

Abstract
Most studies of innate immunity have focused on leukocytes such as neutrophils, macrophages and natural killer cells. However, epithelial cells play key roles in innate defenses that include providing a mechanical barrier to microbial entry, signaling to leukocytes, and directly killing pathogens. Importantly, all of these defenses are highly inducible in response to the sensing of microbial and host products. In healthy lungs, the level of innate immune epithelial function is low at baseline, as indicated by low levels of spontaneous microbial killing and cytokine release, reflecting low constitutive stimulation in the nearly sterile lower respiratory tract when mucociliary clearance mechanisms are functioning effectively. This contrasts with the colon, where bacteria are continuously present and epithelial cells are constitutively activated. While the surface area of the lungs presents a large target for microbial invasion, activated lung epithelial cells that are closely apposed to deposited pathogens are ideally positioned for microbial killing.
Key Terms: Innate immunity: host antimicrobial defenses involving detection of conserved molecular motifs by germline-encoded pattern recognition receptors, and characterized by rapid but transient responses of both leukocytes and parenchymal cells; Adaptive immunity: host antimicrobial defenses involving pathogen detection of specific antigens by somatically recombined receptors, and characterized by clonal expansion of pathogen-specific lymphocytes and immunologic memory; Hemolymph: fluid in the body cavity of insects, homologous to vertebrate blood, with most proteins produced by the fat body, homologous to the vertebrate liver, with functions in both metabolism and immunity; Resistance: the strategy of host survival of infection that is associated with a reduction in pathogen burden; this is contrasted to tolerance, the strategy of generating a host phenotype indifferent to the pathogen burden; Non-typeable Haemophilus influenzae (NTHi): an unencapsulated (hence non-typeable) strain of a Gram-negative bacterial pathogen that is frequently cultured from the lungs of patients with chronic respiratory disease; Pathogen-associated molecular pattern (PAMP): stereotypic molecular motifs conserved across microbial species that are recognized by pattern recognition receptors, triggering innate immune responses; Damage-associated molecular pattern (DAMP): molecular motifs expressed on or released by infected or injured host cells, also known as danger signals or alarmins; Pattern recognition receptors (PRR): membrane-associated, cytosolic or secreted host products that recognize conserved molecular patterns on pathogens, initiating innate immune responses. These include Toll-like receptors (TLR), NOD-like receptors (NLR), RIG-I-like receptors (RLR), class A scavenger receptors (SR-A), and macrophage receptor with collagenous structure (MARCO); Leucine rich repeats (LRR): common molecular sequences of many pattern recognition receptors that generally occur within pathogen recognition domains; Toll/Interleukin-1 receptor adaptors (TIR adaptors): host peptides that are selectively recruited to Toll-like receptors and IL-1 receptor upon ligand binding and are required for signal propagation; Lipopolysaccharide (LPS): cell wall component of Gram-negative bacteria, the lipid A portion of which is recognized by TLR4 in association with MD2 and CD14; Interleukin (IL): a widely expressed and highly variable group of cytokine signaling molecules involved in both innate and adaptive immune responses; Complement: a system of more than thirty proteins activated by three pathways that permeabilize pathogens with the membrane attack complex, opsonize microbes and release fragments with signaling properties such as the anaphylotoxins C3a and C5a; Receptor for advanced glycation end-products (RAGE): a multifunctional member of the immunoglobulin superfamily that recognizes several host immunomodulatory proteins, including HMGB1 and S100, as well as host proteins without immunomodulatory activity that become glycated during aging or inflammation; Interferon (IFN): a subset of cytokines that inhibit viral replication within host cells and activate leukocytes. There are three classes – Type I (α, β, ω, ε, and κ), Type II (γ), and Type III (λ1-3, also known as IL-28A/B and IL-29), each with distinct receptors; Anoikis: apoptosis of epithelial cells induced by detachment from the extracellular matrix; Transcytosis: mechanism of transport across polarized epithelial cells involving endocytois of extracellular macromolecules or particles on one surface, transcellular vesicle trafficking, and exocytosis on the other surface; Secretory leukocyte proteinase inhibitor (SLPI): epithelium-derived protease inhibitor with intrinsic antibacterial activity; Transmigration: paracellular migration of leukocytes or pathogens through epithelial or endothelial barriers and associated basement membranes
Introduction
Microbes entering the bodies of multicellular eukaryotes must first cross an epithelial cell layer. Besides functioning as physical barriers to prevent infection, mammalian epithelial cells are able to sense the presence of microbes and to respond by augmenting their barrier function, signaling to leukocytes, and directly killing pathogens. While signaling to leukocytes has received considerable attention, augmented barrier function and pathogen killing have received less. Conditional pathogen killing by epithelial cells, in particular, is an important aspect of innate resistance to infection that merits further attention in understanding the homeostasis of epithelial surfaces throughout the body and in manipulating innate immunity therapeutically.
It should not come as a surprise that mammalian epithelial cells are capable of highly inducible antimicrobial defenses since innate immune function was first elucidated in insects in which the systemic response to infection is mediated by the release of antimicrobial peptides from epithelial cells of the fat body into the hemolymph (1, 2). However, leukocytes that are specialized for immune functions have traditionally dominated the attention of mammalian immunologists. Furthermore, the initial identification of Toll-like receptor signaling in mammalian biology was in the induction of costimulatory molecules required for adaptive immune responses (3), reinforcing the focus on leukocytes. While the abilities of professional immune cells to detect and kill pathogens are impressive, non-specialist epithelial cells have retained such capabilities through evolution. This suggests that rather than replacing the innate immune function of epithelial cells in higher eukaryotes, leukocytes complement these functions to collaborate in host defense.
Inducible Innate Resistance of Lung Epithelium
Several lines of evidence have pointed to the inducibility of innate immune defenses in lung epithelial cells. First, the epithelial cells of lower eukaryotes show robust inducible defenses, as above, and the barrier epithelia of Drosophila, including those of the trachea, Malpigian tubules, gut and reproductive tract, show specific patterns of antimicrobial peptide expression (1, 2). Second, cultured lung epithelial cells show the inducible expression of antimicrobial polypeptides in vitro, and transgenic overexpression of some of these has been shown to result in increased resistance to infection in vivo (4-8). Third, lung epithelial cells show remarkable structural and molecular plasticity during inflammation, suggesting that lung epithelial defensive functions are similarly plastic (9). Fourth, modest increases in resistance to bacterial infection by the airway route has been found after exposure of the lungs to single innate immune ligands such as endotoxin (10-18), though the role of the epithelium was not isolated experimentally.
To strongly induce lung defenses in vivo, we exposed mice to an aerosolized lysate of the bacterium NTHi, reasoning that this would stimulate the epithelium with a complex mixture of pathogen-associated molecular patterns (PAMPs) in proportions that reflect a natural exposure. This stimulation rapidly resulted in a high level of resistance to a broad array of microbial pathogens (9, 19, 20). In the initial studies (19), resistance to S. pneumoniae reached a maximum 4 hours after stimulation, remained at the maximal level for 24 hours, then gradually declined over several days (Fig. 1A). Host protection was mirrored by augmented microbial killing within the lungs that subsequently declined over several days (Fig. 1B). In subsequent studies, the NTHi lysate was shown to induce resistance to all pathogens tested, including Gram-positive and Gram-negative bacteria, the spore-forming NIAID Class A bioterror agent B. anthracis, the fungus A. fumigatus, and influenza virus (9, 20). In each case, increased host survival was associated with a reduction of lung pathogen burden, indicating that host protection occurs through a resistance mechanism.
Figure 1. Time Course of Induced Innate Resistance in the Lungs.

A. Host survival. Mice were pretreated in groups of 6 with an aerosolized lysate of the bacterium non-typeable H. influenzae (NTHi) to stimulate innate immunity, then challenged as a single group with live aerosolized S. pneumoniae (Spn) (6.1 × 1010 CFU/ml for 60 min). Survival at 7 days is shown as a function of the interval between treatment and challenge (* p = 0.015, † p = 0.002, treated vs. untreated). B. Bacterial Counts in the Lungs. Mice were pretreated in groups of 4 with an aerosolized NTHi lysate at various time points, then challenged as a single group with live aerosolized Spn (2.1 × 1010 CFU/ml for 60 min). Lungs were removed immediately after the aerosol challenge, homogenized, and plated for bacterial culture (mean ± SEM, * p < 0.05 for treated vs untreated). From Clement et al., Am J Respir Crit Care Med, 2008.
The dominant role of the epithelium in stimulated innate resistance of the lungs is supported by the following evidence. First, resistance is local, such that there is no protection against an intraperitoneal or intravenous microbial challenge after aerosol stimulation of the lungs (19). Second, resistance can be induced in mice deficient in neutrophils, macrophages, or mast cells (19), as well as in mice deficient in dendritic cells, natural killer cells, or lymphocytes (S. E. Evans, unpublished data). Thus, neither resident nor recruited leukocytes are required for stimulated innate resistance of the lungs, though it is likely that resident leukocytes amplify the sensing of PAMPs by signaling to epithelial cells locally (see below), and neutrophil recruitment is clearly important for clearing large microbial inocula in mice and in the susceptibility to infection of neutropenic patients (19, 21, 22). Third, important roles for innate immune signaling in lung parenchymal cells have been demonstrated in the control of bacterial and viral infections (23-26). Fourth, efficient direct microbial killing by epithelial cells stimulated with PAMPs or cytokines in vitro has been demonstrated (9, 21, 27), in contrast to dendritic cells and macrophages stimulated in vitro that do not kill bacteria efficiently (9). Fifth, consistent with the functional capability of isolated epithelial cells to kill pathogens, multiple epithelial-derived antimicrobial proteins are found upregulated in lung lining fluid by proteomic analysis after stimulation with microbial products (19). Similarly, multiple antimicrobial proteins are upregulated by gene expression microarray in leukodepleted lungs and isolated epithelial cells after stimulation with microbial products or cytokines (9, 21), though identification of the critical epithelial effector mechanisms has not yet been determined (see below, Epithelial Effector Mechanisms in Stimulated Resistance).
Together, these results indicate that respiratory epithelial cells are capable both of sensing innate stimuli and carrying out effector responses that underlie inducible innate resistance. In Hydra, a simple metazoan that lacks mobile phagocytes or hemolymph, epithelial cells sense microbial infection by a LRR receptor and respond by synthesizing and secreting antimicrobial polypeptides (28). In plants that are even more distantly related to mammals, LRR receptors similarly recognize PAMPs and initiate complex and vigorous resistance responses (29). This conservation indicates that epithelial activation is an ancient system of defense in multicellular eukaryotes. In mammals, in which a complex array of leukocytes collaborate in host defense, epithelial cells engage in bidirectional signaling with leukocytes while retaining the ability to mount a vigorous defense of their own.
Architecture of Epithelial Surfaces
To understand the innate immune activation of epithelial cells, it is important to appreciate their microenvironments. Epithelial surfaces throughout the mammalian body are in constant or episodic contact with microbial pathogens. A variety of physical strategies are utilized to prevent microbial penetration of epithelial layers that would result in deep tissue or bloodstream infection. For example, the skin utilizes an impermeant outer layer of dead keratinized cells. The stomach utilizes acid that makes its lumen inhospitable to microbes and renders the proximal small bowel and interconnected pancreatic and biliary trees nearly sterile. The distal small bowel and the large bowel, however, are in constant proximity to a rich microbial flora (Fig. 2A), albeit separated by a thick mucus gel layer (30, 31). The lungs utilize a strategy of directed airflow to induce impaction and sedimentation, together with mucociliary and cough clearance, to rapidly remove aspirated microbes from the lower respiratory tract (32-34). The importance of mucociliary clearance in the lungs is indicated by the chronic inflammation that occurs in human subjects with ciliary dyskinesia (33, 35) and the progressive lethal lung inflammation that occurs in mice lacking the gel-forming mucin Muc5b (Chistopher M. Evans, personal communication). Other mucosal surfaces such as the mouth, eyes, and urinary tract employ both overlapping and additional strategies.
