

Abstract
Post-translational protein translocation across the bacterial plasma membrane is mediated by the interplay of the SecA ATPase and the protein-conducting SecY channel. SecA consists of several domains, including two nucleotide-binding domains (NBD1 and NBD2), a polypeptide crosslinking domain (PPXD), a helical scaffold domain (HSD), and a helical wing domain (HWD). PPXD, HSD, and NBD2 form a clamp that positions the polypeptide substrate above the channel, so that it can be pushed into the channel by a two-helix finger of the HSD. How the substrate is accommodated in the clamp during translocation is unclear. Here, we report a crystal structure of T. maritima SecA at 1.9 Å resolution. Structural analysis and free energy calculations indicate that the new structure represents an intermediate state during the transition of the clamp from an open to a closed conformation. Molecular dynamics simulations show that closure of the clamp occurs in two phases, an initial movement of PPXD, HSD, and HWD as a unit, followed by a movement of PPXD alone towards NBD2. Simulations in the presence of a polypeptide chain show that the substrate associates with the back of the clamp by dynamic hydrogen bonding, and that the clamp is laterally closed by a conserved loop of the PPXD. Mutational disruption of clamp opening or closure abolishes protein translocation. These results suggest how conformational changes of SecA allow substrate binding and movement during protein translocation.
Keywords: Protein translocation, SecA, crystal structure, molecular dynamics simulation, free energy
Graphical abstract
Introduction
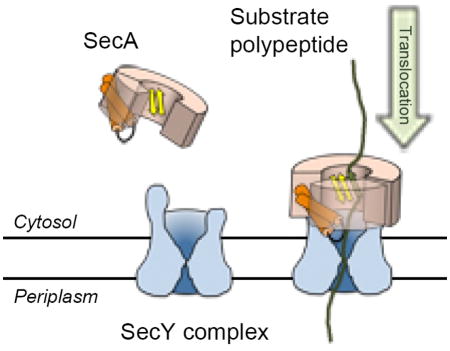
Protein translocation across the eukaryotic endoplasmic reticulum (ER) membrane or the prokaryotic plasma membrane is a decisive step in the biosynthesis of a large class of proteins, which includes secretory and membrane proteins (for reviews, see refs. 1–5). These proteins are targeted to the membrane by hydrophobic sequences, either cleavable signal sequences or non-cleaved trans-membrane (TM) segments. They are then translocated through a protein translocation channel that is formed from a conserved hetero-trimeric membrane protein complex, the Sec61 complex in eukaryotes or the SecY complex in prokaryotes. Translocation can occur either co- or post-translationally. In co-translational translocation, a polypeptide chain is transferred into the channel during its synthesis on a ribosome. In post-translational translocation, a ratcheting mechanism is used by the ER-luminal chaperone BiP in eukaryotes6, and a pushing mechanism is utilized by the SecA ATPase in bacteria3, 7, 8. The mechanism by which SecA pushes polypeptide chains through the SecY channel is only poorly understood.
SecA is composed of five structural domains: nucleotide-binding domains 1 and 2 (NBD1 and NBD2), a polypeptide-cross-linking domain (PPXD), a helical scaffold domain (HSD), and a helical wing domain (HWD)9–15. NBD1 and NBD2 have RecA-like folds and bind the nucleotide at their interface. The HSD consists of a long helix and two shorter ones referred to as the two-helix finger16, 17. The two-helix finger pokes into the cytoplasmic opening of the SecY channel and is thought to move during the ATP hydrolysis cycle, pushing the translocating polypeptide chain into the channel16–18.
Crystal structures of SecA from different bacteria revealed that the PPXD moves relative to the rest of the protein, resulting in distinct conformations of SecA9–11, 13–16. These movements correspond to the opening and closure of a “clamp” that is formed from PPXD, HSD, and NBD2. In crystal structures of SecA in isolation, the clamp is open to varying degrees, with the PPXD at different positions relative to NBD29–11, 13–15. In the most extreme case, the PPXD is far from NBD2 and close to the HWD9, 11, 13–15, 19. In crystal structures of SecA bound to the SecY channel, the clamp is closed, i.e. PPXD is near NBD216. In this conformation, the clamp could position a translocating polypeptide chain above the channel. Indeed, disulfide crosslinking experiments showed that a translocating polypeptide moves through the clamp, passes by the two-helix finger, and is then transferred into the SecY channel17, 20. However, how a polypeptide chain is accommodated inside the clamp during translocation is unclear. A crystal structure of B. subtilis SecA with a small hydrophilic peptide suggested that the substrate forms a short β-strand that interacts with the two β-strands connecting NBD1 and PPXD at the back of the clamp15. However, only the backbones of three amino acids were visible in the density map, and in the crystal structures of the SecA-SecY complex, there is insufficient space for a polypeptide chain, even when in an unfolded conformation. The driving force for clamp closure is also unclear.
Here, we have analyzed conformational changes of the clamp using X-ray crystallography and molecular dynamics simulations. Our results indicate that clamp closure occurs in two distinct phases, an initial movement of PPXD, HSD, and HWD as a unit, and a second movement of PPXD alone. A translocating polypeptide forms transient hydrogen bonds with the two-stranded β-sheet at the back of the clamp. A conserved loop of PPXD occupies the clamp in the crystal structures lacking the substrate. The simulations indicate that, during translocation, the loop moves outwards and interacts with NBD2, thereby stabilizing a laterally closed clamp. Residues involved in this interaction are highly conserved, and their mutagenesis abolishes protein translocation. Taken together, these results suggest a model for how conformational changes of SecA allow substrate binding and movement during protein translocation.
