

Abstract
Apolipoprotein A-I and A-IV are protein constituents of high-density lipoproteins although their functional difference in lipoprotein metabolism is still unclear. To compare anti-atherogenic properties between apoA-I and apoA-4, we characterized both proteins in lipid-free and lipid-bound state. In lipid-free state, apoA4 showed two distinct bands, around 78 and 67 Å on native gel electrophoresis, while apoA-I showed scattered band pattern less than 71 Å. In reconstituted HDL (rHDL) state, apoA-4 showed three major bands around 101 Å and 113 Å, while apoA-I-rHDL showed almost single band around 98 Å size. Lipid-free apoA-I showed 2.9-fold higher phospholipid binding ability than apoA-4. In lipid-free state, BS3-crosslinking revealed that apoA-4 showed less multimerization tendency upto dimer, while apoA-I showed pentamerization. In rHDL state (95:1), apoA-4 was existed as dimer as like as apoA-I. With higher phospholipid content (255:1), five apoA-I and three apoA-4 were required to the bigger rHDL formation. Regardless of particle size, apoA-I-rHDL showed superior LCAT activation ability than apoA-4-rHDL. Uptake of acetylated LDL was inhibited by apoA-I in both lipid-free and lipid-bound state, while apoA-4 inhibited it only lipid-free state. ApoA-4 showed less anti-atherogenic activity with more sensitivity to glycation. In conclusion, apoA-4 showed inferior physiological functions in lipid-bound state, compared with those of apoA-I, to induce more pro-atherosclerotic properties.
Keywords: apolipoprotein A-I, apolipoprotein A-4, fructosylation, recombinant high-density lipoprotein
INTRODUCTION
High-density lipoprotein (HDL) is a complex mixture of lipids, apolipoproteins, and enzymes in a phospholipid bilayer. Apolipoprotein (apo) A-I, a 28-kDa single polypeptide, is a major protein component of HDL (Frank and Marcel, 2000; Jonas, 1998). ApoA-I is responsible for the major functions of HDL, including lipid binding and solubilization, cholesterol removal from peripheral cells, and activation of lecithin:cholesterol acyltransferase (LCAT) (Brouillette et al., 2001). ApoA4, a 46-kDa glycoprotein, is a minor apolipoprotein component of HDL and chylomicron (Mahley et al., 1984), and it functions in LCAT activation (Steinmetz and Utermann, 1985) and CETP activity (Main et al., 1996).
In carbohydrate metabolism, apoA4 improves glucose homeostasis by enhancing insulin secretion as well as inhibits hepatic gluconeogenesis via regulation of nuclear receptor NR1D1 (Li et al., 2014). Unlike other apolipoproteins, apoA4 shows low affinity for plasma lipoproteins (Beisiegel and Utermann, 1979), suggesting that its lipid-free form is more preferable in blood circulation. In a postprandial state, apoA4 is transferred to HDL or a lipoprotein-free fraction in the plasma (Weinberg, 2002). Although it is well known that apoA4 can increase the HDL particle fraction (Goldberg et al., 1990) and facilitate triacylglycerol clearance (VerHague et al., 2013), the biological role of lipid-free apoA4 is unclear.
ApoA-I and apoA4 share several common features in evolution and protein architecture. Both are synthesized in the intestine as preproapolipoprotein and consist of 22-mer amino acid repeats that form amphipathic α-helices. It has been well known that apoA-I, apoA4, and apoE have close structural similarities since they diverged from a common ancestral sequence (Boguski et al., 1984). Specifically, apoA-I and apoA4 both share anti-oxidant and anti-atherosclerotic activity. ApoA-I inhibits Cu2+-mediated LDL oxidation (Cho et al., 2006) and CETP activity (Cho, 2011), whereas apoA4 inhibits lipoprotein peroxidation (Ferretti et al., 2002) and promotes cholesterol efflux via the ABCA1 pathway. Several reports have suggested that apoA4 plays a protective role against atherosclerosis, although the mechanism is still not clear.
The current study was designed to investigate functional and structural differences between apoA-I and apoA4 in regarding to their anti-atherogenic properties in functional and structural correlations. In order to compare physiological functions in lipid-free and lipid-bound states, the apoA-I and apoA4 were individually expressed, purified, and reconstituted in HDL at different molar ratios. Using rHDL containing either apoA-I or apoA4, we carried out several functional evaluations regarding antioxidant and anti-atherosclerotic activities.
METERIALS AND METHODS
Purification of proteins
Human apoA-I was purified from pET30-wildtype apoA-I-vector using the E. coli BL21(DE3) expression system as our previous report (Han et al., 2005). Human recombinant apoA4 containing an N-terminal glycine residue was prepared by cleavage of apoA4 containing an N-terminal His tag and TEV protease cleavage site (His-TEV-apoA4) by TEV protease, similar to a previously described methods (Park et al., 2012; Tubb et al., 2009). His-TEV-apoA4 was expressed as a soluble protein in E. coli BL21(DE3) transformed with pET-22b(+)-His-TEV-apoA4. The E. coli culture pellet was resuspended in lysis buffer (20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 0.1% Triton X-100), after which cells were disrupted by sonication. The supernatant was bound to Ni2+-NTA agarose resin (Cat. No. 30210, Qiagen) at 4°C overnight. The resin was then washed with washing buffer (20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 20 mM imidazole) and eluted by 20–200 mM imidazole gradient. Eluted fractions were analyzed by 10% SDS-PAGE, and fractions containing His-TEV-apoA4 were pooled and dialyzed against reaction buffer (50 mM Tris-HCl, pH 8.0, 100 mM NaCl). To remove the His tag, His-TEV-apoA4 was incubated with His-tagged TEV protease at a 1:2 enzyme/substrate molar ratio in reaction buffer at 25°C overnight. The reaction mixture was subjected to Ni2+-NTA affinity chromatography, and the flow-through fraction containing apoA4 was dialyzed against Tris-buffered saline (20 mM Tris-HCl, pH 7.4, 150 mM NaCl).
