

Abstract
Increasing evidence suggests that inflammatory cytokines play a critical role in tumor initiation and progression. We previously isolated a Cancer Stem Cell-like (CSC) subpopulation in neuroblastoma based on differential expression of the receptor for G-CSF (Granulocyte-Colony Stimulating Factor). Here we demonstrate that G-CSF selectively activates signal transducer and activator of transcription 3 (STAT3) within neuroblastoma CSC subpopulations, promoting their expansion in vitro and in vivo. Exogenous G-CSF enhances tumor growth and metastasis in human xenograft and murine neuroblastoma tumor models. In response to G-CSF, STAT3 transcriptionally activates the G-CSF receptor (encoded by CSF3R), creating a CSC sustaining positive-feedback loop. Blockade of G-CSF/STAT3 signaling loop with either anti-G-CSF antibody or STAT3 inhibitor depletes the CSC subpopulation within tumors, driving correlated tumor regression, blocking metastasis and increasing chemosensitivity. Taken together, these data define G-CSF as a tumorigenic growth factor for neuroblastoma and suggest a comprehensive re-evaluation of the clinical use of G-CSF in these patients. Our data also demonstrate that direct targeting of the G-CSF/STAT3 signaling represents a novel therapeutic approach for neuroblastoma.
Keywords: Neuroblastoma, Cancer stem cells, G-CSF, G-CSF receptor, STAT3 inhibitors
Introduction
Neuroblastoma (NB) accounts for 15% of all pediatric cancer mortality and remains a major clinical challenge. Curative therapy requires intensive chemotherapy, radiation, surgery and biologic therapies, yet yields less than 50% success rate and imparts severe long-term side effects (1, 2). Neuroblastoma arises from the early embryonic neural crest, as this transient neural ectodermal population matures and undergoes a programmed ectoderm-tomesodermal transition (EMT) to obtain their migratory mesenchymal-like phenotype (3). This process is in part controlled by STAT3 activation downstream of FGFs, BMPs and other neural crest specification factors (4, 5). Pro-oncogenic targets of STAT3 include genes involved in metastasis (MMP-2, MMP-9, vimentin), angiogenesis (VEGF-A) and inflammation (IL-10, TGFβ, COX-2) (6). The pro-inflammatory cytokines IL-6 and G-CSF activate STAT3 through phosphorylation via their respective cognate surface receptors. G-CSF is known to promote neuronal stem cell survival (7, 8). Accumulating evidence of similar downstream activation of STAT3 links inflammatory signals to tumor progression and spread (9, 10).
We previously defined the ‘stemness’ of a neuroblastoma subpopulation isolated from primary tissues, xenografts and cell lines, based on expression of the G-CSF receptor (CD114) (11). Understanding the biological impacts of downstream signaling via this receptor on neuroblastoma tumorigenesis and metastasis is a critical question with important clinical implications due to the adjuvant role that ligand G-CSF serves in limiting chemotherapy-induced neutropenia in NB patients. We therefore sought to comprehensively analyze the effects of G-CSF on neuroblastoma CSCs and how this alters overall tumor growth and metastasis. We also hypothesized that blocking the G-CSF/STAT3 signaling pathway can reduce the NB CSCs effects and inhibit NB tumorigenicity and metastasis.
Here we demonstrate that G-CSF specifically activates STAT3 signaling in the receptor positive CSC-like NB subpopulation. G-CSF/STAT3 signaling promotes NB colony formation, proliferation, tumor formation and metastasis in both xenograft and allograft murine models of neuroblastoma. In contrast to the bulk CD114- tumor population, the G-CSF receptor positive (CD114+) cells are remarkably sensitive to STAT3 inhibition. Blocking G-CSF/STAT3 signaling axis either by anti-G-CSF antibody or by pSTAT3 inhibition reverse the pro-tumorigenic effects of G-CSF and leads to tumor regression, metastasis inhibition and chemo-sensitivity. These effects strongly correlate with changes in the percentage of CD114+ subpopulation within the human or murine tumor grafts.
Our findings demonstrate that G-CSF acts as a CSC-specific growth factor for high-risk neuroblastoma. Based on these data we propose a new therapeutic paradigm incorporating anti-G-CSF/STAT3 targeting for NB CSCs combined with standard cytotoxic therapy to block neuroblastoma tumor growth, spread and relapse. Our data also strongly advocate a reevaluation of the clinical utility of G-CSF as growth factor for neutrophil support in neuroblastoma patients.
Materials and Methods
Cell culture and reagents
Human neuroblastoma cell lines (NGP, SH-SY5Y, IMR-32, CHLA-255) and mouse NB cell line, NB975 were routinely maintained and cultured as described previously (11). For the in vitro assays performed in the present study, cells were cultured routinely and then partially starved for at least 16-24 hours by culturing in medium supplemented with 1% FBS unless otherwise stated, before treating with drugs or cytokine. Primary antibodies anti-CD114-PE (554538), anti-CD56-APC (555518), anti-CD24-FITC (553261) and fluorochrome, isotype-matching mAbs for negative controls were purchased from BD Biosciences. DAPI (4′,6-diamidino-2-phenylindole, Invitrogen, D3571) staining was used to exclude dead cells in all flow cytometry analysis. LSRII 5-laser flow cytometer (BD Biosciences) was used to perform flow cytometry experiments followed by analysis using FlowJo vX.0.7 (TreeStar). All flow cytometry assays were performed with antibody isotype controls. Stattic (S7947), etoposide (VP-16, E1383) from Sigma-Aldrich and recombinant human G-CSF (BP000175-GD16) was purchased from Syd Labs, Natick, MA. Anti-G-CSF antibody (MAB414) and matching isotype antibody control (MAB005) was purchased from R&D systems, Minneapolis, MN.
Cell line validation and sources
NGP, IMR-32, and SH-SY5Y were obtained from ATCC. CHLA-255 and mouse NB cell line, NB-975 were obtained from Dr. Metelitsa (Baylor College of Medicine). All cell lines are validated for MYCN and CD56 expression and have been validated by genotyping within the past 12 months. All cell lines used in this article were routinely tested for Mycoplasma on a monthly basis.
Mice
Four to six week-old female inbred athymic immunodeficient Nude mice (Nu/Nu) were purchased from Taconic Biosciences, NY and used for all xenograft studies. Heterozygous GCSF−/+ (B6;129P2-Csf3tm1Ard/J) mice were obtained from Jackson Laboratory and housed in pathogen-free conditions. Mice were inbred and genotyped for colony maintenance. Mice were implanted using our previously described orthotopic xenograft model of neuroblastoma (12). Briefly, 1×106 NB cells were surgically implanted in the sub-renal capsule of mice and tumor growth was monitored bi-weekly by bioluminescent imaging (IVIS Lumina XR System, Caliper Life Sciences, Hopkinton, MA).
Single cell assay
Stable EGFP expressing NGP and SH-SY5Y cell lines were generated by transducing EFSEGFP lentiviral vector (provided by Dr. Michael T. Lewis, Baylor College of Medicine) and flow sorting using MoFlo cell sorter (Beckman Coulter). Stably transduced cells were cultured and sorted again for CD114+ and CD114- subpopulation using anti-CD114 antibody. Single cells were aliquoted in wells of 96-well culture plate by serially diluting sorted cells with medium. Cells were cultured in medium supplemented with 1% FBS and untreated or treated with different doses of G-CSF for 28 days with weekly replenishing of cytokine. Cells and colonies were monitored, analyzed and imaged using Olympus-IX71 inverted florescence microscope with DP2-BSW image acquisition software (Olympus).
Cell cycle analysis
Cell cycle assay was performed using APC BrdU flow kit (BD Biosciences) according to manufacturer's instructions and as described previously (11). Briefly, NB cells were pulsed with 10 μM BrdU for 1 hour followed by surface staining for CD114. DAPI was used for total DNA staining.
Metastatic incidence estimation
Metastatic incidence was estimated using a method described previously with modifications (13). Bone marrow from bilateral femurs of engrafted mice was harvested in cold PBS at the time of necropsy. Bone marrow was mechanically disrupted using a 21 gauge needle and red blood cells were removed using lysis buffer (BD Pharm Lyse, 555899) followed by washing with cold PBS twice. Bone marrow samples from nude mice were used as control. Total RNA was extracted using miRNeasy Mini Kit (217004, Qiagen) and residual DNA was removed by DNase treatment (79254, Qiagen) as specified by manufacturer. cDNA was synthesized using high capacity reverse transcription kit (4368814, Applied Biosystems) and real-time quantitative PCR was performed using Taqman assay for human MYCN (assay ID: Hs03666850_s1, Applied Biosystems). Real-time quantitative PCR was performed in MicroAmp optical 96-well reaction plates using StepOnePlus Real-Time PCR Systems (Applied Biosystems) under the following conditions: incubation at 50°C for 2 min and 95°C for 10 min and 40 cycles of 15 s at 95°C and 1 min at 60°C. All assays were performed in triplicates with efficient controls. Metastatic incidence was calculated by dividing number of bone marrow MYCN+ mice with total number of mice in a treatment cohort.
