

Abstract
Recent studies conducted in hydrogen sulfide (H2S) signaling have revealed potential importance of persulfides (RSSH) in redox biology. The inherent instability of RSSH makes these species difficult to study and sometimes controversial results are reported. In this review article we summarize known knowledge about both small molecule persulfides and protein persulfides. Their fundamental physical and chemical properties such as preparation/formation and reactivity are discussed. The biological implications of persulfides and their detection methods are also discussed.
Introduction
Cysteine is a redox sensitive amino acid residue in proteins. The -SH group of cysteine is electron rich and its d-orbitals enable multiple oxidation states (from −2 to +6) leading to an array of redox modifications that play important roles in regulating protein structures and functions.1,2 A number of endogenous molecules and exogenous chemicals are known to react with cysteines. Among these, the reactions with reactive oxygen species (ROS) and reactive nitrogen species (RNS) have received considerable attention in redox biology.3,4 ROS mainly include superoxide (O2•−), hydrogen peroxide (H2O2), and hydroxyl radical (•OH). These species are generated from incomplete reduction of oxygen during normal respiration in aerobic organisms and by enzymes such as NADPH oxidase. RNS include molecules such as nitric oxide (NO) and peroxynitrite (ONOO−). Both ROS and RNS can react with cysteine residues in the active and allosteric sites of proteins. The resultant post-translational modifications are known to be important redox singling mechanisms. It should be noted that not all cysteines in a protein are equally sensitive to ROS/RNS. Normally the low pKa thiols, often known as “reactive cysteines”, are more susceptible. The low pKa thiols are caused by local environment (such as neighboring positively charged amino acids, hydrogen bonding, and location at the N-terminal end of an α-helix) and ionized to form thiolates under physiological pH. Because of their strong nucleophilic character, thiolates are extremely vulnerable to oxidation. A number of possible thiol derivatives can be produced endogenously and the ones being most well studied include S-nitrosation (-SNO), S-glutathionylation (-SSG), and S-sulfenylation (-SOH).4 These oxidized species can be reduced to form free thiols (-SH) by the antioxidant defense network or converted to further oxidized forms depending on the cellular redox-state. The oxidative posttranslational modification depends on many factors including the reactivity of the individual Cys residue, its surroundings, and the composition of the local redox-environment.
Recently a new cysteine-based posttranslational modification, i.e. S-sulfhydration, has emerged as a hot research area and been linked to redox signaling. S-sulfhydration was initially suggested to be the result of H2S-mediated cysteine modification. As H2S is considered an endogenously generated gasotransmitter,5–7 S-sulfhydration might be a possible way by which H2S alters the functions of a wide range of cellular proteins.8,9 However, recent studies reveal that other reactive sulfur species (RSS) such as sulfane sulfurs or polysulfides can also lead to S-sulfhydration and these species may be even more effective than H2S in S-sulfhydration.10–14 In addition, the detection of S-sulfhydration is still challenging. Some commonly used methods are problematic (vide infra). As such, more work is needed to elucidate how endogenous S-sulfhydration occurs and what their functions/roles are in biology. The products of S-sulfhydrations are cysteine persulfides (R-S-SH), also referred to as perthiols or hydrodisulfides. Unlike their thiol analogs (RSH), the fundamental chemistry and chemical biology of persulfides are still poorly understood. Two reasons contribute to this lack of knowledge: 1) the biological significance of RSSH has only been recently discovered; and 2) persulfides are unstable species, especially in aqueous buffers, which make them difficult to study. Persulfides readily decompose in a pH-dependent manner and possible products include the corresponding thiol, disulfides, polysulfide, and elemental sulfur (S8). H2S can also be a decomposition product. In this review article, we wish to summarize known information about persulfides. The preparation/formation, reactivity, and biological implication of both small molecule persulfides and protein persulfides will be reviewed.
General physical/chemical properties of persulfides
Persulfides have significantly different chemical properties compared to their structurally related thiol analogs (RSH). For example, the difference in S-H bond energies for persulfides versus thiols is significant.14–17 The disulfide bond in persulfides weakens the RSS-H bond by ~22 kcal/mol relative to RS-H. This energy difference can be attributed to the increased stability of the perthiyl radical (RSS•) compared to thiyl radical (RS•). The adjacent sulfur atom can stabilize the odd electron of RSS• via resonance effect while this is unavailable for RS•. The perthiyl radical has an absorbance at 374 nm (ε ~ 1700 M−1cm−1) which can be used to determine the presence of perthiyl in reactions.18
The pKa of persulfides is approximately 6.2, which makes them stronger acids compared to thiols (pKa ~7.6).19 At physiological pH, thiols predominantly exist the protonated forms but persulfides (RSSH) exist as the deprotonated anions (RSS−). Therefore, the conversion of cysteine residues in proteins to persulfides (i.e. S-sulfhydration) should increase the nucleophilicity of the reactive sulfur atom and this may be associated with higher reactivity as reducing agents. It is believed that tautomerization of persulfides exists, leading to the formation of thiosulfoxides (RS(=S)H) (Scheme 1).14 Tautomerization would generate a reactive ‘singlet sulfur’ that would be very electrophilic. However, thiosulfoxides, for the most part, are thought to be thermodynamically uphill compared to the persulfide tautomer. Persulfides belong to the sulfane sulfur family (note: sulfane sulfur is defined as a sulfur bonded sulfur atom formally with six electrons20 – e.g. the terminal sulfur atom of a persulfide is a sulfane sulfur). The characteristic reaction with CN− to form SCN− and RSH has been used as evidence for the existence of a persulfide.
Scheme 1.
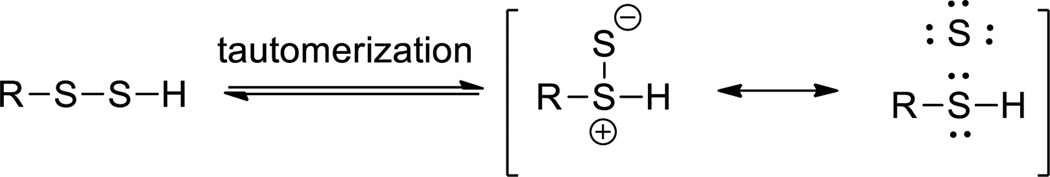
Tautomerization between persulfides and thiosulfoxides
Small molecule persulfides: synthesis and reactions
In 1954, Böhme and Zinner reported the first preparation of aryl- and alkyl-persulfides (R-SSH).21 The key intermediates were acyl disulfides, which were prepared from sulfenyl chloride (RC(=O)SCl) and mercaptans (Scheme 2). Acid (usually HCl) mediated hydrolysis of acyl disulfides provided the persulfide products, which were quantified as the stable tetrasulfide upon iodine oxidation. So far, acid-mediated hydrolysis of acyl disulfides has become a general method for the preparation of small molecule persulfides, while different methods have been developed to prepare the acyl disulfide intermediates. For example, alkyl persulfide has also been synthesized from both dialkyl thiosulfones with thioacid.22 Persulfides can also be prepared from alkoxide-induced displacement of the RSS- anion from methoxycarbonyl disulfides (RSSO2CH3),19 which in turn can be prepared from the condensation of thiol with methoxycarbonyl disulfonyl chloride (Scheme 2).
Scheme 2.

