

An evolutionarily conserved quality control in ribosome biogenesis reveals that two human rRNA base methyltransferases associated with cell differentiation and cancer but, surprisingly, not their RNA-modifying activity are required for small ribosomal subunit biogenesis.
Abstract
At the heart of the ribosome lie rRNAs, whose catalytic function in translation is subtly modulated by posttranscriptional modifications. In the small ribosomal subunit of budding yeast, on the 18S rRNA, two adjacent adenosines (A1781/A1782) are N6-dimethylated by Dim1 near the decoding site, and one guanosine (G1575) is N7-methylated by Bud23-Trm112 at a ridge between the P- and E-site tRNAs. Here we establish human DIMT1L and WBSCR22-TRMT112 as the functional homologues of yeast Dim1 and Bud23-Trm112. We report that these enzymes are required for distinct pre-rRNA processing reactions leading to synthesis of 18S rRNA, and we demonstrate that in human cells, as in budding yeast, ribosome biogenesis requires the presence of the modification enzyme rather than its RNA-modifying catalytic activity. We conclude that a quality control mechanism has been conserved from yeast to human by which binding of a methyltransferase to nascent pre-rRNAs is a prerequisite to processing, so that all cleaved RNAs are committed to faithful modification. We further report that 18S rRNA dimethylation is nuclear in human cells, in contrast to yeast, where it is cytoplasmic. Yeast and human ribosome biogenesis thus have both conserved and distinctive features.
INTRODUCTION
Ribosomes are essential to protein production in all living cells. At the core of the translational machinery reside rRNAs, whose catalytic function in translation is subtly modulated by posttranscriptional modifications (Lafontaine, 2015). Eukaryotic rRNAs are extensively modified by 2′-O methylation and pseudouridylation, carried out, respectively, by box C/D and box H/ACA small nucleolar ribonucleoprotein (RNP) particles (Watkins and Bohnsack, 2012). In addition, conventional stand-alone protein enzymes modify some bases—six on the small and six on the large ribosomal subunit (Sharma and Lafontaine, 2015).
Although the precise involvement of rRNA base modifications in translation is not fully understood, their conservation and conspicuous presence at functional sites suggest that they are important. The 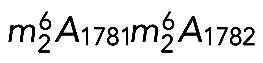 modification (yeast numbering), in the 18S rRNA 3′ minor domain, is on the apical loop of a highly conserved hairpin (helix 45), which folds near the decoding site in the three-dimensional structure of the ribosome (Figure 1). The m7G1575 modification, in the 18S rRNA 3′ major domain, is at a ridge forming a steric block between the P-site and E-site tRNAs at the back of the small subunit head (Figure 1). The
modification (yeast numbering), in the 18S rRNA 3′ minor domain, is on the apical loop of a highly conserved hairpin (helix 45), which folds near the decoding site in the three-dimensional structure of the ribosome (Figure 1). The m7G1575 modification, in the 18S rRNA 3′ major domain, is at a ridge forming a steric block between the P-site and E-site tRNAs at the back of the small subunit head (Figure 1). The 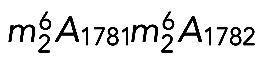 modification is one of the most highly conserved rRNA base modifications (van Knippenberg, 1986; Rife, 2009). With a few exceptions, including organellar ribosomes of Euglena gracilis and Saccharomyces cerevisiae, it is present on all ribosomes inspected to date in all three kingdoms of life (van Knippenberg, 1986; Rife, 2009). In a recent phylogenetic analysis focused on Mollicutes parasitic bacteria, which has undergone massive genome reduction during evolution, investigators defined a minimal set of core ribosome biogenesis and translation factors (Grosjean et al., 2014). They showed that KsgA (the bacterial homologue of Dim1) is part of this set. This is significant because KsgA is one of the very few rRNA modification enzymes to have been retained during Mollicutes genomic erosion (Grosjean et al., 2014). This supports the notion that KsgA is primordial and was presumably present in the last common ancestor (Rife, 2009). The m7G1575 modification, on the other hand, is conserved throughout eukaryotes but is not found in bacteria or Archaea.
modification is one of the most highly conserved rRNA base modifications (van Knippenberg, 1986; Rife, 2009). With a few exceptions, including organellar ribosomes of Euglena gracilis and Saccharomyces cerevisiae, it is present on all ribosomes inspected to date in all three kingdoms of life (van Knippenberg, 1986; Rife, 2009). In a recent phylogenetic analysis focused on Mollicutes parasitic bacteria, which has undergone massive genome reduction during evolution, investigators defined a minimal set of core ribosome biogenesis and translation factors (Grosjean et al., 2014). They showed that KsgA (the bacterial homologue of Dim1) is part of this set. This is significant because KsgA is one of the very few rRNA modification enzymes to have been retained during Mollicutes genomic erosion (Grosjean et al., 2014). This supports the notion that KsgA is primordial and was presumably present in the last common ancestor (Rife, 2009). The m7G1575 modification, on the other hand, is conserved throughout eukaryotes but is not found in bacteria or Archaea.