Cell viability and soft agar assay
Cell viability assay was performed using CellTiter 96® Cell Proliferation Assay (G4000, Promega) according to manufacturer's instructions. Briefly, five thousand cells/well were plated into 96-well plate in 1% FBS supplemented medium and treated with indicated drugs for 24h followed by incubation with dye for 4 h. The plates were analyzed by spectrophotometric absorbance at 490 nm using a microplate reader (DTX-800, Beckman Coulter) with Softmax Pro v6.2.2 software (Molecular Devices, Sunnyvale, CA). The data were analyzed and IC50 were calculated using GraphPad Prism 6 (GraphPad Software, La Jolla, CA). Colony formation soft agar assays were performed using standard conditions as described previously (14). Briefly, soft agar plates with bottom gel of medium supplemented with 10% FBS and 0.5% agar were set-up followed by setting top gel of medium supplemented with 1% FBS and 0.3% agar. NB cells were plated with top agar and the plates were incubated for 15 days. Colonies were stained with Thiazolyl Blue Tetrazolium Bromide (M5655, Sigma-Aldrich) and visualized and counted using Versadoc (BioRad, Quantity1 software). All assays were performed in triplicates and repeated twice with proper controls.
Apoptosis assay
Apoptosis in NB cell lines was analyzed using Annexin V- APC apoptosis detection kit (88-8007, eBiosciences) according to manufacturer's instructions. Briefly, cells were partially starved and treated with indicated doses of drugs for 2 h followed by washing with cold PBS and stained with anti-CD114 antibody. Cells were then washed with cold PBS and resuspend in binding buffer followed by incubation with APC-Annexin V antibody for 15 min. Cells were washed and resuspended in binding buffer followed by analysis on LSRII flow cytometer. DAPI was used for analyzing dead and late apoptotic cells.
Chromatin immunoprecipitation
STAT3 binding sites on CSF3R 5′UTR was analyzed by UCSC genome browser (www.genome.ucsc.edu) and confirmed by TransFac analysis (BioBase, Beverly, MA). ChIP was performed on 0.5×105 flow sorted CD114+ and CD114- NGP cells using ChIP-IT high sensitivity kit (Active Motif, 53040) according to manufacturer's instructions. Samples were sonicated for 20 cycles of 30 sec intervals in a Bioruptor UCD-200 sonicator (Diagenode). ChIP-grade anti-STAT3 (9132), anti-pSTAT3 (Y705) (9131, Cell Signaling, Denvers, MA) and IgG control (12-370, EMD-Millipore) antibodies were used. Input was generated by purifying DNA from the sonicated lysates of each sample.
ChIP-quantitative PCR
Primers were designed for ChIP-qPCR using UCSC genome browser and Primer3 software (www.SimGene.com) and are listed in Supplementary Table S3. Real-time PCR reactions were performed as described above using Power SYBR Green PCR master mix. Input and negative control IgG ChIP samples were also analyzed for each sample. The amount of genomic DNA precipitated with specific antibody was calculated in comparison to the total input DNA used for each immunoprecipitation and fold enrichment above background was calculated by normalizing against control IgG. The qPCR reactions were performed in triplicates for each sample with Input and control IgG.
Reporter assay
The CSF3R 5′UTR and promoter regions were amplified from genomic DNA isolated from NGP NB cell line and cloned upstream of the EGFP gene by replacing existing promoter motifs in the lentiviral STAT3.EGFP reporter plasmid (11). The reporter lentiviral plasmids were packaged and NGP cells were transduced and further reporter assays were performed as described previously (11).
Generation of stable STAT3 knockdown cell lines
Lentiviral shRNA vectors pSIH1-puro-STAT3 (26596, Addgene) and pSIH1-puro-control (26597, Addgene) (15) were used to transduce NB cell lines as described previously (11). Seventy-two hours after transduction, cells were selected by media containing 1 μg/ml puromycin. Stably transduced cell lines were further verified for knockdown efficiencies by Western immunoblotting using STAT3 (4904, Cell signaling) antibody using protocol as described previously (14).
Statistical Analysis
Data values for in vivo experiments are expressed as mean ± SEM and compared using Mann-Whitney (two-tailed nonparametric analysis) test. Fisher's exact test was used to compare metastatic incidence between groups. Student's t-test (two-tailed or one-tailed distribution with unequal variance) was applied to compare the results shown for in vitro experiments unless otherwise stated. Assays were performed in triplicates and repeated.
Results
G-CSF induces colony formation in CD114+ cells
To assess the differential responses of neuroblastoma (NB) subpopulations to G-CSF, we purified G-CSF receptor positive (CD114+) and receptor negative (CD114-) subsets from the NB cell lines SH-SY5Y and NGP using Fluorescence Activated Cell Sorting (FACS). Cell proliferation and colony formation from single cells was measured with and without G-CSF over 28 days. Treatment with G-CSF growth factor significantly increased the cell counts and colony counts generated from CD114+ subpopulation with minimal to no change in colony formation in response to G-CSF in the receptor negative subpopulation (Fig. 1A, B). We note a difference in dose dependence between the NGP (MYCN amplified) and SH-SY5Y (non-amplified) cell lines, possibly due to differences in feedback inhibition or cytokine receptor density (additional data in Supplementary Fig. S1). The NGP response fell off above 10 ng/ml while SH-SY5Y cells continued to respond to higher levels of G-CSF. Cell cycle analysis with G-CSF treatment demonstrated a significant increase in S-phase population within the NGP CD114+ subset in a dose-dependent manner compared to control (Fig. 1C, D). In contrast, no significant changes in the cell cycle phases of the CD114- subpopulation were observed in response to G-CSF (Fig. 1C, D). These data correlated with increased activation of STAT3 as measured by increased pSTAT3 (Y705) levels in the CD114+ cells. No change in pSTAT3 was observed upon G-CSF treatment of CD114- cells (Supplementary Fig. S2 A). These in vitro data prompted a more detailed in vivo analysis of G-CSF on NB tumor subpopulations, tumor growth and metastasis.
Figure 1. Effect of G-CSF on CD114+ and CD114- cells in vitro.
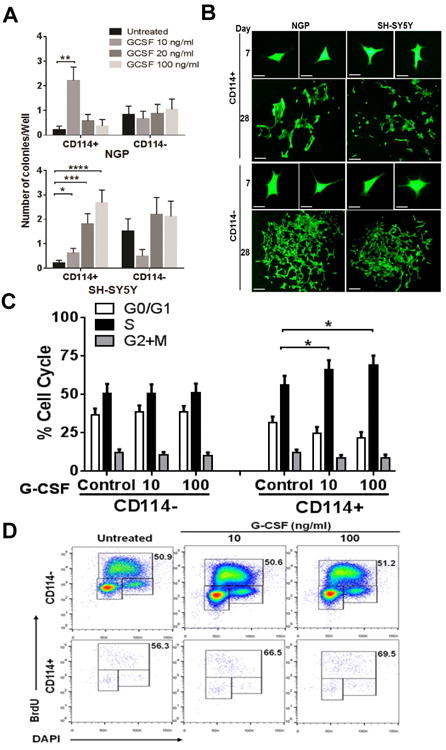
(A) Single cell colony formation assay showing effect of G-CSF treatment on neuroblastoma subpopulations. CD114+ and CD114- cells were flow sorted in to 96-well plates and untreated or treated for 28 days with indicated dose of G-CSF. Each condition is replicated in at least 20 individual wells and data is represented as number of colonies/well (mean ± SEM). No significant effect was observed in CD114- cells under similar culture conditions and G-CSF treatments. (t-test, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Stattic treatment was also performed under similar experimental conditions but even lower doses of Stattic completely blocked the colony formation (data not included). (B) Representative photographs showing colonies developed from CD114+ and CD114- cells of NGP and SH-SY5Y NB cell lines at day 28 in response to G-CSF treatment (Scale bar, 200 μm). Photographs at day 7 scale bar, 100 μm. (C) BrdU cell cycle analysis of NGP NB subpopulations shows increase in S-phase in CD114+ cells in response to G-CSF treatment (10 ng/ml) for 2 h (t-test, p<0.05). CD114- cell cycle remains unaffected from G-CSF. Experiment was repeated in triplicates (mean ± SEM). (D) Representative flow cytometry plots showing effect of G-CSF on cell cycle of NGP NB subpopulations. Values shown in the upper right corner of each dot plot reveal the percentage of the S phase cells under different treatments.