Persulfide generation reactions
So far a limited number of small molecule persulfides have been prepared and some of them are only partially characterized. The structures shown in Scheme 3 are most often seen in literature, which include benzyl/diphenylmethyl/trityl persulfides (1–3)23–36, t-butyl persulfide 436,37, penicillamine-derived persulfides 538,39, and adamantyl persulfide 6.40 In general more sterically hindered persulfides are more stable. For example, adamantyl persulfide can be purified by distillation and stored in a sealed ampule under ambient conditions for several months. Aryl persulfides (Ar-SSH) were also reported41 but these compounds seem to be less stable compared to the ones shown in Scheme 3 and not well characterized.
Scheme 3.

Representative small molecule persulfides
Persulfides are in general transient species. Both acidic and basic conditions were found to promote the decomposition.39 The major decomposition products are polysulfides (RSSnSR). H2S was reported to be a by-product of decomposition in some studies35,39,42 while other studies claimed H2S was not the product.43 Solvents or reaction conditions may determine the decomposition products. Unstable persulfides can serve as potential H2S donors. For example, penicillamine-derived acyl disulfides 7 can release H2S in the presence of cysteine or glutathione (Scheme 4).44 In this process penicillamine persulfide 8 is the key intermediate. Similarly dithioperoxyanhydride 9 was proved to be another type of thiol-activated H2S donor with acyl persulfide 10 and cysteine persulfide proposed to be the active intermediates.45
Scheme 4.

Persulfide intermediates as H2S donors
Persulfides are excellent radical precursors allowing some of their reactions to proceed via free radical mechanisms. For example, the thermal decomposition in organic solvents is initiated by homolytic dissociation of the S-S bond into two thiyl radicals (RS• and HS•), which further react with persulfides to form the final products: H2S, thiol, and polysulfide (Scheme 5).35 Persulfides can also undergo hydrogen transfer (from their RSSH forms) or electron transfer (from their anionic forms RSS−, normally under physiological pH) to generate perthiyl radicals (RSS•) that undergo subsequent reactions. An excellent review by Everett and Wardman summarized some important radical scavenging reactions of persulfides as antioxidants.18
Scheme 5.

Formation of perthiyl radicals
Reactions with phosphines/phosphites
Persulfides can be considered as the thio-analogs of hydroperoxides. Their reactions with tertiary phosphines (PPh3 or PEt3) were studied and compared with that of hydroperoxides.24 Since empty d-orbitals of the sulfur atom may participate in bond formation by accepting electrons from phosphorus, sulfur should be more susceptible to attack by phosphine than oxygen. Such reactions led to the corresponding phosphine sulfide and thiol as the major products (Scheme 6),24 along with small amounts of disulfides and H2S. Both sulfur atoms in persulfide could be attacked by the phosphine. Steric effects in the persulfide and thiophilicity of the phosphines are the factors governing which sulfur atom is attacked. In another study, phosphites (such as P(OEt)3 were found to react with persulfides in a similar way, producing the corresponding thiol and phosphorothionate as the major products.33 In an analogous study the reactions between persulfides and tertiary arsines (Ph3As or Et3As) were tested and the corresponding thiols (RSH) and arsine sulfides were the major products.32 It should be noted that persulfides belong to the sulfane sulfur family (also include polysulfides and elemental sulfur). The reaction of phosphines to form phosphine sulfide is generic for sulfane sulfurs. This reaction has been used to detect sulfane sulfurs in biological samples.46,47
Scheme 6.

Reactions of persulfides with phosphines and phosphites
Reactions with nucleophiles
The reactions between RSSH and nucleophiles have been well studied.25,48,49 Theoretically four pathways are possible when RSSH is treated with a nucleophile: 1) deprotonation of the sulfhydryl group; 2) nucleophile attacking the outer sulfur atom; 3) nucleophile attacking the inner sulfur atom; and 4) deprotonation of the alpha-carbon. The reactions of benzyl persulfide (BnSSH) with various nucleophiles such as hydroxide, sulfite, cyanide, and thiolate, were first studied.49 Products were H2S, benzyl disulfide, benzyl polysulfide, and elemental sulfur (Scheme 7). Benzylthiol (BnSH) was also found for cyanide and thiolate. It was postulated that less nucleophilic reagents (hydroxide and sulfite) preferably attack the inner sulfur atom while the stronger nucleophilic reagents (cyanide and thiolate) may attack both sulfur atoms. Steric hindrance on the persulfide substrates should also be considered. In the case of diphenylmethyl persulfide (Ph2CHSSH), attack on the outer sulfur was usually more predominant. Interestingly a recent work by Pluth et al found that a more steric hindered persulfide-Ph3CSSH did not undergo nucleophilic reactions with nucleophiles (such as thiolate, CN−, I−, Br−, AcO−).30 In organic solvents, these nucleophiles promoted deprotonation of the persulfide to generate Ph3CSS−, which then disproportionated into Ph3CS− and S0. The corresponding thiol (Ph3CSH) and S8 were found to be the final products.
Scheme 7.