Results
Structure of T. maritima SecA at 1.9 Å resolution
We were able to obtain a crystal structure of T. maritima SecA with bound ADP at 1.9Å resolution. Although crystallization was performed in the presence of SecY complex, ADP, and vanadate, the crystals contained only SecA and ADP; nonetheless, the presence of the SecY complex was essential for crystal formation.
In our new structure, T. maritima SecA displays an open conformation, in which the PPXD domain leans against HWD; the substrate-binding clamp formed by PPXD, HSD, and NBD2 is wide open (Figure 1a). Superficially, the structure looks similar to a published structure of T. maritima SecA (PDB accession code: 3JUX)15. However, although the overall R.M.S.D. is 2.06 Å, alignment of the two structures on the basis of the NBD domains reveals that PPXD, HSD, and HWD have moved towards NBD2 by ~12 Å and 14° (Figure 1b). The two β-strands at the back of the clamp, which connect NBD1 and PPXD, serve as the hinge for this movement. The C-terminal half of the long helix of HSD is bent, moving together with PPXD, HSD, and HWD, resulting in a ~6 Å displacement of the end of the helix.
Figure 1. Comparison of a new structure of T. maritima SecA with previous structures from the same species.

(a) A new structure of T. maritima SecA with bound ADP was determined at 1.9 Å resolution. Shown is the main chain with NBD1 colored in blue, NBD2 in cyan, PPXD in green, HWD in orange, and HSD in red. The two β-strands connecting NBD1 and PPXD are highlighted in yellow. Bound ADP is shown in stick representation (in magenta).
(b) Comparison of the new T. maritima SecA-ADP structure (in light purple) with a previous SecA-ADP structure from the same species (PDB accession code: 3JUX, in beige). The alignment is based on NBD1. The hinge for the movement of PPXD, HSD, and HWD is indicated by the yellow circle.
(c) Superposition of the new structure with other T. maritima SecA structures. The alignment is based on NBD1. The PPXDs are shown in different colors: the new structure - light purple; PDB 3JUX - beige; PDB 3DIN - red.
The new T. maritima SecA structure can be compared with previous structures from the same species (Figure 1c). The most open conformation is the previously published structure (PDB accession code 3JUX, shown in beige in Figure 1c; the distance between PPXD-NBD2 and the angle of PPXD-hinge-NBD2 are 36.2 Å and 124°, respectively). The most closed conformation is seen in the SecA-SecY complex crystallized in the presence of ADP and BeFx (PDB accession code 3DIN, shown in red in Figure 1c; the distance between PPXD-NBD2 and the angle of PPXD-hinge-NBD2 are 7 Å and 74°, respectively)16. In the new structure (shown in blue in Figure 1c), the clamp is at an intermediate position (30 Å and 115°, respectively). The three structures from the same species define a path for the clamp from the open to the closed state.
Free energy analysis of clamp movements
We used molecular dynamics simulations to analyze the energetics of conformational transitions of the clamp. When each of the three T. maritima SecA structures was used as the initial state for the simulations, no large conformational changes were observed during the tested time period (~850 ns). The energy difference between the two SecA-ADP structures was estimated to be only 3–4 kcal/mol, as indicated by the significant overlap in the conformational space sampled (cyan and grey dots in Figure 2a). Repeated simulations of ~150 ns for each of the three SecA structures did not produce notably different behavior (Supplemental Figures S1 and S2).
Figure 2. Free energy analysis of clamp movements.

(a) The potential of mean force (PMF) of T. maritima SecA was calculated for clamp closure as a function of the HWD-PPXD distance (x-axis) and the PPXD-hinge-NBD2 angle (y-axis). The PMF is presented as a heat map ranging from dark purple (lowest energy) to bright yellow (highest energy). Also shown are contour lines for the indicated energy values (in kcal/mol). The scale is shown on the right. Structures of T. maritima SecA reported in this study (square), PDB 3JUX (circle), and PDB 3DIN (star) are mapped at corresponding positions. Points shown in cyan (new structure), gray (PDB 3JUX), and pink (SecA in PDB 3DIN) give the regions of conformational space sampled in equilibrium simulations starting from crystal structures. The green line represents the least-free-energy path connecting the open and closed clamp conformations of SecA.
(b) The PPXD-HSD interaction energy was calculated for clamp closure as a function of the HWD-PPXD distance (x-axis) and PPXD-hinge-NBD2 angle (y-axis). PPXD-HSD interaction energy is shown as a heat map ranging from dark purple (lowest energy) to bright yellow (highest energy), with the scale given on the right. Contour lines of constant energy in kcal/mol are also shown. Structures of SecA are mapped into the diagram by symbols as in (a). The minimum-energy path is shown in green.