G-CSF promotes tumorigenicity and metastasis in vivo
We next evaluated the impact of exogenous G-CSF on tumor growth in vivo. One million human NB cells (NGP and SH-SY5Y) were xenotransplanted in athymic immunodeficient nude mice via injection under the renal capsule. This previously well-established orthotopic model of neuroblastoma faithfully recapitulates the highly vascular and aggressive growth and metastatic patterns of human neuroblastoma (11, 12). Cohorts of mice were either treated with G-CSF or vehicle control daily for 21 days, starting the day after tumor cell implantation (Fig. 2A). For these studies human recombinant G-CSF was administered at doses comparable to previous studies in murine tumor models which takes into account the rapid metabolism of GCSF in murine model systems (16, 17). Xenografts derived from the NGP cell line showed significant increase in tumor weights with G-CSF treatment. Notably G-CSF treatment also increased the percentage of CD114+ cells in xenografts by three-fold in comparison to controls. We also observed a significant direct correlation between tumor mass and percentage of CD114+ cells within the tumors (Fig. 2B). Similar results were observed for SH-SY5Y xenografts, where G-CSF treatment substantially increased tumor weight with a corresponding increase in CD114+ percentage compared to vehicle treated controls. Again, we found a highly significant correlation between tumor mass and CD114+ percentage (Fig. 2C). Immunohistochemical analysis also showed a significant increase in pSTAT3 (Y705) staining in tumor xenografts treated with G-CSF in comparison to controls (Supplementary Fig. S5). We next examined the effect of G-CSF on bone marrow metastasis (a common site of neuroblastoma metastasis) by detecting human MYCN mRNA in bone marrow of xenotransplanted mice using quantitative PCR. The MYCN oncogene is a well-characterized driver of both MYCN-amplified and non-amplified neuroblastoma that is not expressed in normal adult tissues (18). Results showed that administration of exogenous G-CSF significantly increased the incidence of bone marrow metastasis by two-fold for the MYCN-amplified NGP xenografts in contrast to vehicle treated controls. MYCN non-amplified SH-SY5Y xenografts, which express lower but readily detectable levels of MYCN, also showed a similar trend of increase in bone marrow metastasis in response to G-CSF (Fig. 2B, C).
Figure 2. G-CSF promotes Neuroblastoma tumorigenicity in vivo.
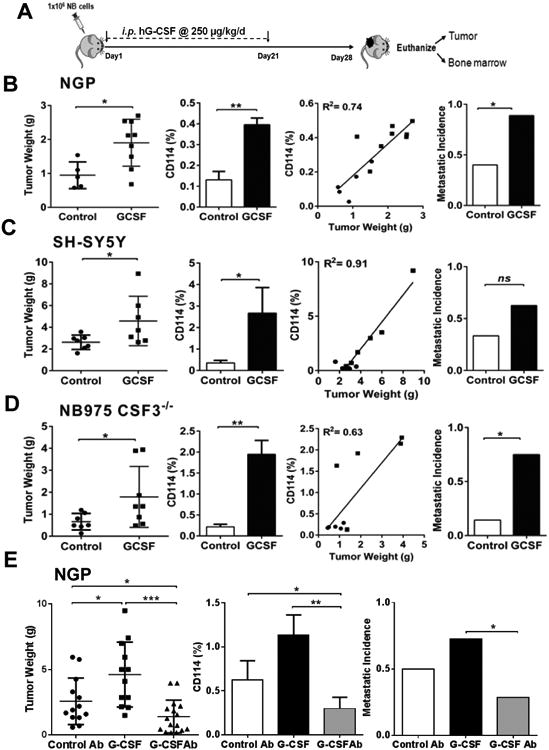
(A) Schematic representation of experimental plan to analyze the effect of G-CSF on neuroblastoma in vivo. One million NB cells were injected under the sub-renal capsule of mouse to develop orthotopic xenografts/allografts by using orthotopic mouse model. Implanted mouse treated with exogenous G-CSF (i.p. injection, 250 μg/kg/day) or vehicle control (5% dextrose water) starting next day from implantation until day 21 followed by necropsy at day 28. (B) NGP xenografts showing significant increase in tumor weights in response to G-CSF treatment compared to controls (p=0.019) with relative increase in the percentage of CD114+ cells in treatment cohorts (p=0.003). Liner regression analysis showing direct correlation between individual tumor mass and percentage of CD114+ cells (●= control, ■=G-CSF treated mice). Detection of human MYCN mRNA by qPCR in bone marrow of xenotransplanted mice shows increase in metastatic incidence in G-CSF treatment cohort (p<0.027) (Mann-Whitney test, *p<0.05, **p<0.01). (C) SH-SY5Y xenografts showing significant increase in tumor weights in response to G-CSF treatment compared to controls (p=0.038) with relative increase in the percentage of CD114+ cells (p=0.03). Liner regression analysis showing direct correlation between individual tumor mass and percentage of CD114+ cells (●= control, ■=G-CSF treated mice). Metastatic incidence analysis shows increase in G-CSF treatment cohort (p=ns) (Mann-Whitney test, *p<0.05). (D) CSF3−/− mice allografted with murine NB cell line NB975 showing significant increase in tumor weights in response to G-CSF treatment compared to controls (p=0.04) and increased the percentage of CD114+ cells (p=0.01). Liner regression analysis showing direct correlation between individual tumor mass and percentage of CD114+ cells (●= control, ■=G-CSF treated mice). G-CSF treatment significantly increases the metastatic incidence (p=0.016) (Mann-Whitney test, *p<0.05, **p<0.01). (E) NGP xenografts treated with anti-G-CSF antibody (i.p. injection, 100 μg/kg/day) showing significant decrease in tumor mass in contrast to exogenous G-CSF (p=0.0001) and isotype control antibody treatment (p=0.04). Treatment plan for all cohorts followed similar to as shown in A. Tumor flow cytometry analysis showing significant decrease in total percentage of CD114+ cells in anti-G-CSF antibody cohort in contrast to G-CSF (p=0.001) and control antibody (p=0.03) cohorts. Metastatic incidence was significantly decreased in anti-G-CSF antibody cohort in comparison to G-CSF treatment (p=0.04). (Mann-Whitney test,*p<0.05, **p<0.01, ***p<0.001).
G-CSF may be produced by both tumor cells and the stromal cells within the tumor microenvironment (19). To evaluate the contribution of stroma-derived G-CSF, we utilized GCSF knockout mice (CSF3−/−). We engrafted a syngeneic murine NB tumor cell line, NB975, which is derived from transgenic TH-MYCN NB tumors and previously shown to contain a small CD114+ subpopulation analogous to human tumors (11). We found that while stromal G-CSF was not required for tumor engraftment, tumor growth and metastasis were markedly enhanced in the presence of exogenous G-CSF (Fig. 2D). Treatment with exogenous G-CSF significantly increased tumor weight with a correlated increase in percentage of G-CSF receptor positive cells within these tumors, derived from murine NB cells in CSF3−/− mice. Here again, exogenous G-CSF significantly increased metastasis compared to controls (Fig. 2D). These data demonstrate, in both xenogeneic (immunodeficient) and syngeneic murine (immunocompetent) NB mouse models, that exogenous G-CSF significantly promotes tumor growth and metastasis. These results are also consistent with our early data defining the increased tumorigenicity and stem cell-like features of CD114+ NB subpopulations found in primary human biopsy specimens and in all NB cell lines and xenografts analyzed to date (11). They also agree with increased downstream pSTAT3 levels in response to G-CSF both in vitro and in vivo (Supplementary Fig. S2 and S5).