Synthesis of rHDL
Reconstituted HDL (rHDL) containing either apoA-I or apoA4 was prepared by the sodium cholate dialysis method (Matz and Jonas, 1982) using POPC:cholesterol: apolipoprotein:sodium cholate at initial molar ratios of 95:5:1:150 and 255:13:1:150 for synthesis of smaller and larger rHDL, respectively.
Compositional analysis of the rHDL
The protein concentration was determined in according to Lowry protein assay (Markwell et al., 1978), using bovine serum albumin as a standard. Analysis of phosphorus (Chen et al., 1956) and free cholesterol (Heider and Boyett, 1978) contents was carried out in accordance with standard procedures.
Isoelectric focusing
Calculation of isoelectric point (pI) was carried out using the Protean module 8.1.4 of DNASTAR Lasergene (USA). Isoelectric focusing (IEF) was carried out using a pre-casted gel with PhastGel IEF 3–9 (17-0543-01; GE Healthcare) and PhastSystem (GE Healthcare, Uppsala, Sweden). The protein bands were visualized by PhastGel Blue R (17-0518-01, GE Healthcare) staining.
BS3 cross-linking
The number of apoA-I molecules per rHDL particle, as well as the self-association properties of lipid-free proteins, were determined by crosslinking with BS3 using the method described previously (Staros, 1982).
Circular dichroism and fluorospectroscopy
The average alpha-helix content of proteins in lipid-free and lipid-bound states were measured by circular dichroism (CD) spectroscopy, using a J-715 Spectropolarimeter (Jasco, Japan). The spectra were obtained from 250-190 nm at 25°C in a 0.1-cm path-length quartz cuvette, using a 1.0-nm bandwidth, a speed of 50 nm/min, and a 4 s response time. The protein samples, which were dialyzed against the TBS to remove any residual fructose, of the lipid-free proteins were diluted to 0.07 mg/ml to avoid self-association of the apolipoproteins (Davidson et al., 1996), while lipid-bound proteins were diluted to 0.1 mg/ml. Four scans were accumulated and averaged. The α-helical content was calculated from the molar ellipticity at 222 nm (Chen et al., 1972).
The wavelengths of maximum fluorescence (WMF) of tryptophan residues in native and glycated apoA-I were determined from uncorrected spectra obtained on a LS55 spectrofluorometer (Perkin-Elmer, USA) using WinLab software package 4.00 (Perkin-Elmer) and a 1 cm path-length suprasil quartz cuvette (Fisher Scientific, USA). The samples were excited at 295 nm to avoid tyrosine fluorescence, and the emission spectra were scanned from 305–400 nm at room temperature.
DMPC clearance assay
Interactions of mutant proteins with DMPC were monitored by a previous method (Pownall et al., 1978) with slight modifications. The molar ratio of protein to DMPC was 2:1 (mol/mol) in a total reaction volume of 760 μl. Measurements were initiated after addition of DMPC and monitored at 325 nm every 2 min using a DU800 spectrophotometer (Beckman Coulter, USA) equipped with a MultiTemp III thermocirculator (Amersham Biosciences, Sweden).
LCAT activation ability
LCAT assay was carried out as described previously (Han et al., 2005) using POPC-rHDL containing apoA-I or apoA4 (POPC: FC:protein, 95:5:1 molar ratio) and human HDL3 (25 μg) as a substrate and enzyme source, respectively. The reaction mixture consisted of radio-labeled cholesterol (1 μCi of [14C]-cholesterol/69 μg of cholesterol/1 mg of apoA-I) in POPC-rHDL with 4% defatted bovine serum albumin and 4 mM β-mercaptoethanol. The reaction was initiated by addition of 25 μl of HDL3 (1 mg/ml) and incubated for 1 h at 37°C. ApoA-I and apoA4 were present in POPC-rHDLs at various concentrations ranging from 1.0 × 10−6 to 2.5 × 10−7 M. The reaction was performed in duplicate, and background values were determined by omitting only HDL3 from the reaction tubes at each substrate concentration. Initial reaction velocities at each substrate concentration were determined by thin layer chromatography (TLC) of the cholesterol and cholesterol esters, and Lineweaver-Burk plots were used to obtain the apparent Km and Vmax values by linear regression.
Glycation of apolipoproteins
Purified lipid-free apoA-I and apoA4 (2 mg/ml) were incubated with 5 mM D-fructose in 200 mM potassium phosphate/0.02% sodium azide buffer (pH 7.4) for up to 90 h in a 5% CO2 atmosphere at 37°C. Extent of advanced glycation was determined from reading the fluorometric intensity at 370 nm (excitation) and 440 nm (emission) as previously described (Park et al., 2010).
Acetylation of LDL
LDL (1.019 < d < 1.063) was purified from human plasma (Blood Bank of Yeungnam University Medical Center, Korea) obtained from healthy male volunteers by ultracentrifugation, according to standard protocol (Havel et al., 1955). Acetylation of LDL (acLDL) was performed using saturated sodium acetate and acetic anhydride according to a previous method (Fraenkal-Conrat, 1957). After acetylation and subsequent dialysis, acLDL protein content was determined and labeled with NBD-cholesterol (molecular probe N-1148, 70 μg of NBD-cholesterol/mg of protein)
Cellular uptake of the acLDL into macrophage
THP-1 cells, a human monocyte cell line, were obtained from the American Type Culture Collection (ATCC, #TIB-202™; USA) and maintained in RPMI-1640 medium (Hyclone, USA) supplemented with 10% fetal bovine serum (FBS) until needed as described previously (Kim et al., 2013). Cells that had undergone ≤ 20 passages were incubated in medium containing phorbol 12-myristate 13-acetate (PMA; final concentration, 150 nM) on 24-well plates for 48 h at 37°C in a humidified incubator (5% CO2 and 95% air) to induce differentiation into macrophages.
