

Abstract
Our understanding of transcriptional gene regulation has dramatically increased over the past decades, and many regulators of gene expression, such as transcription factors, have been analyzed extensively. Additionally, in recent years, deeper insights into the physiological roles of RNA have been obtained. More precisely, splicing, polyadenylation, various modifications, localization and the translation of messenger RNAs (mRNAs) are regulated by their interaction with RNA-binding proteins (RBPs). New technologies now enable the analysis of this regulation at different levels. A technique known as ultraviolet (UV) cross-linking and immunoprecipitation (CLIP) allows us to determine physical protein–RNA interactions on a genome-wide scale. UV cross-linking introduces covalent bonds between interacting RBPs and RNAs. In combination with immunoprecipitation and deep sequencing techniques, tens of millions of short reads (representing bound RNAs by an RBP of interest) are generated and are used to characterize the regulatory network mediated by an RBP. Other methods, such as mass spectrometry, can also be used for characterization of cross-linked RBPs and RNAs instead of CLIP methods. In this review, we discuss experimental and computational methods for the generation and analysis of CLIP data. The computational methods include short-read alignment, annotation and RNA-binding motif discovery. We describe the challenges of analyzing CLIP data and indicate areas where improvements are needed.
Keywords: Next-generation sequencing, cross-linking and immunoprecipitation, posttranscriptional gene regulation, RNA-binding motif discovery
BACKGROUND ON RNA-BINDING PROTEINS
The recognition and binding of certain RNAs by different RNA-binding proteins (RBPs) is essential to maintain the viability of any living cell. RBPs act on many kinds of RNA, such as ribosomal RNA (rRNA), transfer RNA (tRNA), small interfering RNA and microRNA, particularly at different stages of the messenger RNA (mRNA) life-cycle from splicing, polyadenylation, various modifications and subcellular localization to translation [1]. The mechanisms of RBPs regulating selected mRNAs have already been described, but RBPs have a high diversity [2]. Thus, a detailed investigation of the regulatory networks mediated by RBPs is needed to fully understand their posttranscriptional regulatory mechanisms [3]. Consequently, it is crucial to explore the specific effects of particular RBPs on the bound mRNAs. A large-scale study identified >300 RBPs and described a systematic approach that makes use of two different cross-linking protocols (with individual advantages as well as disadvantages) followed by mass spectrometry [4]. Among other findings, the study identified a set of novel RBPs involved in different human diseases, for example, insulin-independent type 2 diabetes or infantile mitochondrial encephalomyopathy. These findings represent a promising starting point for further investigations of these diseases.
The binding between an RNA and RBP involves the recognition of a specific sequence element and also often the identification of a specific secondary structure within the RNA molecule by the RBP [5]. Single-stranded RNA seems to be more accessible for proteins, whereas structural elements such as hairpin loops result in a weaker binding affinity [6]. However, cases in which RBPs have high binding affinity to RNAs forming hairpin loops have also been reported [7]. Because the knowledge of the functional importance of the secondary RNA structures has increased, computational methods for RNA-binding site discovery that take information about the secondary structure into account have recently been developed [8].
The RNA-binding domain (RBD) of an RBP recognizes a region of up to 5 or 6 nt, which determines its specificity and binding affinity to a particular RNA. Additionally, some RBPs increase their specificity for particular RNAs through the presence of more than one RBD in the same RBP [9, 10]. The binding by a single RBD as well as the cooperative binding by multiple RBDs enables the identification of a wide range of RNA molecules (Figure 1).
Figure 1:

RBPs and their different RBDs.
Importance of RBPs in neurological diseases and cancerogenesis
RNAs are crucial for cell viability. Without the regulation, transport and other mechanisms regulated by RBPs, RNAs cannot perform their activities [1]. Genomic aberrations such as single nucleotide mutations, chromosomal translocations or gene amplifications can result in the gain or loss of a function of particular RBPs, thus probably leading to the development of specific disorders [11]. Many RBPs have, for example, been found to be altered in neurological diseases. The insertion of a specific trinucleotide repeat within the 5′ untranslated region (UTR) of the FMR1 gene (fragile X mental retardation 1), e.g. leads to the loss of its function [12]. FMR1 is an RBP that downregulates the translation of proteins that are important in synaptic plasticity in dendrites. If this regulation is interfered with, mental retardation by a decrease in the synaptic plasticity is the result [13]. We have previously described the shift in bound mRNA targets in disease-causing mutants of the RBP FUS (fused in sarcoma) [14]. These FUS mutants cause familial amyotrophic lateral sclerosis, an adult-onset, rapidly progressing and fatal neurodegenerative disorder [15, 16].
Given the important roles of RBPs in the life cycles of all mRNAs, it is not surprising that RBP mutations or altered expression levels have also been described for carcinogenesis. An RBP that affects tumor progression is Sam68. It is involved in alternative splicing and is overexpressed in several cancer types, including breast and prostate cancer. Upon overexpression of Sam68, an additional exon is retained when splicing the mRNA of CD44, which, in turn, has tumorigenic effects [17]. For the aforementioned RBP FUS, we could show an upregulation in liposarcoma [18]. FUS, together with EWSR1 and TAF15, forms a gene family (FET) of abundant ubiquitously expressed RBPs [19]. FET genes are affected by genomic rearrangements, primarily in sarcomas and in leukemia [20, 21].
EXPERIMENTAL TECHNIQUES
Initial method for characterizing RNA-binding sites
The term RNA-recognition element (RRE) is sometimes used for the RNA sequence recognized by the RBP [22] or for the highly conserved protein domain of the RBP that binds to the RNA [23]. In this review, we follow the first convention and refer to the RNA sequence that is recognized by the RBP as the RRE. In the 1990s, an in vitro method for the identification of RREs was developed and widely established [24, 25], called systematic evolution of ligands by exponential enrichment (SELEX). In the first step, a library of synthetic random RNA molecules of a specified length is generated. Next, RNA molecules that bind to the molecule of interest (e.g. an RBP) are enriched with a purification technique such as affinity chromatography. To increase specificity, this step is repeated several times and followed by conventional Sanger sequencing. However, using synthetic random RNA molecules limits the biological significance of the method, as the bound RNAs do not necessarily correspond to naturally occurring RNAs. In contrast, the use of naturally occurring mRNAs extracted from human brain samples instead of a library of random RNA molecules led to the discovery of the RRE for the RBP ELAVL2 (also known as HuB) by an iterative binding and purification strategy [26].
