell death (YO-PRO-1-positive cells). 2-ME significantly increases cell death in adult cardiac myocytes overexpressing BNIP3. Values are means ± SE; n = 3. *P < 0.05. C: cells were infected with 50 MOI of Ad-β-gal or Ad-BNIP3 in the presence of 100 μM 2-ME, 5 μM MitoTEMPO, or 10 μM MnTBAP alone or in combination. Cell death was assessed using YO-PRO-1 staining. MitoTEMPO or MnTBAP significantly reduced 2-ME-mediated cell death in adult cardiac myocytes overexpressing BNIP3. Values are means ± SE; n = 4. *P < 0.05.
Increased oxidative stress promotes BNIP3-mediated cell death in adult myocytes.
Studies have found that increased ROS leads to dimerization and activation of BNIP3 (15, 21). To investigate whether additional oxidative stress is needed to activate the proapoptotic activity of BNIP3 in adult myocytes, we exposed adult myocytes overexpressing BNIP3 to H2O2. As expected, H2O2 alone caused loss of mitochondrial membrane potential (Fig. 7, A and B) and cell death (Fig. 7C) in a significant number of cells. Interestingly, although BNIP3 overexpression alone had no effect on mitochondrial membrane potential and cell viability, addition of H2O2 to these cells significantly augmented loss of mitochondrial membrane potential and cell death in adult myocytes compared with H2O2 alone (Fig. 7). These findings indicate that activation of BNIP3-mediated cell death is dependent on the redox status of the cell.
Fig. 7.

Oxidative stress-mediated cell death is enhanced in adult cardiac myocytes overexpressing BNIP3. Cells were infected with 50 MOI of Ad-β-gal or Ad-BNIP3 for 21 h and then treated with H2O2 (100 μM) for 3 h. To assess loss of Δψm and cell death, cells were stained with TMRM and YO-PRO-1, respectively. A: representative images of adult cardiac myocytes stained with TMRM (red) and Hoechst 3342 (blue). Scale bars = 100 μm. B: quantitation of Δψm loss in adult myocytes. H2O2-mediated loss of Δψm is augmented in cells overexpressing BNIP3. Values are means ± SE; n = 3. *P < 0.05. C: quantitation of cell death (YO-PRO-1). H2O2-mediated cell death is augmented in cells overexpressing BNIP3. Values are means ± SE; n = 3. *P < 0.05.
DISCUSSION
Although BNIP3 is known to play a role in various diseases, its exact function in cells is unclear. This is further complicated by recent data indicating that BNIP3 also regulates mitochondrial turnover under normal conditions (6, 9). Interestingly, cells also differ in their susceptibility to BNIP3-mediated cell death, but the molecular basis for this difference is unknown. Our study reports the first evidence that adult myocytes are resistant to BNIP3-mediated cell death under nonstressed conditions and that this is due to their increased antioxidant defense. MnSOD is important in neutralizing superoxide, and inhibition of MnSOD in the adult myocytes restores the sensitivity to BNIP3-mediated cell death. In contrast, neonatal myocytes express significantly lower levels of MnSOD and are very sensitive to the prodeath activity of BNIP3. Increasing levels of MnSOD in neonatal myocytes restores resistance to BNIP3-mediated cell death. Another key finding of this study is that BNIP3 requires additional oxidative stress for activation of its prodeath activity in adult myocytes. Thus our data suggest that the cellular redox status determines sensitivity to BNIP3-mediated cell death in cardiac myocytes. Our findings also provide some insight into why pediatric patients are more vulnerable to ischemic injury during cardiac surgery.
Antioxidant levels in the myocardium are known to follow a developmental pattern where levels of the major antioxidants, such as catalase, SOD, and glutathione, are lower in newborn than adult hearts (3, 26). Our studies show that the reduced levels of MnSOD lead to increased susceptibility to BNIP3-mediated cell death in neonatal myocytes. We and others previously reported that neonatal myocytes express little or no BNIP3 under normal conditions (2, 10, 33). Instead, during hypoxia, BNIP3 is rapidly upregulated in neonatal myocytes, where it perturbs mitochondrial function and activates cell death. Our data suggest that elevated BNIP3 is detrimental in neonatal myocytes due to the reduced ability to neutralize and defend against oxidants, such as H2O2 and superoxide, that activate BNIP3.