To compare anti-atherosclerotic activity using cells, inhibitory activities of apoA-I-rHDL and apoA4-HDL against acLDL up-take by macrophages were measured as previously reported by our research group (Park et al., 2014). Differentiated and adherent macrophages were then rinsed with warm PBS and incubated with 400 μl of fresh RPMI-1640 medium containing 1% FBS, 50 μl of acLDL (50 μg of protein in PBS), and 50 μl of each HDL [56 μg of protein (final concentration, 4 μM)] for 48 h at 37°C in a humidified incubator. After 48 h of incubation, cells were washed three times in PBS. Fluorescence (Ex = 488 nm, Em = 535 nm) was detected using an LS55 spectrofluorometer with WinLab software equipped with a Plate Reader (L2250140). Cell and fluorescence images were observed and photographed using a Nikon Eclipse TE2000 microscope (Japan). Quantification of fluorescence in cells was carried out via computer-assisted morphometry using the Image Proplus software (version 4.5.1.22; Media Cybernetics, USA).
Data analysis
All data are expressed as the mean ± SD from at least three independent experiments with duplicate samples. Comparisons between results were made by Student’s t-test using a SPSS program (version 12.0; SPSS, Inc., USA). Statistical significance was defined as a p < 0.05.
RESULTS
Structural analysis
The rHDL synthesis was carried out using purified apoA-I and apoA4 (at least 98% purity, Fig. 1A). As phospholipid content increased, WMF of apoA-I showed a 3-nm blue shift in a lipid-free state, whereas apoA4 did not show alteration of WMF (Table 1). Alpha-helical content of apoA-I in rHDL increased at a 95:1 molar ratio and decreased at a 255:1 molar ratio of phospholipids to apoA-I. On the other hand, α-helical content of apoA4 did not significantly change between lipid-free and lipid bound states (Table 1 and Fig. 2). As phospholipid content increased, rHDL size increased for both apoA-I and apoA4. ApoA-I-rHDL showed a similar ratio of POPC:FC:protein to the initial molar ratio from the actual ingredient analysis.
Fig. 1.
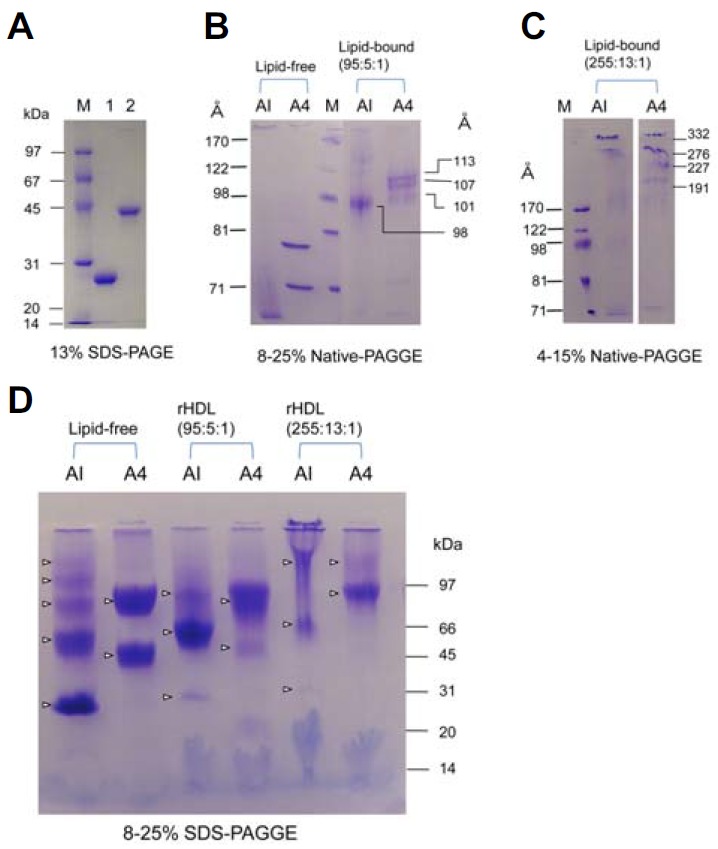
Electrophoretic patterns of apoA-I and apoA4 in lipid-free and lipid-bound states. (A) Protein purity in lipid-free state was assessed by SDS-PAGE. (B) Each protein in lipid-free state and smaller rHDL state (POPC:FC:protein molar ratio 95:5:1) was subjected to 8–25% native gradient gel electrophoresis (GGE) without denaturation of proteins. (C) Larger rHDL (POPC:FC:protein, molar ratio 255:13;1) was electrophoresed on 4–15% native GGE. (D) BS3 crosslinking patterns of apoA-I and apoA4 in lipid-free and rHDL states. The crosslinked products were subjected to 8–25% SDS-PAGE. The crosslinking reaction was carried out in 20 mM phosphate buffer, pH 7.4, with 1.0 mg/ml of each protein. Major products of apoA4 were dimers in lipid-free and rHDL states.
Table 1.