Reactions of persulfides with various nucleophiles
Amines can act as bases and nucleophiles in the presence of persulfides. The reactions between persulfides (mainly PhCH2SSH and Ph2CHSSH) and amines (aniline, pyridine, butylamine, morpholine, etc.) were studied by Tsurugi et al.26 Products were found to be H2S, disulfide, polysulfide, thiol, and S8. It was concluded that weaker bases (pKa < 5.7) behave as nucleophiles while stronger bases (pKa > 8.2) behave as bases. For sterically hindered persulfide like Ph3CSSH, amines mainly acted as bases. Deprotonation and subsequently disproportion led to the thiol and S8 as the major products.30
Reactions with electrophiles
Although persulfides (RSSH) are believed to be more nucleophilic than thiols (RSH) their reactivity toward electrophiles have not been well studied. A large number of thiol (-SH) blocking reagents such as methyl methane sulfonate (MMTS), iodoacetamide (IAM), N-ethylmaleimide (NEM), etc. are known, so all of these compounds should also react with RSSH. However, very few such reaction examples can be found in literature. So far only NEM30, 2,4-dinitrochlorobenene (DNCB)40,50, and iodoacetate27 have been reported to derivatize RSSH (Scheme 8). Surprisingly in many cases the yields of the products formation were not reported, perhaps due to low isolated yields. It was found that the reaction between electrophiles (such as NEM) and RSSH was also rather slow.30 The addition of bases (such as Et3N) could drastically increase the reaction rate. However bases could promote the decomposition of RSSH, presumably leading to decreased product yields.
Scheme 8.

Reactions of persulfides with selected electrophiles
Reactions with iron salts
The redox reactions of persulfides with iron salts were studied and compared with hydroperoxides (ROOH).51 In the absence of oxygen, the reaction of persulfides led to the disappearance of the color of ferric ion. Tetrasulfides were obtained in good yields (Scheme 9). The redox behavior of persulfides is clearly different from that of hydroperoxides. The former acts as a reductant, which easily reduces ferric ion, while the latter is an oxidant.
Scheme 9.

Persulfide reaction with iron (III)
Persulfides as pro-oxidants and antioxidants
The biological relevance of persulfides has been discussed in terms of persulfides being both pro-oxidative and anti-oxidative species (summarized in Scheme 10). The predominant anion form (II), at physiological pH, is expected to be oxidized by the molecular oxygen to form reactive oxygen species (O2•−, H2O2 and HO•).27,52 These reactive oxygen radicals were found to induce DNA damage in the Gates’s Leinamycin model.53,54 Furthermore Fe(III)-cytochrome c mediated-oxidation of II formed the more thermodynamically stable perthiyl radical (III).55 This radical III can lead to the generation of superoxide radical anion via the formation of a putative tetrasulfide radical anion (IV). However, no experimental evidence has been reported supporting the existence of such a species.56 Molecular oxygen can react with radical III to generate perthiyl peroxyl radical species V, VI and VII. The oxidative properties attributed with those species could cause biological damage.57
Scheme 10.

Persulfides as pro-oxidants and antioxidants
The radical scavenging role of persulfides was also reported and based on the ability of RSSH to donate a hydrogen atom.18 The more stable perthiyl radical is responsible for H-atom donation and reductive electron transfer to an oxidant. Therefore, persulfides are stronger antioxidants than thiols. At lower pH RSSH (I) could donate hydrogen to a carbon radical, which is responsible for its anti-oxidant property.19 However the diminished rate of hydrogen transfers to (CH3)2COH radical with increasing pH is evidence that under physiological pH, hydrogen donation by RSSH may not be an efficient process. The formation of more stable radical (III) from the anion (II) via electron donation could be dominant at physiological pH. Therefore RSSH’s antioxidative properties could still be the result of electron donation to oxidizing species. Indeed, Fukuto et al found that GSSH has much stronger reducing ability for ferric-cytochrome c than H2S and GSH.58
Cysteine and glutathione persulfides (CysSSH, GSSH)
The existence of cysteine persulfide (also known as thiocysteine) has been recognized for a long time. CysSSH is suggested to be the product of cystine (CysSSCys) upon reacting Na2S or with pyridoxal and Cu2+.59,60 It is also an intermediate in the cystathionase catalyzed degradation of the substrate cystine.61,62 Synthetically Rao and Gorin reported that Na2S can react with cystine under strong basic conditions to form CysSSH.63 Smith et al reported a method to prepare CysSSH by treating methoxycarbonylcysteine disulfide with potassium hydrosulfide (KSH) (Scheme 11).64 A unique S- to N-carbonyl transfer was also used by Galardon et al to prepare cysteine persulfide analogs (penicillamine persulfides).43 CysSSH appears to be quite unstable. Polysulfides, cysteine, and S8 are found to be the decomposition products. It should be noted that H2S was not found to be the decomposition product but the presence of thiols can lead to H2S formation.43,44 Theoretically the reactivity of CysSSH should be similar as other small molecule persulfides. For example, CysSSH can be trapped by iodoacetate to form the corresponding disulfide derivative.61 Endogenous electrophiles like 8-nitro-cGMP can also trap CysSSH or GSSH.12 Some reactions mentioned here are summarized in Scheme 11.
Scheme 11.

Formation and reactivity of CysSSH and GSSH
Although CysSSH has been known for a long time, its significance in thiol-related redox biology has been recognized only recently. Ida et al. described a mass spectrometric method to quantitate persulfide (RSSH) in cells.12 With this detection method, they demonstrated the high levels (up to > 100 µM) of cysteine- and glutathione-persulfides in cells, tissues, and plasma. They showed that cystathionine-β-synthase (CBS) and cystathionine γ-lyase (CSE) can convert cystine to CysSSH, and subsequently lead to the formation of GSSH and polysulfides. GSSH can also be produced by glutathione reductase (GSR)-mediated glutathione polysulfide reduction. Glutathione (GSH) is a potent antioxidant in cells. However its antioxidant activity is typically mediated by specific enzymes such as GSH-dependent peroxidase. Without this assistance, GSH is relatively inert with low nucleophilicity and reacts poorly with electrophilic oxidants like H2O2. However GSSH has much stronger nucleophilic/antioxidant activity.12,58 The strong H2O2-scavenging activity of GSSH was confirmed by Ida et al.12 Thus the presence of CysSSH and GSSH may provide a primary and potent antioxidant defense in cells. In addition, the high in vivo concentrations of persulfides may indicate that these species are the real players in cellular signaling and regulation.
Persulfides in biosynthesis of sulfur-containing biomolecules
Sulfur is an important element of living systems. Strategies to incorporate sulfur atom into biomolecules involve biosyntheses of iron sulfur clusters, cofactors such as thiamin, molybdopterin, biotin and lipoic acid, and the thio-modification of tRNA via enzymatically generated persulfide species at enzyme active sites. In these processes the persulfide adducts in proteins are generated by cysteine desulfurases (such as NifS, IscS, SufS) and sulfurtransferases (like rhodanese and mercaptopyruvate sulfurtransferase).65,66 Cysteine desulfurases are pyridoxal 5′-phosphate (PLP)-dependent enzymes that catalyze the conversion of Cys to Ala and sulfane sulfur via the formation of a protein-bound persulfide intermediate on a conserved cysteine residue. NifS was the first cysteine desulfurase found to generate an enzyme-based persulfide as a crucial intermediate. NifS provides a sulfur atom for the iron-sulfur clusters in nitrogenase.67 The catalytic reaction of NifS is initiated by Schiff base formation between cysteine and pyridoxal 5′-phosphate (PLP) (Scheme 12). Lys202 and Cys325 residues work together and transfer the sulfur atom from Schiff base adduct to Cys325 to form a persulfide. The terminal sulfur can then be transferred to other acceptor proteins and subsequently produce sulfur-containing molecules.
Scheme 12.