FIGURE 1:

The 18S rRNA m7G and 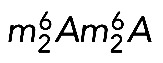 modifications are conserved in S. cerevisiae and Homo sapiens. (A) Secondary structure of the 18S rRNA. The insets illustrate conservation of rRNA sequence and secondary structure near the N7-methylguanosine (m7G) modification introduced by Bud23-Trm112 in yeast and WBSCR22-TRMT112 in humans and in the vicinity of the two N6,N6-dimethyladenosines
modifications are conserved in S. cerevisiae and Homo sapiens. (A) Secondary structure of the 18S rRNA. The insets illustrate conservation of rRNA sequence and secondary structure near the N7-methylguanosine (m7G) modification introduced by Bud23-Trm112 in yeast and WBSCR22-TRMT112 in humans and in the vicinity of the two N6,N6-dimethyladenosines 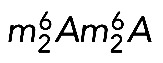 synthesized by Dim1 in yeast and DIMT1L in humans. Modified nucleotide positions are highlighted in red. The 5′, central (C), 3′ major (3′ M), and 3′ minor (3′ m) domains are indicated. (B) Three-dimensional representation of the yeast small subunit (model based on Protein Data Bank entry 3U5B) with posttranscriptional modifications and functional sites highlighted. The decoding site (DCS, in cyan) at the base of helix 44 (in anthracite) and the mRNA entry (green circle) and exit (red circle) sites are indicated. Residues shown as green and red spheres are 2′-O methylated or pseudouridylated, respectively. Bd, body; Bk, beak; H, head; Lf, left foot; Nk, neck; Pt; platform, Rf, right foot; Sh, shoulder.
synthesized by Dim1 in yeast and DIMT1L in humans. Modified nucleotide positions are highlighted in red. The 5′, central (C), 3′ major (3′ M), and 3′ minor (3′ m) domains are indicated. (B) Three-dimensional representation of the yeast small subunit (model based on Protein Data Bank entry 3U5B) with posttranscriptional modifications and functional sites highlighted. The decoding site (DCS, in cyan) at the base of helix 44 (in anthracite) and the mRNA entry (green circle) and exit (red circle) sites are indicated. Residues shown as green and red spheres are 2′-O methylated or pseudouridylated, respectively. Bd, body; Bk, beak; H, head; Lf, left foot; Nk, neck; Pt; platform, Rf, right foot; Sh, shoulder.
In budding yeast, the twin modification N6,N6-dimethyladenosine 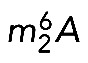 is synthesized by Dim1 and the 7-methylguanosine (m7G) modification by Bud23-Trm112 (Lafontaine et al., 1994; White et al., 2008; Figaro et al., 2012). In bacterial cells lacking ksgA, expression of the DIM1 gene restores 16S rRNA dimethylation and sensitivity to the aminoglycoside antibiotic kasugamycin (Lafontaine et al., 1994), suggesting that Dim1 is active by itself. Yet, in yeast cells, Dim1 activity is inhibited upon depletion of the assembly factor Dim2 (Pno1; Vanrobays et al., 2004), which binds to precursor ribosomes near the dimethylation site (Strunk et al., 2011) and presumably affects its structure. In contrast to Dim1, which does not seem to require any coactivator, Bud23 is the catalytic subunit of a heterodimeric Bud23-Trm112 methyltransferase complex, and it strictly requires its coactivator Trm112 to gain metabolic stability (Figaro et al., 2012; Sardana and Johnson, 2012; Létoquart et al., 2014).
is synthesized by Dim1 and the 7-methylguanosine (m7G) modification by Bud23-Trm112 (Lafontaine et al., 1994; White et al., 2008; Figaro et al., 2012). In bacterial cells lacking ksgA, expression of the DIM1 gene restores 16S rRNA dimethylation and sensitivity to the aminoglycoside antibiotic kasugamycin (Lafontaine et al., 1994), suggesting that Dim1 is active by itself. Yet, in yeast cells, Dim1 activity is inhibited upon depletion of the assembly factor Dim2 (Pno1; Vanrobays et al., 2004), which binds to precursor ribosomes near the dimethylation site (Strunk et al., 2011) and presumably affects its structure. In contrast to Dim1, which does not seem to require any coactivator, Bud23 is the catalytic subunit of a heterodimeric Bud23-Trm112 methyltransferase complex, and it strictly requires its coactivator Trm112 to gain metabolic stability (Figaro et al., 2012; Sardana and Johnson, 2012; Létoquart et al., 2014).