Immunoprecipitation methods for identifying protein–RNA interactions
The first method that used immunoprecipitation for the identification of protein-bound RNA transcripts is known as RNA immunoprecipitation CHIP (RIP-CHIP), which couples immunoprecipitation to microarray analysis [27–29]. The term CHIP signifies that the methods use microarray chips. RIP-CHIP is performed without any treatment of the cells. After cell lysis, the RBP of interest and the bound RNAs are immunoprecipitated. After separation of the RBP and RNA, the purified RNA molecules are characterized by microarray analysis. One of the limitations of RIP-CHIP is that full-length mRNAs are extracted during immunoprecipitation, which does not allow resolving the binding site with single nucleotide resolution. Nevertheless, the results of a RIP-CHIP experiment can be used to identify the RRE for a particular RBP with RNA-binding motif discovery software. Although the RIP-CHIP procedure, like the cross-linking and immunoprecipitation (CLIP) procedures discussed in the next paragraph, has advantages and disadvantages [30], reassortment of mRNA targets is not observed in RIP-CHIP mRNA experiments that use the original experimental protocol [27, 28]. For example, Mili and Steitz [30] used a sonication method optimized for small nuclear ribonucleoproteins, whereas mRNA RIP-CHIP uses mild polysome lysis buffer and no sonication to avoid the shearing of large RNAs and their reassortment [29].
Ultraviolet (UV) CLIP methods [22, 31–33] overcome some drawbacks of SELEX and RIP-CHIP. When coupled to deep sequencing, CLIP allows characterizing RNA–RBP-binding interactions on a genome-wide scale thus revealing starting points for further investigations of targeted RNAs by the RBP. The general idea of CLIP is to retrieve a snapshot of all bound RNAs by an RBP of interest in the cell. Before cell lysis, the cells are exposed to UV light at 254 nm (or 365 nm, depending on the protocol), which results in covalent links of single nucleotides being formed between bound RNA and the RBP [34–36]. In the next step, the cross-linked RNA molecules are purified via immunoprecipitation of the RBP and sequenced. Next-generation sequencing (NGS) is widely available, and a single sequencing run covers thousands of transcripts on a genome-wide scale. The sequence reads that are obtained by CLIP experiments reflect the functional network in which a particular RBP operates. Furthermore, additional information can be gathered by an analysis of the RNA sequences, such as whether the protein regulates only a subset of splice variants for a particular gene. A potential limitation is that the purified RNA molecules only correspond to the bound RNAs by the RBP at the moment of cross-linking. Potentially, therefore, not all possible RNA targets are identified within a single experiment. Besides this, the molecular mechanism of cross-linking is still not fully understood, and low efficiencies of cross-linking specific RBPs to their target RNAs were reported [35]. Low cross-linking efficiencies seem to be associated with specific amino acid compositions of the RBP’s RBD or with the nucleotide distribution within the RRE [37].
Photoactivatable-ribonucleoside-enhanced CLIP (PAR-CLIP) [22] improves the cross-linking compared with standard UV cross-linking by using a photoactivatable nucleoside (e.g. 4-thiouridine), which is incorporated into nascent transcripts. This incorporation leads to more cross-linked sites between the RNA and the protein because of the higher photoreactivity of such nucleosides [38]. This increases the specificity of the method by recovering a larger fraction of RBP-bound RNAs and the removal of less specifically bound RNAs through more stringent washing steps during the immunoprecipitation. Furthermore, the incorporation of 4-thiouridine allows to irradiate cells with 365 nm UV light, which minimizes the risk of unwanted photodamage on the cellular level. The incorporated 4-thiouridine results in a thymidine to cytidine (T–C) conversion, sometimes also called a ‘mutation’, in the cross-linked sites during the following reverse transcription [22]. When sequencing the generated complementary DNA (cDNA) library, one thus obtains sequences with T–C mutations at the cross-linked sites (Figure 2). The presence of such mutations in the sequences obtained represents a useful criterion for distinguishing between truly RBP-bound and nonspecifically bound RNAs. However, the incorporation of photoreactive nucleosides comes with its own issues. The most important limitation is the cytotoxicity, which was observed in some cell lines and tissues following exposure to photoreactive nucleosides [40] such as 4-thiouridine. This cytotoxicity makes it difficult to investigate RBPs in some settings with PAR-CLIP. A detailed protocol for users who are new to PAR-CLIP that guides one through all steps of the technique is given in [41].
Figure 2:

Process from the RNA–RBP interaction to the RNA sequence using PAR-CLIP. First, the mRNA with incorporated 4-thiouridines is bound within the binding pocket of the RBP. Next, the cell is irradiated with UV light at 365 nm, thereby cross-linking single nucleotides from the RNA to amino acids of the RBP. The RNA–protein complex is then extracted by cell lysis, and the mRNA portion that is not protected by the binding pocket is cleaved by RNase T1. Afterward, immunoprecipitation and proteinase K are used to extract and then separate RNA and RBP. Adapters are ligated to the 3′ and 5′ ends of the free mRNA to make the short RNA fragments accessible for reverse transcription, when conversions occur. These conversions can be seen as T–C mutations on the sequence level (adapted from [39]).
In summary, CLIP can be used for a first investigation of the RBP’s regulatory network and indicate potential target genes that are important in this network [3]. Additionally, integrating the results of different global protein–RNA analysis techniques (e.g. RIP-CHIP and PAR-CLIP) can be helpful to overcome the different technical as well as systematic limitations of individual methods. Such data integration provided more detailed insights for an RBP of interest in recent studies [42, 43].
BIOINFORMATIC METHODS AND SOFTWARE FOR ANALYZING (PAR-) CLIP NGS DATASETS
Because NGS data and, in particular, (PAR-) CLIP data can be challenging to analyze [44, 45], we discuss the individual steps and available methods in detail in the next sections.