Generation of NifS persulfide
IscS is biologically similar as NifS.68,69 IscS is needed for the synthesis of 4-thiouridine (s4U) in tRNA as well as the incorporation of sulfur into the thiazole ring of thiamine.70,71 The synthesis of s4U in tRNA is initiated by the transfer of cysteine sulfur to IscS, forming an IscS persulfide (like NifS-persulfide formation). The IscS persulfide then transfers the sulfhydryl sulfur to Cys465 of Thil (a rhodanese-like sulfurtransferase), forming a Thil persulfide.70 In the presence of ATP-Mg, the activated uridine residue reacts with Thil-persulfide (Cys456-SSH) to form a disulfide intermediate and expel AMP. Another active site cysteine (Cys344) then attacks the Thil-tRNA disulfide to produce s4U in tRNA and form an enzymic disulfide bond, which can be reduced for another round of catalysis.72 (Scheme 13).
Scheme 13.

Enzymatic formation of Thil persulfide and the sulphur transfer into tRNA
Persulfide residues are also found in the active sites of rhodanese and similar enzymes (Scheme 14). Thiosulfate (SSO32-) is the common substrate. The formation of persulfide was confirmed by X-ray crystallography.73 The reaction between persulfide and cyanide (CN−) to form thiocyanate (SCN−) accounts for detoxification of rhodaneses.74–76
Scheme 14.

Generation of rhodanese persulfide and the reaction with cyanide
Protein persulfide formation and H2S-related signaling
Persulfide formation on specific proteins (also known as S-sulfhydration or S-persulfidation) is a recently defined oxidative posttranslational modification that leads to either activation or inhibition of protein activity by regulating protein structures and ultimately their functions. This concept was formulized along with the characterization of H2S as a signaling molecule and S-sulfhydration is believed to be involved in H2S-based signal transduction. In this regard, the mechanism behind S-sulfhydration is still poorly understood. Some hypotheses have been proposed. It is well accepted that H2S cannot directly react with protein cysteine residues (-SH) to form SSH in the reducing intracellular environment. The oxidative forms of cysteine, for example, sulfenic acid (-SOH), disulfide (-SS-), nitrosothiol (-SNO), could react with H2S to form persulfides. Hydrogen polysulfides (H2Sn), formed by two-electron oxidation of H2S are possible cellular sulfhydration reagents. Other cellular sulfane sulfur species accumulated with H2S biosynthesis or sulfur catabolism may also play a role in S-sulfhydration.77 Nevertheless a large number of S-sulfhydrated proteins have been identified so far and sulfhydration-mediated functional changes have been studied. For example sulfhydration induced by transfection of HEK293 cells with CSE was studied by Snyder et al.78 Three S-sulfhydrated proteins were characterized: glyceraldehyde-3-phosphate dehydrogenase (GAPDH), β-tubulin, and actin. Sulfhydration altered actin-dependent cytoskeletal rearrangements in HEK293 cells and enhanced actin polymerization. In another study the same group found that S-sulfhydration of the p65 subunit of nuclear factor κB (NF-κB) facilitated its translocation to the nucleus and increased interaction with ribosomal protein S3 (rps3), resulting in enhanced transcription of anti-apoptotic gene.79 Wang et al reported that S-sulfhydration of Kelch-like ECH-associated protein-1 (Keap1) at Cys151 stimulated its dissociation from nuclear factor erythroid 2-related factor 2 (Nrf2).80 This process enables Nrf2 to translocate to the nucleus and regulates the expression of cytoprotective genes. Tonks et al suggested that S-sulfhydration of protein tyrosine phosphatase 1B (PTP1B) inhibited its activity, thus regulating the endoplasmic reticulum (ER) stress response.81 Yang et al discovered that S-sulfhydration at Cys611 and Cys614 located in the DNA binding domain (DBD) of androgen receptor (AR) inhibited its transactivation.82 It was proposed that S-sulfhydration of AR might shatter zinc-sulfur clusters and cause a structural modification of AR, resulting in inhibition of the formation of abnormal AR dimerization and its DNA binding. Table 1 summarizes selected S-sulfhydrated protein examples. It is expected that more S-sulfhydrated proteins will be discovered.
Table 1.
Examples of sulfhydrated proteins: outcomes and detection methods
| Proteins | Outcomes of sulfhydration |
Detection methods |
|---|---|---|
| GAPDH78,83 | activation of GAPDH | aMBS, bTS, cLC-MS |
| Actin78 | activation of actin polymerization | MBS |
| Kir6.1 subunit of KATP84 | reducing ATP binding, activation | MBS |
| Hsp7083 | N/A | TS |
| Protein disulfide isomerase12 | N/A | TS |
| Aldo-keto reductase12 | N/A | TS |
| Phosphoglycerate kinase12 | N/A | TS |
| NF-κB79,85 | activation of RPS3 binding, increasing transcription of genes | MBS, LC-MS |
| Keap180,86 | activation of Nrf2 | MBS |
| Parkin87 | activation of parkin ligase | MBS, LC-MS |
| Androgen receptor82 | inhibition of its dimerization | MBS |
| MEK188 | activation of PARP-1 | MBS |
| eNOS89 | Activation of its dimerization | MBS |
| Ca2+ TRP channels90 | activation of Ca2+ influx | MBS |
| IKCa84 | activation of IKCa | MBS |
| P66Shc91 | Inhibition of PKCβII binding | MBS |
| Cu/Zn-SOD92 | inhibition of its oxidation-induced aggregation | Cyanolysis, dMaldi-MS |
| Papain58 | Inhibition of activity | Cyanolysis |
| Platelet proteins93 | activation of its activity | MBS |
MBS: modified biotin switch assay,
TS: tag-switch assay,
LC-MS/MS: liquid-chromatography mass spectrometry,
Maldi MS: matrix-assisted laser desorption/ionization mass spectrometry
Methods for the detection of protein persulfides
Persulfide residues in proteins seem to have better stability than those in small molecules, presumably due to steric hindrance of second order decomposition pathways. Nevertheless the detection of protein persulfides is still challenging, especially in intact cells. One should consider similar reactivity of other sulfur species when applying certain methods in analysis, such as being a nucleophile similar to thiols (-SH) and being an electrophile similar to S-nitrosothiols (-SNO) and sulfenic acids (-SOH). This section summarizes currently available methods for identifying protein persulfides.
Cyanolysis
Cyanolysis was the first method that was used in the identification of protein sulfides. For example, the presence of persulfides in the active sites of xanthine oxidase94,95 and aldehyde oxidase96 was confirmed by cyanolysis. In this method, thiocyanate (SCN−) is formed by nucleophilic attack of cyanide anion (CN−) on the terminal sulfur (Scheme 15). The thiocyanate formed can be determined spectrophotometrically following reaction with ferric nitrate reagent.97 However, the nucleophilic attack of CN− may also occur on the inner sulfur, releasing the terminal sulfur as sulfide (HS−). This competing reaction may cause underestimation of the persulfides in proteins. Another problem is that cyanolysis cannot distinguish persulfides and polysulfides as polysulfides can also react with CN− to form SCN−. As such, this method may also cause overestimation of persulfides. Moreover this method is usually used for individual/purified proteins, and cannot be used for identifying persulfide-containing proteins in complex systems like cell lysates.
Scheme 15.