Eukaryotic ribosome biogenesis is a complicated multistep process. Ribosome production is initiated in the nucleolus, where long precursor rRNAs are synthesized and initial RNA processing, RNA modification, and RNP assembly reactions take place (Woolford and Baserga, 2013; Fernandez-Pevida et al., 2015; Henras et al., 2015). Ribosome biogenesis then proceeds in the nucleoplasm and is finalized in the cytoplasm, where protruding subunit structures are formed and important proofreading steps occur (Lafontaine, 2015). Remarkably, both 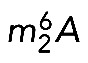 formation and m7G synthesis are late events in small-subunit biogenesis, whereas Dim1 and Bud23-Trm112 association with preribosomes are early steps occurring on nascent nucleolar transcripts (Grandi et al., 2002; Figaro et al., 2012; Létoquart et al., 2014). This implies that the methylation activities of Dim1 and Bud23-Trm112 are specifically delayed, presumably until subunit assembly has achieved sufficient maturation and a modifiable substrate has been produced. Characterization of catalytically defective variants of Dim1 and Bud23 has led to the conclusion that the modifications catalyzed by these enzymes are not required for yeast cell growth, at least under optimal laboratory conditions (Lafontaine et al., 1998; White et al., 2008; Létoquart et al., 2014). This was unexpected, as Dim1 is essential to cell growth and bud23Δ and trm112Δ cells are strongly impaired in growth, particularly at low temperature (Lafontaine et al., 1994; White et al., 2008; Figaro et al., 2012; Létoquart et al., 2014). It turns out that the presence of Dim1 and Bud23-Trm112 within precursor ribosomes, rather than their RNA-methylating catalytic activity, is required for pre-rRNA processing reactions leading to synthesis of small-subunit 18S rRNA (Lafontaine et al., 1995, 1998; White et al., 2008). On the basis of these observations, it is suggested that in yeast, Dim1 and Bud23 participate in robust quality control mechanisms that make binding of the methyltransferase to precursor rRNAs a prerequisite to processing (Lafontaine et al., 1995; White et al., 2008), thus committing all cleaved RNAs to modification.
formation and m7G synthesis are late events in small-subunit biogenesis, whereas Dim1 and Bud23-Trm112 association with preribosomes are early steps occurring on nascent nucleolar transcripts (Grandi et al., 2002; Figaro et al., 2012; Létoquart et al., 2014). This implies that the methylation activities of Dim1 and Bud23-Trm112 are specifically delayed, presumably until subunit assembly has achieved sufficient maturation and a modifiable substrate has been produced. Characterization of catalytically defective variants of Dim1 and Bud23 has led to the conclusion that the modifications catalyzed by these enzymes are not required for yeast cell growth, at least under optimal laboratory conditions (Lafontaine et al., 1998; White et al., 2008; Létoquart et al., 2014). This was unexpected, as Dim1 is essential to cell growth and bud23Δ and trm112Δ cells are strongly impaired in growth, particularly at low temperature (Lafontaine et al., 1994; White et al., 2008; Figaro et al., 2012; Létoquart et al., 2014). It turns out that the presence of Dim1 and Bud23-Trm112 within precursor ribosomes, rather than their RNA-methylating catalytic activity, is required for pre-rRNA processing reactions leading to synthesis of small-subunit 18S rRNA (Lafontaine et al., 1995, 1998; White et al., 2008). On the basis of these observations, it is suggested that in yeast, Dim1 and Bud23 participate in robust quality control mechanisms that make binding of the methyltransferase to precursor rRNAs a prerequisite to processing (Lafontaine et al., 1995; White et al., 2008), thus committing all cleaved RNAs to modification.