Neuroblastoma cell lines and tumors produce elevated levels of G-CSF in comparison to respective controls, as demonstrated by ELISA assay (Supplementary Fig. S3). Other studies suggest that the tumor-derived or endogenous G-CSF also influences the tumor microenvironment and metastatic potential (19). Therefore, we evaluated the effect of direct inhibition of endogenous or tumor-derived G-CSF on NB xenografts. Here, NGP NB xenografts were treated with anti-G-CSF antibody or isotype matched control antibody, starting the day after tumor cell implantation and tumor growth was compared to treatment with G-CSF in an independent experiment (Fig. 2E). Remarkably, we found significant reduction in tumor growth, metastasis and percentage of CD114+ cells after treatment with anti-G-CSF antibody. Taken together these in vivo data support our hypotheses that tumor-derived G-CSF is vital for NB CSC maintenance and that exogenous G-CSF promotes the expansion of NB CSCs, which leads to overall tumor proliferation and metastasis.
STAT3 activation is critical for G-CSF/GCSFR functions in neuroblastoma
In neutrophil and myeloid lineages, STAT3 transcriptional activation is the primary consequence of G-CSFR ligation. We therefore evaluated the specificity of the G-CSF/STAT3 pathway using a small molecule inhibitor (Stattic, Sigma), which blocks STAT3 dimerization and prevents subsequent STAT3 dependent transcriptional responses (20). Cellular proliferation of multiple NB cell lines (i.e. NGP, SH-SY5Y, IMR-32, CHLA-255) was markedly inhibited by the STAT3 inhibitor at low μM levels (p<0.0001) (Fig. 3A and Supplementary Table S1). Colony formation in soft agar was also reduced more than 80% in all tested cell lines with similarly low doses (Fig. 3B, C). Interestingly, when apoptosis in response to STAT3 inhibition was evaluated on FACS purified NB subpopulations from NGP and SH-SY5Y cell lines, we observed differential sensitivity of the CD114+ and CD114- subpopulations. CD114+ cells were highly sensitive to STAT3 inhibition while the CD114- cells were relatively resistant (Fig. 3D). BrdU-based cell cycle analysis further validated these findings, demonstrating STAT3 inhibition selectively increased the sub-G0/G1 population (apoptotic cells) and decreased the S-phase percentage within the CD114+ subpopulation. STAT3 inhibition also blocks the cell cycle progression within the CD114- subset at G1-S checkpoint and increases the population of cells in G1 phase (Fig. 3E, F). Intracellular staining of pSTAT3 (Y705) by phospho-flow assay in the NGP NB cell line shows that Stattic reduces the percentage of CD114+ cells and levels of pSTAT3 (Y705)+ cells in a dose dependent manner either alone or combined with G-CSF or with anti-G-CSF antibody. (Supplementary Fig. S2 B, C). We also found that anti-G-CSF antibody neutralizes the effects of G-CSF (Supplementary Fig. S2 C).
Figure 3. Effect of STAT3 inhibition on Neuroblastoma in vitro.
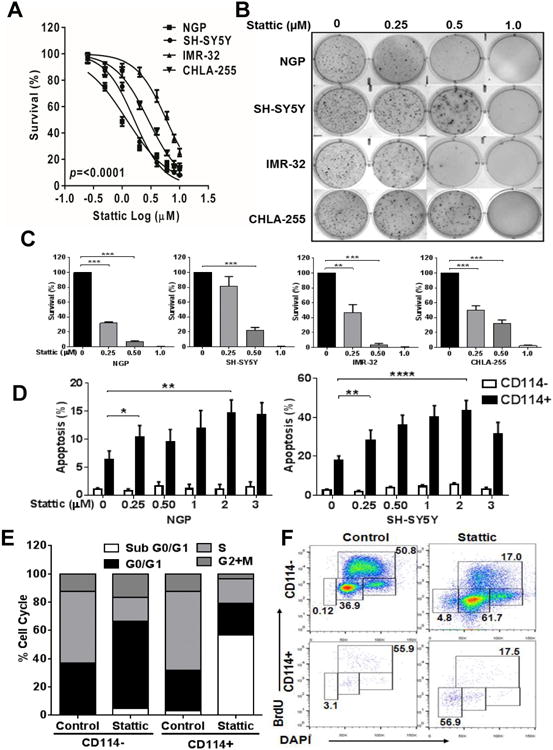
(A) Cell viability assay of different NB cell lines (NGP, SH-SY5Y, IMR-32, CHLA-255) in response to various concentrations of STAT3 inhibitor (Stattic) treatment for 24 h. Cell viability was measured using MTS assay. Experiment was repeated two times with six replicates for each condition and represented as mean ± SD. (B) Representative pictures of colony formation assay for different NB cell lines treated with various doses of Stattic (μM). (C) Quantitation of relative inhibition of colony formation is shown as mean ± SD (t-test, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). (D) Measurement of apoptosis in CD114+ and CD114- NB subpopulations of NGP and SH-SY5Y cell lines using Annexin-V staining along with CD114 staining in response to increasing doses of Stattic for 2 h. Data are represented as means ± SD for three independent experiments. DMSO was used as control in “0” groups. (t-test, *p<0.05, **p<0.01, ****p<0.0001). (E) Cell cycle analysis using BrdU incorporation assay in NGP NB subpopulations with 1μM Stattic treatment for 2 h. Data is represented as relative percentage and representative of experiment repeated three times. (F) Representative flow cytometry analysis plots for cell cycle analysis on NGP NB subpopulations in response to Stattic treatment (1μM). Values shown on each dot plot are representing the percentage of cells.
STAT3 inhibition inhibits NB tumorigenicity and sensitizes NB to chemotherapy
We next evaluated the hypothesis that dual treatment with a genotoxic chemotherapy agent combined with the STAT3 inhibitor would have a combined inhibitory effect on NB tumorigenesis by targeting both tumor subpopulations as proposed for other CSC models (21, 22). Addition of the STAT3 inhibitor markedly sensitizes different NB cell lines to etoposide (VP-16, a topoisomerase inhibitor used extensively in neuroblastoma treatment regimens) as determined by MTS proliferation assays (Fig. 4A and Supplementary Table S1). We further tested the efficacy of this combination in vivo using orthotopic xenografts of NGP and SH-SY5Y according to the treatment regimen outlined in Fig. 4B. For both cell lines the STAT3 inhibitor alone significantly reduces tumor burden with an overall decrease in total percentage of CD114+ cells and metastatic incidence.
Figure 4. STAT3 inhibition sensitizes neuroblastoma to chemotherapy.
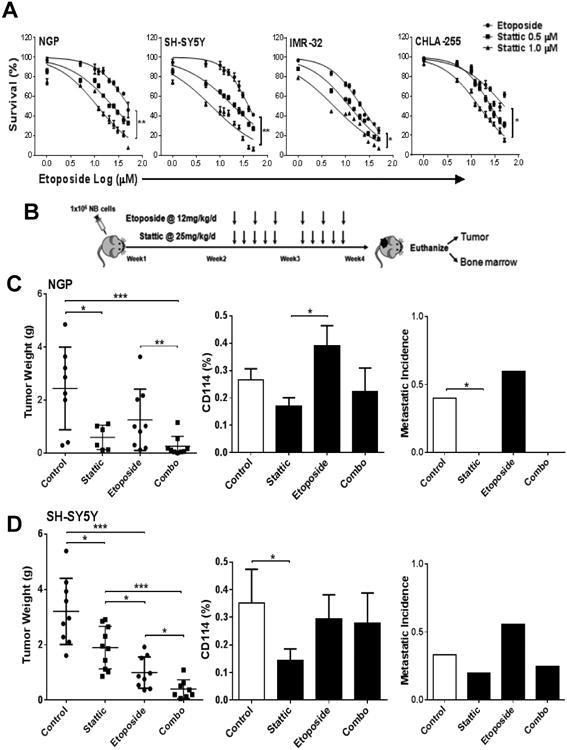
(A) Cell viability assay of different NB cell lines (NGP, SH-SY5Y, IMR-32, CHLA-255) were tested by MTS assay in response to etoposide alone and in combination of 0.5 μM and 1.0 μM Stattic treatment for 24 h. Experiments were repeated two times with six replicates for each condition and represented as mean ± SD. (B) Schematic representation of experimental plan to analyze the in vivo effect of STAT3 inhibitor and dual treatment strategy on neuroblastoma. Orthotopic mouse model was used to develop xenografts of NB NGP and SH-SY5Y cell lines and treated with either etoposide (12 mg/kg/day, i.p. injection, three times/week) and Stattic (25 mg/kg/day, i.p. injection, five days/week) alone or combined. Implanted mouse were treated two weeks post-implantation for two weeks. Vehicle (DMSO) was used as control in all experiments. (C) NGP xenografts showing significant decrease in tumor weights in response to Stattic treatment (p=0.03), etoposide treatment and combo treatment (p=0.001) in comparison to controls. Combo treatment also significantly reduced tumor burden in comparison to etoposide alone (p=0.008). (Mann-Whitney test, *p<0.05, **p<0.01, ***p<0.001). The percentage of CD114+ cells determined by flow cytometry analysis on individual tumors showed as mean ± SEM. Human MYCN mRNA was detected by qPCR in bone marrow of xenotransplanted mice. (D) SH-SY5Y xenografts showing significant decrease in tumor weights in response to Stattic treatment (p=0.02), etoposide treatment (p=0.0002) and combo treatment (p=<0.0001) in comparison to controls. Combo treatment also significantly reduced tumor burden in comparison to etoposide (p=0.02) and Stattic alone (p=0.0002). (Mann-Whitney test (*p<0.05, **p<0.01, ***p<0.001). The percentage of CD114+ cells determined by flow cytometry analysis on individual tumors showed as mean ± SEM. Detection of human MYCN mRNA by qPCR in bone marrow of xenotransplanted mice shows trend of reduction of metastasis with Stattic (p=ns).