Figure 2. Architecture and Innate Immune Tone of Mucosal Surfaces.
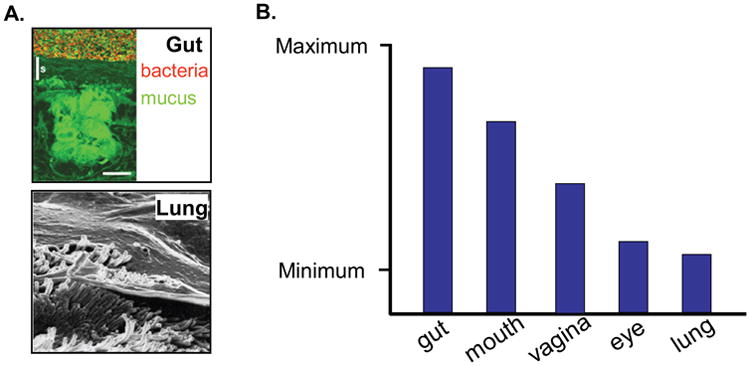
A. Architecture of Colon and Lung. The architecture of the mouse colon on the top, showing the epithelium with intracellular mucin (intense green), extracellular mucin (faint green), and overlying bacteria (red). The mucus gel layer is thick (up to 500 μm), and is adherent to the epithelium. From Johansson MEV, PNAS 2008, 105:15064. The architecture of the airway is illustrated on the bottom, showing a thin mucus layer (5 μm in distal airways to 50 μm in proximal airways) overlying a periciliary liquid layer ∼7 μm in depth. Aspirated microbes become entrapped in the mucus gel layer and are rapidly swept out of the lungs by ciliary action, keeping the lungs nearly sterile. B. Baseline Innate Immune Tone of Mucosal Surfaces. The epithelial resistance of mucosal surfaces of the mammalian body are postulated to be related to exposure to microbial products. The tone of the lung and gut are illustrated in relative terms with regard to each other and to the minimal and maximal points of the scale; the tone of all other surfaces is speculative.
These distinct epithelial microenvironments with markedly different baseline exposure to microbial products reveal differences in the baseline induction of innate immune defenses, with the most informative contrast being between the lungs that are nearly sterile and the distal bowel that is heavily colonized with microbes (Fig. 2B). In the lungs, innate immune activation (“tone”) is low, as indicated by the low level of microbial killing within the lungs of mice at baseline (9, 19, 20), or by unstimulated lung epithelial cells in vitro (9, 21). From this low baseline, innate resistance is highly inducible (Fig. 3). In the distal bowel, in contrast, innate immune tone is substantial at baseline, as indicated by rapid bacterial killing within an isolated ileal loop (36). Bacterial killing can be reduced by prior exposure of mice to antibiotics that reduce commensal microbes, and can be restored by exposure to TLR agonists. Thus, diverse epithelial cells appear to be capable of a range of innate immune tone determined by local microbial signals, and modulated by innate and adaptive immune cells (37). Consistent with the ability of epithelial cells to maintain tone, we have found no tachyphylaxis of resistance despite repetitive stimulation of the lungs (9, 20), similar to the tachyphylaxis of inflammation but not of resistance described by others (38).
Epithelial Sensing of Innate Stimuli
For the induction of microbial resistance, lung epithelial cells must sense stimuli directly from microbes or indirectly from non-epithelial host cells and extracellular molecules. Evidence exists that lung epithelial cells are highly responsive to both categories, with the relative importance of individual stimuli in inducing resistance in vivo against specific pathogens a subject of ongoing study.
Direct Sensing of Microbial Products
Jawed vertebrates have two distinct means of detecting the presence of pathogens, innate and adaptive immunity, distinguished fundamentally by the nature of their receptors. Adaptive immune recognition relies upon antigen receptors expressed by T and B lymphocytes that are encoded by somatically recombined gene segments, resulting in an immense library of receptors for precise epitopes. The clonal distribution of highly specific antigen receptors allows for expansion of pathogen-appropriate lymphocyte populations and for immunologic memory, but limits the number of pathogens that can be detected by individual cells and requires prior exposure to the pathogen (39). In contrast to the highly refined epitope-sensing of adaptive immune receptors, innate immune receptors rely on recognition of conserved molecular features common to multiple microorganisms (PAMPs). Germline-encoded pattern recognition receptors (PRRs) bind PAMPs, allowing recognition of a large number of different microorganisms, though the broad conservation of these structural motifs does not generally allow discernment of pathogenic from non-pathogenic microorganisms (40-44). PAMPs may be either surface associated or internal elements of microbes, and are best suited to inducing efficient innate responses when they are invariant across many species, critical to microbial metabolic or virulence processes, and not present in host products (39). Additionally, some PPRs identify host molecules that are expressed in response to infection or host molecules that have been modified in the course of infection, known as “danger signals” or damage-associated molecular patterns (DAMPs). PRRs can be soluble, bound to cell surface or endosomal membranes, or cytosolic in distribution. Recognition of PAMPs and DAMPs by PRRs activates intracellular signaling cascades, leading to the expression of effector molecules involved in microbial defense, inflammation and modulation of adaptive immunity (45). The past two decades have witnessed the identification of several distinct classes of PRRs.
Toll-Like Receptors (TLRs)
TLRs were the first class of innate immune receptors identified, and remain the best characterized. TLRs are highly conserved class I transmembrane proteins, consisting of an ectodomain with multiple leucine-rich repeats (LRR) for pattern recognition, a single membrane-spanning α-helix, and a Toll/interleukin-1 receptor (TIR) domain for intracellular signaling. The structure and function of TLRs are described in detail in a number of recent reviews (3, 43, 46, 47). Upon ligand binding, signal transduction occurs via receptor-specific recruitment of cytosolic TIR adaptor protein combinations. In concert with one or more of the four others, MyD88 is involved in more TLR signaling than any other TIR adaptors (42-44, 48, 49). The MyD88-independent signaling events observed from TLR3 and TLR4 utilize the TIR adaptor Trif, with or without TRAM (43, 48, 49). Numerous recent reviews describe the current understanding of TLR downstream signaling (3, 43, 46, 47).
The primary ligand for TLR4 and its co-receptor CD14 is a complex of the soluble host protein MD2 with the lipid A moiety of bacterial lipopolysaccharide (LPS), allowing detection of Gram negative pathogens. Many Gram positive bacteria, parasites, and some Gram negative bacteria can be recognized by TLR2-dependent binding of lipopeptides, such as peptidoglycan, lipotechoic acid, and atypical LPS. TLR2 generally functions as a heterodimer with TLR1 or TLR6, with TLR2/1 recognizing triacylated lipopeptides (e.g., Pam3CSK4) and TLR2/6 recognizing diacylated lipopeptides (e.g., Pam2CSK4, MALP2). Fungal zymosan has been described to be a ligand for TLR2/6, as well. A highly conserved motif of flagellin that spans many bacterial species is recognized with high affinity by a well defined TLR5 binding site. Four TLRs recognize microbial nucleic acids. TLR3 recognizes double stranded RNA, and can be stimulated by synthetic mimetic copolymers, such as poly inosine:poly cytosine (poly I:C). TLRs 7 and 8 recognize U-rich (i.e., non-mammalian) single stranded RNA, as well as imidazoquinolones such as imiquimod. TLR9 detects DNA with unmethylated CpG motifs, which differ from mammalian DNA that is typically methylated. A number of host danger signals, such as heat shock proteins, are also protein ligands for TLRs (3, 43, 46, 47).
Insight into the roles of TLR in defense against pneumonia is provided by experiments in TLR-deficient mice. Mice spontaneously deficient in TLR4 show increased susceptibility to H. influenzae and E. coli pneumonia associated with impaired pathogen clearance (50, 51). Dual deficiency in TLR4 and CD14 was also found to increase susceptibility to RSV (52), consistent with the observation that TLR4 is a co-receptor for RSV fusion protein. TLR5 deficient mice have increased susceptibility to lung pathogens including Legionella pneumophila (53). Interestingly, mice deficient in both TLR2 and TLR4 do not demonstrate hypersusceptibility to P. aeruginosa (54), even though mutations of pseudomonal flagellin that prevent TLR5 binding result in impaired bacterial clearance and host survival (55). Surprisingly, TLR3 deficiency was reported to confer a survival advantage in an influenza virus pneumonia model (56), presumably through prevention of an excessive host response. However, the finding that intranasal pretreatment with TLR3 agonists protects against influenza pneumonia highlights the requirement for precise regulation of TLR-dependent responses in microbial defense (57).
It has been apparent for nearly a decade that microbial products could stimulate innate responses from the respiratory epithelium, and it has been suspected that TLRs contributed to those responses since they were first identified in mammals (4). While there are 13 known TLRs in humans, most is known about TLRs 1-9 and their murine orthologs (3, 47). PCR investigations of primary cells and immortalized cell lines indicate that TLRs 1-9 are all expressed by human and mouse lung epithelial cells (3, 5, 58-60). Cultured respiratory epithelial cells respond to stimulation with TLR agonists by expression of proinflammatory and antimicrobial mediators (60-62). In vivo, LPS has been administered intranasally and by aerosol to protect against bacterial and fungal lung infections, either by enhancing innate defenses or by attenuating lung injury associated with infection (10, 17, 19). Intratracheal and intraperitoneal administration of CpG ODNs (TLR9 ligand) enhances survival of lung infection by a number of pathogens, including M. avium and K. pneumoniae (13, 16). Treatment of mice with the TLR2/6 agonist MALP-2 induces cytokine production, reduces the pathogen burden, and enhances host survival after challenge with S. pneumoniae (18). Mice pretreated with TLR3-stimulating poly I:C or liposomal preparations of TLR9-stimulating CpG ODNs display enhanced survival after challenge with several strains of influenza (57).
Given increasing evidence of cooperative signaling by PRRs (63-65), synergistic combinations of TLR agonists or combinations of TLR agonists and ligands for other PRRs may provide even greater protection than single ligands alone. Consistent we this, we have found that certain combinations of synthetic TLR agonists greatly outperform individual ligands in terms of both induced pathogen killing and enhanced host survival (our unpublished results). Similarly, by exposing mice to multiple TLR ligands in an aerosolized bacterial lysate, we have found robust survival benefits across a broad spectrum of pathogens (9, 19, 20). The importance of TLRs in general and MyD88 in particular to this response is demonstrated by the complete loss of protection when the aerosolized bacterial lysate is delivered to MyD88-deficient mice, but not to Trif-deficient mice (our unpublished results). The importance of epithelial MyD88 is indicated by the insensitivity of MyD88-deficient mice transplanted with wildtype bone marrow to intranasal LPS (53, 66), and their impaired cytokine production and decreased survival when challenged with P. aeruginosa in the lungs (23).