Detection of protein persulfides by cyanolysis
2,4-Dinitrothiophenol (DNTP) based methods
The presence of persulfides in proteins can also be detected by persulfide-mediated nucleophilic aromatic substituted. 1-Fluoro-2,4-dinitrobenzene (FDNB) was a probe based on such a reaction.98 As shown in Scheme 16, this method involves derivatization of protein SSH with FDNB to form a dinitrobenzene disulfide, which is then treated with dithiothreitol (DTT) to release DNTP. The concentrations of DNTP can be measured by spectroscopic methods and reflect the original persulfide concentrations. This technique can distinguish persulfides from thiols (-SH), thioethers, and polysulfides. However it is again limited to individual and purified proteins. Other thiol-alkylation reagents can also be used in similar derivatization-cleavage sequences to identify protein persulfides. For example, N-(iodoacetaminoethyl)-1-naphthylamine-5-sulfonic acid (1,5-I-AEDANS) was used to identify NifS-bound persulfide.99 This reagent blocked NifS-SSH to form a disulfide product, which was then cleaved by DTT to give NifS-SH and a small molecule thiol 11. Methylation of 11 provided a stable product 12 for quantification.
Scheme 16.

Trapping protein persulfides by aromatic substitution with FDNB and alkylation with 1,5-I-AEDANS
Modified biotin switch assay
In order to identify and isolate S-sulfhydrated proteins in cell lysates a modified biotin-switch assay was reported by Snyder et al in 2009.78 The goal of this method is to label protein persulfide residues with detectable molecules like biotin. As shown in Scheme 17-A, the first step is to block protein free thiols (-SH) with methyl methane sulfonate (MMTS). It was claimed that persulfides did not react with MMTS but could be tagged by a thiol specific biotinylating agent, biotin-HPDP (N-[(6-biotinamido) hexyl]-3’-(2’-pyridyldithio) propionamide) which seems improbable. This method allows capture and identification of S-sulfhydrated proteins by immunoblotting or LC/MS analysis. Using this method, proteins in mouse liver were analyzed and 25–50% of hepatic proteins were found to be sulfhydrated. The three most prominent proteins were glyceraldehyde-3-phosphate dehydrogenase (GAPDH), β-tubulin, and actin.78 Despite these interesting findings, the specificity of the method is questioned as it is hard to justify the selectivity of MMTS for thiols (-SH) vs persulfides (-SSH). It is normally accepted that SSH should also be reactive for thiol-blocking reagents. Carroll et al proved that protein persulfides could react very effectively with commonly used thiol-blocking reagents including IAM, NEM, MMTS, 5,5'-dithiobis-(2-nitrobenzoic acid) (DTNB), N-acetylcysteine pyridyldisulfide (NACP).100 These results suggest that protein persulfides behave quite differently from small molecule persulfides in reactions with electrophiles (see Scheme 8 and related discussions). Perhaps protein persulfides are more stable and their self-decomposition is less likely to happen. In another study Tonks et al used a different biotin switch strategy for analyzing S-sulfhydrated proteins.81 They first blocked both thiols and persulfides with iodoacetic acid (IAA). Then the disulfide adducts formed on persulfides were reduced by dithiothreitol (DTT) to give free thiol groups, and subsequently labeled with iodoacetamide-linked biotin (IAP) (Scheme 17-B). Using this method protein tyrosine phosphatase 1B (PTP1B) was found to be reversibly inactivated by H2S, resulting in regulating the endoplasmic reticulum (ER) stress response. More recently, Snyder et al reported a similar method employing fluorescent maleimide to detect and label persulfides in proteins.79 As such, the proteins of interest were immunoprecipitated and treated with a fluorescent derivative of maleimide, which selectively labeled both free thiols and persulfides. One portion of the reaction mix was then treated with DTT to release the maleimide moiety, resulting in a decrease in fluorescence. The samples were run on a polyacrylamide gel and scanned to detect the fluorescent proteins. The decrease in fluorescence could be quantified using an image analysis software.
Scheme 17.