(c) Binding SecY to SecA (right panel) interferes with the interaction between PPXD and HSD/HWD seen in the open clamp conformation of SecA in isolation (left panel). PPXD is colored light purple and pink in the open and closed conformations, respectively. HSD and HWD domains are shown in cyan, and the TM6/7 and TM8/9 loops of SecY in red and yellow, respectively. PPXD loop 319-327 binds to HSD in the open conformation (in blue) and moves outwards in the SecY-bound state (shown in magenta).
Next we calculated the free energy (Potential of Mean Force, PMF) as a function of the PPXD-HWD distance and PPXD-hinge-NBD2 angle (Figure 2a). The PMF is lowest (2 –3 kcal/mol) for the open clamp conformation (3JUX) and highest for the closed clamp conformation (3DIN; 15–16 kcal/mol). The free energy for the new intermediate conformation is about 5–6 kcal/mol.
Based on the free-energy plot, we calculated the least free-energy pathway21 (Figure 2a), which represents the most probable transition path between two states in our reduced coordinate space. Along this pathway, the PPXD, HSD, and HWD initially move together towards NBD2 for about one third of the distance. PPXD then separates from HSD and HWD, and continues to move alone towards NBD2 for the rest of the distance. This pathway is supported by calculations of the interaction energy between PPXD and HSD (Figure 2b). A significant drop in the interaction energy is observed at the point where the PPXD separates from the HWD and starts to move independently towards NBD2. In the open clamp conformation, the PPXD associates with both HSD and HWD with buried interfaces of 651.4 Å2 and 116.2 Å2, respectively. These interaction surfaces must be broken for the second phase of clamp movement. Both energy calculations indicate that the new structure of SecA-ADP is an intermediate in the transition from the open to the closed clamp conformation.
The transition from the intermediate to the closed conformation is likely caused by interactions of SecA with the SecY channel. Indeed, the loop between TM6 and TM7 of SecY inserts into the interface between PPXD and HSD/HWD and disrupts the interaction between the SecA domains (Figure 2c). In addition, loop 319-327 of PPXD, which associates with the HSD in the open conformation, rotates outwards and interacts with the loop between TM8 and TM9 of SecY in the closed clamp conformation (Figure 2c). The interactions between the PPXD and the HSD/HWD are thus replaced by interactions between SecA and SecY. These features and the energetic estimates suggest that binding of SecA to SecY is necessary to drive the conformational changes between the open and closed clamp states.
Molecular dynamics simulation of substrate binding to SecA
To understand how SecA interacts with a substrate polypeptide, we performed molecular dynamic simulations of T. maritima SecA in the presence of a 30-alanine substrate polypeptide. The substrate was initially placed as an extended chain into the closed clamp as seen in the SecA-SecY structure (PDB accession code: 3DIN) using the B. subtilis SecA-peptide structure15 to guide its placement within the clamp (Supplemental Figure S3a, left panel). After simulation run for 570 ns, we analyzed both the conformations of the substrate and of SecA.
Throughout the simulations, the substrate polypeptide formed 1–3 hydrogen bonds with the two-stranded β-sheet at the back of the clamp (Figure 3a), similar to what was observed in a structure of B. subtilis SecA with a bound peptide (PDB accession code: 3JV2)15. In agreement with the experimental observations, the region that associates with the back of the clamp adopts a β-strand conformation, in which the strand runs parallel to the exposed strand (residue 264-269) of the β-sheet (Figure 3a). The hydrogen bonds between substrate and the two β-strands form and break repeatedly during the simulations, suggesting that each segment of a polypeptide chain interacts only transiently during translocation (Supplemental Figure S3c). The dynamic interaction between peptide substrate and the two β-strands was also observed if the substrate was initially placed with its N-terminus in a bent conformation (Supplemental Figure S3b and S3c) or if B. subtilis SecA was used instead of T. maritima SecA (Supplemental Figure S4). These results support the idea that β-strand augmentation may allow sequence-independent substrate binding to the clamp of SecA15.
Figure 3. Molecular dynamics simulation of SecA with a peptide inside the clamp.

(a) A 30-amino acid peptide was placed inside the closed clamp of T. maritima SecA (see Figure S3a). Shown is the position of the main chain of the peptide (blue line) after 50 ns of the simulation run. A short segment of the peptide forms a β-strand (dark blue arrow) that interacts through hydrogen bonds (in yellow) with the β-sheet at the back of the clamp (cyan arrows). The right panel shows a magnified view of the augmented β-sheet.
(b) Peptide binding to the clamp moves the C-loop (residues 360-370) from the interior (right panels) towards the outside (left panels), where it interacts with NBD2. The C-loop in the two states is shown in magenta and orange, respectively. The upper panels show space-filling models, the lower ones show the secondary structure of the main chain. The left-most panel shows a magnified view of the interaction between the C-loop and NBD2 with the residues involved in stick presentation. The substrate peptide is in shown in ribbon presentation in blue.