NOD-Like Receptors (NLRs)
The NLR family is defined by proteins that share a C-terminal LRR domain that interacts with PAMPs, a central nucleotide oligomerization domain (NOD), and one of three N-terminal signaling domains (67). Humans express at least 23 of these proteins, with most apparently restricted to leukocytes, but the best studied NLRs, NOD1 and NOD2, are both expressed by lung epithelial cells (62, 67). Unlike membrane-associated TLRs, NLRs are cytosolic in distribution. NOD1 recognizes γ-D-glutamyl-meso-diaminopimelic acid present in the peptidoglycan of Gram negative and some Gram positive bacteria (67), whereas NOD2 binds the muramyl dipeptide universally present in bacterial peptidoglycan (68). Activation of signaling via NOD1 or NOD2 results in MAPK and NF-κB-dependent production of proinflammatory mediators, though the details of this cascade are less well characterized than for the TLRs. Mice deficient in these NLRs display increased susceptibility to gastrointestinal bacterial infections, including those caused by H. pylori and L. monocytogenes (69-71). In the lungs, NLRs are critical to the host response to S. pneumoniae, P. aeruginosa, M. catarrhalis, C. pneumoniae, and M. tuberculosis (5, 72-74).
Another important subfamily of NLRs are those that induce activation of the inflammasome, a molecular complex that activates caspases to convert pro-interleukin (IL)-1β and pro-IL-18 (and, possibly pro-IL-33) into their mature forms. The NLRs known to contribute to this response are NALP1, NALP2, NALP3, Ipaf and NAIP (75). These proteins primarily recognize danger signals, including host inflammatory mediators and crystals, but also detect product of microbial pathogens. For example, the NALP3 inflammasome can be activated by MDP, viral DNA, and bacterial toxins (75), while Ipaf appears to sense bacterial flagellin (76). The observation that IL-1β mRNA increases in the lungs of mice by almost 100-fold after treatment with an aerosolized bacterial lysate to induce resistance suggests a role for inflammasome activation (9). Furthermore, NLRs demonstrate synergistic signaling with TLRs (69).
RIG-I-Like Receptors (RLRs)
The RLRs are cytosolic PRRs that have been demonstrated to be involved in TLR-independent sensing of viruses, and the associated production of type I interferons (77). The RLR family is presently composed of two members – retinoic acid induced gene-1 (RIG-I) and melanoma differentiation associated gene 5 (MDA5). RIG-I detects noncapped 5′-triphosphate RNA (78), and appears essential to detecting ssRNA viruses (77). RIG-I-deficient mice demonstrate impaired antiviral responses and increased susceptibility to paramyxoviruses, influenza, varicella, and Japanese encephalitis virus (79). The primary ligand for MDA5 is dsRNA (78), and recent work has shown that it can detect poly I:C in a TLR-independent manner (79). MDA5 deficiency increases susceptibility to several picornaviruses (79). Consequently, the therapeutic activation of these RLRs may promote antiviral defense, though their role in induced epithelial resistance has not been addressed.
Additional Innate Receptors
Besides these innate immune receptor families, additional cellular products participate in microbial recognition. For example, the glycosphingolipid lactosylceramide is found on the apical surface of lung epithelial cells and detects fungal β-glucans (80, 81). Other PRRs, such as class A scavenger receptors (e.g., MARCO and SR-AI/II) appear to participate in lung defense, but their expression and function in lung epithelium are unresolved.
Indirect Sensing by Host Signaling
Besides receptors for microbial products themselves, epithelial cells possess mechanisms for detecting the presence of microbes indirectly by sensing the release of molecular constituents from injured neighboring cells (e.g., ATP, adenosine, urate, HMGB1), the activation of extracellular fluid phase proteins (e.g., complement and coagulation cascades), the degradation of extracellular matrix macromolecules (e.g., hyaluronan, elastin), and the secretion of inflammatory signals from leukocytes and other parenchymal cells (e.g., cytokines, eicosanoids).
Host Cell Products
In normal lung function, ATP is released by mechanical stretch into lung lining fluid where it performs important homeostatic functions such as regulation of secretion, surface liquid depth, and ciliary beat frequency (82). However, ATP can also be released in large quantities by cell injury, and in this setting ATP and its metabolites are powerful mediators of inflammation. In the gut, ATP that is mostly released from microbes drives TH17 differentiation in the lamina propria (83), and ATP activates dendritic cells in asthmatic airway inflammation (84). Whether epithelial resistance is induced by ATP together with leukocyte recruitment and activation is not known. Adenosine, generated from ATP and released from cells directly, is an important mediator of inflammation in the lungs as indicated by the lethal pulmonary phenotype of adenosine deaminase null mice and the dependence of allergic and other airway inflammatory disorders upon adenosine (85). Adenosine signals through four G-protein coupled receptors that have both pro-inflammatory and anti-inflammatory properties, are expressed on airway epithelial cells as well as immune cells, and interact with PRR signaling in activating leukocytes (85, 86). Adenosine signaling modulates survival in mouse models of bacterial infection, though whether this is through induced resistance or attenuation of excessive inflammation in sepsis is not clear (86). Urate and calcium pyrophosphate are products of purine metabolism that activate inflammation through the NALP3 inflammasone. NALP3 activation is important for resistance of mice to fungal infection (87), and urate contributes to inflammation during bleomycin-induced lung injury through NALP3 (88), but whether urate contributes to lung resistance to microbial infection is unknown. Besides the purines, multiple cellular macromolecules can signal to neighboring cells when released during injury, and have been termed “alarmins” (89). These include heat shock proteins, the chromatin-associated protein HMGB1, members of the calcium-binding S100 family, and antimicrobial peptides such as cathelicidin. These have been examined mostly for inflammatory rather than resistance properties, and data on their functions in epithelial cells are scant.
Proteolytic Cascades (Complement, Coagulation, Kinins)
Besides effector functions in microbial lysis and opsonization (see below), complement promotes resistance by signaling to leukocytes and parenchymal cells. Airway epithelial cells express receptors for the C3a and C5a anaphylatoxins (90), and stimulation by C3a is required for robust epithelial mucous metaplasia in allergic airway inflammation (91, 92). Allergic inflammation is thought to represent an antiparasitic defense (32), and whether C3a or C5a signaling to epithelial cells is similarly required for full induction of antimicrobial resistance is not yet know. The coagulation cascade and platelets are also activated during infection, and interact bidirectionally with the complement system (93, 94). Research on interactions of the coagulation system with lung epithelial cells has focused primarily on mesothelial cells of the pleural space in fibrinolysis, permeability changes of the alveolar epithelium in diffuse lung injury, and occlusion of distal airways in asthma (95-97), while the role of the coagulation system in inducible resistance is mostly unknown. The kallikrein-kinin cascade is co-activated with the complement and coagulation cascades, generating potent inflammatory mediators whose role in inducible resistance is yet unknown, and activating cathelicidin (98) that likely participates in inducible resistance (see below).
Extracellular Matrix Products
The hydrolysis of extracellular matrix components releases fragments with inflammatory signaling properties. Prominent among these in the lungs are the glycosaminoglycan hyaluronan that releases fragments that activate TLR4 (99), and fibronectin that binds latent TGF-β (100), though their roles in microbial resistance within the lungs are not known. In addition to signaling themselves, matrix macromolecules provide nucleation sites for the growth of urate and calcium pyrophosphate crystals (see above), and they are subject to non-enzymatic glycation that is recognized by RAGE that has inflammatory activity both alone and in partnership with TLRs (101).
Cytokines and Other Inflammatory Mediators
Cytokines are generated to some degree by all mammalian cells, but most prominently by leukocytes. A subset of leukocyte-generated cytokines induces resistance of epithelial cells to infection (102). First described among these were the Type I interferons that were found in the 1950's to induce epithelial resistance to viral infection (103, 104). In the 1960's, Type II interferon (IFN-γ) was discovered, and more recently Type III interferons have also been found to be important in epithelial resistance. Besides viruses, interferons also induce resistance to bacterial and fungal pathogens. In the lungs, resistance to F. tularensis induced by prior administration of a TLR4 agonist was highly dependent upon IFN-γ (15). Other cytokines that induce resistance in epithelial cells are IL-22 and IL-17. IL-22 is produced both by lymphocytes and by NK cells, with the latter providing an innate source for mucosal immunity (105, 106). IL-22 acts primarily on epithelial cells, markedly increasing the production of antimicrobial proteins (21, 105, 107). Importantly, IL-22 induces microbial killing by mouse airway epithelial cells in vitro, and this depends to a large degree upon lipocalin-2 (21). IL-17 plays important roles in mucosal defense against extracellular lung pathogens (108, 109), and upregulates the production of antimicrobial proteins by lung epithelial cells (107, 110). IL-22 and IL-17 can be co-expressed and can cooperatively enhance the expression of antimicrobial proteins and cytokines by keratinocytes and airway epithelial cells (21, 107). Many other signaling molecules such as eicosanoids, biogenic amines, and proteases also mediate inflammatory signaling, but their roles in induced resistance of epithelial cells are less well defined than those of cytokines.
Epithelial Effector Mechanisms of Inducible Resistance
Resistance of a multicellular eukaryotic host to infection can be due either to prevention of microbial entry or induction of microbial killing. Epithelial cells participate in both effector mechanisms.
Barrier Function
Epithelia throughout the body serve as mechanical barriers to microbial entry. However, microbes have developed numerous strategies for crossing this barrier by passing between cells, entering and passing through cells, and simply killing cells to eliminate the barrier. Conversely, since it is comprised of living cells, the epithelial barrier is capable of plasticity in its ability to resist microbial penetration. Indeed, epithelial barrier functions are modulated both by pathogens and as part of the host response, presenting a dynamic situation during infection. The different strategies utilized by microbial pathogens to breach epithelial barriers have been discussed in several recent reviews (98, 111-114). Here we focus on inducible barrier function.
Paracellular Transit
Cell-cell junctions and cell-matrix adhesions are critical structures for maintaining the epithelial barrier. It is not surprising that these structures as well as the associated adaptors and regulators are among the most commonly targeted host factors by microbial pathogens to either disrupt the barrier or gain entry into the host cells. For example, transmigration of leukocytes to mucosal surfaces in response to microbial pathogens requires reorganization of the epithelial junctions. In the lungs, this process involves TLR2 signaling resulting in calcium fluxes that activate calpains, which are cysteine proteases that cleave epithelial junctional proteins (115). Conversely, migration of S. pneumoniae and H. influenzae across polarized respiratory epithelial cells was shown to be accompanied by disruption of the epithelial barrier mediated by TLR2, p38 MAPK and TGF-β signaling (116). Whether these two phenomena are connected is not known, though it seems that the immune signaling activated by these pathogens promotes their migration across the epithelial barrier at the same it facilitates leukocyte transmigration. In intestinal epithelial cells, TLR2 signaling protects tight junction assembly (117) and gap junction intercellular communication against damage associated with inflammation (118). There is also evidence that E. hirae cell wall fractions such as lipotechoic acid protects against intestinal barrier impairment by regulation of tight junctions via TLR2 signaling (119).
Transcellular Transit
Pathogens at a variety of mucosal surfaces enter epithelial cells to invade underlying tissues (22, 98, 112). This pathogenic mechanism is counteracted by epithelial exfoliation. In the distal mammalian gut that is continuously exposed to microbes (Fig. 2A), the epithelium is short-lived (turnover time 5 days), and anoikis likely plays a role in preventing penetration by bacteria that have entered epithelial cells, and is antagonized by pathogens (120). In the lungs that are only intermittently exposed to microbes (Fig. 2B), the epithelium is long-lived (turnover time 180 days), and epithelial shedding only occurs during infection or injury (121). In the bladder, uropathogenic E. coli invade epithelial cells and establish intracellular reservoirs that serve as sources for recurrent infections. Normal turnover of epithelial cells is slow (∼ 40 weeks), and stimulating epithelial turnover with protamine sulfate can eradicate infection (122). Further, TLR4 signaling activated by bacterial LPS suppresses the invasion of uropathogenic type 1 fimbriated E. coli and K. pneumoniae into bladder epithelial cells (123), reducing penetration of the epithelium by these pathogens. In the lungs, B. anthracis presents an interesting case. Spores of B. anthracis are capable of crossing the lung epithelium by transcytosis without causing apparent disruption to the epithelium or eliciting a strong inflammatory response (124, 125). This capability may be a reason for the lack of symptoms during early stages of inhalational anthrax.