Compositional properties and spectroscopic data of apoA-I and apoA-4 in lipid-free and lipid-bound states
| (Initial molar ratio) | apoA-I | apoA-4 | ||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Lipid-free | rHDL (95:5:1) | rHDL (255:13:1) | Lipid-free | rHDL (95:5:1) | rHDL (255:13:1) | |
| WMF (nm) | 347 ± 1 | 344 ± 1 | 344 ± 1 | 351 ± 1 | 352 ± 1 | 351 ± 1 |
| α-helicity (%) | 55 ± 5 | 73 ± 4 | 41 ± 1 | 45 ± 1 | 47 ± 7 | 39 ± 11 |
| (α-helicity after glycation) | (27.5) | (37.9) | (41.1) | (30.6) | (19.5) | (28.0) |
| Size (Å) | 61–74 | 98 | 163, 184, 209 | 67 and 79 | 101, 107, 113 | 191, 227, 276 and 332 |
| Determind molar composition | ND | 95 ± 1:5:1 | 224 ± 4:13:1 | ND | 62 ± 7:5:1 | 200 ± 8:13:1 |
At calculation of α-helicity, protein concntration is 0.07 mg/ml at lipid-free state and 0.1 mg/ml at lipid-bound states Glycation condition; final 250 mM of fructose treat and incubation during 72 h at 5% CO2 containing incubator.
Fig. 2.
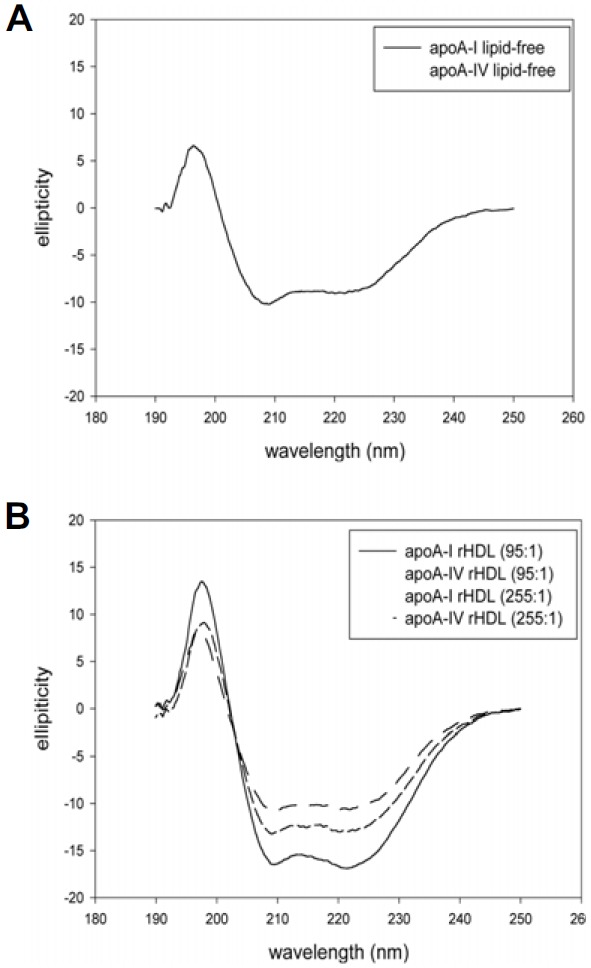
Circular dichroic spectra of apoA-I and apoA-4 under native state in lipid-free state (A) and lipid-bound state (B).
Electrophoretic properties of apoA-I and apoA4
The purities of apoA-I (28 kDa) and apoA4 (46 kDa) were higher than 97%, as shown in Fig. 1A. In native gel electrophoresis, apoA-I showed a smeared band pattern less than 71 Å in a lipid-free state, whereas apoA4 showed two distinct bands around 78 and 67 Å. In a lipid-bound state at a lower POPC: cholesterol:apoA-I molar ratio (95:5:1), apoA-I-rHDL showed a relatively more homogeneous band pattern at 98 Å, whereas apoA4-rHDL showed three major bands with weaker band intensity (101, 107, and 113 Å) as shown in Fig. 1B. At a higher POPC:cholesterol: apoA-I molar ratio (255:5:1), apoA-I exhibited bands at 163, 184, and 209 Å, whereas apoA4-rHDL showed larger bands at 191, 227, 276, and 332 Å (Fig. 1C). Calculated isoelectric points (pI) of mature apoA-I (243 amino acids) and apoA4 (376 amino acids) were 5.24 and 5.13, respectively.
BS3-crosslinking
As shown in Fig. 1D, BS3-crosslinking using lipid-free apoA-I resulted in good multimeric formation up to pentamer, whereas lipid-free apoA4 showed only monomer and dimer formation. At a lower molar ratio (95:5:1), apoA-I-rHDL and apoA4-rHDL contained two molecules per particle. At a higher molar ratio, apoA-I-rHDL contained dimeric to pentameric apoA-I, whereas apoA4-rHDL contained dimeric and trimeric apoA-I (Fig. 1D).
Phospholipid binding ability
DMPC clearance assay revealed that lipid-free apoA4 showed much weaker phospholipid-binding ability, whereas apoA-I showed more potent binding ability (Fig. 3). After 60 min of incubation, 100% of DMPC was cleared by apoA-I (T1/2 = 11 ± 1 min), whereas apoA4 showed 42% clearance (T1/2 = 62 ± 10 min).
Fig. 3.