The simulations also showed significant changes in SecA, particularly for the loop comprising residues 360-370 (C-loop; Figure 3b). In the closed conformation of the clamp (PDB accession code: 3DIN), the C-loop is located in the center of the clamp (Figure 3b). However, in the simulations with a translocating polypeptide, the loop is moved outwards and forms new interactions with NBD2. These interactions are electrostatic and involve residue R367 of PPXD and residues E485, E488 of NBD2 (Figure 3b). The net number of hydrogen bonds between the C-loop and NBD2 increases as well. All residues involved in the lateral closure of the clamp are conserved among SecA proteins from different bacterial species (Supplemental Figure S5). The C-loop-NBD2 interactions likely stabilize the closed state of the clamp, preventing the translocating polypeptide from exiting the clamp laterally. Consistent with this assumption, the C-loop stayed inside the clamp when the simulations were performed in the absence of substrate polypeptide (data not shown). Furthermore, the displacement of the conserved C-loop from the clamp and the formation of the exact same interactions between PPXD and NBD2 were also observed in simulations of a B. subtilis SecA – substrate complex (not shown).
To assess whether the interaction between the C-loop of PPXD and NBD2 is important for protein translocation, we mutated the conserved residue R367 to Glu and repeated the molecular dynamics simulations with a translocating peptide. A significant lateral opening of the clamp was observed (Figure 4a and Supplemental Figure S6), which resulted in increased dissociation of the substrate (not shown). Simulation of a triple mutant (E485R, E488R, and R367E) showed that the C-loop remained inside the clamp (Figure 4a). The closed conformation of the clamp in the triple mutant was stabilized by interactions between D350 of PPXD with R485 and R488 of NBD2, as well as by interactions between the backbone of loop 347 - 351 of PPXD with R485 of NBD2. R367E moved away from NBD2 in both mutants (Supplemental Figure S6), but the formation of the additional interactions between PPXD and NBD2 in the triple mutant locked the C-loop inside the clamp. We also observed changes in the HWD during the simulations with all SecA proteins, which are likely caused by the fact that several helices are no longer constrained when the PPXD has moved away from the HWD. Taken together, the simulations identified SecA mutants that either cannot properly close (R367E) or open (E485R, E488R, and R367E) their clamps.
Figure 4. Analysis of SecA mutants defective in clamp opening or closure.

(a) Comparison of the molecular dynamics simulations of wild type SecA (C-loop residues 360-370 in orange), a SecA mutant (R367E) defective in maintaining a closed clamp conformation (C-loop in blue), and a SecA mutant defective in clamp opening (R367E/E485R/E488R; C-loop in magenta). The alignment is based on NBD2. The left panel shows a magnified view of the interaction between the C-loop and NBD2, with residue 367 in all SecA proteins and E485/E488 in wild type SecA shown in stick presentation.
(b) Translocation assays were performed by incubating 35S-methionine labeled proOmpA (pOA) with wild type or mutant SecA and proteoliposomes containing SecY complex. The reactions were performed in the presence or absence of ATP. Proteinase K was added and material protected from proteolysis was analyzed by SDS-PAGE and autoradiography. The left most lane shows 20% of the input material.
(c) A translocation intermediate was generated with proOmpA containing a disulfide bridge at its the C-terminus in the presence of His-tagged SecA and proteoliposomes containing SecY complex and Ni-NTA lipids. The samples were incubated with imidazole and either wild type or mutant SecA, followed by addition of DTT as indicated by arrows. Samples taken at the indicated time points were treated with proteinase K and analyzed by SDS-PAGE and autoradiography. IM, intermediate; FL, full-length protein.
Translocation activity of clamp mutants
To experimentally test the role of the C-loop-NBD2 interaction, we performed in vitro translocation experiments. Radioactively labeled proOmpA (pOA), a post-translational substrate, was incubated with proteoliposomes containing SecY complex and either wild type SecA or SecA that carried the R367E mutation. Whereas wild type SecA translocated the substrate in the presence of ATP, the mutant showed only low activity and seemed to accumulate some translocation intermediates (Figure 4b). Because some SecA mutants are primarily affected in the initiation of translocation18, i.e. the assembly of substrate, SecA, and SecY complex, we performed a second assay in which we tested whether the SecA mutant can move a polypeptide when starting with a previously established translocation intermediate. The translocation intermediate was generated with proOmpA containing a disulfide bridge that prevents the C-terminus from moving into the vesicles. It was formed in the presence of N-terminal His-tag-substituted SecA (HisN-SecA) and proteoliposomes containing Ni-NTA lipids18. Then, imidazole and either wild type SecA or SecA R367E were added to replace His-SecA. With wild type SecA, the translocation intermediate was stable over time (Figure 4c, lanes 1–3). Upon addition of dithiothreitol (DTT) to reduce the disulfide bridge in proOmpA, the C-terminus of proOmpA was translocated, resulting in proteolytically protected full-length protein (lanes 4–6). In contrast, with the R367E mutant, the intermediate was not stable and its conversion into fully translocated product was inefficient (Figure 4c, lanes 7–12). Thus, a SecA mutant with defective clamp closure not only fails to initiate translocation, but also cannot sustain translocation when starting from an intermediate. Interestingly, the triple SecA mutant (E485R, E488R, and R367E), which is defective in clamp opening according to our molecular dynamics simulations, was totally inactive in both translocation assays (Figures 4b, 4c). Thus, clamp interactions need to be balanced to allow both its closing and its opening during translocation.