Epithelial Killing
Lung pathogens release a wide array of toxins and exoenzymes capable of killing lung epithelial cells and allowing microbial penetration (22, 98, 112). Further, viral injury of lung epithelium allows bacterial penetration that appears to be an important cause of mortality (126). Innate immune stimulation activates NF-κB and kinase cascades that are anti-apoptic and proliferative (3, 22), partially counteracting toxic effects of pathogens. When breaches in the airway epithlium do occur, the growth factor heregulin that is normally sequestered on the apical surface gains access to its receptors on the basolateral surface of the remaining cells, initiating a tissue repair response that includes changes in cell shape to rapidly cover the surface, proliferation to reestablish cell number, and promotion of antimicrobial defenses (127).
In summary, the strategies utilized by microbial pathogens to breach epithelial barrier functions are varied, with modulation of epithelial barrier functions having effects on both the capacity of the host to clear microbes and the ability of pathogens to invade the host.
Microbial Killing
Another important mechanism by which the lung epithelium promotes host survival is through pathogen killing. One means to achieve this is through the engagement of professional immune cells. In response to infection or injury, epithelial cells elaborate abundant proinflammatory and leukocyte chemotactic cytokines, including TNF, IL-1β, IL-6, IL-8 (or its murine orthologs), GM-CSF, CXCL5, and leukotrienes, to recruit neutrophils, monocytes, macrophages, and NK cells (128-130). They also express inducible adhesion factors that facilitate leukocyte influx (34, 129). Beyond sounding an alert, the epithelium also sculpts the nature of the adaptive immune response. For example, following exposure to certain viruses, helminthes, allergens, and TLR agonists, the lung epithelium produces thymic stromal lymphopeoitin (TSLP), IL-25 (also known as IL-17E), GM-CSF, IL-1β, IL-25, IL-33, promoting selective dendritic cell recruitment and TH2 deviation of the adaptive immune response (130, 131). Besides leukocytes, epithelial cells secrete microbicidal products into the airway lining fluid, and secretion of these products can be increased in response to infection or therapeutic stimulation. These epithelial effectors primarily limit pathogen survival through disruption of the cell walls, sequestration of iron and other nutrients, and by providing decoy targets for essential microbial metabolic and pathogenic processes (5, 128, 132, 133).
Small Antimicrobial Peptides
Predominant among the therapeutically inducible bacteriostatic and bactericidal products of the epithelium are the small cationic antimicrobial peptides, a diverse family of amphipathic, gene-encoded immune effectors. Hundreds of these peptides have been identified in eukaryotes, with tremendous species-specific variation, but human lung epithelium primarily expresses members of just two groups: defensins and cathelicidins (5, 134-140).
Defensins are defined by unique cysteine motifs resulting in three disulfide bonds, and are subdivided into categories based on tertiary structure (α, β and θ). α- and β-defensins are both expressed by neutrophils, but only β-defensins are expressed by the lung epithelium. Human β-defensin 1 (HBD-1) is constitutively elaborated into the airway lining fluid, and is be further induced by inflammatory stimuli in mechanically ventilated newborns (141, 142). HBD-2, -3, and -4 do not appear to be expressed at baseline, but are induced by epithelial cell exposure to pathogens, TLR agonists, or inflammatory cytokines (140, 141, 143, 144). Mice express numerous β-defensins in the lungs, many of them inducible by bacterial and viral infection, and they demonstrate broad antimicrobial activity, including against influenza virus and fungi (145, 146). In our mouse model of stimulation with an aerosolized NTHi lysate, we observe induced expression of several β-defensins (9).
Cathelicidins, like many antimicrobial effectors, are expressed as propeptides that require cleavage of an N-terminal cathelin domain to gain antimicrobial activity. Humans express only one cathelicidin propeptide (CAMP or hCAP-18) which is cleaved to the active antimicrobial peptide, LL-37 (135). Lung epithelial LL-37 elaboration can be stimulated by TLR agonists, and production is at least partially vitamin D dependent (147). In the mouse, lung transcripts roughly double after treatment with NTHi lysate (9). There is abundant evidence that cathelicidins promote pathogen clearance in pneumonia (138, 148), so induced production, activation and secretion may contribute to induced resistance.
Small cationic antimicrobial peptides exert direct antimicrobial effects on Gram-positive, Gram-negative, fungal and viral pathogens (140). The mechanisms of pathogen killing remain incompletely elucidated, though most literature suggests that the primary mechanism relates to microbial permeabilization (132, 149). This may occur by creation of membrane-spanning pores or by detergent-like disruption of pathogen membranes. In fact, antimicrobial peptides may use different mechanisms on different pathogen membranes. Further, there are antimicrobial peptides that kill bacteria without detectable lysis, apparently breaching the membrane to bind critical metabolic targets (139). The importance of pathogen killing by small cationic peptides to host defense is demonstrated by the impaired pathogen clearance from the lungs and decreased survival of mutant mice deficient in cathelicidin or defensin when challenged with a number of bacterial species (128, 147, 150). Further substantiating their importance in lung defense, forced overexpression of LL37 in the airway epithelium of mice enhances protection against bacterial pneumonia and sepsis (151, 152). Many antimicrobial peptides demonstrate pathogen killing effects against bacteria, fungi, protozoa and some virus when tested in vitro (139), and virtually all peptides with a net positive charge and a few hydrophobic residues show some antimicrobial activity in non-physiologic dilute media (137). However, in vivo testing reveals that mammalian antimicrobial peptides have evolved enhanced activity against the pathogens most often encountered in the niche where they are expressed.
Stimulated epithelial production of antimicrobial peptides are also important to lung defenses through their immunomodulatory properties, complementing the actions of epithelium-derived cytokines. For example, hBD2 is chemotactic for mast cells, whereas hBD3 and 4 recruit monocytes and macrophages (153-156). Similarly, LL37 recruits neutrophils, monocytes, mast cells and T cells, but not dendritic cells (157, 158). The antimicrobial peptides also exert indirect chemotactic effects, as they enhance secretion of proinflammatory and chemotactic cytokines from local leukocytes (139, 155). Conversely, as shown in keratinocyte and leukocyte models, LL37 attenuates TLR4-dependent cytokine secretion, indicating that antimicrobial peptide also play a role in immune regulation.
Large Antimicrobial Proteins
Besides small cationic antimicrobial peptides, several larger proteins elaborated by the epithelium promote resistance, as recently reviewed (102). Among these, lysozyme is expressed in the greatest abundance. Lysozyme was among the first antimicrobial effectors identified in pulmonary secretions and hydrolyzes β1-4 glycosidic bonds in peptidiglycan, disrupting Gram-positive bacteria (159). Increased lysozyme is measureable in lung lining fluid after treatment of mice with an aerosolized NTHi lysate (19). Similarly, lactoferrin is induced by many infectious and inflammatory stimuli, including NTHi lysate (9, 19). Lactoferrin has been presumed to exert its antimicrobial effects through sequestration of iron from pathogens, though it appears to be directly bactericidal, as well (5, 159).
Another group of inducible epithelial antimicrobial products is the surfactant collectins. Collectins are comprised of an N-terminal cystein-rich domain, a collagen domain, a coiled coil neck domain, and a carbohydrate recognition domain (CRD, or C-type lectin domain) (160). As innate immune molecules, they function as soluble PRRs, with surfactant proteins A (SpA) and D (SpD) the best characterized. The surfactant collectins recognize conserved sugar patterns present on respiratory pathogens. Collectin binding of microbes can result in pathogen opsonization and neutralization through agglutination, while modulating dendritic and T cell responses (161, 162). We find SpD greatly increased in lung lining fluid following treatment with NTHi lysate (19). However, in some cases, the binding of collectins to microbes may actually promote adhesion and facilitate infection, as in pneumocystis pneumonia (163). Thus, their therapeutic augmentation may have some practical limitation.
Lipocalin-2 is an epithelium-expressed protein that binds iron siderophores, is important in defense against K. pneumoniae pneumonia (164, 165), and appears to be particularly important to induced resistance. Lipocalin-2 is markedly upregulated by IL-22, and its deficiency greatly reduces in vitro microbial killing by mouse tracheal epithelial cells after stimulation with IL-22 (21). We find increased lipocalin-2 gene expression together with increased protein in lung lavage fluid after stimulation of mouse lungs with aerosolized NTHi lysate (9, 19). S100 protein family members, including calgranulins A and B, are expressed by airway epithelial cells, and have been found to exert antimicrobial effects on a host of respiratory pathogens in a manner independent of their calcium binding domains (5, 102). These are also increased by NTHi lysate (9, 19). A family of proteins known as palate-lung-nasal-clone (PLUNC), comprised of short (SPLUNC1 and 2) and long (LPLUNC) members, shares features with many of the other antimicrobial effectors. There is evidence they contribute to lung epithelial defenses (166, 167), though we do not find them increased by NTHi lysate aerosol. In addition to its leukocyte chemotactic activity, CCL20 (also known as LARC or MIP-3α) shares structural homology with β-defensins and has intrinsic antibacterial activity against Gram negative pathogens (168). CCL20 mRNA increases in the lungs by over 200-fold after NTHi lysate treatment (9). Secretory leukocyte proteinase inhibitor (SLPI) and elafin are constitutively expressed in the airways, and are further induced from the epithelium in response to infection. While they appear to limit host injury following infection through protease inhibition, both also exert direct toxic effects on bacterial species (5), and we observe the induction of SLPI and numerous elafin-related protease inhibitors after treatment with NTHi lysate (9, 19). Complement promotes microbial clearance by inducing lysis through the membrane attack complex and by opsonization, as well as by signaling to leukocytes and parenchymal cells (see above). Local production of complement may be particularly important (169), and complement gene expression is increased during induced resistance of the lungs (9, 19). The significance of complement activation is suggested by the numerous mechanisms employed by pathogens to evade its effects (170).
Reactive Oxygen Species
The airway epithelium also generates reactive oxygen species at bactericidal concentrations in response to infection or therapeutic stimulation (171-173). The hydrogen peroxide source for reactive oxygen species is primarily generated in the airway via the dual NADPH oxidase/peroxidase (Duox) system, with Duox 1 constitutively expressed and Duox 2 induced by inflammatory cytokines and infection (174). The bactericidal effects of hydrogen peroxide are greatly enhanced by the presence of peroxidases, and the airway epithelium produces abundant lactoperoxidase (175, 176). Nitric oxide is also produced both constitutively and inducibly in the lung epithelium, and contributes importantly to microbial killing, though the relative importance of epithelial versus macrophage production within the lungs is not known (177).
Just as the respiratory epithelium exhibits synergistic PRR signaling, antimicrobial effector molecules expressed by the lungs display synergistic killing. Subinhibitory concentrations of lysozyme and lactoferrin both enhance the killing of bacteria by LL37 and hBD2 (5, 139); combinations of lactoferrin, SLPI, and lysozyme show synergistic killing (178); and antimicrobial proteins interact with reactive oxygen species, as above.
Microbial Evasion of Epithelial Resistance
Not surprisingly, in view of the efficacy of induced epithelial resistance in suppressing microbial colonization and invasion, pathogens have evolved a wide variety of mechanisms to subvert induced resistance. Many of the signaling pathways and effector mechanisms described above are targeted by pathogens, and this subject has recently been reviewed (98, 170, 179, 180).