Quality and adapter clipping
Raw NGS reads may contain parts of the adapter sequences that have been used for cDNA library preparation and sequencing. Therefore, after sequencing, all known adapters that were used have to be identified and removed from all reads. This can be achieved by searching for (inexact) matches between partial or the full adapter sequences and parts of a read. This identifies undesirable adapter sequences at the ends or even within the reads, which can then be removed. Furthermore, the 5′ and 3′ ends of reads often have low base quality scores from the sequencing run. As this results in incorrect base calls and may affect their alignment to the reference genome, one strategy is to remove (clip) such regions from the reads. Read aligners such as MAQ [46] or SOAP [47] provide such functionality. However, the clipped reads cannot be directly exported, and thus these methods cannot be used solely for this purpose within a pipeline. Alternatively, read clippers such as cutadapt [48] or Trimmomatic [49] can be used as stand-alone tools. The performance and accuracy of these tools was evaluated using Illumina NGS data [50], which revealed that trimming increases the quality of subsequent read alignment, assembly or single nucleotide polymorphism calling. A potential problem of this approach is, however, that the removal of such low-quality regions can generate short reads that are uninformative and cannot be unambiguously mapped to a reference genome.
An alternative strategy is to correct errors caused by low base quality scores in the read sequences, such as false base calls (mismatches), deletions or insertions. The benefit of a correction is that the retained reads are longer, and thereby can be used to improve assembly and mapping quality. Compared with deleting information, the general concept of correcting reads to decrease the number of mismatches relative to the reference is thus more powerful. Tools such as SHREC [51] or Quake [52] identify different types of errors such as amplification or sequencing errors within genomic DNA sequence reads and return corrected reads that can subsequently be mapped less ambiguously to a reference genome. Software for correcting sequencing errors in RNA reads, such as SEECER, has also been published [53]. The algorithm of SEECER tries to distinguish between single nucleotide variants and sequencing errors within sets of reads that cover the same genomic area. However, SEECER is not suitable for analyzing PAR-CLIP data, as T–C mutations would be ‘corrected’ and would no longer be available as a quality criterion for subsequent analyses.
Short-read mappers
The next step after read quality improvement is to map the obtained reads against a reference genome to infer the genomic origin of a read. There are two different ways of addressing this problem. The first one is de novo assembly. This approach assembles the reads based on partial sequence overlaps into longer continuous pieces without considering a reference genome sequence. The second approach attempts to map the sequence reads to a position within a reference genome. Here, we focus on the second case, as CLIP methods are commonly used for identifying posttranscriptional regulation in organisms with an already existing genome sequence, such as the human genome sequence, version 19 [54].
A common computational approach to search for matches of a query sequence within a large reference sequence is the use of prefix or suffix trees. For this purpose, the reference sequence is preprocessed and saved as a prefix/suffix tree. This subsequently enables the efficient identification of all exact matches to the query sequence in linear time relative to the length of the reference sequence [55]. However, the maximum memory requirements for prefix/suffix trees (or even arrays) increase quadratically with the length of the reference sequence. The Burrows–Wheeler Transformation (BWT) reduces the use of memory to a linear scale [56] by generating an index for the prefix/suffix tree (Figure 3) [57]. A widely used algorithm for short-read mapping based on the BWT is the Burrows–Wheeler Aligner (BWA) [58]. Other commonly used aligners, such as Bowtie [59] and Bowtie2 [60], also use the BWT.
Figure 3:

Application of the BWT algorithm to the string ‘AGGCT$’, where ‘$’ marks the end of the string and is lexicographically smaller than all other characters. First, a suffix array is created; afterward, it is sorted lexicographically. After the BWT has been performed, only the last column of characters and the order of the starting positions (each has the same length as the string) within the original string are saved. This reduces the memory requirements to a linear scale relative the length of the string.
For finding inexact matches of a read in the reference sequence, an adaptation of the common backward search of the BWT has been realized in both BWA and Bowtie. It is necessary to allow mismatches when using these algorithms on PAR-CLIP data because of the presence of T–C mutations in the reads. Bowtie incorporates a strategy for read alignment, which evaluates mismatches based on base-calling quality: Unless the alignment of a query sequence is found without a mismatch at a particular position, a mismatch can be introduced. Mismatches can only be introduced if the base-calling quality of a mispairing base is low, as these are most likely sequencing errors. Mismatches are introduced until the sum of the base quality scores for all incorporated mismatches exceeds a given threshold. This approach has a constraint, as it follows a greedy strategy: it finds a valid alignment if one exists, but this might not be the best alignment.
An important property of RNA sequencing data is that the reads may span exon–exon junctions, which makes it difficult to align them to a reference genome sequence. This is because inserts of several hundred bases within the reference sequence can occur relative to the read because of the presence of intron sequences (Figure 4). Bowtie and BWA outperform previous read mappers, such as MAQ and SOAP, for contiguous reference alignment in terms of speed and accuracy (Table 1). However, they are not designed for the identification of exon–exon junctions but for the alignment of continuous sequences with only smaller deletions relative to a reference, such as sequence reads of genomic DNA. Thus, other methods were introduced to align RNA-seq reads across exon–exon junctions [45]. Some are restricted to the identification of already known and annotated splice junctions [66, 67], whereas others have the ability to identify de novo splice junctions [68].
Figure 4:

Example of the two types of RNA-seq reads. Read 1 is fully contained within Exon I. Read 2 contains an exon–exon junction, so that the first part of the read matches Exon I, but the second part matches Exon II.
Table 1.