As expected, etoposide alone decreases the tumor burden but does not significantly reduce the CD114+ population or metastasis. In fact, genotoxic chemotherapy appears to moderately increase the relative CD114+ percentage in the xenografts. This observation is consistent with the enrichment of CD114+ subpopulations demonstrated previously in patient biopsies examined before and after intensive chemotherapy and correlates with increased pSTAT3 staining in tumors treated with etoposide (Supplementary Fig. S5) (11). Combination treatment of the STAT3 inhibitor with etoposide further reduced tumor burden, and decreased metastatic incidence compared to etoposide alone or controls, and partially reversed the chemotherapy induced increase in CD114 percentage in the tumors (Fig 4C, D). Furthermore, immunohistochemical analysis showed an increase of Cleaved Caspase-3 (apoptosis marker) staining in tumor xenografts treated with Stattic and Combo in comparison to controls (Supplementary Fig. S4). These tumor responses correlated with decreased pSTAT3 (Y705) staining in tumor xenografts treated with Stattic and Combo in comparison to controls or etoposide alone (Supplementary Fig. S5). Taken together, these pre-clinical in vitro and in vivo data support the clinical testing of a dual treatment strategy of combining STAT3 inhibition (targeting the G-CSF receptor positive CSC-like subpopulation) with standard cytotoxic chemotherapy (targeting the bulk tumor) for high risk neuroblastoma.
STAT3 responsive genes are differentially regulated by G-CSF in neuroblastoma subpopulations
Transcriptional targets of STAT3 include genes and microRNAs controlling stemness and differentiation (6, 23, 24). STAT3 opposes p53-mediated effects on cell cycle, apoptosis and inflammation through direct and indirect mechanisms (9, 25, 26). To evaluate the STAT3 specific molecular responses downstream of G-CSF signaling in the CD114+ subpopulation, we used a low-density qPCR array to profile JAK/STAT3 pathway genes (Fig. 5A). We found that G-CSF treatment strongly modulated 38 STAT3 pathway genes and that STAT3 inhibition reversed this effect (Fig. 5B). Subsequently, the expression pattern of key genes were independently validated by qPCR, confirming specific regulation of STAT3 targets via GCSF/G-CSF receptor ligation in CD114+ cells as compared to CD114- cells which are relatively unresponsive to G-CSF, and moderately responsive to STAT3 inhibition (Fig. 5C, D). Scatter plot analysis further demonstrated that G-CSF treatment up-regulates the key STAT3 pathway genes while STAT3 inhibition reverses this effect (Supplementary Fig. S6A, B, C). Ingenuity pathway analyses shows that up-regulation of STAT3 in CD114+ cells directly and indirectly inhibit TP53 (Supplementary Fig. S6D). Gene ontology analysis demonstrates the expected association of STAT3 activation with genes involved in proliferation, stemness and anti-apoptotic responses (Supplementary Table S2) (27).
Figure 5. G-CSF influences STAT3 target genes.
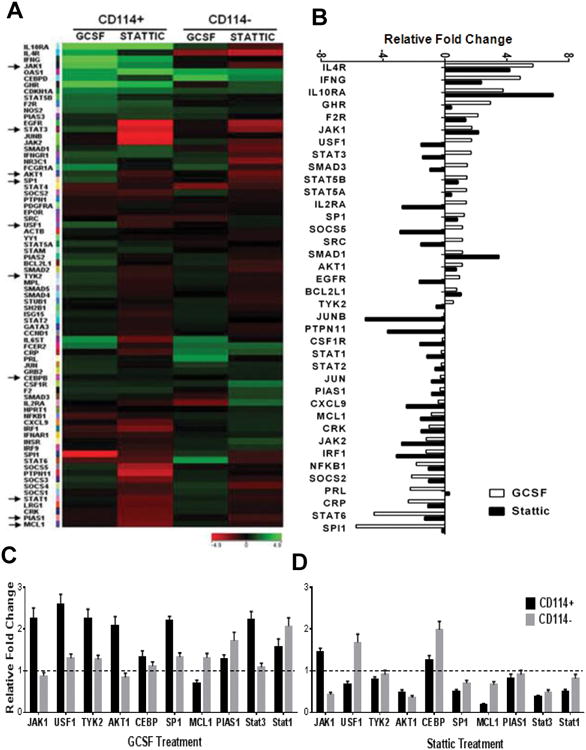
(A) JAK/STAT pathway genes were analyzed using a low-density qPCR based array (SABiosciences) in NB subpopulations from NGP cell line. Heatmap is generated by comparing untreated to G-CSF (20 ng/ml) or Stattic (1μM) treatment for 2 h for respective subpopulations using GeneSpring GX. Arrow marks on left indicate the genes further validated by qPCR as shown in (C, D). (B) Water-fall plot showing effect of G-CSF (20 ng/ml) and Stattic (1μM) treatment for 2 h on individual genes in CD114+ cells in comparison to CD114- cells. (C) Key genes in CD114+ and CD114- NB subpopulations with G-CSF treatment (20 ng/ml for 2 h) were validated by qPCR in triplicates and represented here as mean ± SEM. (D) Key genes in CD114+ and CD114- NB subpopulations with Stattic treatment (1μM for 2 h) were validated by qPCR in triplicates and represented here as mean ± SEM.
G-CSF receptor is a direct transcriptional target of STAT3 in neuroblastoma
Our findings suggest that G-CSF increases both STAT3 activation and percentage of CD114 positive NB cells. We hypothesized that STAT3 may directly regulate expression of the gene coding for CD114 (CSF3R) in neuroblastoma as was previously demonstrated for murine neutrophil precursors (28). First, we analyzed CSF3R mRNA expression by qPCR in FACS isolated subpopulations from multiple NB cell lines. There is a significant increase in expression in response to exogenous G-CSF in CD114+ cells with no increase in CD114- tumor cells. This increase in CSF3R mRNA was blocked by STAT3 inhibition, either alone or combined with GCSF treatment, suggesting pSTAT3 directly regulates CSF3R expression in CD114+ cells (Fig. 6A).
Figure 6. STAT3 directly regulates CSF3R expression.