Diagnostic and Therapeutic Implications
From a diagnostic standpoint, epithelial innate immune mechanisms are increasingly recognized as contributing to inflammatory diseases of the gastrointestinal tract, skin and airways. The contributions of these mechanisms to resistance to microbial infection are less well understood, though spontaneously occurring mutations in humans dovetail informatively with targeted mutations in mice. For example, deficiency of IFN-γ signaling leads to increased susceptibility to mycobacterial infections of the lungs whereas deficiencies of Type I or III IFN signaling lead to increased susceptibility to viral infections (181), and deficiency of MyD88 leads to pyogenic bacterial infections (182). The contribution of the epithelium to these susceptibilities is being uncovered by hematopoietic stem cell transplantation in humans and tissue-specific studies in mice (183).
From a therapeutic standpoint, innate immunity is being targeted to attenuate inflammatory diseases, provide adjuvant activity in vaccines, deviate the immune system in allergic diseases, and prevent tumor promotion (184, 185). The lung epithelia of human subjects have not been stimulated therapeutically to prevent or treat infection, though topical TLR7/8 agonists are currently in use for treatment of viral and parasitic infections of the skin and genitourinary tract, and TLR 9 agonists are being developed for treatment of hepatitis C (184, 185). The rapid, broad and high level of resistance that can be transiently induced in the lungs of mice suggests that human populations transiently at high risk of pneumonia might benefit from treatment. Such situations include cancer patients being treated with myeloablative chemotherapy, the general population during an epidemic for which a vaccine is not available, or a bioterror attack in which the agent is not known or the population cannot be fully protected with existing therapies (186).
Conclusion
The efficacy of the lung epithelium in microbial killing has been neglected relative to its roles in signaling to leukocytes and acting as a mechanical barrier. This neglect is due in part to the requirement for stimulation before the antimicrobial capabilities of the lung epithelium become apparent, and in part to being overshadowed by the dazzling array of antimicrobial activities displayed by the wide variety of mammalian leukocytes. Better understanding of inducible lung epithelial innate resistance is likely to lead to insight into the variable individual susceptibility to infection and the ability to manipulate resistance therapeutically.
Summary Points.
Inducible innate immune resistance to microbial infection in the lungs is mediated primarily by epithelial cells.
Lung epithelial cells are conditional pathogen killing machines that require stimulation for efficient effector function.
Lung epithelial cells are capable both of directly sensing microbes and of responding to signals from leukocytes and other host cells to increase resistance.
While the surface area of the lungs presents a large target for microbial invasion, lung epithelial cells that are closely apposed to deposited pathogens are ideally positioned for microbial killing when activated.
The full diagnostic and therapeutic implications of the inducibility of innate immunity in lung epithelium remain to be determined.
Future Issues.
The necessary and sufficient pathways for epithelial sensing of innate immune stimuli to confer resistance against specific pathogens remain to be fully elucidated.
The necessary and sufficient epithelial effector mechanisms in the mediation of resistance to specific pathogens remain to be fully elucidated.
The relative importance of direct pathogen sensing by epithelial cells and positive and negative signaling from host cells and extracellular macromolecules in the achievement of homeostasis remain to be determined.
Whether stimulation of innate resistance of lung epithelial cells can have clinical therapeutic value remains to be determined.
Acknowledgments
The authors thank their many colleagues who contributed directly or indirectly to this work, and apologize to those whose work we overlooked or couldn't cite due to space limitations. Research in the Evans laboratory was supported by a NIH grant RR02419 and a Physician-Scientist Award from the M. D. Anderson Cancer Center; research in the Xu laboratory was supported by NIH grant AI061555 to Y. Xu, a career development award to Y. Xu through the Region VI Center for Biodefense and Emerging Infections, NIH U54 AI057156 (to D. Walker), start-up funds from Texas A&M Health Science Center and an award from the Hamill Foundation; research in the Dickey laboratory was supported by grants from the NIH (HL72984, HL094848, AI82226, RR02419), the Cystic Fibrosis Foundation (08G0), and the George and Barbara Bush Endowment for Innovative Cancer Research.
Common Acronyms
- PAMP
pathogen associated molecular pattern
- PRR
pattern recognition receptor
- NTHi
non-typeable H. influenzae
- TLR
Toll-like receptor
- LPS
lipopolysaccharide
Footnotes
Disclosure Statement: S.E.E., M.J.T, and B.F.D. own stock in Pulmotect, Inc., which is developing methods for stimulating innate immunity within the lungs to prevent and treat pneumonia. B.F.D serves on the Board of Pulmotect, Inc. Y.X. does not have any financial interest in the subject matter discussed in the manuscript.
Contributor Information
Scott E. Evans, Email: sevans@mdanderson.org.
Yi Xu, Email: yxu@ibt.tamhsc.edu.
Michael J. Tuvim, Email: mtuvin@mdanderson.org.
Burton F. Dickey, Email: bdickey@mdanderson.org.
References
- 1.Ferrandon D, Imler JL, Hetru C, Hoffmann JA. The Drosophila systemic immune response: sensing and signalling during bacterial and fungal infections. Nat Rev Immunol. 2007;7:862–74. doi: 10.1038/nri2194. [DOI] [PubMed] [Google Scholar]
- 2.Lemaitre B. The road to Toll. Nat Rev Immunol. 2004;4:521–7. doi: 10.1038/nri1390. [DOI] [PubMed] [Google Scholar]
- 3.Gay NJ, Gangloff M. Structure and function of Toll receptors and their ligands. Annu Rev Biochem. 2007;76:141–65. doi: 10.1146/annurev.biochem.76.060305.151318. [DOI] [PubMed] [Google Scholar]
- 4.Diamond G, Legarda D, Ryan LK. The innate immune response of the respiratory epithelium. Immunol Rev. 2000;173:27–38. doi: 10.1034/j.1600-065x.2000.917304.x. [DOI] [PubMed] [Google Scholar]
- 5.Bartlett JA, Fischer AJ, McCray PB., Jr Innate immune functions of the airway epithelium. Contrib Microbiol. 2008;15:147–63. doi: 10.1159/000136349. [DOI] [PubMed] [Google Scholar]
- 6.Rogan MP, Geraghty P, Greene CM, O'Neill SJ, Taggart CC, McElvaney NG. Antimicrobial proteins and polypeptides in pulmonary innate defence. Respir Res. 2006;7:29. doi: 10.1186/1465-9921-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ganz T. Epithelia: not just physical barriers. Proc Natl Acad Sci U S A. 2002;99:3357–8. doi: 10.1073/pnas.072073199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Travis SM, Singh PK, Welsh MJ. Antimicrobial peptides and proteins in the innate defense of the airway surface. Curr Opin Immunol. 2001;13:89–95. doi: 10.1016/s0952-7915(00)00187-4. [DOI] [PubMed] [Google Scholar]
- 9.Evans SE, Scott BL, Clement CG, Pawlik J, Bowden MG, et al. Stimulation of lung innate immunity protects mice broadly against bacterial and fungal pneumonia. Am J Respir Cell Molec Biol 2009 ePub ahead of print. [Google Scholar]
- 10.Jean D, Rezaiguia-Delclaux S, Delacourt C, Leclercq R, Lafuma C, et al. Protective effect of endotoxin instillation on subsequent bacteria-induced acute lung injury in rats. Am J Respir Crit Care Med. 1998;158:1702–8. doi: 10.1164/ajrccm.158.6.9709122. [DOI] [PubMed] [Google Scholar]
- 11.Cluff CW, Baldridge JR, Stover AG, Evans JT, Johnson DA, et al. Synthetic toll-like receptor 4 agonists stimulate innate resistance to infectious challenge. Infect Immun. 2005;73:3044–52. doi: 10.1128/IAI.73.5.3044-3052.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee PH, Ohtake T, Zaiou M, Murakami M, Rudisill JA, et al. Expression of an additional cathelicidin antimicrobial peptide protects against bacterial skin infection. Proc Natl Acad Sci U S A. 2005;102:3750–5. doi: 10.1073/pnas.0500268102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng JC, Moore TA, Newstead MW, Zeng X, Krieg AM, Standiford TJ. CpG oligodeoxynucleotides stimulate protective innate immunity against pulmonary Klebsiella infection. J Immunol. 2004;173:5148–55. doi: 10.4049/jimmunol.173.8.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karaolis DK, Newstead MW, Zeng X, Hyodo M, Hayakawa Y, et al. Cyclic di-GMP stimulates protective innate immunity in bacterial pneumonia. Infect Immun. 2007;75:4942–50. doi: 10.1128/IAI.01762-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lembo A, Pelletier M, Iyer R, Timko M, Dudda JC, et al. Administration of a synthetic TLR4 agonist protects mice from pneumonic tularemia. J Immunol. 2008;180:7574–81. doi: 10.4049/jimmunol.180.11.7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Standiford TJ, Deng JC. Immunomodulation for the prevention and treatment of lung infections. Semin Respir Crit Care Med. 2004;25:95–108. doi: 10.1055/s-2004-822309. [DOI] [PubMed] [Google Scholar]
- 17.Empey KM, Hollifield M, Garvy BA. Exogenous heat-killed Escherichia coli improves alveolar macrophage activity and reduces Pneumocystis carinii lung burden in infant mice. Infect Immun. 2007;75:3382–93. doi: 10.1128/IAI.00174-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reppe K, Tschernig T, Luhrmann A, van Laak V, Grote K, et al. Immunostimulation with Macrophage-Activating Lipopeptide-2 Increased Survival in Murine Pneumonia. Am J Respir Cell Mol Biol. 2008 doi: 10.1165/rcmb.2008-0071OC. [DOI] [PubMed] [Google Scholar]
- 19.Clement CG, Evans SE, Evans CM, Hawke D, Kobayashi R, et al. Stimulation of lung innate immunity protects against lethal pneumococcal pneumonia in mice. Am J Respir Crit Care Med. 2008;177:1322–30. doi: 10.1164/rccm.200607-1038OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tuvim MJ, Evans SE, Clement CG, Dickey BF, Gilbert BE. Augmented lung inflammation protects against influenza A pneumonia. PLoS ONE. 2009;4:e4176. doi: 10.1371/journal.pone.0004176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–81. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mizgerd JP. Acute lower respiratory tract infection. N Engl J Med. 2008;358:716–27. doi: 10.1056/NEJMra074111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hajjar AM, Harowicz H, Liggitt HD, Fink PJ, Wilson CB, Skerrett SJ. An essential role for non-bone marrow-derived cells in control of Pseudomonas aeruginosa pneumonia. Am J Respir Cell Mol Biol. 2005;33:470–5. doi: 10.1165/rcmb.2005-0199OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sadikot RT, Zeng H, Joo M, Everhart MB, Sherrill TP, et al. Targeted immunomodulation of the NF-kappaB pathway in airway epithelium impacts host defense against Pseudomonas aeruginosa. J Immunol. 2006;176:4923–30. doi: 10.4049/jimmunol.176.8.4923. [DOI] [PubMed] [Google Scholar]