Selection of publicly available read alignment tools
| Aligning tool | Identifies exon junctions | Novel splice junction | Useful for CLIP data | Algorithm type | References |
|---|---|---|---|---|---|
| Bowtie | No | – | Yes, but it is not able to map reads that span exon junctions | BWT | [59, 60] |
| BWA | No | – | Yes, but it is not able to map reads that span exon junctions | BWT | [58] |
| GSNAP | Yes | No | Yes | Seed and extend | [61] |
| MapSplice | Yes | Yes | Yes | Seed and extend | [62] |
| MAQ | No | – | Yes, but it is not able to map reads that span exon junctions | Seed and extend | [46] |
| SOAPsplice | Yes | Yes | Yes | BWT | [63] |
| Subjunc | Yes | Yes | Yes | Seed and vote | [64] |
| TopHat | Yes | Yes | No | BWT | [65] |
A method called Subjunc [64] has been designed to identify exon–exon junctions within RNA-seq data. It applies a seed-and-vote principle to align short-read datasets, in contrast to the commonly used seed-and-extend principle (e.g. MapSplice [62]). The seed-and-vote principle does not use a single seed mapped with high quality to a reference genome sequence to extend it to both sides. Instead, a single read is split into many slightly overlapping ‘subreads’ of ∼10–25 bases, and each subread is aligned without errors to the reference sequence. Some subreads may match perfectly to different parts within the genome (so-called multireads). Multireads that map more often than a given threshold (e.g. 10 times) are excluded. For every read, the two genomic regions with most aligned subreads are determined as potential exons, and this is called the ‘voting’ step of the algorithm. Subsequently, the genome sequence between these two putative exons is scanned for a donor site (GT) and an acceptor site (AG). Next, L1 and L2, the distances of the outermost uniquely mapping subreads to the donor or acceptor sites, respectively, are determined (Figure 5). If the overall distance L between these outmost uniquely matching subreads equals the sum of L1 and L2, this supports the presence of an exon–exon junction within this read. In the second step of Subjunc, a validation of all putative junctions is performed. The following criteria have to be fulfilled: A read mapping across a splice junction must have more matching bases with the reference than the best continuous mapping to any genomic location for this read. Additionally, this splice junction has to be supported by at least one more read. If all criteria are met, the putative splice junction is accepted.
Figure 5:

This figure shows the first step of the read alignment tool Subjunc. Donor (GT) and acceptor sites (AG) between two putative exons are searched, and the length of the genomic mapping (L1 + L2) is compared with the overall distance between the outmost mapping subreads (L) (adapted from [64]).
Another method that identifies splice junctions de novo is TopHat [65]. This includes a two-step procedure, where all reads are initially aligned to the reference genome with Bowtie. All mapped reads are assembled with the assembly module of the read aligner MAQ [46], which identifies read-covered regions as putative exons. The remaining reads—the so-called ‘initially unmapped’ reads—could originate from spliced transcripts, as Bowtie does not support read alignments across larger introns. Next, the algorithm tries to map the initially unmapped reads across pairs of the putative exons determined above. An update of TopHat, called TopHat-Fusion [69], has been designed to identify gene fusions in a similar fashion. However, TopHat is not ideal for the alignment of CLIP reads, as entire and rather short reads are less likely to map to two neighboring exons than subreads, though the latter approach may also lead to more false-positive findings.
Annotation of mapped reads
Many genes and their functions are annotated and curated in public databases, such as the UCSC Genome Browser database [70], the HUGO Gene Nomenclature Committee [71] or DAVID [72]. This information allows one to connect the aligned reads and genomic regions to functional annotations of genes or other genomic elements. Stand-alone software such as HOMER [73] directly annotates gene names and gene functions for genomic regions covered by aligned read sequences. Additionally, for every read, HOMER can report whether it is part of an intron or exon, or if it lies within the 3′ or 5′ UTR of a gene.
Processing PAR-CLIP data
What we have described so far are more or less standard procedures for preprocessing NGS datasets. In the following, we discuss specific analysis methods for PAR-CLIP data that allow us to determine a certain RRE that is recognized by an RBP and to characterize the posttranscriptional effects of an RBP.
An important step after the preprocessing of NGS data produced by PAR-CLIP is the clustering of reads and subsequent assessment of the clusters, regarding, for example, the presence of an RRE. Clustering can, for instance, be performed by hierarchical bottom-up clustering [74]. In single-linkage hierarchical clustering, clusters of reads are determined, in which every read of a cluster overlaps with at least one other read of the same cluster by at least a prespecified minimum length. The resulting clusters correspond to pileups of reads in certain regions on the reference genome and therefore candidate binding sites for RBPs.
The resulting clusters can be analyzed to determine whether they include valid RREs: here, the presence of T–C mutations can be used as a criterion of cluster quality for PAR-CLIP data [22]. A genome region covered by a cluster of PAR-CLIP reads is assumed to encode a transcript bound by the RBP if a certain percentage of all reads of the cluster, e.g. 20%, contain T–C mutations [22]. Only regions with a thymidine inside or in the vicinity of the RRE will be identified by this method, which is almost always the case. Optionally, if there is a reason to assume that the regions will not contain thymidines, different photoactivatable nucleosides can be incorporated, such as 6-thioguanosine [22]. These will generate other mutations on the sequence level. If the data have been generated by CLIP methods without the use of photoactivatable nucleosides, the read coverage can be used as a criterion of the cluster quality. However, this is not as stringent as the T–C criterion, as read clusters can also represent highly abundant but nonspecifically bound transcripts. The publicly available PARalyzer algorithm [75] is specifically designed to generate such read pileups for PAR-CLIP reads. It makes use of a kernel density estimate classifier to generate read pileups representing genomic regions that are targeted by an RBP. It additionally uses the information given by the T–C mutations of each aligned read as a quality measurement.
A motif finder for RREs
The genome sequence covered by a read cluster can be used as input for RRE discovery methods. Notably, a ‘cluster sequence’ usually contains only one RRE, although several RREs within nearby clusters may also occur [42]. Many RBPs containing more than one RBD of the same type have been identified during recent years. Some RBDs of the same type identify slightly different RNA motifs. For instance, zinc finger domains of the same protein identify distinct RREs [76]. The cooperative binding of multiple RBDs that recognize similar sequences allows realizing a wide range of binding specificities and affinities to different mRNAs.
Several motif-finding methods have been developed to identify an RRE from a set of sequences that share at least one RRE (Table 2). Some of these methods search for a single motif within a given set of sequences, whereas others can infer multiple RREs from a set of sequences. Some methods are trained with already published RREs, whereas others identify de novo RREs. The mcast software [79], provided by the online available MEME suite [82], is able to identify multiple RREs within a set of sequences. Although it was originally conceived for detecting instances of DNA-binding motifs in a sequence or, more precisely, for the identification of transcription factor binding sites in promoter regions, it is also applicable to find known RREs in a set of sequences. The scoring scheme of mcast allows the program to determine instances of multiple but already known motifs within a given sequence. This is achieved by scoring matches of known motifs within the sequences, allowing gaps between these matches and the assembly of these to clusters of matches using a nontraditional hidden Markov model (HMM) (called the Meta-MEME model). The HMM is called nontraditional because it has no transition probabilities; instead, arbitrary transition costs are defined between any two states. The Meta-MEME model is built from known motifs from a motif database. Each motif has a state within the model for its forward strand and also a second state for the reverse complement of the motif, which is relevant for the analysis of DNA motifs. Afterward, the Viterbi algorithm is applied to find instances of a known motif in a particular input sequence. As only few RREs have so far been described, this restricts the use of mcast for this problem.