Congenital heart disease in newborns and infants often requires corrective surgery for survival into adulthood. Such surgery involves cardiopulmonary bypass, which can lead to ischemic injury. It is also known that the younger the age of the patients, the more vulnerable they are to ischemic injury during surgical repair of the heart (12). Thus our data suggest that potential upregulation of BNIP3 during ischemia combined with the inability to neutralize excessive ROS production may be a key factor in the increased susceptibility to ischemic injury in pediatric patients. Additional studies are necessary for a full understanding of the underlying mechanisms of the increased susceptibility, so that therapies can be developed to limit the myocardial injury in pediatric patients.
In contrast to neonatal myocytes, BNIP3 is constitutively expressed in the adult heart (5, 10). Interestingly, recent findings suggest that BNIP3 plays an important role in mitochondrial maintenance in adult myocardium (6). A similar function for BNIP3 has been reported in liver, where loss of BNIP3 leads to an increase in mitochondrial mass and the presence of dysfunctional mitochondria (9). Clearly, there is an increased need to turn over aged and defective mitochondria in aging tissues, whereas this is less critical for homeostasis in the neonatal heart. The need to turn over mitochondria on a regular basis in adult cells would explain why BNIP3 is constitutively expressed in the adult, but not neonatal, heart. Adult myocytes are resistant to BNIP3-mediated cell death in the absence of additional oxidative stress, and our data demonstrate that there is still activation of autophagy in these cells by BNIP3. Autophagy can protect cells against activation of cell death by clearing dysfunctional mitochondria (20), and we previously found that inhibition of autophagy in fibroblasts and HL-1 cells increases BNIP3-mediated cell death (10, 34). Interestingly, in this study we observed that inhibition of autophagy in adult myocytes did not result in increased susceptibility to BNIP3-mediated cell death, suggesting that the enhanced autophagy is not a major contributor to resistance to BNIP3 under these conditions. It also suggests that the function of BNIP3-mediated autophagy in adult cardiac myocytes might differ from that in cells that are susceptible to BNIP3-mediated cell death.
Our data demonstrate clearly that oxidative stress plays a critical role in determining cell susceptibility to BNIP3-mediated cell death. There are no pharmacological inhibitors that specifically target only MnSOD, and the inhibitor that we used in this study also inhibits Cu,ZnSOD in the cytosol (16). Similarly, MnTBAP also mimics both isoforms of SOD (7). Although BNIP3 and MnSOD are localized to the mitochondria and mitochondrial ROS are involved in activating cell death in myocytes overexpressing BNIP3, it is possible that the cytosolic Cu,ZnSOD also contributes to the resistance to BNIP3-mediated cell death in the adult myocytes. We recognize that this is a limitation of our study and that future studies using tissue-specific MnSOD conditional knockout mice are needed to examine the susceptibility of BNIP3 in adult myocytes.
Upregulation of BNIP3 in neonatal myocytes during ischemia appears to be a detrimental event that contributes to loss of myocytes. Thus the possibility of targeting BNIP3 to limit ischemic injury in patients undergoing cardiac surgery is very intriguing. Although BNIP3 represents an interesting potential therapeutic target to prevent activation of cell death, this protein also appears to be important in maintaining mitochondrial mass in cells. Thus further studies are necessary to enhance our understanding of how BNIP3 regulates cell death and mitochondrial autophagy in the immature and adult myocardium.