- 25.Diamond G, Russell JP, Bevins CL. Inducible expression of an antibiotic peptide gene in lipopolysaccharide-challenged tracheal epithelial cells. Proc Natl Acad Sci U S A. 1996;93:5156–60. doi: 10.1073/pnas.93.10.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shornick LP, Wells AG, Zhang Y, Patel AC, Huang G, et al. Airway epithelial versus immune cell Stat1 function for innate defense against respiratory viral infection. J Immunol. 2008;180:3319–28. doi: 10.4049/jimmunol.180.5.3319. [DOI] [PubMed] [Google Scholar]
- 27.Hertz CJ, Wu Q, Porter EM, Zhang YJ, Weismuller KH, et al. Activation of Toll-like receptor 2 on human tracheobronchial epithelial cells induces the antimicrobial peptide human beta defensin-2. J Immunol. 2003;171:6820–6. doi: 10.4049/jimmunol.171.12.6820. [DOI] [PubMed] [Google Scholar]
- 28.Bosch TC, Augustin R, nton-Erxleben F, Fraune S, Hemmrich G, et al. Uncovering the evolutionary history of innate immunity: the simple metazoan Hydra uses epithelial cells for host defence. Dev Comp Immunol. 2009;33:559–69. doi: 10.1016/j.dci.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 29.Boller T, He SY. Innate immunity in plants: an arms race between pattern recognition receptors in plants and effectors in microbial pathogens. Science. 2009;324:742–4. doi: 10.1126/science.1171647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johansson ME, Phillipson M, Petersson J, Velcich A, Holm L, Hansson GC. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A. 2008;105:15064–9. doi: 10.1073/pnas.0803124105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Linden SK, Sutton P, Karlsson NG, Korolik V, McGuckin MA. Mucins in the mucosal barrier to infection. Mucosal Immunol. 2008;1:183–97. doi: 10.1038/mi.2008.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evans CM, Kim K, Tuvim MJ, Dickey BF. Mucus hypersecretion in asthma: causes and effects. Curr Opin Pulm Med. 2009;15:4–11. doi: 10.1097/MCP.0b013e32831da8d3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knowles MR, Boucher RC. Mucus clearance as a primary innate defense mechanism for mammalian airways. J Clin Invest. 2002;109:571–7. doi: 10.1172/JCI15217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin TR, Frevert CW. Innate immunity in the lungs. Proc Am Thorac Soc. 2005;2:403–11. doi: 10.1513/pats.200508-090JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mall MA. Role of Cilia, Mucus, and Airway Surface Liquid in Mucociliary Dysfunction: Lessons from Mouse Models. J Aerosol Med. 2008 doi: 10.1089/jamp.2007.0659. [DOI] [PubMed] [Google Scholar]
- 36.Brandl K, Plitas G, Mihu CN, Ubeda C, Jia T, et al. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature. 2008;455:804–7. doi: 10.1038/nature07250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Izcue A, Coombes JL, Powrie F. Regulatory lymphocytes and intestinal inflammation. Annu Rev Immunol. 2009;27:313–38. doi: 10.1146/annurev.immunol.021908.132657. [DOI] [PubMed] [Google Scholar]
- 38.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–8. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 39.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–26. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 40.Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524–5. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- 41.Boldrick JC, Alizadeh AA, Diehn M, Dudoit S, Liu CL, et al. Stereotyped and specific gene expression programs in human innate immune responses to bacteria. Proc Natl Acad Sci U S A. 2002;99:972–7. doi: 10.1073/pnas.231625398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kapetanovic R, Cavaillon JM. Early events in innate immunity in the recognition of microbial pathogens. Expert Opin Biol Ther. 2007;7:907–18. doi: 10.1517/14712598.7.6.907. [DOI] [PubMed] [Google Scholar]
- 43.Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816–25. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 44.Kopp E, Medzhitov R. Recognition of microbial infection by Toll-like receptors. Curr Opin Immunol. 2003;15:396–401. doi: 10.1016/s0952-7915(03)00080-3. [DOI] [PubMed] [Google Scholar]
- 45.Zhang G, Ghosh S. Toll-like receptor-mediated NF-kappaB activation: a phylogenetically conserved paradigm in innate immunity. J Clin Invest. 2001;107:13–9. doi: 10.1172/JCI11837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev. 2008;226:10–8. doi: 10.1111/j.1600-065X.2008.00701.x. [DOI] [PubMed] [Google Scholar]
- 47.Beutler BA. TLRs and innate immunity. Blood. 2009;113:1399–407. doi: 10.1182/blood-2008-07-019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–64. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 49.Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–3. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 50.Lee JS, Frevert CW, Matute-Bello G, Wurfel MM, Wong VA, et al. TLR-4 pathway mediates the inflammatory response but not bacterial elimination in E. coli pneumonia. Am J Physiol Lung Cell Mol Physiol. 2005;289:L731–8. doi: 10.1152/ajplung.00196.2005. [DOI] [PubMed] [Google Scholar]
- 51.Wang X, Moser C, Louboutin JP, Lysenko ES, Weiner DJ, et al. Toll-like receptor 4 mediates innate immune responses to Haemophilus influenzae infection in mouse lung. J Immunol. 2002;168:810–5. doi: 10.4049/jimmunol.168.2.810. [DOI] [PubMed] [Google Scholar]
- 52.Kurt-Jones EA, Popova L, Kwinn L, Haynes LM, Jones LP, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 53.Gribar SC, Richardson WM, Sodhi CP, Hackam DJ. No longer an innocent bystander: epithelial toll-like receptor signaling in the development of mucosal inflammation. Mol Med. 2008;14:645–59. doi: 10.2119/2008-00035.Gribar. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramphal R, Balloy V, Huerre M, Si-Tahar M, Chignard M. TLRs 2 and 4 are not involved in hypersusceptibility to acute Pseudomonas aeruginosa lung infections. J Immunol. 2005;175:3927–34. doi: 10.4049/jimmunol.175.6.3927. [DOI] [PubMed] [Google Scholar]
- 55.Ramphal R, Balloy V, Jyot J, Verma A, Si-Tahar M, Chignard M. Control of Pseudomonas aeruginosa in the lung requires the recognition of either lipopolysaccharide or flagellin. J Immunol. 2008;181:586–92. doi: 10.4049/jimmunol.181.1.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, et al. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2006;2:e53. doi: 10.1371/journal.ppat.0020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wong JP, Christopher ME, Viswanathan S, Karpoff N, Dai X, et al. Activation of toll-like receptor signaling pathway for protection against influenza virus infection. Vaccine. 2009;27:3481–3. doi: 10.1016/j.vaccine.2009.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muir A, Soong G, Sokol S, Reddy B, Gomez MI, et al. Toll-like receptors in normal and cystic fibrosis airway epithelial cells. Am J Respir Cell Mol Biol. 2004;30:777–83. doi: 10.1165/rcmb.2003-0329OC. [DOI] [PubMed] [Google Scholar]
- 59.Schleimer RP. Glucocorticoids suppress inflammation but spare innate immune responses in airway epithelium. Proc Am Thorac Soc. 2004;1:222–30. doi: 10.1513/pats.200402-018MS. [DOI] [PubMed] [Google Scholar]
- 60.Sha Q, Truong-Tran AQ, Plitt JR, Beck LA, Schleimer RP. Activation of airway epithelial cells by toll-like receptor agonists. Am J Respir Cell Mol Biol. 2004;31:358–64. doi: 10.1165/rcmb.2003-0388OC. [DOI] [PubMed] [Google Scholar]
- 61.Koff JL, Shao MX, Ueki IF, Nadel JA. Multiple TLRs activate EGFR via a signaling cascade to produce innate immune responses in airway epithelium. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1068–75. doi: 10.1152/ajplung.00025.2008. [DOI] [PubMed] [Google Scholar]
- 62.Uehara A, Fujimoto Y, Fukase K, Takada H. Various human epithelial cells express functional Toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol Immunol. 2007;44:3100–11. doi: 10.1016/j.molimm.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 63.Merlo A, Calcaterra C, Menard S, Balsari A. Cross-talk between toll-like receptors 5 and 9 on activation of human immune responses. J Leukoc Biol. 2007;82:509–18. doi: 10.1189/jlb.0207100. [DOI] [PubMed] [Google Scholar]
- 64.Powell JD, Boodoo S, Horton MR. Identification of the molecular mechanism by which TLR ligation and IFN-gamma synergize to induce MIG. Clin Dev Immunol. 2004;11:77–85. doi: 10.1080/10446670410001670535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–90. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 66.Noulin N, Quesniaux VF, Schnyder-Candrian S, Schnyder B, Maillet I, et al. Both hemopoietic and resident cells are required for MyD88-dependent pulmonary inflammatory response to inhaled endotoxin. J Immunol. 2005;175:6861–9. doi: 10.4049/jimmunol.175.10.6861. [DOI] [PubMed] [Google Scholar]
- 67.Shaw MH, Reimer T, Kim YG, Nunez G. NOD-like receptors (NLRs): bona fide intracellular microbial sensors. Curr Opin Immunol. 2008;20:377–82. doi: 10.1016/j.coi.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. 2003;4:702–7. doi: 10.1038/ni945. [DOI] [PubMed] [Google Scholar]
- 69.Bourhis LL, Werts C. Role of Nods in bacterial infection. Microbes Infect. 2007;9:629–36. doi: 10.1016/j.micinf.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 70.Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–74. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 71.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–4. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 72.Divangahi M, Mostowy S, Coulombe F, Kozak R, Guillot L, et al. NOD2-deficient mice have impaired resistance to Mycobacterium tuberculosis infection through defective innate and adaptive immunity. J Immunol. 2008;181:7157–65. doi: 10.4049/jimmunol.181.10.7157. [DOI] [PubMed] [Google Scholar]
- 73.Opitz B, Puschel A, Schmeck B, Hocke AC, Rosseau S, et al. Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J Biol Chem. 2004;279:36426–32. doi: 10.1074/jbc.M403861200. [DOI] [PubMed] [Google Scholar]
- 74.Shimada K, Chen S, Dempsey PW, Sorrentino R, Alsabeh R, et al. The NOD/RIP2 pathway is essential for host defenses against Chlamydophila pneumoniae lung infection. PLoS Pathog. 2009;5:e1000379. doi: 10.1371/journal.ppat.1000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–65. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 76.Miao EA, Andersen-Nissen E, Warren SE, Aderem A. TLR5 and Ipaf: dual sensors of bacterial flagellin in the innate immune system. Semin Immunopathol. 2007;29:275–88. doi: 10.1007/s00281-007-0078-z. [DOI] [PubMed] [Google Scholar]
- 77.Kawai T, Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann N Y Acad Sci. 2008;1143:1–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- 78.Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–7. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- 79.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–5. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 80.Evans SE, Hahn PY, McCann F, Kottom TJ, Pavlovic ZV, Limper AH. Pneumocystis cell wall beta-glucans stimulate alveolar epithelial cell chemokine generation through nuclear factor-kappaB-dependent mechanisms. Am J Respir Cell Mol Biol. 2005;32:490–7. doi: 10.1165/rcmb.2004-0300OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hahn PY, Evans SE, Kottom TJ, Standing JE, Pagano RE, Limper AH. Pneumocystis carinii cell wall beta-glucan induces release of macrophage inflammatory protein-2 from alveolar epithelial cells via a lactosylceramide-mediated mechanism. J Biol Chem. 2003;278:2043–50. doi: 10.1074/jbc.M209715200. [DOI] [PubMed] [Google Scholar]