Table 2.
Available software for RNA-binding motif discovery
| Tool | De novo identification | Allows for multiple RREs per sequence set | Algorithm | References |
|---|---|---|---|---|
| cERMIT | Yes | Yes | k-mer ranking | [77] |
| mCarts | Yes | No | HMM | [78] |
| Mcast | No | Yes | Nontraditional HMM | [79] |
| PhyloGibbs | Yes | Yes | Gibbs sampling coupled with simulated annealing | [80] |
| RNAcontext | Yes | Yes | Adaptation of logistical function | [81] |
As CLIP experiments characterize RNA-binding sites on a genome-wide scale, de novo RRE identification tools have also been developed during recent years and will be discussed here. The mCarts software package [78] is an example of a de novo identification method, which determines new RRE motifs from a set of input sequences. The input corresponds to a set of sequences, each sequence of which contains a particular RRE (the positive set) and a set of sequences without this RRE (the negative set). The read sequences of a PAR-CLIP experiment can be assigned to a positive and negative set using the cluster quality criteria discussed earlier. The HMM used in mCarts is more generic than the one in mcast. It contains only states that represent a motif or a background signal. Transitions between these states are given by a probability combining the distance to the previous motif, the accessibility within a possible secondary structure and the conservation of this motif within different species. The positive and negative sets are used for parameterization of the HMM. Afterward, the trained HMM is able to identify motifs on a genome-wide scale by applying the Viterbi algorithm, e.g. using all exonic regions of annotated genes as input. mCarts can find multiple instances of one RRE, which occur in all sequences of the positive set, but a potential limitation is that it would not find different RREs.
RNAcontext [81] also identifies de novo motifs by considering the secondary structure of an RNA molecule. This method also uses two distinct sets of sequences as input (‘positive’ and ‘negative’). From these, the algorithm derives a structural annotation for each sequence. This means that possible secondary structures of a sequence are predicted and scored with publicly available software [76]. Second, the parameters for the motif identification model are estimated. An interesting aspect of the parameter estimation is that information on the RBPs’ binding affinity is included, which is determined with RNAcompete [83]. The latter assures that the model depends not only on a positional weight of a certain base but also on the context of the secondary structure and the binding affinity, which are both included in the complete model. The output of mCarts and RNAcontext are motifs of a few bases in length, which may also include ambiguous base characters (Figure 6) and the location of motif instances in the input sequences. The motifs represent the RREs of the RBP of interest.
Figure 6:

Example output of RNA-binding motif discovery tools. It shows a pattern of four bases with more or less clearly assigned bases, where Position 1 can be either an adenosine or a cytidine, but the second base must be a cytidine.
SAMPLE PIPELINE FOR ANALYZING PAR-CLIP DATA
After discussing the different programs available for each data processing step, we will now describe an exemplary PAR-CLIP analysis pipeline (Figure 7). It starts with the preprocessing of the NGS data for the mRNAs bound by an RBP of interest. First, quality and adapter clipping is performed with cutadapt. Next, the reads are mapped to the publicly available human genome version 19 and against rRNA, tRNA and other noncoding RNA databases provided by the UCSC genome browser. To this end, we use BWA with one mismatch at most per aligned read, which means that we only allow for a single T–C mutation. Allowing for more than one T–C mutation is possible but would reduce the fraction of unambiguously alignable reads. Afterward, reads are clustered with single-linkage clustering and scored based on the percentage of T–C mutations per cluster. This procedure identifies the genomic regions that are most extensively covered by mRNA reads. The putative RBP-bound sequences identified in this manner are returned in a plain text format and can be used for further analyses. Motif identification within this positive set of sequences containing a binding motif is then performed using the MEME suite (Figure 7).
Figure 7:
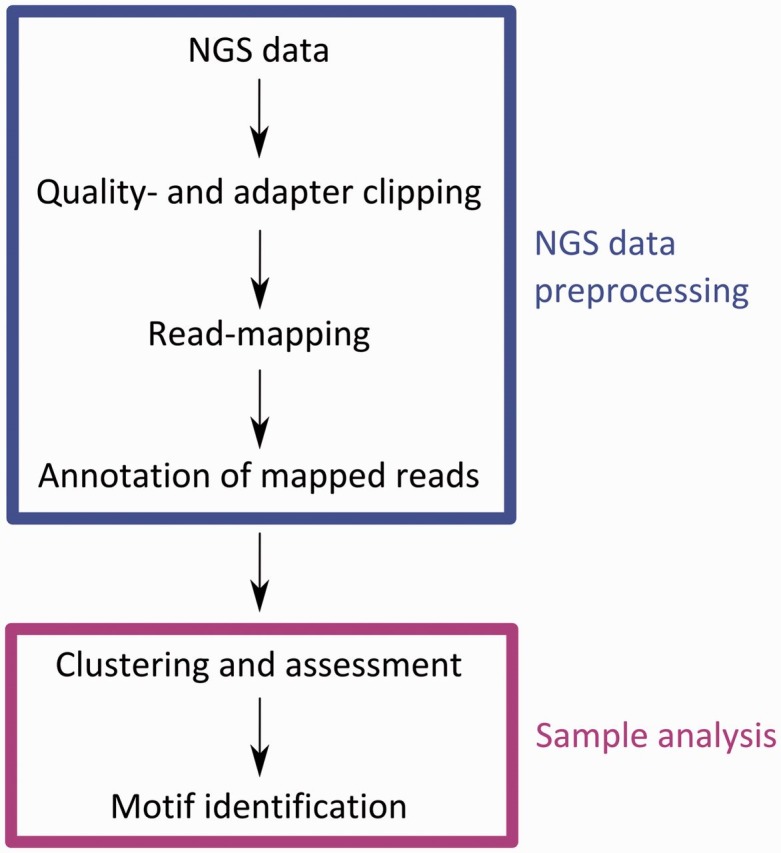
Scheme for NGS data preprocessing and analysis of PAR-CLIP data. The upper box represents steps contained in the preprocessing of NGS data. Steps within the lower box are performed to identify the binding motifs of a particular RBP investigated by a PAR-CLIP experiment.