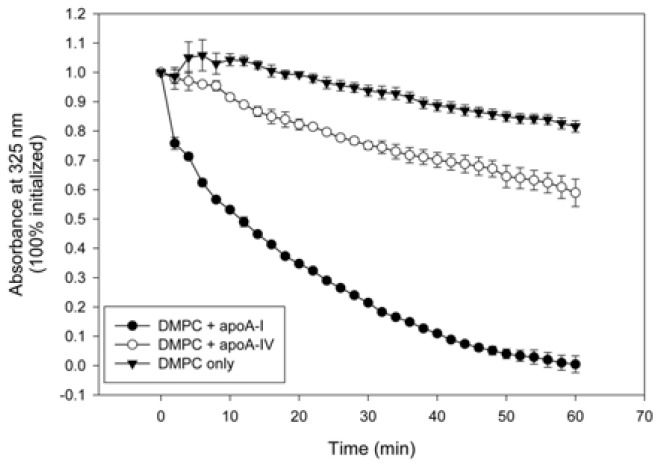
Phospholipid-binding ability of apoA-I and apoA4. Kinetics of interaction at the same molar ratio of proteins with DMPC multilamellar liposomes. Absorbance at 325 nm was monitored at 24.5°C with 2-min intervals.
LCAT activation
For apoA-I-rHDL, smaller rHDL (molar ratio 95:5:1, POPC: FC:apoA-I) substrate showed the highest LCAT activation activeity (around 47% conversion rate), whereas larger rHDL (255:13:1) showed a 10% conversion rate (Fig. 4). For apoA4-rHDL, smaller rHDL showed 22% LCAT activation ability, whereas larger rHDL showed almost loss of LCAT activation. This result suggests that apoA-I has stronger LCAT activation ability than apoA-4 and smaller rHDL showed higher LCAT activation activity than larger rHDL in both apoA-I and apoA-4.
Fig. 4.
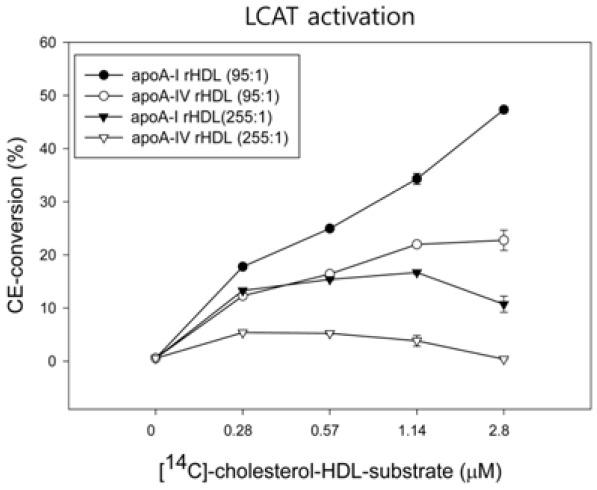
Kinetics of LCAT reaction with POPC-rHDL substrates. The rHDLs were prepared at a molar ratio of 95:5:1:150 (POPC:FC: apoA-I:Na-cholate) with 4-[14C]-cholesterol. After the reaction, esterified products (CE) were isolated via thin-layer chromatography and quantitated via scintillation counting.
Inhibition of acLDL phagocytosis
ApoA-I and apoA4 in a lipid-free state showed weaker inhibition of acLDL phagocytosis, as shown in Fig. 5. In a native state, apoA-I-rHDL exhibited stronger inhibitory ability than apoA4-rHDL at both 95:1 and 255:1 (POPC:protein) molar ratios, although apoA-I-rHDL at 95:1 inhibited phagocytosis more effectively than at 255:1.
Fig. 5.
Inhibition of acLDL phagocytosis into macrophages by apoA-I and apoA-4. Differentiated THP-1 cells were incubated with 50 μl of each apoA-I or apoA-4 in lipid-free and rHDL states (50 μg of protein) for 48 h with or without 50 μl of acLDL. Cellular uptake of acLDL was inhibited by apoA-I and apoA-4 as visualized by fluorescence detection (Ex = 588 nm, Em = 605 nm).
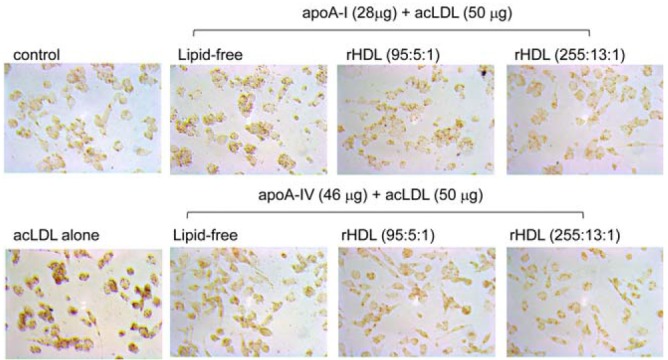
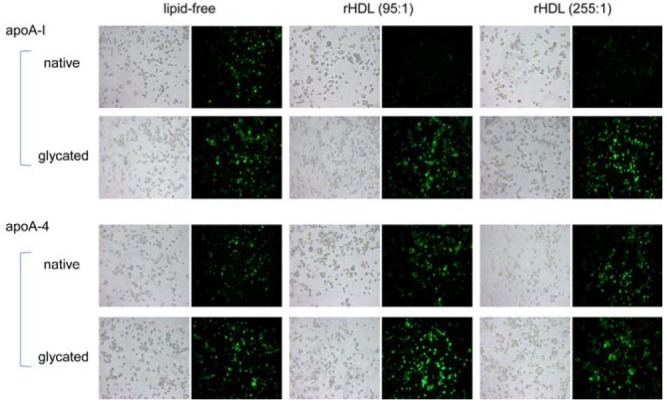
Glycated apoA-I or apoA4 in both lipid-free and rHDL states showed increased phagocytosis compared to native state (Fig. 6), suggesting that the fructosylation reduced anti-atherosclerotic activity of the apolipoproteins and rHDL. More interestingly, glycated apoA4-rHDL at a molar ratio of 95:1 induced greater uptake of acLDL than at 255:1, whereas glycated apoA-I-rHDL at 95:1 showed more potent inhibitory activity than at 255:1. These results suggest that smaller apoA-I-rHDL exerted strong anti-atherosclerotic activity in native and glycated states, whereas smaller apoA4-rHDL did not.