Discussion
Our results provide insight into the mechanism by which SecA translocates polypeptides through the SecY channel. Specifically, they elucidate the role of SecA’s clamp. Structural studies and molecular dynamics simulations indicate that the clamp moves from its open to its closed state in two phases. In a first phase, PPXD, HSD, and HWD move as a unit towards NBD2. Our new crystal structure at 1.9 Å resolution corresponds to an early intermediate during this phase. In the next phase, the interactions between PPXD and HSD/HWD are broken, and the PPXD alone continues to move towards NBD2. Our analysis shows that the second phase requires most of the free energy input. The disruption of the PPXD – HSD/HWD interfaces is likely triggered by binding of SecA to SecY. The insertion of the TM6/7 loop of SecY into the interface between PPXD and HSD/HWD is probably responsible for overcoming the energy barrier of clamp closure. A partial separation of PPXD from HWD and HSD has been observed in some structures of B. subtilis SecA in the absence of SecY10, 15. However, these conformational changes were probably caused by crystal contacts, as the N-terminal part of the long helix of HSD and a short helix of NBD2 from a neighboring molecule were inserted into the PPXD – HWD/HSD interface.
Based on these considerations, we suggest that clamp closure occurs when SecA binds to substrate and SecY channel. Our previous and current experiments (ref. 18 and Figure 4c) indicate that SecA can bind to substrate that is already located inside the SecY channel. Thus, in this case, clamp closure occurs around the translocating polypeptide chain. How the polypeptide chain would enter the clamp during initiation of translocation, when SecA, substrate, and SecY channel all come together and assemble into a complex, remains to be clarified.
Cross-linking experiments have shown that a substrate polypeptide moves through the clamp during protein translocation20. However, how substrate is accommodated inside the clamp was unknown, particularly because the closed clamp in crystal structures of SecA-SecY complexes did not leave space for a polypeptide substrate. We now show by molecular dynamic simulations that a conserved loop of SecA (called C-loop) is displaced from the interior of the clamp and forms interactions with NBD2 that stabilize the closed conformation of the clamp and prevents lateral exit of the translocating polypeptide chain.
The translocating polypeptide forms a short β-strand that interacts through a few hydrogen bonds with the two-stranded β-sheet at the back of the clamp15. Our simulations show that the interaction is weak, resulting in the continuous association and dissociation of polypeptide segments. During translocation, successive segments of a polypeptide chain would interact in a sequence-independent manner (Figure 5). The interaction needs to be relatively weak to allow substrate movement, but it must be strong enough to hold the polypeptide chain in the clamp above the channel. The actual translocation of the polypeptide chain is performed by the two-helix finger, whose movements are triggered by the ATP-hydrolysis cycle (Figure 5). A previous crystal structure supports the idea that the β-sheet at the back of the clamp can interact with extended polypeptide segments and cause them to form an additional β-strand15. However, in contrast to the molecular dynamics simulations, the clamp was not completely closed, likely because the structure was determined in the absence of SecY complex.
Figure 5. Model for SecA-mediated protein translocation.

SecA starts out in the cytosol with an open clamp. Upon binding to the SecY channel, PPXD moves and closes the clamp. The substrate polypeptide is embraced by the clamp and forms a short β-strand that interacts with the two-stranded β-sheet at the back of the clamp. During translocation, the two-helix finger of SecA moves towards the channel and drags the polypeptide chain with it. It then resets to grab the next polypeptide segment. The polypeptide chain can also slide back and forth through the SecA-SecY complex and only occasionally gets a push from the two-helix finger (“push-and-slide” model)18.
It is intriguing that the lateral opening of the clamp is aligned with the lateral gate of the SecY channel (Supplemental Figure S7). This suggests that a translocating polypeptide can laterally enter or exit both SecA and SecY at the same time. An obvious scenario in which this could happen is during the integration of a nascent membrane protein into the lipid phase: a TM segment located inside the SecY channel would exit its lateral gate into the surrounding lipid, and at the same time, the cytosolic domain of the translocating polypeptide would laterally exit the clamp of SecA. SecA is in fact required for the biosynthesis of some membrane proteins24, but whether it is involved in the actual membrane integration is unknown. Another, not mutually exclusive, possibility is that the alignment of the lateral gates is important for initiation of translocation of a secretory protein; the intercalation of the signal sequence into the lateral gate of SecY may be coordinated with the lateral entry of the following polypeptide segment into the clamp.
Methods
Protein crystallization and structure determination
T. maritima SecA-SecY complex with a His-tag on the C-terminus of SecA was prepared as previously described16. One microliter of the protein solution at 15 mg/ml was mixed with an equal volume of well solution containing 100 mM imidazole, pH 7.0, and 50% methyl-2,4-pentanediol. Crystals were obtained at 22°C in three days by the hanging-drop vapor diffusion method, and were directly frozen in liquid nitrogen. Diffraction data were collected at beamline 24ID-C at the Argonne National Laboratory, and were processed using the Denzo and Scalepack as part of the HKL2000 software package25. The structure was determined by molecular replacement using PHASER in PHENIX software suite, using a published T. maritima SecA structure (PDB accession code 3JUX) as search model15, 26. Only SecA and bound ADP were present in the structure. The model was modified in Coot27, 28, and refined with Phenix.refine26, 29. The final refined atomic model contained residues 2-816 of T. maritima SecA followed by three amino acids of the tag. It also contained ADP, Mg2+, and water molecules. Figures showing structures were prepared in PyMOL30. All software packages were accessed through SBGrid31.