Modified biotin switch assays for S-sulfhydration detection
Tag-switch assay
Another method suitable for proteomic studies of protein S-sulfhydration is the tag-switch assay.12,83 A two-step sequence can selectively label persulfides. In the first step, free thiols (-SH) and persulfide (-SSH) are blocked by methylsulfonyl benzothiazole (MSBT) to give the corresponding thioether (-S-BT) and disulfide (-SS–BT) adducts. Benzothiazole-containing disulfides are highly reactive towards certain carbon nucleophiles. Therefore a tag-switch step using cyanoacetate-biotin conjugate can convert the benzothiazole disulfides to biotin tags. Benzothiazole thioesters are not affected in this step. Finally the biotinylated proteins can be captured by streptavidin agarose beads for pulldown and detected by Western blotting or LC/MS. The selectivity of this method was confirmed by Gpx3 and BSA, two established protein-SSH models.83 The method was also applied in identification of endogenous S-sulfhydrated proteins in A549 cells.12 The proteomic analysis was able to identify a series of sulfhydrated proteins including protein disulfide isomerase, heat shock proteins, aldo-keto reductase, GAPDH, enolase, and phosphoglycerate kinase.
Summary
The discovery that various persulfide species (i.e. CysSSH, GSSH, protein-SSH, etc) are formed in living organisms and exert specific roles in redox signaling is rather attractive. This review presents some general descriptions of persulfide formation and reactivity from small molecules to proteins. Despite recent advances in understanding persulfide chemistry and chemical biology, a number of important questions remain unanswered, such as what the intracellular targets are and to what extent such reactions can impact signaling. The mechanisms that confer sulfhydration specificity (i.e. what proteins are sulfhydrated and why) also needs to be addressed as well as mechanisms of the reversal of sulfhydration back to the thiol species. Further understanding of biological roles of persulfides will require a better understanding of their fundamental chemistry (which is currently underdeveloped). Chemical tools that allow easy and reliable access to persulfides (both in small molecules and proteins) should be critical for this field. Novel and ‘easy to use’ detection methods for protein S-sulfhydration are also necessary. There is little doubt that the general area of investigation of persulfide chemistry and chemical biology will be a topic of significant research interest for many years to come.
Supplementary Material
{kind=link}
{kind=link}
Scheme 18.