- 82.Davis CW, Lazarowski E. Coupling of airway ciliary activity and mucin secretion to mechanical stresses by purinergic signaling. Respir Physiol Neurobiol. 2008;163:208–13. doi: 10.1016/j.resp.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008;455:808–12. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- 84.Idzko M, Hammad H, van Nimwegen M, Kool M, Willart MA, et al. Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med. 2007;13:913–9. doi: 10.1038/nm1617. [DOI] [PubMed] [Google Scholar]
- 85.Zhou Y, Schneider DJ, Blackburn MR. Adenosine signaling and the regulation of chronic lung disease. Pharmacol Ther. 2009;123:105–16. doi: 10.1016/j.pharmthera.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hasko G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7:759–70. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009;459:433–6. doi: 10.1038/nature07965. [DOI] [PubMed] [Google Scholar]
- 88.Gasse P, Riteau N, Charron S, Girre S, Fick L, et al. Uric acid is a danger signal activating NALP3 inflammasome in lung injury inflammation and fibrosis. Am J Respir Crit Care Med. 2009;179:903–13. doi: 10.1164/rccm.200808-1274OC. [DOI] [PubMed] [Google Scholar]
- 89.Akashi-Takamura S, Miyake K. TLR accessory molecules. Curr Opin Immunol. 2008;20:420–5. doi: 10.1016/j.coi.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 90.Drouin SM, Kildsgaard J, Haviland J, Zabner J, Jia HP, et al. Expression of the complement anaphylatoxin C3a and C5a receptors on bronchial epithelial and smooth muscle cells in models of sepsis and asthma. J Immunol. 2001;166:2025–32. doi: 10.4049/jimmunol.166.3.2025. [DOI] [PubMed] [Google Scholar]
- 91.Drouin SM, Corry DB, Hollman TJ, Kildsgaard J, Wetsel RA. Absence of the complement anaphylatoxin C3a receptor suppresses Th2 effector functions in a murine model of pulmonary allergy. J Immunol. 2002;169:5926–33. doi: 10.4049/jimmunol.169.10.5926. [DOI] [PubMed] [Google Scholar]
- 92.Dillard P, Wetsel RA, Drouin SM. Complement C3a regulates Muc5ac expression by airway Clara cells independently of Th2 responses. Am J Respir Crit Care Med. 2007;175:1250–8. doi: 10.1164/rccm.200701-049OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–7. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 94.Gushiken FC, Han H, Li J, Rumbaut RE, Afshar-Kharghan V. Abnormal platelet function in C3-deficient mice. J Thromb Haemost. 2009;7:865–70. doi: 10.1111/j.1538-7836.2009.03334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shetty S, Padijnayayveetil J, Tucker T, Stankowska D, Idell S. The fibrinolytic system and the regulation of lung epithelial cell proteolysis, signaling, and cellular viability. Am J Physiol Lung Cell Mol Physiol. 2008;295:L967–75. doi: 10.1152/ajplung.90349.2008. [DOI] [PubMed] [Google Scholar]
- 96.Neyrinck AP, Liu KD, Howard JP, Matthay MA. Protective mechanisms of activated protein C in severe inflammatory disorders. Br J Pharmacol. 2009 doi: 10.1111/j.1476-5381.2009.00251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Irvin CG, Bates JH. Physiologic dysfunction of the asthmatic lung: what's going on down there, anyway? Proc Am Thorac Soc. 2009;6:306–11. doi: 10.1513/pats.200808-091RM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gallo RL, Nizet V. Innate barriers against infection and associated disorders. Drug Discov Today Dis Mech. 2008;5:145–52. doi: 10.1016/j.ddmec.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jiang D, Liang J, Noble PW. Hyaluronan in tissue injury and repair. Annu Rev Cell Dev Biol. 2007;23:435–61. doi: 10.1146/annurev.cellbio.23.090506.123337. [DOI] [PubMed] [Google Scholar]
- 100.Sheppard D. Transforming growth factor beta: a central modulator of pulmonary and airway inflammation and fibrosis. Proc Am Thorac Soc. 2006;3:413–7. doi: 10.1513/pats.200601-008AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–35. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 102.Kolls JK, McCray PB, Jr, Chan YR. Cytokine-mediated regulation of antimicrobial proteins. Nat Rev Immunol. 2008;8:829–35. doi: 10.1038/nri2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ank N, Paludan SR. Type III IFNs: new layers of complexity in innate antiviral immunity. Biofactors. 2009;35:82–7. doi: 10.1002/biof.19. [DOI] [PubMed] [Google Scholar]
- 104.Billiau A, Matthys P. Interferon-gamma: a historical perspective. Cytokine Growth Factor Rev. 2009;20:97–113. doi: 10.1016/j.cytogfr.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 105.Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. IL-22 increases the innate immunity of tissues. Immunity. 2004;21:241–54. doi: 10.1016/j.immuni.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 106.Cella M, Fuchs A, Vermi W, Facchetti F, Otero K, et al. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature. 2009;457:722–5. doi: 10.1038/nature07537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, et al. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–9. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–27. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wu Q, Martin RJ, Rino JG, Breed R, Torres RM, Chu HW. IL-23-dependent IL-17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect. 2007;9:78–86. doi: 10.1016/j.micinf.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kao CY, Chen Y, Thai P, Wachi S, Huang F, et al. IL-17 markedly up-regulates beta-defensin-2 expression in human airway epithelium via JAK and NF-kappaB signaling pathways. J Immunol. 2004;173:3482–91. doi: 10.4049/jimmunol.173.5.3482. [DOI] [PubMed] [Google Scholar]
- 111.Guttman JA, Finlay BB. Tight junctions as targets of infectious agents. Biochim Biophys Acta. 2009;1788:832–41. doi: 10.1016/j.bbamem.2008.10.028. [DOI] [PubMed] [Google Scholar]
- 112.Sousa S, Lecuit M, Cossart P. Microbial strategies to target, cross or disrupt epithelia. Curr Opin Cell Biol. 2005;17:489–98. doi: 10.1016/j.ceb.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 113.Marsh M, Helenius A. Virus entry: open sesame. Cell. 2006;124:729–40. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gonzalez-Mariscal L, Garay E, Lechuga S. Virus interaction with the apical junctional complex. Front Biosci. 2009;14:731–68. doi: 10.2741/3276. [DOI] [PubMed] [Google Scholar]
- 115.Chun J, Prince A. TLR2-induced calpain cleavage of epithelial junctional proteins facilitates leukocyte transmigration. Cell Host Microbe. 2009;5:47–58. doi: 10.1016/j.chom.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Beisswenger C, Coyne CB, Shchepetov M, Weiser JN. Role of p38 MAP kinase and transforming growth factor-beta signaling in transepithelial migration of invasive bacterial pathogens. J Biol Chem. 2007;282:28700–8. doi: 10.1074/jbc.M703576200. [DOI] [PubMed] [Google Scholar]
- 117.Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology. 2007;132:1359–74. doi: 10.1053/j.gastro.2007.02.056. [DOI] [PubMed] [Google Scholar]
- 118.Ey B, Eyking A, Gerken G, Podolsky DK, Cario E. TLR2 mediates gap junctional intercellular communication through connexin-43 in intestinal epithelial barrier injury. J Biol Chem. 2009 doi: 10.1074/jbc.M901619200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Miyauchi E, Morita H, Okuda J, Sashihara T, Shimizu M, Tanabe S. Cell wall fraction of Enterococcus hirae ameliorates TNF-alpha-induced barrier impairment in the human epithelial tight junction. Lett Appl Microbiol. 2008;46:469–76. doi: 10.1111/j.1472-765X.2008.02332.x. [DOI] [PubMed] [Google Scholar]
- 120.Kim M, Ogawa M, Fujita Y, Yoshikawa Y, Nagai T, et al. Bacteria hijack integrin-linked kinase to stabilize focal adhesions and block cell detachment. Nature. 2009;459:578–82. doi: 10.1038/nature07952. [DOI] [PubMed] [Google Scholar]
- 121.Rawlins EL, Hogan BL. Ciliated epithelial cell lifespan in the mouse trachea and lung. Am J Physiol Lung Cell Mol Physiol. 2008;295:L231–L4. doi: 10.1152/ajplung.90209.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mysorekar IU, Hultgren SJ. Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc Natl Acad Sci U S A. 2006;103:14170–5. doi: 10.1073/pnas.0602136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Song J, Bishop BL, Li G, Duncan MJ, Abraham SN. TLR4-initiated and cAMP-mediated abrogation of bacterial invasion of the bladder. Cell Host Microbe. 2007;1:287–98. doi: 10.1016/j.chom.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Russell BH, Liu Q, Jenkins SA, Tuvim MJ, Dickey BF, Xu Y. In vivo demonstration and quantification of intracellular Bacillus anthracis in lung epithelial cells. Infect Immun. 2008;76:3975–83. doi: 10.1128/IAI.00282-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Russell BH, Vasan R, Keene DR, Koehler TM, Xu Y. Potential dissemination of Bacillus anthracis utilizing human lung epithelial cells. Cell Microbiol. 2008;10:945–67. doi: 10.1111/j.1462-5822.2007.01098.x. [DOI] [PubMed] [Google Scholar]
- 126.Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. 2008;198:962–70. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Vermeer PD, Einwalter LA, Moninger TO, Rokhlina T, Kern JA, et al. Segregation of receptor and ligand regulates activation of epithelial growth factor receptor. Nature. 2003;422:322–6. doi: 10.1038/nature01440. [DOI] [PubMed] [Google Scholar]
- 128.Bals R, Hiemstra PS. Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur Respir J. 2004;23:327–33. doi: 10.1183/09031936.03.00098803. [DOI] [PubMed] [Google Scholar]
- 129.Hippenstiel S, Opitz B, Schmeck B, Suttorp N. Lung epithelium as a sentinel and effector system in pneumonia--molecular mechanisms of pathogen recognition and signal transduction. Respir Res. 2006;7:97. doi: 10.1186/1465-9921-7-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hammad H, Lambrecht BN. Dendritic cells and epithelial cells: linking innate and adaptive immunity in asthma. Nat Rev Immunol. 2008;8:193–204. doi: 10.1038/nri2275. [DOI] [PubMed] [Google Scholar]
- 131.Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell-derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev. 2008;226:172–90. doi: 10.1111/j.1600-065X.2008.00713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. 2005;3:238–50. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 133.Cole AM, Liao HI, Stuchlik O, Tilan J, Pohl J, Ganz T. Cationic polypeptides are required for antibacterial activity of human airway fluid. J Immunol. 2002;169:6985–91. doi: 10.4049/jimmunol.169.12.6985. [DOI] [PubMed] [Google Scholar]