In bacteria, KsgA is both important for cell growth at low temperature and required for pre-rRNA processing (Connolly et al., 2008). Its binding sites on 30S subunits overlap with that of initiation factor 3 (IF3; Xu et al., 2008). This prevents incompletely matured subunits from entering the translation cycle before methylation (Xu et al., 2008; Boehringer et al., 2012). Structural studies further demonstrated that KsgA binding prevents helix 44 from adopting its conformation in mature 30S, thus blocking decoding site (DCS) formation and subunit joining (Boehringer et al., 2012). Like KsgA, yeast Dim1 binds at the DCS (Granneman et al., 2010; Strunk et al., 2011), preventing de facto, together with other assembly factors, premature translation initiation (reviewed in Lafontaine, 2015).
In eukaryotes other than yeast and in Homo sapiens in particular, the precise functions of Dim1 and Bud23-Trm112 in ribosome biogenesis have not yet been fully explored. In A. thaliana, Dim1 is required for organized root growth, epidermal patterning, and chloroplast development at low temperature (Tokuhisa et al., 1998; Wieckowski and Schiefelbein, 2012), and WBSCR22 (known as RID2 in plants) is required for reactivation of cell division during dedifferentiation, a process essential to organ regeneration and wound healing (Ohbayashi et al., 2011). In human cells, WBSCR22 (MERM1) is required for ribosome biogenesis (Wild et al., 2010; Ounap et al., 2013; Tafforeau et al., 2013; Haag et al., 2015) and is known as a breast cancer biomarker and metastasis promoter, a therapeutic target in myeloma, and an enhancer of glucocorticoid receptor function that is regulated during lung inflammation and cancer (Jangani et al., 2014). It is also one of the numerous genes associated with the Williams–Beuren neurodevelopmental syndrome (Doll and Grzeschik, 2001; Nakazawa et al., 2011; Tiedemann et al., 2012).
In the present study we characterized the human homologues of Dim1 and Bud23-Trm112. Our work led us to conclude that the functions of DIMT1L and WBSCR22-TRMT112 in pre-rRNA processing and modification can be uncoupled, that DIMT1L is required for earlier processing steps than WBSCR22-TRMT112, and that in human cells, dimethylation takes place in the nucleus, whereas it is reported to be cytoplasmic in yeast.
RESULTS
DIMT1L is a nucleolar protein, and WBSCR22 and TRMT112 are nucleocytoplasmic
To gain initial insight into the functions of DIMT1L, WBSCR22, and TRMT112, we determined the subcellular distribution of the endogenous proteins by fluorescence microscopy in HeLa cells expressing the nucleolar antigen fibrillarin as a green fluorescent fusion (FIB-GFP; Figure 2A). DIMT1L was detected almost exclusively in the nucleoli, with weak nucleoplasmic staining (Figure 2A). WBSCR22 and TRMT112 were observed in the nucleoplasm, with for TRMT112 a clear exclusion from the nucleolus. In addition, TRMT112 was found to decorate a perinuclear “basket” polarized at one end of the nucleus (arrows; Figure 2A), which was even more evident in nonmanipulated HeLa cells, in which FIB-GFP is not stably expressed (Figure 2B). The TRMT112 perinuclear “basket” was always detected on the same side of the nucleus as the Golgi and lysosomes, showing partial colocalization with these structures (Figure 2C). Some WBSCR22 signal was consistently detected outside the nucleus, suggesting that this protein might also be part of such a perinuclear structure (Figure 1B). The nucleolar and nuclear localization of DIMT1L and WBSCR22 was confirmed in cells expressing Flag-tagged constructs (see later discussion; unpublished data).
FIGURE 2:

Subcellular distribution of DIMT1L, WBSCR22, and TRMT112. (A–C) DIMT1L is a nucleolar protein. WBSCR22 and TRMT112 localize to the nucleoplasm, with nucleolar exclusion for TRMT112, and to a polarized perinuclear structure (white arrows), overlapping partially with the Golgi and lysosomes. Cells were counterstained with DAPI for DNA labeling. Image sections were captured in confocal mode with a Yokogawa spinning disk on a Zeiss Axiovert microscope at 40× magnification. Scale bar, 10 μm. (A) HeLa cells stably expressing the nucleolar protein fibrillarin fused to GFP (FIB-GFP) were processed for immunofluorescence with a primary antibody specific to DIMT1L, WBSCR22, or TRMT112 and a red fluorescent secondary antibody. The intrinsic GFP fluorescence was used to visualize fibrillarin. (B) HeLa cells decorated with a WBSCR22- or TRMT112-specific primary antibody, revealed with a green fluorescent secondary antibody. (C) HeLa cells incubated with either Lyso- or Golgi-tracker and decorated with a TRMT112-specific primary antibody, revealed with a red fluorescent secondary antibody.