Molecular Dynamics simulations
Starting models were derived from crystal structures of (1) T. maritima SecA bound to ADP (3JUX), (2) T. maritima SecA bound to ADP.BeFx (3DIN), and (3) the new SecA structure reported here. Additionally, two systems incorporating a 30-aminoacid substrate peptide were constructed starting from (1) T. maritima SecA bound to ADP.BeFx (3DIN) and (2) B. subtilis SecA bound to a short peptide (3JV2). For all systems, SecA was solvated in TIP3P water sufficient to provide 15–30 Å between periodic images of the protein and ionized with 150 mM of KCl. System sizes ranged from 114,000–144,000 atoms. Simulations of T. maritima SecA were carried out at 353 K, which is the organism’s optimal growth temperature, while those of B. subtilis SecA were carried out at 310 K. All simulations were run using the CHARMM36 force field32, while setup and analysis were performed in VMD33. Initial equilibration runs of 10–50 ns were performed using the MD software NAMD34.
Long-time simulations of the new structure of SecA as well of T. maritima SecA bound to ADP.BeFx (3DIN) and T. maritima SecA bound to ADP (3JUX) were carried out using the special-purpose Anton supercomputer at Pittsburgh Supercomputing Center35 for approximately 150 ns or 850 ns each. The two systems containing a 30-amino-acid substrate peptide were run for 140 ns or 570 ns on Anton. Additionally, two mutants of the T. maritima SecA bound to substrate peptide, R367E and the triple mutant E485R/E488R/R367E, were modified in silico and also simulated for 570 ns each. Approximately 5.6 μs of equilibrium simulations were run in total.
Free energy analysis
The two-dimensional potential of mean force (PMF) for SecA as a function of two reaction coordinates, namely HWD-PPXD distance and NBD2-hinge-PPXD angle, was calculated using the adaptive biasing forces (ABF) method36 in NAMD37. Initial states of SecA at different separation distances and angles were generated using targeted MD, in which SecA was driven from its conformation in 3JUX to that in 3DIN over the course of a 15-ns simulation. To increase the efficiency of sampling, the 2D parameter space was broken up into 24 windows, with those along the distance ranging from 26–49 Å in 6-Å increments (four windows) and those along the angle ranging from 65–135° in 10–15° increments (six windows). Convergence of the PMF in each window was assumed when the system could diffuse freely along both reaction coordinates. A total of ~600 ns was used to obtain the PMF.
In vitro protein translocation assays
SecA and SecYEG were purified as described before20. SecYEG was reconstituted into proteoliposomes containing E. coli polar lipids as described before18. pOA-CC(61) (E.coli pOA with residues 176-297 deleted, containing two cysteines at position 167 and 205, inserted into the pGEM3G vector 38) was synthesized in vitro in rabbit reticulocyte lysate in the presence of 35S-methionine. The samples were precipitated with ammonium sulfate and resuspended in one volume of 20 mM Hepes pH 6.8, 6 M urea. To test for translocation, 200 nM SecA was mixed with 100 nM SecYEG in proteoliposomes, 5 mM ATP, and pOA-CC(61) (diluted 1:50) in buffer containing 20 mM Hepes, 50 mM KCl, 5 mM MgCl2. For reactions lacking ATP, 0.4 U/ml hexokinase and 20 mM glucose were added instead of ATP. Reactions were incubated for 10 min at 37°C, followed by the addition of 0.2 mg/ml proteinase K (Pierce) for 30 min on ice. Protected fragments were precipitated by 10% TCA and analyzed by SDS-PAGE and autoradiography.
Translocation assays starting with an intermediate were performed as follows. A translocation intermediate was generated by mixing 200 nM SecA-HisDN20 (Ref. 18), 100 nM SecYEG reconstituted into proteoliposomes containing 90% E. coli polar lipids and 10% Ni-NTA-DGS, pOA-CC(61) (diluted 1:5) in buffer containing 20 mM Hepes, pH7.5, 50 mM KCl, 5 mM MgCl2, and 400 nM sodium tetrathionate. After 15 min at 37°C, the samples were incubated with 400 nM SecA mutants and 250 mM imidazole for 20 min at 37°C. Translocation of the C-terminus of pOA-CC(61) was then started by the addition of 50 mM DTT. Samples were taken at the indicated time points, treated with 0.2 mg/ml proteinase K for 30 min on ice, precipitated with 10% TCA, and analyzed by SDS-PAGE followed by autoradiography.
Supplementary Material
Table 1.