Tag-switch assay
Acknowledgements
This work is supported by an American Chemical Society-Teva USA Scholar Grant and NIH (R01GM088226 and R01HL116571).
Notes and references
- 1.Reddie KG, Carroll KS. Curr. Opin. Chem. Biol. 2008;12(6):746–754. doi: 10.1016/j.cbpa.2008.07.028. [DOI] [PubMed] [Google Scholar]
- 2.Wani R, Nagata A, Murray BW. Front Pharmacol. 2014;5(224):1–8. doi: 10.3389/fphar.2014.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chung HS, Wang SB, Venkatraman V, Murray CI, Van Eyk JE. Circ. Res. 2013;112(2):382–392. doi: 10.1161/CIRCRESAHA.112.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paulsen CE, Carroll KS. Chem. Rev. 2013;113(7):4633–4679. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szabo C. Nature reviews. Drug discovery. 2007;6(11):917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 6.Li L, Rose P, Moore PK. Annu. Rev. Pharmacol. Toxicol. 2011;51:169–187. doi: 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]
- 7.Kabil O, Banerjee R. J. Biol. Chem. 2010;285(29):21903–21907. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paul BD, Snyder SH. Nat. Rev. Mol. Cell Biol. 2012;13(8):499–507. doi: 10.1038/nrm3391. [DOI] [PubMed] [Google Scholar]
- 9.Wang R. Physiol. Rev. 2012;92(2):791–896. doi: 10.1152/physrev.00017.2011. [DOI] [PubMed] [Google Scholar]
- 10.Kimura H. Antioxid. Redox Signal. 2015;22(5):347–349. doi: 10.1089/ars.2014.6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kimura H. Molecules. 2014;19(10):16146–16157. doi: 10.3390/molecules191016146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y, Suematsu M, Motohashi H, Fujii S, Matsunaga T, Yamamoto M, Ono K, Devarie-Baez NO, Xian M, Fukuto JM, Akaike T. Proc. Natl. Acad. Sci. U. S. A. 2014;111(21):7606–7611. doi: 10.1073/pnas.1321232111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toohey JI. Anal. Biochem. 2011;413(1):1–7. doi: 10.1016/j.ab.2011.01.044. [DOI] [PubMed] [Google Scholar]
- 14.Ono K, Akaike T, Sawa T, Kumagai Y, Wink DA, Tantillo DJ, Hobbs AJ, Nagy P, Xian M, Lin J, Fukuto JM. Free Radical Biol. Med. 2014;77:82–94. doi: 10.1016/j.freeradbiomed.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wardman P, Candeias LP, Everett SA, Tracy M. Int. J. Radiat. Biol. 1994;65(1):35–41. doi: 10.1080/09553009414550051. [DOI] [PubMed] [Google Scholar]
- 16.Everett SA, Schoeneich C, Stewart JH, Asmus KD. J. Phys. Chem. 1992;96(1):306–314. [Google Scholar]
- 17.Benson SW. Chem. Rev. 1978;78(1):23–35. [Google Scholar]
- 18.Everett SA, Wardman P. Methods Enzymol. 1995;251:55–69. doi: 10.1016/0076-6879(95)51110-5. [DOI] [PubMed] [Google Scholar]
- 19.Everett SA, Folkes LK, Wardman P, Asmus KD. Free Radical Res. 1994;20(6):387–400. doi: 10.3109/10715769409145638. [DOI] [PubMed] [Google Scholar]
- 20.Toohey JI. Biochem. J. 1989;264(3):625–632. doi: 10.1042/bj2640625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Böhme H, Zinner G. Justus Liebigs Annalen der Chemie. 1954;585(1):142–149. [Google Scholar]
- 22.Tsurugi J, Abe Y, Kawamura S. Bull. Chem. Soc. Jpn. 1970;43(6):1890–1892. [Google Scholar]
- 23.Nakabayashi T, Tsurugi J, Yabuta T. J. Org. Chem. 1964;29(5):1236–1238. [Google Scholar]
- 24.Tsurugi J, Nakabayashi T, Ishihara T. J. Org. Chem. 1965;30(8):2707–2710. [Google Scholar]
- 25.Kawamura S, Kitao T, Nakabayashi T, Horii T, Tsurugi J. J. Org. Chem. 1968;33(3):1179–1181. [Google Scholar]
- 26.Tsurugi J, Abe Y, Nakabayashi T, Kawamura S, Kitao T, Niwa M. J. Org. Chem. 1970;35(10):3263–3266. [Google Scholar]
- 27.Chatterji T, Keerthi K, Gates KS. Bioorg. Med. Chem. Lett. 2005;15(17):3921–3924. doi: 10.1016/j.bmcl.2005.05.110. [DOI] [PubMed] [Google Scholar]
- 28.Kawamura S, Horii T, Tsurugi J. Bull. Chem. Soc. Jpn. 1971;44(10):2878–2879. [Google Scholar]
- 29.Franzi R, Geoffroy M, Bernardinelli G. Mol. Phys. 1984;52(4):947–954. [Google Scholar]
- 30.Bailey TS, Zakharov LN, Pluth MD. J. Am. Chem. Soc. 2014;136(30):10573–10576. doi: 10.1021/ja505371z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakabayashi T, Horii T, Kawamura S, Tsurugi J. Annu. Rep. Radiat. Cent. Osaka Prefect. 1968;9:64–67. [Google Scholar]
- 32.Tsurugi J, Horii T, Nakabayashi T, Kawamura S. J. Org. Chem. 1968;33(11):4133–4135. [Google Scholar]
- 33.Nakabayashi T, Tsurugi J, Kawamura S, Kitao T, Ui M, Nose M. J. Org. Chem. 1966;31(12):4174–4178. [Google Scholar]
- 34.Nakabayashi T, Kawamura S, Kitao T, Tsurugi J. J. Org. Chem. 1966;31(3):861–864. [Google Scholar]
- 35.Nakabayashi T, Tsurugi J. J. Org. Chem. 1963;28(3):813–816. [Google Scholar]
- 36.Derbesy G, Harpp DN, Rather B, Carroll G. Sulfur Lett. 1992;14:199–204. [Google Scholar]
- 37.Aycock DF, Jurch GR. J. Org. Chem. 1979;44(4):569–572. [Google Scholar]
- 38.Heimer NE, Field L. J. Org. Chem. 1984;49(8):1446–1449. [Google Scholar]
- 39.Heimer NE, Field L, Neal RA. J. Org. Chem. 1981;46(7):1374–1377. [Google Scholar]
- 40.Heimer NE, Field L, Waites JA. J. Org. Chem. 1985;50(21):4164–4166. [Google Scholar]
- 41.Tsurugi J, Kawamura S, Horii T. J. Org. Chem. 1971;36(24):3677–3680. [Google Scholar]
- 42.Tsurugi J, Nakabayashi T. J. Org. Chem. 1959;24(6):807–810. [Google Scholar]
- 43.Artaud I, Galardon E. Chembiochem. 2014;15(16):2361–2364. doi: 10.1002/cbic.201402312. [DOI] [PubMed] [Google Scholar]
- 44.Zhao Y, Bhushan S, Yang C, Otsuka H, Stein JD, Pacheco A, Peng B, Devarie-Baez NO, Aguilar HC, Lefer DJ, Xian M. ACS Chem. Biol. 2013;8(6):1283–1290. doi: 10.1021/cb400090d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roger T, Raynaud F, Bouillaud F, Ransy C, Simonet S, Crespo C, Bourguignon M-P, Villeneuve N, Vilaine J-P, Artaud I, Galardon E. Chem Bio Chem. 2013;14(17):2268–2271. doi: 10.1002/cbic.201300552. [DOI] [PubMed] [Google Scholar]
- 46.Hannestad U, Margheri S, Sörbo B. Anal. Biochem. 1989;178(2):394–398. doi: 10.1016/0003-2697(89)90659-3. [DOI] [PubMed] [Google Scholar]
- 47.Liu C, Zhang F, Munske G, Zhang H, Xian M. Free Radical Biol. Med. 2014;76:200–207. doi: 10.1016/j.freeradbiomed.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kawamura S, Otsuji Y, Nakabayashi T, Kitao T, Tsurugi J. J. Org. Chem. 1965;30(8):2711–2714. [Google Scholar]
- 49.Kawamura S, Nakabayashi T, Kitao T, Tsurugi J. J. Org. Chem. 1966;31(6):1985–1987. [Google Scholar]
- 50.Nakabayashi T, Kawamura S, Horii T, Hamada M. Chem. Lett. 1976;5(8):869–874. [Google Scholar]
- 51.Kawamura S, Abe Y, Tsurugi J. J. Org. Chem. 1969;34(11):3633–3635. [Google Scholar]
- 52.Chatterji T, Gates KS. Bioorg. Med. Chem. Lett. 2003;13(7):1349–1352. doi: 10.1016/s0960-894x(03)00103-3. [DOI] [PubMed] [Google Scholar]
- 53.Mitra K, Kim W, Daniels JS, Gates KS. J. Am. Chem. Soc. 1997;119(48):11691–11692. [Google Scholar]