WBSCR22 and TRMT112 form a heterodimeric methyltransferase complex
In budding yeast, the 18S rRNA m7G1575 methylation is carried out by a heterodimeric complex consisting of Bud23 and Trm112 (Figaro et al., 2012). In this complex, Bud23 acts as a catalytic subunit and Trm112 as a coactivator, conferring metabolic stability to Bud23. Determination of the crystal structure of yeast Bud23-Trm112 established that the interaction between Bud23 and Trm112 involves formation of a conserved β-zipper, masking, in the water-based cell environment, a naturally unfavorable large hydrophobic surface on Bud23 (Létoquart et al., 2014).
The considerable primary sequence conservation between Bud23 and WBSCR22 and between Trm112 and TRMT112 and their distribution in the same subcellular compartments are compatible with their acting as a complex in human cells also. Consistently, we found polyhistidine-tagged WBSCR22 to copurify efficiently with TRMT112 when the proteins were coexpressed in bacteria (Figure 3, A–C, lane 5). As control, no such copurification is seen in cells expressing only untagged TRMT112 (Figure 3, A–C, lanes 6–9). WBSCR22 is largely insoluble and could not be stably isolated in the absence of TRMT112 coexpression (unpublished data).
FIGURE 3:
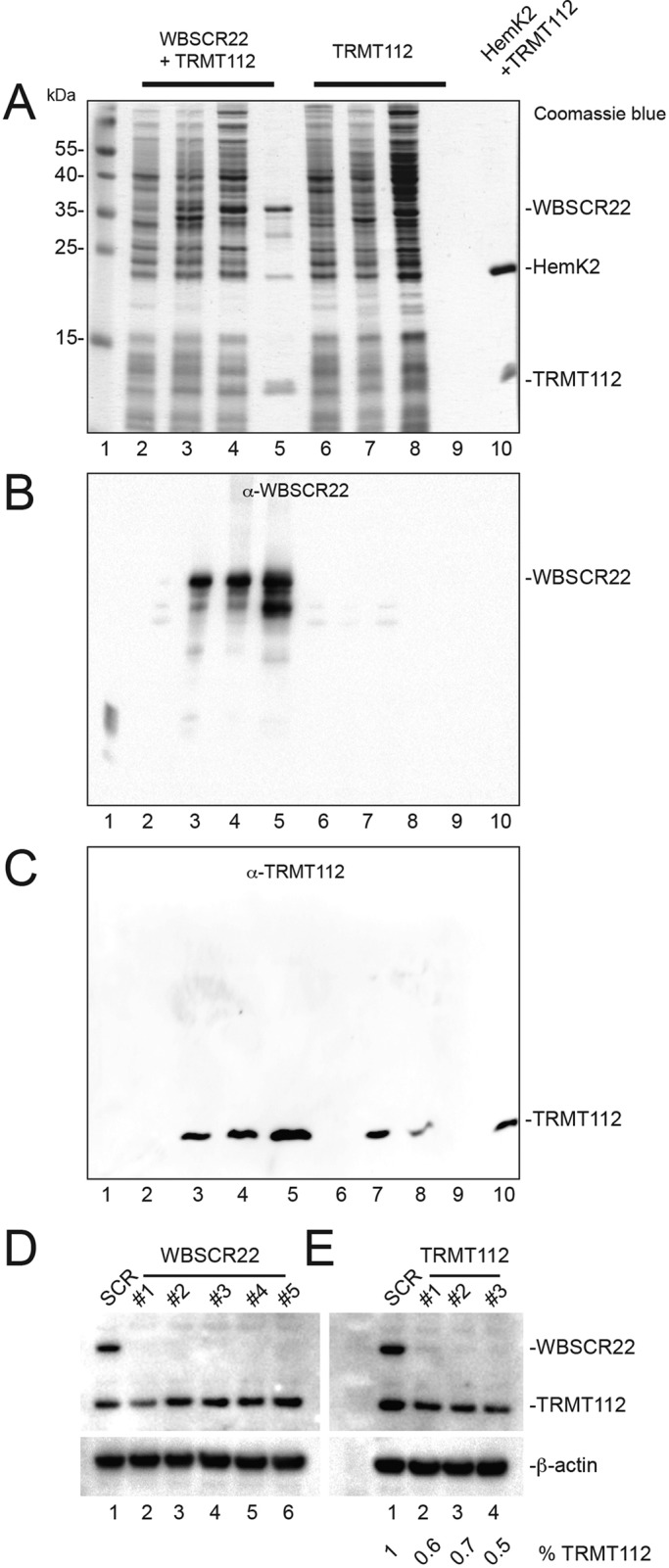
WBSCR22 and TRMT112 form a complex, and TRMT112 is required for the metabolic stability of WBSCR22. (A–C) WBSCR22 and TRMT112 interact directly. Constructs encoding polyhistidine-tagged WBSCR22 and nontagged TRMT112 were overexpressed in bacterial cells, total extracts were purified over a nickel column, and eluates were tested by Coomassie blue staining (A) and Western blotting with antibodies against WBSCR22 (B) or TRMT112 (C). Lane 1, molecular weight marker; 2–5, cells coexpressing His6-WBSCR22 and TRMT112; 6–9, cells expressing only untagged TRMT112. Lanes 2 and 6, total extract from noninduced cells; 3 and 7, total extract from IPTG-induced cells; 4 and 8, supernatant loaded on Ni-NTA column; 5 and 9, 50 mM imidazole eluates, revealing copurification of WBSCR22 and TRMT112 in extracts prepared from bacterial cells coexpressing His6-WBSCR22 and TRMT112, or the absence of any purification from cells expressing only the construct encoding untagged TRMT112. As a control, lane 10 shows the purified Hemk2-TRMT112 complex (Figaro et al., 2008). (D) Depletion of WBSCR22 is efficient at the protein level. HeLa cells were treated for 3 d with one of five different siRNAs (#1–#5) targeting WBSCR22. Total protein was analyzed by Western blotting with antibodies against TRMT112 and WBSCR22. β-Actin was used as loading control. (E) TRMT112 is required for WBSCR22 metabolic stability. HeLa cells were treated for 3 d with an siRNA (#1, #2, or #3) targeting TRMT112. Total protein extracts were processed as in C. The residual level of TRMT112 was estimated by densitometry to be between 50 and 70%.