Data collection and refinement statistics
| T. maritima SecA bound to ADP | |
|---|---|
| Data collection | |
| Resolution limit (Å) | 1.897 |
| Space group | P 21 21 21 |
| Cell dimensions | |
| a, b, c (Å) | 64.404, 110.16, 115.863 |
| α, β, γ (°) | 90, 90, 90 |
| Total reflections | 364622 |
| Rsym | 0.086 (0.059) |
| Mean I/σI | 13.21 (1.24) |
| Completeness (%) | 98.8 (97.0) |
| Redundancy | 3.0 (2.8) |
| Wavelength (Å) | 0.97914 |
| Refinement | |
| Resolution range (Å) | 39.92 – 1.897 |
| Rwork/Rfree (%) | 17.86/22.64 |
| Number of reflections | |
| Total | 65151 |
| Rfree | 3300 |
| No. non-hydrogen atoms | 7090 |
| Protein | 6598 |
| Ligand/ion | 28 |
| Average B-factor | 28.54 |
| Protein | 28.32 |
| Ligand/ion | 18.36 |
| Solvent | 32.23 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.009 |
| Bond angles (°) | 1.11 |
| Ramachandran analysis (%) | |
| Favored | 98 |
| Allowed | 1.7 |
| Outliers | 0 |
Statistics for the highest-resolution shell are shown in parentheses. Rsym=ΣhklΣi|Ii(hkl)−I(hkl)|/ΣhklΣi|Ii(hkl)|, where I(hkl) is the integrated intensity of the reflection. R=Σ|Fobs−Fcalc|/ΣFobs, where Fobs and Fcalc are observed and calculated structure factors, respectively. Rfree was calculated with 5% of the reflections.
Highlights.
The clamp of SecA undergoes open to closed conformations and recognizes substrate polypeptides
Closure of SecA clamp occurs in two phases, and a new structure of an intermediate state is reported
Substrate associates with the back of the clamp by dynamic hydrogen bonding
SecA clamp is laterally closed by a conserved loop that is essential for protein translocation
Conformational changes of SecA clamp allows substrate binding and movement during translocation
Acknowledgments
We thank the staff at the Advanced Photon Source of the Northeastern Collaborative Access Team (NE-CAT) beamlines for help with data collection. NE-CAT is supported by a grant from the National Institute of General Medical Sciences (P41 GM103403) from the National Institutes of Health (NIH). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. NAMD simulations were carried out using the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by NSF grant number OCI-1053575. Anton computer time was provided by the National Resource for Biomedical Supercomputing and the Pittsburgh Supercomputing Center through NIH Grant RC2GM093307, using a machine donated by DE Shaw Research. Y.C. was supported by an HHMI-Helen Hay Whitney Foundation fellowship. B.W.B. was the recipient of a fellowship from the Boehringer Ingelheim Fonds. The work in the laboratory of T.A.R. was supported by NIH grant GM052586. T.A.R. is a Howard Hughes Medical Institute Investigator. J.C.G. is supported by NIH grant K22-AI100927.
Footnotes
Accession code
Coordinates for the new crystal structure of T. maritima SecA with bound ADP at 1.9Å resolution have been deposited at the RSCB Protein Data Bank (PDB) with the accession code 4YS0.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rapoport TA. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature. 2007;450:663–669. doi: 10.1038/nature06384. [DOI] [PubMed] [Google Scholar]
- 2.Park E, Rapoport TA. Mechanisms of Sec61/SecY-mediated protein translocation across membranes. Annual review of biophysics. 2012;41:21–40. doi: 10.1146/annurev-biophys-050511-102312. [DOI] [PubMed] [Google Scholar]
- 3.Driessen AJ, Nouwen N. Protein translocation across the bacterial cytoplasmic membrane. Annu Rev Biochem. 2008;77:643–667. doi: 10.1146/annurev.biochem.77.061606.160747. [DOI] [PubMed] [Google Scholar]
- 4.Mori H, Ito K. The Sec protein-translocation pathway. Trends Microbiol. 2001;9:494–500. doi: 10.1016/s0966-842x(01)02174-6. [DOI] [PubMed] [Google Scholar]
- 5.Denks K, Vogt A, Sachelaru I, Petriman NA, Kudva R, Koch HG. The Sec translocon mediated protein transport in prokaryotes and eukaryotes. Mol Membr Biol. 2014;31:58–84. doi: 10.3109/09687688.2014.907455. [DOI] [PubMed] [Google Scholar]
- 6.Matlack KE, Misselwitz B, Plath K, Rapoport TA. BiP acts as a molecular ratchet during posttranslational transport of prepro-alpha factor across the ER membrane. Cell. 1999;97:553–564. doi: 10.1016/s0092-8674(00)80767-9. [DOI] [PubMed] [Google Scholar]
- 7.Economou A, Wickner W. SecA promotes preprotein translocation by undergoing ATP-driven cycles of membrane insertion and deinsertion. Cell. 1994;78:835–843. doi: 10.1016/s0092-8674(94)90582-7. [DOI] [PubMed] [Google Scholar]
- 8.Vrontou E, Economou A. Structure and function of SecA, the preprotein translocase nanomotor. Biochim Biophys Acta. 2004;1694:67–80. doi: 10.1016/j.bbamcr.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 9.Hunt JF, Weinkauf S, Henry L, Fak JJ, McNicholas P, Oliver DB, et al. Nucleotide control of interdomain interactions in the conformational reaction cycle of SecA. Science. 2002;297:2018–2026. doi: 10.1126/science.1074424. [DOI] [PubMed] [Google Scholar]
- 10.Osborne AR, Clemons WM, Jr, Rapoport TA. A large conformational change of the translocation ATPase SecA. Proc Natl Acad Sci U S A. 2004;101:10937–10942. doi: 10.1073/pnas.0401742101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zimmer J, Li W, Rapoport TA. A novel dimer interface and conformational changes revealed by an X-ray structure of B. subtilis SecA. J Mol Biol. 2006;364:259–265. doi: 10.1016/j.jmb.2006.08.044. [DOI] [PubMed] [Google Scholar]