GRANTS
S. Miyamoto is supported by National Heart, Lung, and Blood Institute Grant R01 HL-097037. Å. B. Gustafsson is supported by an American Heart Association Established Investigator Award and National Heart, Lung, and Blood Institute Grants R01 HL-087023, R01 HL-101217, and P01 HL-085577.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.L. and A.B.G. developed the concept and designed the research; Y.L., D.A.K., R.A.H., M.Q.C., H.-Y.L., and S.M. performed the experiments; Y.L., D.A.K., R.A.H., M.Q.C., H.-Y.L., and S.M. analyzed the data; Y.L., D.A.K., and A.B.G. interpreted the results of the experiments; Y.L., D.A.K., M.Q.C., and S.M. prepared the figures; Y.L. and A.B.G. drafted the manuscript; Y.L., D.A.K., M.Q.C., and A.B.G. edited and revised the manuscript; Y.L., D.A.K., R.A.H., M.Q.C., H.-Y.L., S.M., and A.B.G. approved the final version of the manuscript.
ACKNOWLEDGMENTS
Present address of Y. Lee: Department of Health and Exercise Science, University of West Florida, Pensacola, FL.
REFERENCES
- 1.Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29: 2570–2581, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci USA 97: 9082–9087, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cabigas EB, Liu J, Boopathy AV, Che PL, Crawford BH, Baroi G, Bhutani S, Shen M, Wagner MB, Davis ME. Dysregulation of catalase activity in newborn myocytes during hypoxia is mediated by c-Abl tyrosine kinase. J Cardiovasc Pharmacol Ther 20: 93–103, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daido S, Kanzawa T, Yamamoto A, Takeuchi H, Kondo Y, Kondo S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res 64: 4286–4293, 2004. [DOI] [PubMed] [Google Scholar]
- 5.Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, Li H, Kirshenbaum LA, Hahn HS, Robbins J, Jones WK, Dorn GW. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest 117: 2825–2833, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dorn GW., 2nd Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res 3: 374–383, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gauuan PJ, Trova MP, Gregor-Boros L, Bocckino SB, Crapo JD, Day BJ. Superoxide dismutase mimetics: synthesis and structure-activity relationship study of MnTBAP analogues. Bioorg Med Chem 10: 3013–3021, 2002. [DOI] [PubMed] [Google Scholar]
- 8.Giatromanolaki A, Koukourakis MI, Sowter HM, Sivridis E, Gibson S, Gatter KC, Harris AL. BNIP3 expression is linked with hypoxia-regulated protein expression and with poor prognosis in non-small cell lung cancer. Clin Cancer Res 10: 5566–5571, 2004. [DOI] [PubMed] [Google Scholar]
- 9.Glick D, Zhang W, Beaton M, Marsboom G, Gruber M, Simon MC, Hart J, Dorn GW 2nd, Brady MJ, Macleod KF. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol 32: 2570–2584, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ 14: 146–157, 2007. [DOI] [PubMed] [Google Scholar]
- 11.Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 287: 19094–19104, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hasegawa T, Yamaguchi M, Yoshimura N, Okita Y. The dependence of myocardial damage on age and ischemic time in pediatric cardiac surgery. J Thorac Cardiovasc Surg 129: 192–198, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Hilal-Dandan R, Means CK, Gustafsson AB, Morissette MR, Adams JW, Brunton LL, Heller Brown J. Lysophosphatidic acid induces hypertrophy of neonatal cardiac myocytes via activation of Gi and Rho. J Mol Cell Cardiol 36: 481–493, 2004. [DOI] [PubMed] [Google Scholar]
- 14.Holley AK, Dhar SK, Xu Y, St Clair DK. Manganese superoxide dismutase: beyond life and death. Amino Acids 42: 139–158, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoshino A, Matoba S, Iwai-Kanai E, Nakamura H, Kimata M, Nakaoka M, Katamura M, Okawa Y, Ariyoshi M, Mita Y, Ikeda K, Ueyama T, Okigaki M, Matsubara H. p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. J Mol Cell Cardiol 52: 175–184, 2012. [DOI] [PubMed] [Google Scholar]
- 16.Huang P, Feng L, Oldham EA, Keating MJ, Plunkett W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature 407: 390–395, 2000. [DOI] [PubMed] [Google Scholar]
- 17.Imura H, Caputo M, Parry A, Pawade A, Angelini GD, Suleiman MS. Age-dependent and hypoxia-related differences in myocardial protection during pediatric open heart surgery. Circulation 103: 1551–1556, 2001. [DOI] [PubMed] [Google Scholar]