In yeast cells, Trm112 interacts with, and stabilizes, at least four methyltransferase partners: Bud23, Mtq2, Trm9, and Trm11 (Figaro et al., 2012; Létoquart et al., 2014). Trm112 is in limiting amounts in cells, and its multiple partners compete with each other in order to gain metabolic stability (Figaro et al., 2012). In humans, TRMT112 also interacts with several methyltransferases, as illustrated here for HemK2 (the human homologue of Mtq2), with which it interacts directly (Figure 3, lane 10; Figaro et al., 2008), and elsewhere for ALKBH8 (the homologue of Trm9; Fu et al., 2010; Songe-Moller et al., 2010).
Further evidence of the existence of a WBSCR22-TRMT112 complex was provided by the observation that TRMT112 is strictly required for the metabolic stability of WBSCR22 (Figure 3E). Depleting TRMT112 to 50–70% of its wild-type level with any of three small interfering RNAs (siRNAs; #1, #2, or #3) indeed led to loss of WBSCR22 detection by Western blot (Figure 3E, lanes 2–4, and Supplemental Figure S1). As a control, a nontargeting siRNA (SCR) was used throughout this work (Figure 3E, lane 1). Of interest, depleting TRMT112 to only ∼40% of its normal cellular level is sufficient to promote near-total loss of WBSCR22. This is consistent with the idea that TRMT112 interacts with multiple competing partners in human cells, similar to yeast.
DIMT1L and WBSCR22-TRMT112 are required for distinct early pre-rRNA processing steps leading to 18S rRNA synthesis
In budding yeast, Dim1 and Bud23-Trm112 are required for pre-rRNA processing (Lafontaine et al., 1995; White et al., 2008; Figaro et al., 2012; Sardana and Johnson, 2012). This prompted us to test the involvement of DIMT1L and WBSCR22-TRMT112 in this process in human cells.
Mature rRNAs are produced from long precursors by multistep processing (Mullineux and Lafontaine, 2012; see Supplemental Figure S2 for a summary of the pre-rRNA processing pathway in human cells). Pre-rRNA processing was analyzed in detail by quantitative Northern blotting (“steady-state analysis”) in primary cells (WI-38) and three cell lines (HeLa, RKO, and HCT116; Supplemental Figure S3 and Figure 4) and by metabolic labeling (“dynamic analysis”) in HeLa cells (Figure 5).