- 12.Papanikolau Y, Papadovasilaki M, Ravelli RB, McCarthy AA, Cusack S, Economou A, et al. Structure of dimeric SecA, the Escherichia coli preprotein translocase motor. J Mol Biol. 2007;366:1545–1557. doi: 10.1016/j.jmb.2006.12.049. [DOI] [PubMed] [Google Scholar]
- 13.Sharma V, Arockiasamy A, Ronning DR, Savva CG, Holzenburg A, Braunstein M, et al. Crystal structure of Mycobacterium tuberculosis SecA, a preprotein translocating ATPase. Proc Natl Acad Sci U S A. 2003;100:2243–2248. doi: 10.1073/pnas.0538077100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vassylyev DG, Mori H, Vassylyeva MN, Tsukazaki T, Kimura Y, Tahirov TH, et al. Crystal structure of the translocation ATPase SecA from Thermus thermophilus reveals a parallel, head-to-head dimer. J Mol Biol. 2006;364:248–258. doi: 10.1016/j.jmb.2006.09.061. [DOI] [PubMed] [Google Scholar]
- 15.Zimmer J, Rapoport TA. Conformational flexibility and peptide interaction of the translocation ATPase SecA. J Mol Biol. 2009;394:606–612. doi: 10.1016/j.jmb.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zimmer J, Nam Y, Rapoport TA. Structure of a complex of the ATPase SecA and the protein-translocation channel. Nature. 2008;455:936–943. doi: 10.1038/nature07335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Erlandson KJ, Miller SB, Nam Y, Osborne AR, Zimmer J, Rapoport TA. A role for the two-helix finger of the SecA ATPase in protein translocation. Nature. 2008;455:984–987. doi: 10.1038/nature07439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bauer BW, Shemesh T, Chen Y, Rapoport TA. A “push and slide” mechanism allows sequence-insensitive translocation of secretory proteins by the SecA ATPase. Cell. 2014;157:1416–1429. doi: 10.1016/j.cell.2014.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim DM, Zheng H, Huang YJ, Montelione GT, Hunt JF. ATPase active-site electrostatic interactions control the global conformation of the 100 kDa SecA translocase. J Am Chem Soc. 2013;135:2999–3010. doi: 10.1021/ja306361q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bauer BW, Rapoport TA. Mapping polypeptide interactions of the SecA ATPase during translocation. Proc Natl Acad Sci U S A. 2009;106:20800–20805. doi: 10.1073/pnas.0910550106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ensing B, Laio A, Parrinello M, Klein ML. A recipe for the computation of the free energy barrier and the lowest free energy path of concerted reactions. The journal of physical chemistry B. 2005;109:6676–6687. doi: 10.1021/jp045571i. [DOI] [PubMed] [Google Scholar]
- 22.Natale P, den Blaauwen T, van der Does C, Driessen AJ. Conformational state of the SecYEG-bound SecA probed by single tryptophan fluorescence spectroscopy. Biochemistry. 2005;44:6424–6432. doi: 10.1021/bi047488r. [DOI] [PubMed] [Google Scholar]
- 23.Robson A, Gold VA, Hodson S, Clarke AR, Collinson I. Energy transduction in protein transport and the ATP hydrolytic cycle of SecA. Proc Natl Acad Sci U S A. 2009;106:5111–5116. doi: 10.1073/pnas.0809592106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deitermann S, Sprie GS, Koch HG. A dual function for SecA in the assembly of single spanning membrane proteins in Escherichia coli. J Biol Chem. 2005;280:39077–39085. doi: 10.1074/jbc.M509647200. [DOI] [PubMed] [Google Scholar]
- 25.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 26.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 29.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC.
- 31.Morin A, Eisenbraun B, Key J, Sanschagrin PC, Timony MA, Ottaviano M, et al. Collaboration gets the most out of software. eLife. 2013:e01456. doi: 10.7554/eLife.01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Best RB, Zhu X, Shim J, Lopes PE, Mittal J, Feig M, et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone phi, psi and side-chain chi(1) and chi(2) dihedral angles. Journal of chemical theory and computation. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. Journal of molecular graphics. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 34.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. Journal of computational chemistry. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaw DE, Dror RO, Salmon JK, Grossman JP, Mackenzie KM, Bank JA, et al. Millisecond-scale molecular dynamics simulations on Anton. Proceedings of the Conference on High Performance Computing Networking, Storage and Analysis; 2009. p. 39. [Google Scholar]
- 36.Darve E, Pohorille A. Calculating free energies using average force. The Journal of Chemical Physics. 2001;115 [Google Scholar]
- 37.Henin J, Fiorin G, Chipot C, Klein ML. Exploring Multidimensional Free Energy Landscapes Using Time-Dependent Biases on Collective Variables. Journal of chemical theory and computation. 2010;6:35–47. doi: 10.1021/ct9004432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Erlandson KJ, Or E, Osborne AR, Rapoport TA. Analysis of polypeptide movement in the SecY channel during SecA-mediated protein translocation. J Biol Chem. 2008;283:15709–15715. doi: 10.1074/jbc.M710356200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.