- 54.Gates KS. Chem. Res. Toxicol. 2000;13(10):953–956. doi: 10.1021/tx000089m. [DOI] [PubMed] [Google Scholar]
- 55.Prütz WA. Int. J. Radiat. Biol. 1992;61(5):593–602. doi: 10.1080/09553009214551401. [DOI] [PubMed] [Google Scholar]
- 56.Kende I, Pickering TL, Tobolsky AV. J. Am. Chem. Soc. 1965;87(24):5582–5586. [Google Scholar]
- 57.Sevilla MD, Becker D, Yan M. Int. J. Radiat. Biol. 1990;57(1):65–81. doi: 10.1080/09553009014550351. [DOI] [PubMed] [Google Scholar]
- 58.Francoleon NE, Carrington SJ, Fukuto JM. Arch. Biochem. Biophys. 2011;516(2):146–153. doi: 10.1016/j.abb.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 59.Cavallini D, De Marco C, Mondovi B. Arch. Biochem. Biophys. 1960;87(2):281–288. doi: 10.1016/0003-9861(60)90173-9. [DOI] [PubMed] [Google Scholar]
- 60.Villarejo M, Westley J. J. Biol. Chem. 1963;238(12):4016–4020. [PubMed] [Google Scholar]
- 61.Flavin M. J. Biol. Chem. 1962;237:768–777. [PubMed] [Google Scholar]
- 62.Cavallini D, De Marco C, Mondovi B, Mori BG. Enzymologia. 1960;22:161–173. [PubMed] [Google Scholar]
- 63.Rao GS, Gorin G. J. Org. Chem. 1959;24(6):749–753. [Google Scholar]
- 64.Smith DJ, Venkatraghavan V. Synth. Commun. 1985;15(10):945–950. [Google Scholar]
- 65.Mueller EG. Nat. Chem. Biol. 2006;2(4):185–194. doi: 10.1038/nchembio779. [DOI] [PubMed] [Google Scholar]
- 66.Mihara H, Esaki N. Appl. Microbiol. Biotechnol. 2002;60:12–23. doi: 10.1007/s00253-002-1107-4. [DOI] [PubMed] [Google Scholar]
- 67.Zheng L, White RH, Cash VL, Jack RF, Dean DR. Proc. Natl. Acad. Sci. U. S. A. 1993;90(7):2754–2758. doi: 10.1073/pnas.90.7.2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zheng L, Cash VL, Flint DH, Dean DR. J. Biol. Chem. 1998;273(21):13264–13272. doi: 10.1074/jbc.273.21.13264. [DOI] [PubMed] [Google Scholar]
- 69.Takahashi Y, Nakamura M. J Biochem. 1999;126(5):917–926. doi: 10.1093/oxfordjournals.jbchem.a022535. [DOI] [PubMed] [Google Scholar]
- 70.Palenchar PM, Buck CJ, Cheng H, Larson TJ, Mueller EG. J. Biol. Chem. 2000;275(12):8283–8286. doi: 10.1074/jbc.275.12.8283. [DOI] [PubMed] [Google Scholar]
- 71.Kambampati R, Lauhon CT. J. Biol. Chem. 2000;275(15):10727–10730. doi: 10.1074/jbc.275.15.10727. [DOI] [PubMed] [Google Scholar]
- 72.Nilsson K, Lundgren HK, Hagervall TG, Bjork GR. J. Bacteriol. 2002;184(24):6830–6835. doi: 10.1128/JB.184.24.6830-6835.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ploegman JH, Drent G, Kalk KH, Hol WG, Heinrikson RL, Keim P, Weng L, Russell J. Nature. 1978;273(5658):124–129. doi: 10.1038/273124a0. [DOI] [PubMed] [Google Scholar]
- 74.Westley J. Adv. Enzymol. Relat. Areas Mol. Biol. 1973;39(327–368) doi: 10.1002/9780470122846.ch5. [DOI] [PubMed] [Google Scholar]
- 75.Knowles CJ. Bacteriol. Rev. 1976;40(3):652–680. doi: 10.1128/br.40.3.652-680.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nandi DL, Horowitz PM, Westley J. Int. J. Biochem. Cell. B. 2000;32(4):465–473. doi: 10.1016/s1357-2725(99)00035-7. [DOI] [PubMed] [Google Scholar]
- 77.Iciek M, Wlodek L. Pol J Pharmacol. 2001;53(3):215–225. [PubMed] [Google Scholar]
- 78.Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH. Sci. Signal. 2009;2(96):ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R, Kim S, Snyder SH. Mol. Cell. 2012;45(1):13–24. doi: 10.1016/j.molcel.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang G, Zhao K, Ju Y, Mani S, Cao Q, Puukila S, Khaper N, Wu L, Wang R. Antioxid. Redox Signal. 2013;18(15):1906–1919. doi: 10.1089/ars.2012.4645. [DOI] [PubMed] [Google Scholar]
- 81.Krishnan N, Fu C, Pappin DJ, Tonks NK. Sci. Signal. 2011;4(203):ra86. doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhao K, Li S, Wu L, Lai C, Yang G. J. Biol. Chem. 2014;289(30):20824–20835. doi: 10.1074/jbc.M114.559518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang D, Macinkovic I, Devarie-Baez NO, Pan J, Park CM, Carroll KS, Filipovic MR, Xian M. Angew. Chem. Int. Ed. 2014;53(2):575–581. doi: 10.1002/anie.201305876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK, Barodka VM, Gazi FK, Barrow RK, Wang R, Amzel LM, Berkowitz DE, Snyder SH. Circ. Res. 2011;109(11):1259–1268. doi: 10.1161/CIRCRESAHA.111.240242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Du J, Huang Y, Yan H, Zhang Q, Zhao M, Zhu M, Liu J, Chen SX, Bu D, Tang C, Jin H. J. Biol. Chem. 2014;289(14):9741–9753. doi: 10.1074/jbc.M113.517995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guo C, Liang F, Shah Masood W, Yan X. Eur. J. Pharmacol. 2014;725:70–78. doi: 10.1016/j.ejphar.2014.01.009. [DOI] [PubMed] [Google Scholar]
- 87.Vandiver MS, Paul BD, Xu R, Karuppagounder S, Rao F, Snowman AM, Ko HS, Lee YI, Dawson VL, Dawson TM, Sen N, Snyder SH. Nat. Commun. 2013;4:1626. doi: 10.1038/ncomms2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhao K, Ju Y, Li S, Altaany Z, Wang R, Yang G. EMBO Rep. 2014;15(7):792–800. doi: 10.1002/embr.201338213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Altaany Z, Ju Y, Yang G, Wang R. Sci. Signal. 2014;7(342):ra87. doi: 10.1126/scisignal.2005478. [DOI] [PubMed] [Google Scholar]
- 90.Liu Y, Yang R, Liu X, Zhou Y, Qu C, Kikuiri T, Wang S, Zandi E, Du J, Ambudkar IS, Shi S. Cell Stem Cell. 2014;15(1):66–78. doi: 10.1016/j.stem.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xie Z-Z, Shi M-M, Xie L, Wu Z-Y, Li G, Hua F, Bian J-S. Antioxid. Redox Signal. 2014;21(18):2531–2542. doi: 10.1089/ars.2013.5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.de Beus MD, Chung J, Colón W. Protein Sci. 2004;13(5):1347–1355. doi: 10.1110/ps.03576904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Grambow E, Mueller-Graf F, Delyagina E, Frank M, Kuhla A, Vollmar B. Platelets. 2014;25(3):166–174. doi: 10.3109/09537104.2013.786823. [DOI] [PubMed] [Google Scholar]
- 94.Massey V, Edmondson D. J. Biol. Chem. 1970;245(24):6595–6598. [PubMed] [Google Scholar]
- 95.Edmondson D, Massey V, Palmer G, Beacham LM, 3rd, Elion GB. J. Biol. Chem. 1972;247(5):1597–1604. [PubMed] [Google Scholar]
- 96.Branzoli U, Massey V. J. Biol. Chem. 1974;249(14):4346–4349. [PubMed] [Google Scholar]
- 97.Sörbo B. Biochim. Biophys. Acta. 1957;24:324–329. doi: 10.1016/0006-3002(57)90201-9. [DOI] [PubMed] [Google Scholar]
- 98.Sawahata T, Neal RA. Anal. Biochem. 1982;126(2):360–364. doi: 10.1016/0003-2697(82)90528-0. [DOI] [PubMed] [Google Scholar]
- 99.Zheng L, White RH, Cash VL, Dean DR. Biochemistry. 1994;33(15):4714–4720. doi: 10.1021/bi00181a031. [DOI] [PubMed] [Google Scholar]
- 100.Pan J, Carroll KS. ACS Chem. Biol. 2013;8(6):1110–1116. doi: 10.1021/cb4001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.