Fig. 6.
Inhibition of acLDL phagocytosis into macrophages by apoA-I and apoA-4. Differentiated THP-1 cells were incubated with 50 μl of each apoA-I or apoA-4 in lipid-free and rHDL states (50 μg of protein) for 48 h with or without 50 μl of acLDL. The extent of cellular uptake of the LDL by macrophages was then compared by oil red O staining. The cells were then photographed using a Nikon Eclipse TE2000 microscope (Japan) at ×600 magnification.
Fructosylation of protein
ApoA-I and apoA4 in a lipid-free state showed resistance to fructose-mediated glycation during 72 hr of incubation, as shown in Supplementary Fig. 1. In an rHDL state, both apoA-I-rHDL and apoA4-rHDL showed increased glycation in a time-dependent manner. A smaller molar ratio (95:5:1) of apoA-I-rHDL and apoA-4-rHDL resulted less glycation extent compared to larger molar ratio (255:13:1). Larger apoA4-rHDL (255:13:1) showed greater sensitivity to glycation than smaller apoA4-rHDL (95:5:1).
DISCUSSION
It is well known that HDL exerts beneficial effects on serum due to its strong antioxidant, anti-inflammatory, anti-atherosclerotic, and anti-senescence activities (Cho, 2009). However, HDL consists of heterogeneous subspecies with different apolipoprotein composition and particle size, which means physiological functions of HDL can vary among populations. ApoA-I and apoA4 share similar structural features. They both lack a cysteine residue in their primary structures and consist of 22 amino acid homologous repeats in their helix domain (Tubb et al., 2008). However, the two molecules have different molecular weights in addition to five isoleucines in apoA4 and none in apoA-I.
In the current study, we demonstrated functional similarities and differences between apoA-I and apoA4, in lipid-free state and lipid-bound state with different rHDL particle sizes. Specifically, in native gel electrophoresis, they showed different electromobilities in lipid-free state (Fig. 1B) and rHDL state (Fig. 1C). ApoA4 showed less BS3-crosslinking tendency (Fig. 1D), weaker phospholipid-binding ability (Fig. 3), and lower LCAT activation (Fig. 4) activity as compared to those of apoA-I. In a native state, apoA-I-rHDL showed greater inhibition of acLDL phagocytosis into THP-1 cells than apoA4-rHDL (Fig. 5). In a glycated state, cells treated with smaller apoA4-rHDL (95:1) showed highest uptake of acLDL. Lipid-free apoA-I showed potent inhibition against cupric ion-mediated oxidation (data not shown), which is in agreement with our previous report (Han et al., 2005) that found lipid-free apoA-I exerts antioxidant activity.
In coronary artery patients (CAD) patients, plasma level of lipid-free apoA-I is elevated while that of apoA4 is significantly reduced compared to healthy controls (Ezeh et al., 2003). In rat models, apoA4 is a major component of HDL and exerts antioxidant activity against lipid oxidation (Qin et al., 1998). Both showed hinge domain movement with rHDL in the presence of LDL (Supplementary Figs. 2A and 2C) as well as potent antioxidant ability in a lipid-free state. However, apoA-I and apoA4 with larger rHDL was more resistant to particle rearrangement (Supplementary Figs. 2B and 2D) with less α-helical content compared to the lipid-free state (Table 1), indicating that α-helices were completely stretched for formation of larger rHDL.
ApoA4 showed a 42% reduction in phospholipid-binding ability compared to apoA-I. Furthermore, apoA4 showed 35% and 22% reductions in POPC composition at molar ratios of 95:1 and 255:1 (POPC:apoA4), as shown in Table 1. Similarly, Calabresi group reported that recombinant apoA4 expressed in E. coli shows less phospholipid-binding ability than plasma apoA4 (Gomaraschi et al., 2010). Davidson group also reported that recombinant apoA4 shows less DMPC-binding ability (Pearson et al., 2004). These reports suggest that glycosylation is required to maintain phospholipid-binding ability of native apoA4.
Since apoA4 contains a Trp at the 32nd position (Trp32) in its mature sequence, WMF did not change between lipid-free and lipid-bound states, indicating no movement of Trp upon lipid association. WMF of apoA-I-rHDL showed 3-nm blue shift, suggesting that Trp108 moved to hydrophobic phase for lipid association.
For fructosylation sensitivity, apoA4 in a lipid-free state was slightly more sensitive to fructosylation than apoA-I. Regardless of particle size, all rHDLs showed greater glycation than in a lipid-free state. However, apoA-I-rHDL (95:1) and apoA4-rHDL (255:1) showed the highest glycation and proteolytic sensitivity (data not shown), suggesting that glycation sensitivity was not dependent on α-helical content or rHDL size. Generally, proteins with greater glycation showed higher proteolytic sensitivity, suggesting that fructosylation may expose the cleavage site. Fructosyllysine formation by glycation contributed to multimeric formation, as previously suggested (McPherson et al., 1988).
The results of acLDL phagocytosis (Fig. 6) confirmed that apoA-I-rHDL at 95:1 showed the strongest anti-atherosclerotic activity. In conclusion, ApoA4 showed less anti-atherogenic activity with more sensitivity to glycation. ApoA4 revealed inferior physiological functions in lipid-bound state compared with those of apoA-I to induce more pro-atherosclerotic properties.