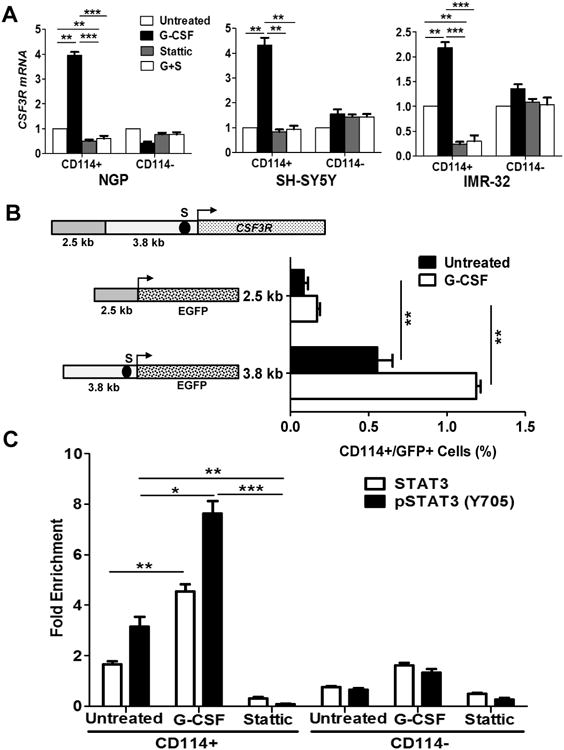
(A) Relative CSF3R expression analysis in NB subpopulations of NGP, SH-SY5Y, and IMR-32 in response to G-CSF (20 ng/ml), Stattic (1μM) and combination treatment (G+S; represents G-CSF + Stattic) for 2 hr. Data are mean ± SEM of three replicates of experiment repeated twice. (t-test, *p<0.05, **p<0.01, ***p<0.001). (B) Schematic representation of CSF3R gene promoter showing 3.8 kb long 5′ untranslated region (5′ UTR) and 2.5 kb long promoter region. A potential STAT3 binding site was determined in 5′ UTR region at -200 bp (S) from translation start site (arrow). EGFP reporter driven by 5′UTR contacting STAT3 binding site (S) shows significant increase in percentage of CD114+/GFP+ cells in response to G-CSF treatment (20 ng/ml for 2 h) in contrast to untreated or reporter driven by promoter region. CHIP-qPCR primers were designed for STAT3 binding site (S). (C) CHIP-qPCR analysis showing direct binding enrichment of STAT3 and pSTAT3 (Y705) at S site of CSF3R 5′ UTR. NGP NB subpopulation CD114+ cells showing enriched binding of STAT3 by 2.2 fold (p=0.007) pSTAT3 by 3.1 fold (p=0.002) in comparison to CD114- cells. G-CSF treatment (20ng/ml for 2 h) in CD114+ cells further enriched the binding of STAT3 by 2.7 fold (p<0.001) and pSTAT3 (Y705) by 2.4 fold (p<0.01) in comparison to baseline untreated while Stattic treatment (1μM for 2 h) blocks the binding of STAT3 or pSTAT3. Data are mean ± SEM of three replicates of experiment repeated twice. (t-test *p<0.05, **p<0.01, ***p<0.001).
Next, we analyzed the CSF3R 5′UTR (3.8 kb) and promoter (2.5 kb) regions for putative STAT3 binding sites and found a canonical STAT3 binding site TTCCCGTAA in the 5′UTR, located 200 bp (S) upstream from the translation start site (Fig. 6B). The promoter and 5′ UTR regions were cloned to drive Green Fluorescent Protein expression using an EGFP reporter plasmid. G-CSF treatment in cells transduced with 5′UTR driven reporter shows significant increase in percentage of CD114+/GFP+ cells in contrast to untreated or reporter driven by promoter region (Fig. 6B). Finally, chromatin immunoprecipitation with quantitative-PCR (ChIP-qPCR) confirmed direct binding of STAT3 and pSTAT3 (Y705) to the STAT3 transcriptional response element (S) upstream of the translational start site for CSF3R in neuroblastoma tumor subpopulations (Fig. 6C). Importantly, we demonstrate that G-CSF treatment, which we have shown activates pSTAT3, significantly enhanced pSTAT3 binding in CD114+ cells. Stattic treatment dramatically reduced both STAT3 and pSTAT3 binding to the CSF3R binding site. Minimal STAT3 or pSTAT3 enrichment was observed in the CD114- tumor population (Fig. 6C). Consistent with STAT3 transcriptional activation of the G-CSF receptor and inhibition by Stattic, STAT3 knockdown with shRNA reduced the percentage of CD114+ cells in different NB cell lines tested (Supplementary Fig. S7). These data confirm that in neuroblastoma, STAT3 directly regulates CD114 expression in response to G-CSF treatment in CD114 receptor positive cells.
Taken together these data establish a positive feedback loop between G-CSF binding to its receptor (CD114) and STAT3-mediated up-regulation of this receptor in CD114+ CSC-like NB subpopulations (Fig. 7). Blockade of G-CSF/STAT3 signaling pathway, either by pSTAT3 inhibition or by anti-G-CSF antibody, specifically down-regulates expression of the G-CSF receptor and limits pSTAT3 signaling events that are involved in neural crest development and in CSC-driven neuroblastoma tumorigenesis. Our data also suggest that STAT3 mediated anti-apoptotic pathways likely promote the survival and expansion of the CD114+ tumorigenic subpopulation (e.g. the observed marked sensitivity of CD114+ cells to pSTAT3 inhibitors compared to CD114- cells (Figure 3D). Therefore, we propose that a G-CSF/G-CSFR/STAT3 feedback loop influences the survival and metastatic behavior of the NB cancer stem cell subpopulation and represents an important CSC-specific therapeutic target.
Figure 7. Model representing positive feedback loop of G-CSF/STAT3 signaling axis in neuroblastoma cancer stem cells.
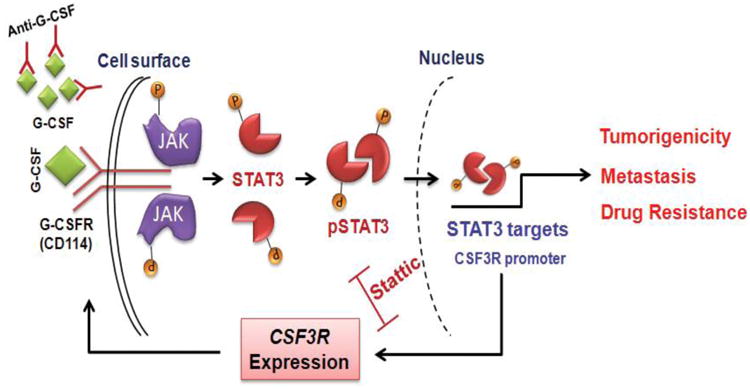
Schematic representation of conclusions drawn from current study showing upregulation of JAK/STAT3 pathway in response to binding of G-CSF to G-CSFR. Active STAT3 translocate to nucleus and upregulate the expression of key target genes that are know to be involved in tumorigenicity, metastasis and drug resistance. CSF3R (gene coding for G-CSFR) is also a direct transcriptional target of STAT3. Binding of STAT3 on CSF3R promoter further upregulate surface expression of the G-CSFR (CD114), making a feedback loop mechanism for tumor progression in NB. STAT3 inhibition and anti-G-CSF antibody are shown to block this positive feedback loop and therefore decrease the cancer stem cell functions in NB.
Discussion
G-CSF is an inflammatory cytokine and neutrophil growth factor expressed from multiple cell types. G-CSF activates pSTAT3, a transcription factor with essential functions for iPSCs (induced pluripotent stem cells) reprogramming and for maintenance of ESC (embryonic stem cell) pluripotency (29-31). Here, we demonstrate a novel function of G-CSF promoting the proliferation and metastasis of neuroblastoma. This primarily occurs through the G-CSF dependent activation of pSTAT3 signaling in CD114+ CSC-like tumor subpopulation. In addition, this tumorigenic subpopulation is highly sensitive to blockade of G-CSF signaling, as well as to STAT3 inhibition. Our data confirm that G-CSF is a) an important growth factor for neuroblastoma, b) markedly affects tumor metastasis, and c) suggest that inhibition of the GCSF/STAT3 signaling axis may be an effective therapeutic approach.
Our results demonstrate a direct effect of G-CSF promoting the expansion and activity of the G-CSF receptor positive CSC-like subpopulation in both xenogeneic and syngeneic neuroblastoma murine tumor models. Previous studies of the effect of G-CSF on other murine tumor models demonstrated pro-tumorigenic effects through indirect immune-mediated processes. In breast and lung carcinoma models, exogenous or tumor-derived G-CSF promotes expansion of myeloid-derived suppressor cell (MDSC) subsets within the circulation and tumor microenvironment (19, 32). These MDSCs in turn secrete pro-angiogenic factors and down-regulate pro-apoptotic TRAIL and MPO leading to tumor progression and metastatic spread to the lung (33, 34). These results are consistent with our findings that G-CSF promotes overall tumorigenicity and metastasis. We demonstrated for the first time a direct effect of GCSF in expanding a unique neuroblastoma cancer stem cell sub-population (<1% of total NB cells) to promote neuroblastoma tumorigenicity and metastasis.
Our data also demonstrate maintenance of a positive feedback loop between G-CSF receptor and STAT3-mediated transcription of the G-CSF receptor gene (CSF3R). Thus G-CSF-mediated STAT3 signaling actively maintains and increases surface expression of the G-CSF receptor (CD114) in this tumorigenic subpopulation to further enhance G-CSF derived NB tumorigenicity and metastasis. Neutralizing G-CSF or inhibiting pSTAT3 can disrupt this positive feedback loop leading to the loss of NB CSC subpopulation. This correlated with decreased tumor growth, decreased metastasis, and increased chemosensitivity. Therefore, G-CSF-mediated activation of STAT3 significantly influences the survival and stem cell-like behavior of the receptor positive subpopulation. Conversely inhibition of STAT3 or blockade of G-CSF signaling represents a novel strategy for CSC-specific therapy for neuroblastoma. Previous studies demonstrated the role of STAT3 in IL-6 mediated drug resistance in neuroblastoma (35) and showed STAT3 inhibition sensitizes colorectal cancer and nasopharyngeal carcinoma to both radiation and chemotherapy (36, 37).