- 134.Bals R, Wang X, Wu Z, Freeman T, Bafna V, et al. Human beta-defensin 2 is a salt-sensitive peptide antibiotic expressed in human lung. J Clin Invest. 1998;102:874–80. doi: 10.1172/JCI2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bals R, Wang X, Zasloff M, Wilson JM. The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc Natl Acad Sci U S A. 1998;95:9541–6. doi: 10.1073/pnas.95.16.9541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bals R, Wang X, Meegalla RL, Wattler S, Weiner DJ, et al. Mouse beta-defensin 3 is an inducible antimicrobial peptide expressed in the epithelia of multiple organs. Infect Immun. 1999;67:3542–7. doi: 10.1128/iai.67.7.3542-3547.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hancock RE, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24:1551–7. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 138.Hiemstra PS. The role of epithelial beta-defensins and cathelicidins in host defense of the lung. Exp Lung Res. 2007;33:537–42. doi: 10.1080/01902140701756687. [DOI] [PubMed] [Google Scholar]
- 139.Lai Y, Gallo RL. AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009;30:131–41. doi: 10.1016/j.it.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schutte BC, McCray PB., Jr [beta]-defensins in lung host defense. Annu Rev Physiol. 2002;64:709–48. doi: 10.1146/annurev.physiol.64.081501.134340. [DOI] [PubMed] [Google Scholar]
- 141.Singh PK, Jia HP, Wiles K, Hesselberth J, Liu L, et al. Production of beta-defensins by human airway epithelia. Proc Natl Acad Sci U S A. 1998;95:14961–6. doi: 10.1073/pnas.95.25.14961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Schaller-Bals S, Schulze A, Bals R. Increased levels of antimicrobial peptides in tracheal aspirates of newborn infants during infection. Am J Respir Crit Care Med. 2002;165:992–5. doi: 10.1164/ajrccm.165.7.200110-020. [DOI] [PubMed] [Google Scholar]
- 143.Harder J, Meyer-Hoffert U, Teran LM, Schwichtenberg L, Bartels J, et al. Mucoid Pseudomonas aeruginosa, TNF-alpha, and IL-1beta, but not IL-6, induce human beta-defensin-2 in respiratory epithelia. Am J Respir Cell Mol Biol. 2000;22:714–21. doi: 10.1165/ajrcmb.22.6.4023. [DOI] [PubMed] [Google Scholar]
- 144.Lehrer RI, Ganz T. Antimicrobial peptides in mammalian and insect host defence. Curr Opin Immunol. 1999;11:23–7. doi: 10.1016/s0952-7915(99)80005-3. [DOI] [PubMed] [Google Scholar]
- 145.Jiang Y, Wang Y, Kuang Y, Wang B, Li W, et al. Expression of mouse beta-defensin-3 in MDCK cells and its anti-influenza-virus activity. Arch Virol. 2009;154:639–47. doi: 10.1007/s00705-009-0352-6. [DOI] [PubMed] [Google Scholar]
- 146.Wang Y, Jiang Y, Gong T, Cui X, Li W, et al. High-Level Expression and Novel Antifungal Activity of Mouse Beta Defensin-1 Mature Peptide in Escherichia coli. Appl Biochem Biotechnol. 2009 doi: 10.1007/s12010-009-8566-3. [DOI] [PubMed] [Google Scholar]
- 147.Liu PT, Stenger S, Li H, Wenzel L, Tan BH, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770–3. doi: 10.1126/science.1123933. [DOI] [PubMed] [Google Scholar]
- 148.Herr C, Shaykhiev R, Bals R. The role of cathelicidin and defensins in pulmonary inflammatory diseases. Expert Opin Biol Ther. 2007;7:1449–61. doi: 10.1517/14712598.7.9.1449. [DOI] [PubMed] [Google Scholar]
- 149.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–95. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 150.Moser C, Weiner DJ, Lysenko E, Bals R, Weiser JN, Wilson JM. beta-Defensin 1 contributes to pulmonary innate immunity in mice. Infect Immun. 2002;70:3068–72. doi: 10.1128/IAI.70.6.3068-3072.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bals R, Weiner DJ, Meegalla RL, Wilson JM. Transfer of a cathelicidin peptide antibiotic gene restores bacterial killing in a cystic fibrosis xenograft model. J Clin Invest. 1999;103:1113–7. doi: 10.1172/JCI6570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bals R, Weiner DJ, Moscioni AD, Meegalla RL, Wilson JM. Augmentation of innate host defense by expression of a cathelicidin antimicrobial peptide. Infect Immun. 1999;67:6084–9. doi: 10.1128/iai.67.11.6084-6089.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Niyonsaba F, Iwabuchi K, Matsuda H, Ogawa H, Nagaoka I. Epithelial cell-derived human beta-defensin-2 acts as a chemotaxin for mast cells through a pertussis toxin-sensitive and phospholipase C-dependent pathway. Int Immunol. 2002;14:421–6. doi: 10.1093/intimm/14.4.421. [DOI] [PubMed] [Google Scholar]
- 154.Territo MC, Ganz T, Selsted ME, Lehrer R. Monocyte-chemotactic activity of defensins from human neutrophils. J Clin Invest. 1989;84:2017–20. doi: 10.1172/JCI114394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yang D, Biragyn A, Kwak LW, Oppenheim JJ. Mammalian defensins in immunity: more than just microbicidal. Trends Immunol. 2002;23:291–6. doi: 10.1016/s1471-4906(02)02246-9. [DOI] [PubMed] [Google Scholar]
- 156.Yang D, Chertov O, Oppenheim JJ. Participation of mammalian defensins and cathelicidins in anti-microbial immunity: receptors and activities of human defensins and cathelicidin (LL-37) J Leukoc Biol. 2001;69:691–7. [PubMed] [Google Scholar]
- 157.De Y, Chen Q, Schmidt AP, Anderson GM, Wang JM, et al. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med. 2000;192:1069–74. doi: 10.1084/jem.192.7.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Niyonsaba F, Iwabuchi K, Someya A, Hirata M, Matsuda H, et al. A cathelicidin family of human antibacterial peptide LL-37 induces mast cell chemotaxis. Immunology. 2002;106:20–6. doi: 10.1046/j.1365-2567.2002.01398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ganz T. Antimicrobial polypeptides. J Leukoc Biol. 2004;75:34–8. doi: 10.1189/jlb.0403150. [DOI] [PubMed] [Google Scholar]
- 160.van de Wetering JK, van Golde LM, Batenburg JJ. Collectins: players of the innate immune system. Eur J Biochem. 2004;271:1229–49. doi: 10.1111/j.1432-1033.2004.04040.x. [DOI] [PubMed] [Google Scholar]
- 161.Haagsman HP, Hogenkamp A, van Eijk M, Veldhuizen EJ. Surfactant collectins and innate immunity. Neonatology. 2008;93:288–94. doi: 10.1159/000121454. [DOI] [PubMed] [Google Scholar]
- 162.Pastva AM, Wright JR, Williams KL. Immunomodulatory roles of surfactant proteins A and D: implications in lung disease. Proc Am Thorac Soc. 2007;4:252–7. doi: 10.1513/pats.200701-018AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Vuk-Pavlovic Z, Mo EK, Icenhour CR, Standing JE, Fisher JH, Limper AH. Surfactant protein D enhances Pneumocystis infection in immune-suppressed mice. Am J Physiol Lung Cell Mol Physiol. 2006;290:L442–9. doi: 10.1152/ajplung.00112.2005. [DOI] [PubMed] [Google Scholar]
- 164.Chan YR, Liu JS, Pociask DA, Zheng M, Mietzner TA, et al. Lipocalin 2 is required for pulmonary host defense against Klebsiella infection. J Immunol. 2009;182:4947–56. doi: 10.4049/jimmunol.0803282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, et al. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–21. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 166.Bingle CD, Gorr SU. Host defense in oral and airway epithelia: chromosome 20 contributes a new protein family. Int J Biochem Cell Biol. 2004;36:2144–52. doi: 10.1016/j.biocel.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 167.Bingle L, Barnes FA, Lunn H, Musa M, Webster S, et al. Characterisation and expression of SPLUNC2, the human orthologue of rodent parotid secretory protein. Histochem Cell Biol. 2009 doi: 10.1007/s00418-009-0610-4. [DOI] [PubMed] [Google Scholar]
- 168.Starner TD, Barker CK, Jia HP, Kang Y, McCray PB., Jr CCL20 is an inducible product of human airway epithelia with innate immune properties. Am J Respir Cell Mol Biol. 2003;29:627–33. doi: 10.1165/rcmb.2002-0272OC. [DOI] [PubMed] [Google Scholar]
- 169.Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol. 2004;5:981–6. doi: 10.1038/ni1113. [DOI] [PubMed] [Google Scholar]
- 170.Rooijakkers SH, van Strijp JA. Bacterial complement evasion. Mol Immunol. 2007;44:23–32. doi: 10.1016/j.molimm.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 171.Conner GE, Wijkstrom-Frei C, Randell SH, Fernandez VE, Salathe M. The lactoperoxidase system links anion transport to host defense in cystic fibrosis. FEBS Lett. 2007;581:271–8. doi: 10.1016/j.febslet.2006.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Gerson C, Sabater J, Scuri M, Torbati A, Coffey R, et al. The lactoperoxidase system functions in bacterial clearance of airways. Am J Respir Cell Mol Biol. 2000;22:665–71. doi: 10.1165/ajrcmb.22.6.3980. [DOI] [PubMed] [Google Scholar]
- 173.Moskwa P, Lorentzen D, Excoffon KJ, Zabner J, McCray PB, Jr, et al. A novel host defense system of airways is defective in cystic fibrosis. Am J Respir Crit Care Med. 2007;175:174–83. doi: 10.1164/rccm.200607-1029OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Harper RW, Xu C, Eiserich JP, Chen Y, Kao CY, et al. Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS Lett. 2005;579:4911–7. doi: 10.1016/j.febslet.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 175.Klebanoff SJ, Clem WH, Luebke RG. The peroxidase-thiocyanate-hydrogen peroxide antimicrobial system. Biochim Biophys Acta. 1966;117:63–72. doi: 10.1016/0304-4165(66)90152-8. [DOI] [PubMed] [Google Scholar]
- 176.Wijkstrom-Frei C, El-Chemaly S, Ali-Rachedi R, Gerson C, Cobas MA, et al. Lactoperoxidase and human airway host defense. Am J Respir Cell Mol Biol. 2003;29:206–12. doi: 10.1165/rcmb.2002-0152OC. [DOI] [PubMed] [Google Scholar]
- 177.Vazquez-Torres A, Stevanin T, Jones-Carson J, Castor M, Read RC, Fang FC. Analysis of nitric oxide-dependent antimicrobial actions in macrophages and mice. Methods Enzymol. 2008;437:521–38. doi: 10.1016/S0076-6879(07)37026-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Singh PK, Tack BF, McCray PB, Jr, Welsh MJ. Synergistic and additive killing by antimicrobial factors found in human airway surface liquid. Am J Physiol Lung Cell Mol Physiol. 2000;279:L799–805. doi: 10.1152/ajplung.2000.279.5.L799. [DOI] [PubMed] [Google Scholar]
- 179.Unterholzner L, Bowie AG. The interplay between viruses and innate immune signaling: recent insights and therapeutic opportunities. Biochem Pharmacol. 2008;75:589–602. doi: 10.1016/j.bcp.2007.07.043. [DOI] [PubMed] [Google Scholar]
- 180.Chai LY, Netea MG, Vonk AG, Kullberg BJ. Fungal strategies for overcoming host innate immune response. Med Mycol. 2009;47:227–36. doi: 10.1080/13693780802209082. [DOI] [PubMed] [Google Scholar]
- 181.Zhang SY, Boisson-Dupuis S, Chapgier A, Yang K, Bustamante J, et al. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev. 2008;226:29–40. doi: 10.1111/j.1600-065X.2008.00698.x. [DOI] [PubMed] [Google Scholar]
- 182.von Bernuth H, Picard C, Jin Z, Pankla R, Xiao H, et al. Pyogenic bacterial infections in humans with MyD88 deficiency. Science. 2008;321:691–6. doi: 10.1126/science.1158298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Moilanen P, Korppi M, Hovi L, Chapgier A, Feinberg J, et al. Successful Hematopoietic Stem Cell Transplantation From an Unrelated Donor in a Child With Interferon Gamma Receptor Deficiency. Pediatr Infect Dis J. 2009 doi: 10.1097/INF.0b013e318195092e. [DOI] [PubMed] [Google Scholar]
- 184.Ulevitch RJ. Therapeutics targeting the innate immune system. Nat Rev Immunol. 2004;4:512–20. doi: 10.1038/nri1396. [DOI] [PubMed] [Google Scholar]
- 185.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. 2007;13:552–9. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 186.Hackett CJ. Innate immune activation as a broad-spectrum biodefense strategy: prospects and research challenges. J Allergy Clin Immunol. 2003;112:686–94. doi: 10.1016/S0091-6749(03)02025-6. [DOI] [PMC free article] [PubMed] [Google Scholar]