Acknowledgments
This work was supported by the Mid-carrier Researcher Program (2014-11049455) and Basic Science Research Program (2010-020910) through the National Research Foundation of Korea (NRF).
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Beisiegel U, Utermann G. An apolipoprotein homolog of rat apolipoprotein A-IV in human plasma. Isolation and partial characterization. Eur. J. Biochem. 1979;93:601–608. doi: 10.1111/j.1432-1033.1979.tb12860.x. [DOI] [PubMed] [Google Scholar]
- Boguski MS, Elshourbagy N, Taylor JM, Gordon JI. Rat apolipoprotein A-IV contains 13 tandem repetitions of a 22-amino acid segment with amphipathic helical potential. Proc. Natl. Acad. Sci. 1984;81:5021–5025. doi: 10.1073/pnas.81.16.5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouillette CG, Anantharamaiah GM, Engler JA, Borhani DW. Structural models of human apolipoprotein A-I: a critical analysis and review. Biochim. Biophys. Acta. 2001;1531:40–46. doi: 10.1016/s1388-1981(01)00081-6. [DOI] [PubMed] [Google Scholar]
- Chen PS, Toribara TY, Warner H. Microdetermination of phosphorus. Anal. Chem. 1956;28:1756–1758. [Google Scholar]
- Chen YH, Yang JT, Martinez HM. Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion. Biochemistry. 1972;11:4120–4131. doi: 10.1021/bi00772a015. [DOI] [PubMed] [Google Scholar]
- Cho KH. Biomedicinal implications of high-density lipoprotein: its composition, structure, functions, and clinical applications. BMB Rep. 2009;42:393–400. doi: 10.5483/bmbrep.2009.42.7.393. [DOI] [PubMed] [Google Scholar]
- Cho KH. Enhanced delivery of rapamycin by V156K-apoA-I high-density lipoprotein inhibits cellular proatherogenic effects and senescence and promotes tissue regeneration. J. Gerontol. A. Biol. Sci. Med. Sci. 2011;66:1274–1285. doi: 10.1093/gerona/glr169. [DOI] [PubMed] [Google Scholar]
- Cho KH, Park SH, Han JM, Kim HC, Choi YK, Choi I. ApoA-I mutants V156K and R173C promote anti-inflammatory function and antioxidant activities. Eur. J. Clin. Invest. 2006;36:875–882. doi: 10.1111/j.1365-2362.2006.01737.x. [DOI] [PubMed] [Google Scholar]
- Davidson WS, Hazlett T, Mantulin WW, Jonas A. The role of apolipoprotein A-I domains in lipid binding. Proc. Natl. Acad. Sci. 1996;93:13605–13610. doi: 10.1073/pnas.93.24.13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezeh B, Haiman M, Alber HF, Kunz B, Paulweber B, Lingenhel A, Kraft HG, Weidinger F, Pachinger O, Dieplinger H, et al. Plasma distribution of apoA-IV in patients with coronary artery disease and healthy controls. J. Lipid Res. 2003;44:1523–1529. doi: 10.1194/jlr.M300060-JLR200. [DOI] [PubMed] [Google Scholar]
- Ferretti G, Bacchetti T, Bicchiega V, Curatola G. Effect of human Apo AIV against lipid peroxidation of very low density lipoproteins. Chem. Phys. Lipids. 2002;114:45–54. doi: 10.1016/s0009-3084(01)00201-8. [DOI] [PubMed] [Google Scholar]
- Fraenkal-Conrat H. Methods for investigating essential groups for enzyme activity. Meth. Enzymol. 1957;4:247–269. [Google Scholar]
- Frank PG, Marcel YL. Apolipoprotein A-I: structure-function relationships. J. Lipid Res. 2000;41:853–872. [PubMed] [Google Scholar]
- Goldberg IJ, Scheraldi CA, Yacoub LK, Saxena U, Bisgaier CL. Lipoprotein ApoC-II activation of lipoprotein lipase. Modulation by apolipoprotein A-IV. J. Biol. Chem. 1990;265:4266–4272. [PubMed] [Google Scholar]
- Gomaraschi M, Putt WE, Pozzi S, Iametti S, Barbiroli A, Bonomi F, Favari E, Bernini F, Franceschini G, Talmud PJ, et al. Structure and function of the apoA-IV T347S and Q360H common variants. Biochem. Biophys. Res. Commun. 2010;393:126–130. doi: 10.1016/j.bbrc.2010.01.099. [DOI] [PubMed] [Google Scholar]
- Han JM, Jeong TS, Lee WS, Choi I, Cho KH. Structural and functional properties of V156K and A158E mutants of apolipoprotein A-I in the lipid-free and lipid-bound states. J. Lipid Res. 2005;46:589–596. doi: 10.1194/jlr.M400468-JLR200. [DOI] [PubMed] [Google Scholar]
- Havel RJ, Eder HA, Bragdon JH. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J. Clin. Invest. 1955;34:1345–1353. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heider JG, Boyett RL. The picomole determination of free and total cholesterol in cells in culture. J. Lipid Res. 1978;19:514–518. [PubMed] [Google Scholar]
- Jonas A. Regulation of lecithin cholesterol acyltransferase activity. Prog. Lipid Res. 1998;37:209–234. doi: 10.1016/s0163-7827(98)00007-1. [DOI] [PubMed] [Google Scholar]