FIGURE 4:

DIMT1L, WBSCR22, and TRMT112 are required for distinct pre-rRNA processing steps, and the pre-rRNA processing defects are conserved in different cell types and do not depend on p53. WI-38, RKO, and HCT116 cells were treated for 3 d with a specific siRNA targeting DIMT1L, WBSCR22, or TRMT112. Paired HCT116 cell lines expressing p53 or not were used (+/+ and –/–). Total RNA was extracted, resolved on denaturing agarose gel, transferred to a nylon membrane, and hybridized with probes. The probes used were as follows: (I, II) LD1844, (III) LD1827, and (IV) LD1828. The detected pre-rRNA species are indicated to the right and schematized. (V, VI) The mature 28S and 18S rRNAs stained with ethidium bromide. For each sample, the 28S/18S ratio was calculated from Agilent bioanalyzer electropherograms. All RNA species were quantified with a Phosphorimager normalized with respect to the nontargeting control (SCR), and their abundances represented as a heatmap using the color code indicated to the right. The sequences of the siRNAs used are listed in Supplemental Table S3 (WBSCR22#1, TRMT112#2, and DIMT1L#2). Note that in the heatmap for RKO cells, the signal for lane 8 was corrected for loading.
FIGURE 5:

Dynamic analysis of pre-rRNA processing defects in cells depleted of DIMT1L, WBSCR22, or TRMT112. (I) HeLa cells were depleted of WBSCR22, TRMT112, or DIMT1L for 3 d by means of a specific siRNA, pulse labeled for 30 min with l-(methyl-3H)-methionine, chased with an excess of cold methionine, and collected at different time points over a 4-h period. Total RNA was extracted, resolved on an agarose gel, transferred to a GeneScreen membrane, and then exposed by fluorography. (II, III) The ethidium bromide–stained mature 28S and 18S rRNAs. The sequences of the siRNAs used are listed in Supplemental Table S3 (WBSCR22#1, TRMT112#2, and DIMT1L#2).
In our initial analysis, conducted on HeLa cells, we found that depletion of DIMT1L led to accumulation of the 43S and 26S pre-rRNAs (Supplemental Figure S3, lanes 10–14; see quantitation on the heatmap). Such an accumulation is a sign of disconnection between the early cleavages at sites A0 and 1, which normally occur largely concomitantly (Supplemental Figure S2; discussed in Mullineux and Lafontaine, 2012). In addition, long, truncated forms of 18S precursors (asterisks) were detected. Despite this inhibition of processing, the overall effect on mature rRNA accumulation was limited in HeLa cells, as shown by the largely unaffected 28S/18S ratio (Supplemental Figure S3; values remained close to 1.0). Of interest, the processed spacer fragment +1-01, destined for rapid clearance, also accumulated to some extent upon DIMT1L depletion, indicating that functional coupling might occur between RNA-processing complexes and degradation machineries.
HeLa cells depleted of WBSCR22 showed a mild reduction in 30S, indicative of reduced cleavage at site 2, and significant accumulation of 18S-E (Supplemental Figure S3, lanes 2–6), revealing inhibition of cleavage at site 3. In addition, long and short truncated forms of 18S precursors were detected (single and double asterisks). The long forms appeared similar in length to those observed upon DIMT1L depletion, but they accumulated to lower levels (Supplemental Figure S3, compare lanes 1–6 and 10–14). On WBSCR22 depletion, the amount of mature 18S rRNA was markedly reduced and the 28S/18S rRNA ratio consequently increased (from 1.0 to up to 1.7). TRMT112 depletion led to processing phenotypes largely similar to those observed upon WBSCR22 depletion (moderate 30S reduction and accumulation of 18S-E); Supplemental Figure S3, lanes 7–9). Again, this is consistent with the conclusion that TRMT112 is required for WBSCR22 metabolic stability and that a WBSCR22-TRMT112 complex is formed (Figure 3).
The efficiencies of DIMT1L, WBSCR22, and TRMT112 depletion were assessed at the mRNA and protein levels by quantitative reverse transcription (RT)-PCR (qRT-PCR; Supplemental Figure S1B) and Western blotting (Figure 3, D and E; see later discussion of Figures 7A and 8A; also unpublished data). For WBSCR22 and TRMT112, the residual level of mRNA was 1–3% and 1–5%, respectively; for DIMT1L, it was 8–25% (Supplemental Figure S1B). For DIMT1L and WBSCR22, the residual level of protein was close to or below the detection level; for TMRT112, it was ∼40% (Figure 3, D and E; see later discussion of Figures 7A and 8A).