Increasing evidence suggests that STAT3-mediated inflammatory processes are critical for tumorigenesis (38-40). In addition, downstream effectors of chronic inflammation such as TNFα, IL-10, and other inflammatory cytokines such as G-CSF and IL-6 can activate immune responses which indirectly promote cellular proliferation and limit apoptosis (41). Specifically, Akt, Wnt/β-catenin, and the STAT3 signaling pathways are all linked to tumor promotion and survival of malignant stem cell populations (42-44).
These findings further support the novel concept that aggressive developmental malignancies such as neuroblastoma co-opt immune-mediated inflammatory signaling pathways to drive progression and metastasis as suggested for colorectal, pancreatic and hepatic cancers (45). Importantly, we note administration of anti-GCSF antibody which inhibits signaling, led to decreased tumor growth and metastatic spread, while exogenous G-CSF induces expansion of the entire tumor mass of which only a small percentage is G-CSF receptor positive (<1% typically). In addition, in vitro experiments confirmed that CD114+ subpopulation was at least 5-10 times more sensitive to pSTAT3 inhibition than CD114- bulk tumor cells. Doses of Stattic that had minimal effect on the bulk tumor population strongly induced apoptosis in the <1% CSC population and lead to marked growth inhibition of the entire cell line both in vitro and in vivo (Fig. 3, 4).
Since we demonstrate G-CSF signaling is restricted to the CSC-like receptor positive cells, these observations suggest that CD114+ cells provide a paracrine stimulus to the bulk tumor population that indirectly promotes proliferation and survival. Alternatively, tumor expansion may be secondary to the noted expansion of the small CD114+ population and subsequent increase in daughter cell production. In either case, we note that tumor growth and tumor response to anti-G-CSF antibody or STAT3 inhibitor appears to correlate well to changes in the percentage of CD114+ subpopulation within the tumor. Inhibition of the G-CSF/STAT3 pathway by anti-GCSF or G-CSF receptor antibodies, or blockade of STAT3 signaling may well represent a novel neo-adjuvant to standard chemotherapy. In addition to STAT3 inhibition, JAK inhibition may also provide therapeutic efficacy by blocking phosphorylation of STAT3. Both JAK and STAT3 inhibitors are under clinical development (46). Novel non-genotoxic therapeutic strategies for infants and children with neuroblastoma are urgently needed. These might include combining STAT3 blockade with transcriptional inhibition of MYCN (e.g. with CDK7 (47) or BRD4 (48).
Finally, G-CSF is currently extensively used in pediatric and adult oncology as an adjuvant to chemotherapy to limit therapy-related neutropenia (49). In high-risk NB, it is typically administered after each cycle of chemotherapy to reduce treatment related toxicity although there is very limited evidence that this improves overall survival (50). Our data argue for a reconsideration of G-CSF use in neuroblastoma, as well as a careful re-evaluation of the risk/benefit ratio for G-CSF and other pro-inflammatory cytokines in cancer therapy.
Supplementary Material
Acknowledgments
Grant Support: JMS was supported with grants from the NIH (R01 CA174808), American Cancer Society, Alex's Lemonade Stand Foundation, Gillson-Longenbaugh Foundation, Children's Neuroblastoma Research Foundation. LSM was supported with funding from the NIH (RO1 CA116548). ESK received funding from the ST. St. Baldrick's Foundation and a Texas Children's Hospital Department of Surgery Seed Grant.
The authors thank Drs. David Poplack, Malcolm Brenner and Jeffrey Rosen for their comments and help with this article and Linjie Guo for characterizing NB975 cell line. The authors also would like to thank Amos Gaikwad, Tatiana Goltsova and Rena Mao at the flow-cytometry and pathology core facilities at Texas Children's Hospital and Baylor College of Medicine for their outstanding technical assistance and support.
Grant Support: This work was supported by grants from NIH (R01 CA174808), Alex's Lemonade Stand Foundation, Gilson-Longenbaugh Foundation, and the Children's Neuroblastoma Research Foundation to JMS. Support to LSM from the NIH (RO1 CA116548) and support to ESK from the St. Baldrick's Foundation, Texas Children's Hospital Department of Surgery Seed Funding.
Footnotes
Competing Financial Interests: The authors declare no competing financial interests.
Conflicts of Interest: The authors disclose no potential conflicts of interest.
Authors' Contributions: S.A. designed and performed experiments, developed methodology, acquired and analyzed data, wrote the paper. A.L. performed experiments. Z.C. performed experiments, acquired data. M.J.H. analyzed data. L.S.M. analyzed data, wrote the paper. E.S.K. designed experiments, analyzed data, wrote the paper. J.M.S. conceived the study, designed experiments, analyzed the data and wrote the paper.
References
- 1.Armenian SH, Robison LL. Childhood cancer survivorship: an update on evolving paradigms for understanding pathogenesis and screening for therapy-related late effects. Curr Opin Pediatr. 2013;25:16–22. doi: 10.1097/MOP.0b013e32835b0b6a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:289–97. doi: 10.1200/JCO.2008.16.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Louis CU, Shohet JM. Neuroblastoma: molecular pathogenesis and therapy. Annual review of medicine. 2015;66:49–63. doi: 10.1146/annurev-med-011514-023121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nichane M, Ren X, Bellefroid EJ. Self-regulation of Stat3 activity coordinates cell-cycle progression and neural crest specification. The EMBO journal. 2010;29:55–67. doi: 10.1038/emboj.2009.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strobl-Mazzulla PH, Bronner ME. Epithelial to mesenchymal transition: New and old insights from the classical neural crest model. Semin Cancer Biol. 2012 doi: 10.1016/j.semcancer.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carpenter RL, Lo HW. STAT3 Target Genes Relevant to Human Cancers. Cancers (Basel) 2014;6:897–925. doi: 10.3390/cancers6020897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diederich K, Sevimli S, Dorr H, Kosters E, Hoppen M, Lewejohann L, et al. The role of granulocyte-colony stimulating factor (G-CSF) in the healthy brain: a characterization of G-CSF-deficient mice. J Neurosci. 2009;29:11572–81. doi: 10.1523/JNEUROSCI.0453-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schneider A, Kruger C, Steigleder T, Weber D, Pitzer C, Laage R, et al. The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. The Journal of clinical investigation. 2005;115:2083–98. doi: 10.1172/JCI23559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rokavec M, Oner MG, Li H, Jackstadt R, Jiang L, Lodygin D, et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. The Journal of clinical investigation. 2014;124:1853–67. doi: 10.1172/JCI73531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei W, Tweardy DJ, Zhang M, Zhang X, Landua J, Petrovic I, et al. STAT3 Signaling Is Activated Preferentially in Tumor-Initiating Cells in Claudin-Low Models of Human Breast Cancer. Stem cells. 2014;32:2571–82. doi: 10.1002/stem.1752. [DOI] [PubMed] [Google Scholar]
- 11.Hsu DM, Agarwal S, Benham A, Coarfa C, Trahan DN, Chen Z, et al. G-CSF receptor positive neuroblastoma subpopulations are enriched in chemotherapy-resistant or relapsed tumors and are highly tumorigenic. Cancer research. 2013;73:4134–46. doi: 10.1158/0008-5472.CAN-12-4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patterson DM, Shohet JM, Kim ES. Preclinical models of pediatric solid tumors (neuroblastoma) and their use in drug discovery. Curr Protoc Pharmacol. 2011;Chapter 14 doi: 10.1002/0471141755.ph1417s52. Unit 14 7. [DOI] [PubMed] [Google Scholar]
- 13.Van Maerken T, Ferdinande L, Taildeman J, Lambertz I, Yigit N, Vercruysse L, et al. Antitumor activity of the selective MDM2 antagonist nutlin-3 against chemoresistant neuroblastoma with wild-type p53. Journal of the National Cancer Institute. 2009;101:1562–74. doi: 10.1093/jnci/djp355. [DOI] [PubMed] [Google Scholar]