CONCLUSIONS AND OUTLOOK
Improvements for short-read mapping
Read mapping is not only one of the most important but also one of the most time-consuming steps in the processing of CLIP sequencing data. The results of the overall analysis notably depend on the mapping, as false-positive as well as false-negative alignments will be used in all subsequent steps. Some ideas of how to improve short-read mappers that are also able to map longer reads of up to some hundreds of base pairs are outlined below. This will be of interest not only for analyzing CLIP data but also for all RNA sequencing data. Additionally, a study of four human and mouse RNA-seq datasets has shown that 26 current read-mapping protocols still have various issues with real data [84]. Such problems are exon–exon junction discovery, alignment yield or basewise accuracy, which indicates that improvements in this field are required.
The biggest challenge in aligning mRNA reads is that these often span splice junctions and a specific alignment method for this problem is required. TopHat addresses this by further processing of initially unaligned reads and searching for exon–exon junctions. Additionally, one could align the initially unaligned reads against expressed sequence tags that were obtained by earlier transcriptome sequencing using an accurate read alignment tool, such as Subread [64]. Although this would not discover all expressed variants, it would further reduce the amount of reads that could not be mapped during the first step. This computational effort of mapping a single read against two exons of a reference could thus be avoided for a larger subset of the initially unmapped reads. The computational difficulty of mapping spliced reads is caused by the unknown position of the splice junction. Furthermore, as previously outlined, additional information could be considered when aligning reads. For instance, special handling of the T–C mutations in the PAR-CLIP data instead of treating them like standard sequencing errors would improve read mapping. This could optimize the balance between allowing multiple T–C mutations per read and obtaining unique read mapping to a reference sequence. To this end, simultaneous error correction and read alignment may allow us to align a larger portion of the reads. Current error correction approaches could be improved so that they would be applicable not only to NGS data generated with ‘pure’ CLIP protocols but also to PAR-CLIP data with T–C mutations.
Conclusion on motif identification
The tools for RRE discovery discussed in this review differ in their abilities. Mcast can identify multiple different RREs within the input sequences but does not perform a de novo identification, whereas mCarts can identify a single RRE multiple times in a de novo fashion. However, it may be that different RRE motifs are bound by a single RBP and affect its binding affinity to a particular mRNA [42]. Inferring multiple RRE motifs is therefore an important issue for de novo identification of RREs that would give deeper insights into RBP’s modes of action. The consideration of further information such as secondary structure accessibility or the binding affinity of a given RNA sequence are also useful for validating newly found RREs. Possibly, the currently most suitable tools are RNAcontext and cERMIT [77], as they provide most of the desired functionalities for RRE identification.
Developing a functional analysis/annotation tool
Functional analysis is highly complex and can best be improved by consideration of multiple sources of information, such as existing gene annotations, gene expression profiles and physical binding information [85]. To improve the annotation of candidate target genes identified by PAR-CLIP analysis, databases for gene disease correlations or metabolic pathways can be analyzed. This determines whether multiple genes regulated by the analyzed RBP are part of the same disease or pathway. Clustering algorithms can be used for grouping target genes based on the similarities of, for instance, disease relevance or pathway affiliations. Multidimensional clustering methods, such as biclustering or subspace clustering [86], can be used to integrate multiple heterogeneous data types into the analysis. Such approaches result in a powerful tool when trying to assess the physiological functions of any given RBP.
Key points.
CLIP experiments reveal insights into the posttranscriptional regulation network mediated by particular RBPs.
Many roles of RBPs in disease-relevant pathways can sometimes be postulated or even confirmed by CLIP, but standard computational methods for the analysis of CLIP data have not yet been established.
Algorithmic improvements of different analysis stages, such as read alignment or functional annotation and analysis, may allow us to more fully assess the outcomes of a CLIP experiment.
Acknowledgements
The authors acknowledge Christina Kratsch for her previous work on our in-house pipeline for the analysis of NGS and PAR-CLIP data. They also thank Yao Pan and Johannes Dröge for their critical comments.
FUNDING
This work was supported by the Düsseldorf School of Oncology (funded by the Comprehensive Cancer Center Düsseldorf/Deutsche Krebshilfe and the Medical Faculty HHU Düsseldorf). The authors additionally acknowledge funding by Heinrich Heine University, Düsseldorf, and the Elterninitiative Kinderkrebsklinik e.V., Düsseldorf. Jessica I. Hoell was funded by Deutsche Forschungsgemeinschaft (DFG) through grant HO 5456/3-1.