- Kim JY, Kim H, Jung BJ, Kim NR, Park JE, Chung DK. Lipoteichoic acid isolated from Lactobacillus plantarum suppresses LPS-mediated atherosclerotic plaque inflammation. Mol. Cells. 2013;35:115–124. doi: 10.1007/s10059-013-2190-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Xu M, Wang F, Kohan AB, Haas MK, Yang Q, Lou D, Obici S, Davidson WS, Tso P. Apolipoprotein A-IV Reduces Hepatic Gluconeogenesis through the Nuclear Receptor NR1D1. J. Biol. Chem. 2014;289:2396–2404. doi: 10.1074/jbc.M113.511766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, Innerarity TL, Rall SC, Weisgraber KH. Plasma lipoproteins: apolipoprotein structure and function. J. Lipid Res. 1984;25:1277–1294. [PubMed] [Google Scholar]
- Main LA, Ohnishi T, Yokoyama S. Activation of human plasma cholesteryl ester transfer protein by human apolipoprotein A-IV. Biochim. Biophys. Acta. 1996;1300:17–24. doi: 10.1016/0005-2760(95)00228-6. [DOI] [PubMed] [Google Scholar]
- Markwell MA, Haas SM, Bieber LL, Tolbert NE. A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal. Biochem. 1978;87:206–210. doi: 10.1016/0003-2697(78)90586-9. [DOI] [PubMed] [Google Scholar]
- Matz CE, Jonas A. Micellar complexes of human apolipoprotein A-I with phosphatidylcholines and cholesterol prepared from cholate-lipid dispersions. J. Biol. Chem. 1982;257:4535–4540. [PubMed] [Google Scholar]
- McPherson JD, Shilton BH, Walton DJ. Role of fructose in glycation and cross-linking of proteins. Biochemistry. 1988;27:1901–1907. doi: 10.1021/bi00406a016. [DOI] [PubMed] [Google Scholar]
- Park KH, Jang W, Kim KY, Kim JR, Cho KH. Fructated apolipoprotein A-I showed severe structural modifycation and loss of beneficial functions in lipid-free and lipid-bound state with acceleration of atherosclerosis and senescence. Biochem. Biophys. Res. Commun. 2010;392:295–300. doi: 10.1016/j.bbrc.2009.12.179. [DOI] [PubMed] [Google Scholar]
- Park JY, Park JH, Jang W, Hwang IK, Kim IJ, Kim HJ, Cho KH, Lee ST. Apolipoprotein A-IV is a novel substrate for matrix metalloproteinases. J. Biochem. 2012;151:291–298. doi: 10.1093/jb/mvr137. [DOI] [PubMed] [Google Scholar]
- Park KH, Kim JM, Cho KH. Elaidic acid (EA) generates dysfunctional high-density lipoproteins and consumption of EA exacerbates hyperlipidemia and fatty liver change in zebrafish. Mol. Nutr. Food Res. 2014;58:1537–1545. doi: 10.1002/mnfr.201300955. [DOI] [PubMed] [Google Scholar]
- Pownall HJ, Massey JB, Kusserow SK, Gotto AM. Kinetics of lipid-protein interactions: interaction of apolipoprotein A-I from human plasma high density lipoproteins with phosphatidylcholines. Biochemistry. 1978;17:1183–1188. doi: 10.1021/bi00600a008. [DOI] [PubMed] [Google Scholar]
- Pearson K, Saito H, Woods SC, Lund-Katz S, Tso P, Phillips MC, Davidson WS. Structure of human apolipoprotein A-IV: a distinct domain architecture among exchangeable apolipoproteins with potential functional implications. Biochemisty. 2004;43:10719–10729. doi: 10.1021/bi048978m. [DOI] [PubMed] [Google Scholar]
- Qin X, Swertfeger DK, Zheng S, Hui DY, Tso P. Apolipoprotein AIV: a potent endogenous inhibitor of lipid oxidation. Am. J. Physiol. 1998;274:H1836–H1840. doi: 10.1152/ajpheart.1998.274.5.H1836. [DOI] [PubMed] [Google Scholar]
- Staros JV. N-hydroxysulfosuccinimide active esters: bis(N-hydroxysulfosuccinimide) esters of two dicarboxylic acids are hydrophilic, membrane-impermeant, protein cross-linkers. Biochemistry. 1982;21:3950–3955. doi: 10.1021/bi00260a008. [DOI] [PubMed] [Google Scholar]
- Steinmetz A, Utermann G. Activation of lecithin: cholesterol acyltransferase by human apolipoprotein A-I. J. Biol. Chem. 1985;260:2258–2264. [PubMed] [Google Scholar]
- Tubb MR, Silva RA, Fang J, Tso P, Davidson WS. A three-dimensional homology model of lipid-free apolipoprotein A-IV using cross-linking and mass spectrometry. J. Biol. Chem. 2008;283:17314–17323. doi: 10.1074/jbc.M800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubb MR, Smith LE, Davidson WS. Purification of recombinant apolipoproteins A-I and A-IV and efficient affinity tag cleavage by tobacco etch virus protease. J. Lipid Res. 2009;50:1497–1504. doi: 10.1194/jlr.D900003-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VerHague MA, Cheng D, Weinberg RB, Shelness GS. Apolipoprotein A-IV expression in mouse liver enhances triglyceride secretion and reduces hepatic lipid content by promoting very low density lipoprotein particle expansion. Arterioscler. Thromb. Vasc. Biol. 2013;33:2501–2508. doi: 10.1161/ATVBAHA.113.301948. [DOI] [PubMed] [Google Scholar]
- Weinberg RB. Apolipoprotein A-IV polymorphisms and diet-gene interactions. Curr. Opin. Lipidol. 2002;13:125–134. doi: 10.1097/00041433-200204000-00003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.