- 14.Shohet JM, Ghosh R, Coarfa C, Ludwig A, Benham AL, Chen Z, et al. A genome-wide search for promoters that respond to increased MYCN reveals both new oncogenic and tumor suppressor microRNAs associated with aggressive neuroblastoma. Cancer research. 2011;71:3841–51. doi: 10.1158/0008-5472.CAN-10-4391. [DOI] [PubMed] [Google Scholar]
- 15.Huang S, Sinicrope FA. Sorafenib inhibits STAT3 activation to enhance TRAIL-mediated apoptosis in human pancreatic cancer cells. Molecular cancer therapeutics. 2010;9:742–50. doi: 10.1158/1535-7163.MCT-09-1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deindl E, Zaruba MM, Brunner S, Huber B, Mehl U, Assmann G, et al. G-CSF administration after myocardial infarction in mice attenuates late ischemic cardiomyopathy by enhanced arteriogenesis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2006;20:956–8. doi: 10.1096/fj.05-4763fje. [DOI] [PubMed] [Google Scholar]
- 17.Hermesh T, Moran TM, Jain D, Lopez CB. Granulocyte colony-stimulating factor protects mice during respiratory virus infections. PloS one. 2012;7:e37334. doi: 10.1371/journal.pone.0037334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Spring Harbor perspectives in medicine. 2013;3:a014415. doi: 10.1101/cshperspect.a014415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waight JD, Hu Q, Miller A, Liu S, Abrams SI. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PloS one. 2011;6:e27690. doi: 10.1371/journal.pone.0027690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–42. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 21.Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13:727–38. doi: 10.1038/nrc3597. [DOI] [PubMed] [Google Scholar]
- 22.Vermeulen L, de Sousa e Melo F, Richel DJ, Medema JP. The developing cancer stem-cell model: clinical challenges and opportunities. Lancet Oncol. 2012;13:e83–9. doi: 10.1016/S1470-2045(11)70257-1. [DOI] [PubMed] [Google Scholar]
- 23.Yang J, van Oosten AL, Theunissen TW, Guo G, Silva JC, Smith A. Stat3 activation is limiting for reprogramming to ground state pluripotency. Cell stem cell. 2010;7:319–28. doi: 10.1016/j.stem.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao Q, Li YY, He WF, Zhang ZZ, Zhou Q, Liu X, et al. Interplay between microRNAs and the STAT3 signaling pathway in human cancers. Physiol Genomics. 2013;45:1206–14. doi: 10.1152/physiolgenomics.00122.2013. [DOI] [PubMed] [Google Scholar]
- 25.Niu G, Wright KL, Ma Y, Wright GM, Huang M, Irby R, et al. Role of Stat3 in regulating p53 expression and function. Mol Cell Biol. 2005;25:7432–40. doi: 10.1128/MCB.25.17.7432-7440.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu Y, Zhang K, Li C, Yao Y, Tao D, Liu Y, et al. Piwil2 suppresses p53 by inducing phosphorylation of signal transducer and activator of transcription 3 in tumor cells. PloS one. 2012;7:e30999. doi: 10.1371/journal.pone.0030999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tang Y, Luo Y, Jiang Z, Ma Y, Lin CJ, Kim C, et al. Jak/Stat3 signaling promotes somatic cell reprogramming by epigenetic regulation. Stem cells. 2012;30:2645–56. doi: 10.1002/stem.1225. [DOI] [PubMed] [Google Scholar]
- 28.Steinman RA, Tweardy DJ. Granulocyte colony-stimulating factor receptor mRNA upregulation is an immediate early marker of myeloid differentiation and exhibits dysfunctional regulation in leukemic cells. Blood. 1994;83:119–27. [PubMed] [Google Scholar]
- 29.Kim SY, Kang JW, Song X, Kim BK, Yoo YD, Kwon YT, et al. Role of the IL-6-JAK1-STAT3-Oct-4 pathway in the conversion of non-stem cancer cells into cancer stem-like cells. Cell Signal. 2013;25:961–9. doi: 10.1016/j.cellsig.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin L, Liu A, Peng Z, Lin HJ, Li PK, Li C, et al. STAT3 is necessary for proliferation and survival in colon cancer-initiating cells. Cancer research. 2011;71:7226–37. doi: 10.1158/0008-5472.CAN-10-4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Oosten AL, Costa Y, Smith A, Silva JC. JAK/STAT3 signalling is sufficient and dominant over antagonistic cues for the establishment of naive pluripotency. Nature communications. 2012;3:817. doi: 10.1038/ncomms1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:6742–7. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan B, Wei JJ, Yuan Y, Sun R, Li D, Luo J, et al. IL-6 cooperates with G-CSF to induce protumor function of neutrophils in bone marrow by enhancing STAT3 activation. J Immunol. 2013;190:5882–93. doi: 10.4049/jimmunol.1201881. [DOI] [PubMed] [Google Scholar]
- 34.Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21248–55. doi: 10.1073/pnas.1015855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ara T, Nakata R, Sheard MA, Shimada H, Buettner R, Groshen SG, et al. Critical role of STAT3 in IL-6-mediated drug resistance in human neuroblastoma. Cancer research. 2013;73:3852–64. doi: 10.1158/0008-5472.CAN-12-2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spitzner M, Roesler B, Bielfeld C, Emons G, Gaedcke J, Wolff HA, et al. STAT3 inhibition sensitizes colorectal cancer to chemoradiotherapy in vitro and in vivo. International journal of cancer Journal international du cancer. 2014;134:997–1007. doi: 10.1002/ijc.28429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pan Y, Zhou F, Zhang R, Claret FX. Stat3 inhibitor Stattic exhibits potent antitumor activity and induces chemo- and radio-sensitivity in nasopharyngeal carcinoma. PloS one. 2013;8:e54565. doi: 10.1371/journal.pone.0054565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bass AJ, Wang TC. An inflammatory situation: SOX2 and STAT3 cooperate in squamous cell carcinoma initiation. Cell Stem Cell. 2013;12:266–8. doi: 10.1016/j.stem.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 39.Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, et al. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer research. 2011;71:5020–9. doi: 10.1158/0008-5472.CAN-11-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell. 2010;39:493–506. doi: 10.1016/j.molcel.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Lerma Barbaro A, Perletti G, Bonapace IM, Monti E. Inflammatory cues acting on the adult intestinal stem cells and the early onset of cancer (Review) Int J Oncol. 2014;45:959–68. doi: 10.3892/ijo.2014.2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ji J, Wang XW. Clinical implications of cancer stem cell biology in hepatocellular carcinoma. Semin Oncol. 2012;39:461–72. doi: 10.1053/j.seminoncol.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Natsume A, Kinjo S, Yuki K, Kato T, Ohno M, Motomura K, et al. Glioma-initiating cells and molecular pathology: implications for therapy. Brain Tumor Pathol. 2011;28:1–12. doi: 10.1007/s10014-010-0011-3. [DOI] [PubMed] [Google Scholar]
- 44.Baumgart S, Chen NM, Siveke JT, Konig A, Zhang JS, Singh SK, et al. Inflammation-induced NFATc1-STAT3 transcription complex promotes pancreatic cancer initiation by KrasG12D. Cancer discovery. 2014;4:688–701. doi: 10.1158/2159-8290.CD-13-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dibra D, Mishra L, Li S. Molecular mechanisms of oncogene-induced inflammation and inflammation-sustained oncogene activation in gastrointestinal tumors: An underappreciated symbiotic relationship. Biochimica et biophysica acta. 2014;1846:152–60. doi: 10.1016/j.bbcan.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiong A, Yang Z, Shen Y, Zhou J, Shen Q. Transcription Factor STAT3 as a Novel Molecular Target for Cancer Prevention. Cancers. 2014;6:926–57. doi: 10.3390/cancers6020926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159:1126–39. doi: 10.1016/j.cell.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schnepp RW, Maris JM. Targeting MYCN: a good BET for improving neuroblastoma therapy? Cancer discovery. 2013;3:255–7. doi: 10.1158/2159-8290.CD-13-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ozkaynak MF, Krailo M, Chen Z, Feusner J. Randomized comparison of antibiotics with and without granulocyte colony-stimulating factor in children with chemotherapy-induced febrile neutropenia: a report from the Children's Oncology Group. Pediatr Blood Cancer. 2005;45:274–80. doi: 10.1002/pbc.20366. [DOI] [PubMed] [Google Scholar]
- 50.Russell H, Shohet JM. Pediatric oncology: G-CSF counteracts chemotherapy toxicity in neuroblastoma. Nat Rev Clin Oncol. 2011;8:6–8. doi: 10.1038/nrclinonc.2010.195. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.