References
- 1.Hieronymus H, Silver PA. A systems view of mRNP biology. Genes Dev. 2004;18:2845–60. doi: 10.1101/gad.1256904. [DOI] [PubMed] [Google Scholar]
- 2.Glisovic T, Bachorik JL, Yong J. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008;582:1977–86. doi: 10.1016/j.febslet.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ascano M, Gerstberger S, Tuschl T. Multi-disciplinary methods to define RNA–protein interactions. regulatory networks. Curr Opin Genet Dev. 2013;23:20–8. doi: 10.1016/j.gde.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castello A, Fischer B, Eichelbaum K. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012;149:1393–406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- 5.Wan Y, Kertesz M, Spitale RC. Understanding the transcriptome through RNA structure. Nat Rev Genet. 2011;12:641–55. doi: 10.1038/nrg3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, Quon G, Lipshitz HD. Predicting in vivo binding sites of RNA-binding proteins using mRNA secondary structure. RNA. 2010;16:1096–107. doi: 10.1261/rna.2017210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aviv T, Lin Z, Ben-Ari G. Sequence-specific recognition of RNA hairpins by the SAM domain of Vts1p. Nat Struct Mol Biol. 2006;13:168–76. doi: 10.1038/nsmb1053. [DOI] [PubMed] [Google Scholar]
- 8.Kazan H, Morris Q. RBPmotif: a web server for the discovery of sequence and structure preferences of RNA-binding proteins. Nucleic Acids Res. 2013;41:W180–6. doi: 10.1093/nar/gkt463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat Rev Mol Cell Biol. 2007;8:479–90. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doolittle RF. The multiplicity of domains in proteins. Ann Rev Biochem. 1995;64:287–314. doi: 10.1146/annurev.bi.64.070195.001443. [DOI] [PubMed] [Google Scholar]
- 11.Lukong KE, Chang KW, Khandjian EW. RNA-binding proteins in human genetic disease. Trends Genet. 2008;24:416–25. doi: 10.1016/j.tig.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 12.Penagarikano O, Mulle JG, Warren ST. The pathophysiology of fragile X syndrome. Annu Rev Genomics Hum Genet. 2007;8:109–29. doi: 10.1146/annurev.genom.8.080706.092249. [DOI] [PubMed] [Google Scholar]
- 13.McLennan Y, Polussa J, Tassone F. Fragile X syndrome. Curr Genomics. 2011;12:216. doi: 10.2174/138920211795677886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoell JI, Larsson E, Runge S. RNA targets of wild-type, mutant FET family proteins. Nat Struct Mol Biol. 2011;18:1428–31. doi: 10.1038/nsmb.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwiatkowski TJ, Bosco D, Leclerc A. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–8. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 16.Vance C, Rogelj B, Hortobágyi T. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–11. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wurth L. Versatility of RNA-binding proteins in cancer. Comp Funct Genomics. 2012;2012:178525. doi: 10.1155/2012/178525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spitzer JI, Ugras S, Runge S. mRNA and protein levels of FUS, EWSR1, and TAF15 are upregulated in liposarcoma. Genes Chromosomes Cancer. 2011;50:338–47. doi: 10.1002/gcc.20858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu C, Orozco C, Boyer J. BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 2009;10:R130. doi: 10.1186/gb-2009-10-11-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ichikawa H, Shimizu K, Hayashi Y. An RNA-binding protein gene, TLS/FUS, is fused to ERG in human myeloid leukemia with t (16; 21) chromosomal translocation. Cancer Res. 1994;54:2865–8. [PubMed] [Google Scholar]
- 21.Singer S, Socci ND, Ambrosini G. Gene expression profiling of liposarcoma identifies distinct biological types/subtypes and potential therapeutic targets in well-differentiated and dedifferentiated liposarcoma. Cancer Res. 2007;67:6626–36. doi: 10.1158/0008-5472.CAN-07-0584. [DOI] [PubMed] [Google Scholar]
- 22.Hafner M, Landthaler M, Burger L. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–41. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stefl R, Skrisovska L, Allain FH. RNA sequence- and shape-dependent recognition by proteins in the ribonucleoprotein particle. EMBO Rep. 2005;6:33–8. doi: 10.1038/sj.embor.7400325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–22. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 25.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–10. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 26.Gao F-B, Carson CC, Levine T. Selection of a subset of mRNAs from combinatorial 3'untranslated region libraries using neuronal RNA-binding protein Hel-N1. Proc Natl Acad Sci USA. 1994;91:11207–11. doi: 10.1073/pnas.91.23.11207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hendrickson DG, Hogan DJ, Herschlag D. Systematic identification of mRNAs recruited to argonaute 2 by specific microRNAs and corresponding changes in transcript abundance. PLoS One. 2008;3:e2126. doi: 10.1371/journal.pone.0002126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Townley-Tilson WH, Pendergrass SA, Marzluff WF. Genome-wide analysis of mRNAs bound to the histone stem-loop binding protein. RNA. 2006;12:1853–67. doi: 10.1261/rna.76006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tenenbaum SA, Carson CC, Lager PJ. Identifying mRNA subsets in messenger ribonucleoprotein complexes by using cDNA arrays. Proc Natl Acad Sci USA. 2000;97:14085–90. doi: 10.1073/pnas.97.26.14085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mili S, Steitz JA. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA. 2004;10:1692–4. doi: 10.1261/rna.7151404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Licatalosi DD, Mele A, Fak JJ. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–9. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Konig J, Zarnack K, Rot G. iCLIP–transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. J Vis Exp. 2011;50:2638. doi: 10.3791/2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ule J, Jensen KB, Ruggiu M. CLIP identifies nova-regulated RNA networks in the brain. Science. 2003;302:1212–15. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 34.Mayrand S, Pederson T. Nuclear ribonucleoprotein particles probed in living cells. Proc Natl Acad Sci USA. 1981;78:2208–12. doi: 10.1073/pnas.78.4.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayrand S, Setyono B, Greenberg JR. Structure of nuclear ribonucleoprotein: identification of proteins in contact with poly (A)+ heterogeneous nuclear RNA in living HeLa cells. J Cell Biol. 1981;90:380–4. doi: 10.1083/jcb.90.2.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Möller K, Brimacombe R. Specific cross-linking of proteins S7 and L4 to ribosomal RNA, by UV irradiation of Escherichia coli ribosomal subunits. Mol Gen Genet. 1975;141:343–55. doi: 10.1007/BF00331455. [DOI] [PubMed] [Google Scholar]
- 37.Klass DM, Scheibe M, Butter F. Quantitative proteomic analysis reveals concurrent RNA–protein interactions and identifies new RNA-binding proteins in Saccharomyces cerevisiae. Genome Res. 2013;23:1028–38. doi: 10.1101/gr.153031.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meisenheimer KM, Koch TH. Photocross-linking of nucleic acids to associated proteins. Crit Rev Biochem Mol Biol. 1997;32:101–40. doi: 10.3109/10409239709108550. [DOI] [PubMed] [Google Scholar]