- 18.Kennedy AS, Harrison GH, Mansfield CM, Zhou XJ, Xu JF, Balcer-Kubiczek EK. Survival of colorectal cancer cell lines treated with paclitaxel, radiation, and 5-FU: effect of TP53 or hMLH1 deficiency. Int J Cancer 90: 175–185, 2000. [PubMed] [Google Scholar]
- 19.Kubasiak LA, Hernandez OM, Bishopric NH, Webster KA. Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc Natl Acad Sci USA 99: 12825–12830, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kubli DA, Gustafsson AB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res 111: 1208–1221, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kubli DA, Quinsay MN, Huang C, Lee Y, Gustafsson AB. Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am J Physiol Heart Circ Physiol 295: H2025–H2031, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kubli DA, Ycaza JE, Gustafsson AB. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem J 405: 407–415, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Landes T, Emorine LJ, Courilleau D, Rojo M, Belenguer P, Arnaune-Pelloquin L. The BH3-only Bnip3 binds to the dynamin Opa1 to promote mitochondrial fragmentation and apoptosis by distinct mechanisms. EMBO Rep 11: 459–465, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma X, Godar RJ, Liu H, Diwan A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy 8: 297–309, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin RC, Liu Q, Wo JM, Ray MB, Li Y. Chemoprevention of carcinogenic progression to esophageal adenocarcinoma by the manganese superoxide dismutase supplementation. Clin Cancer Res 13: 5176–5182, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Maulik N, Baker JE, Engelman RM, Das DK. Postnatal developmental profiles of antioxidant enzymes in heart. Ann NY Acad Sci 793: 439–448, 1996. [DOI] [PubMed] [Google Scholar]
- 27.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 451: 1069–1075, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13: 619–624, 2007. [DOI] [PubMed] [Google Scholar]
- 29.Okami J, Simeone DM, Logsdon CD. Silencing of the hypoxia-inducible cell death protein BNIP3 in pancreatic cancer. Cancer Res 64: 5338–5346, 2004. [DOI] [PubMed] [Google Scholar]
- 30.Papandreou I, Krishna C, Kaper F, Cai D, Giaccia AJ, Denko NC. Anoxia is necessary for tumor cell toxicity caused by a low-oxygen environment. Cancer Res 65: 3171–3178, 2005. [DOI] [PubMed] [Google Scholar]
- 31.Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, Cho DH, Choi B, Lee H, Kim JH, Mizushima N, Oshumi Y, Jung YK. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem 280: 20722–20729, 2005. [DOI] [PubMed] [Google Scholar]
- 32.Ray R, Chen G, Vande VC, Cizeau J, Park JH, Reed JC, Gietz RD, Greenberg AH. BNIP3 heterodimerizes with Bcl-2/Bcl-XL and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem 275: 1439–1448, 2000. [DOI] [PubMed] [Google Scholar]
- 33.Regula KM, Ens K, Kirshenbaum LA. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res 91: 226–231, 2002. [DOI] [PubMed] [Google Scholar]
- 34.Rikka S, Quinsay MN, Thomas RL, Kubli DA, Zhang X, Murphy AN, Gustafsson AB. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ 18: 721–731, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts DJ, Tan-Sah VP, Ding EY, Smith JM, Miyamoto S. Hexokinase-II positively regulates glucose starvation induced autophagy through TORC1 inhibition. Mol Cell 53: 521–533, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sowter HM, Ferguson M, Pym C, Watson P, Fox SB, Han C, Harris AL. Expression of the cell death genes BNip3 and NIX in ductal carcinoma in situ of the breast: correlation of BNip3 levels with necrosis and grade. J Pathol 201: 573–580, 2003. [DOI] [PubMed] [Google Scholar]
- 37.Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol 27: 6229–6242, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vande VC, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol 20: 5454–5468, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yasuda M, Theodorakis P, Subramanian T, Chinnadurai G. Adenovirus E1B-19K/BCL-2 interacting protein BNIP3 contains a BH3 domain and a mitochondrial targeting sequence. J Biol Chem 273: 12415–12421, 1998. [DOI] [PubMed] [Google Scholar]