- 39.König J, Zarnack K, Luscombe NM. Protein–RNA interactions: new genomic technologies and perspectives. Nat Rev Genet. 2012;13:77–83. doi: 10.1038/nrg3141. [DOI] [PubMed] [Google Scholar]
- 40.Lozzio CB, Wigler PW. Cytotoxic effects of thiopyrimidines. J Cell Physiol. 1971;78:25–31. doi: 10.1002/jcp.1040780105. [DOI] [PubMed] [Google Scholar]
- 41.Spitzer J, Landthaler M, Tuschl T. Rapid creation of stable mammalian cell lines for regulated expression of proteins using the gateway® recombination cloning technology and Flp-In T-REx® lines. Methods Enzymol. 2012;529:99–124. doi: 10.1016/B978-0-12-418687-3.00008-2. [DOI] [PubMed] [Google Scholar]
- 42.Ascano M, Jr, Mukherjee N, Bandaru P. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492:382–6. doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mukherjee N, Corcoran DL, Nusbaum JD. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol Cell. 2011;43:327–39. doi: 10.1016/j.molcel.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Korf I. Genomics: the state of the art in RNA-seq analysis. Nat Methods. 2013;10:1165–6. doi: 10.1038/nmeth.2735. [DOI] [PubMed] [Google Scholar]
- 45.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–8. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li R, Li Y, Kristiansen K. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–14. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 48.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–12. [Google Scholar]
- 49.Rice P, Longden I, Bleasby A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–7. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 50.Del Fabbro C, Scalabrin S, Morgante M. An extensive evaluation of read trimming effects on illumina NGS data analysis. PLoS One. 2013;8:e85024. doi: 10.1371/journal.pone.0085024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schroder J, Schroder H, Puglisi SJ. SHREC: a short-read error correction method. Bioinformatics. 2009;25:2157–63. doi: 10.1093/bioinformatics/btp379. [DOI] [PubMed] [Google Scholar]
- 52.Kelley DR, Schatz MC, Salzberg SL. Quake: quality-aware detection and correction of sequencing errors. Genome Biol. 2010;11:R116. doi: 10.1186/gb-2010-11-11-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Le HS, Schulz MH, McCauley BM. Probabilistic error correction for RNA sequencing. Nucleic Acids Res. 2013;41:e109. doi: 10.1093/nar/gkt215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lander ES, Linton LM, Birren B. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 55.Knuth DE, Morris J, James H, Pratt VR. Fast pattern matching in strings. SIAM J Comput. 1977;6:323–50. [Google Scholar]
- 56.Lam TW, Sung W-K, Tam S-L. Compressed indexing and local alignment of DNA. Bioinformatics. 2008;24:791–7. doi: 10.1093/bioinformatics/btn032. [DOI] [PubMed] [Google Scholar]
- 57. Burrows M, Wheeler DJ. A block-sorting lossless data compression algorithm. Technical Report 124, Digital Equipment Corporation, Palo Alto, CA, 1994.
- 58.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Langmead B, Trapnell C, Pop M. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–81. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang K, Singh D, Zeng Z. MapSplice: accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res. 2010;38:e178. doi: 10.1093/nar/gkq622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang S, Zhang J, Li R. SOAPsplice: genome-wide ab initio detection of splice junctions from RNA-Seq data. Front Genet. 2011;2:46. doi: 10.3389/fgene.2011.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liao Y, Smyth GK, Shi W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013;41:e108. doi: 10.1093/nar/gkt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marioni JC, Mason CE, Mane SM. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–17. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mortazavi A, Williams BA, McCue K. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 68.De Bona F, Ossowski S, Schneeberger K. Optimal spliced alignments of short sequence reads. Bioinformatics. 2008;24:i174–80. doi: 10.1093/bioinformatics/btn300. [DOI] [PubMed] [Google Scholar]
- 69.Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meyer LR, Zweig AS, Hinrichs AS. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res. 2013;41:D64–9. doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gray KA, Daugherty LC, Gordon SM. Genenames. org: the HGNC resources in 2013. Nucleic Acids Res. 2013;41:D545–52. doi: 10.1093/nar/gks1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dennis G, Jr, Sherman BT, Hosack DA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 73.Heinz S, Benner C, Spann N. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kaufman L, Rousseeuw PJ. Finding Groups in Data: An Introduction to Cluster Analysis. Hoboken, NJ: John Wiley & Sons; 2009. [Google Scholar]
- 75.Corcoran DL, Georgiev S, Mukherjee N. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol. 2011;12:R79. doi: 10.1186/gb-2011-12-8-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ding Y, Lawrence CE. A statistical sampling algorithm for RNA secondary structure prediction. Nucleic Acids Res. 2003;31:7280–301. doi: 10.1093/nar/gkg938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Georgiev S, Boyle AP, Jayasurya K. Evidence-ranked motif identification. Genome Biol. 2010;11:R19. doi: 10.1186/gb-2010-11-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang C, Lee K-Y, Swanson MS. Prediction of clustered RNA-binding protein motif sites in the mammalian genome. Nucleic Acids Res. 2013;41:6793–807. doi: 10.1093/nar/gkt421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bailey TL, Noble WS. Searching for statistically significant regulatory modules. Bioinformatics. 2003;19:ii16–ii25. doi: 10.1093/bioinformatics/btg1054. [DOI] [PubMed] [Google Scholar]
- 80.Siddharthan R, Siggia ED, van Nimwegen E. PhyloGibbs: a Gibbs sampling motif finder that incorporates phylogeny. PLoS Comput Biol. 2005;1:e67. doi: 10.1371/journal.pcbi.0010067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kazan H, Ray D, Chan ET. RNAcontext: a new method for learning the sequence and structure binding preferences of RNA-binding proteins. PLoS Comput Biol. 2010;6:e1000832. doi: 10.1371/journal.pcbi.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bailey TL, Boden M, Buske FA. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–8. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ray D, Kazan H, Chan ET. Rapid systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat Biotechnol. 2009;27:667–70. doi: 10.1038/nbt.1550. [DOI] [PubMed] [Google Scholar]
- 84.Engström PG, Steijger T, Sipos B. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat Methods. 2013;10:1185–91. doi: 10.1038/nmeth.2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mitra K, Carvunis A-R, Ramesh SK. Integrative approaches for finding modular structure in biological networks. Nat Rev Genet. 2013;14:719–32. doi: 10.1038/nrg3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Van Mechelen I, Bock H-H, De Boeck P. Two-mode clustering methods: astructuredoverview. Stat Methods Med Res. 2004;13:363–94. doi: 10.1191/0962280204sm373ra. [DOI] [PubMed] [Google Scholar]