FIGURE 7:

The methyltransferase function of DIMT1L is not required for pre-rRNA processing. Protein levels and pre-rRNA processing in HEK293 cells stably expressing, via an siRNA-resistant mRNA (siRNAr), a construct encoding the wild-type or a catalytically deficient (Y131G) Flag-tagged version of DIMT1L in the presence or absence of endogenous DIMT1L. The allele encoding catalytically deficient DIMT1L and, as controls, the wild-type construct and the empty plasmid were integrated at the same genomic locus. Expression of the recombinant constructs was under the control of an inducible Tet promoter (bent arrow) and was induced by incubating the cells in 0.2 μg/ml tetracycline. Expression of the endogenous DIMT1L gene was suppressed by incubating the cells for 3 d with siRNA DIMT1L#2 (see Supplemental Table S3). As control, a nontargeting siRNA (SCR, scramble) was used. (A) Western blot analysis with an antibody against DIMT1L. The antibody detects the endogenous DIMT1L protein, displaying the expected molecular weight of 35 kDa, and the Flag-tagged recombinant variants, detected at 38 kDa. β-Actin was used as loading control. The anti-DIMT1L antibody detects a faint nonspecific band (asterisk) above Flag-tagged DIMT1L. Hybridizing the membrane with an anti-Flag antibody revealed only recombinant proteins of the expected size (unpublished data). (B) Northern blot analysis. Total RNA treated as described in Figure 4 was probed with oligonucleotide LD1827. The abundances of the various RNAs detected were established by Phosphorimager quantification and normalized with respect to the nontargeting SCR control and are presented as a heatmap with the color code indicated to the right.
FIGURE 8:

The methyltransferase function of WBSCR22 is not required for pre-rRNA processing. Protein levels and pre-rRNA processing in HEK293 cells stably expressing, via an siRNA-resistant mRNA (siRNAr), a construct encoding the wild-type or a catalytically deficient (G63E, D82K, and D63E/D82K) Flag-tagged version of WBSCR22 in the presence or absence of endogenous WBSCR22. The constructs encoding catalytically deficient WBSCR22 and, as controls, the wild-type construct and the empty plasmid were integrated at the same genomic locus. Expression of the recombinant constructs was under the control of an inducible Tet promoter (bent arrow) and was induced by incubating the cells in 0.2 μg/ml tetracycline. Expression of the endogenous WBSCR22 gene was suppressed by incubating the cells for 3 d with siRNA WBSCR22#1 (see Supplemental Table S3). As control, a nontargeting siRNA (SCR, scramble) was used. (A) Western blot analysis with an antibody against WBSCR22. The antibody detects the endogenous WBSCR22 protein, displaying the expected molecular weight of 32 kDa, and the Flag-tagged recombinant variants, detected at 35 kDa. β-Actin was used as loading control. Hybridizing the membrane with an anti-Flag antibody revealed only the recombinant variants of the expected size (unpublished data). (B) Northern blot analysis. Total RNA treated as described in Figure 4 was probed with oligonucleotide LD1827. The abundances of the various RNAs detected were established by Phosphorimager quantification and normalized with respect to the nontargeting SCR control and are presented as a heatmap with the color code indicated to the right.
It is noteworthy that neither DIMT1L nor WBSCR22-TRMT112 depletion appeared to have any significant effect on the processing steps leading to synthesis of large-subunit rRNAs, as concluded from the largely unaltered levels of 5.8S rRNA, its precursors (5.8S+40, 7S, and 12S), and 28S rRNA (Supplemental Figure S3 and unpublished data).
Our pre-rRNA processing analysis was then extended to primary lung fibroblasts (WI-38) and two colon carcinoma cell lines (RKO, HCT116), with, for each target, one siRNA that proved effective in HeLa cells. The results were largely similar to those described for HeLa cells (Figure 4). In WI-38, RKO, and HCT116 cells, DIMT1L depletion also led to 26S accumulation (Figure 4, lanes 4, 8, 12, and 16). In these cells, and in contrast to HeLa, DIMT1L depletion further led to accumulation of 21S and 21S-C pre-rRNAs and to reduced levels of 18S-E and 18S rRNA (the 28S/18S ratio increased from 1.0 to up to 1.8; Figure 4). In WI-38, RKO, and HCT116 cells, WBSCR22 or TRMT112 depletion led to striking accumulation of 18S-E, as seen in HeLa cells (Figure 4, lanes 2/3, 6/7, 10/11, and 14/15). The long and short forms of truncated 18S rRNA precursors (single and double asterisks) observed in HeLa cells depleted of DIMT1L, WBSCR22, or TRMT112 were also detected in the other cell types tested.
The nucleolus is a long-known cancer biomarker and a recently demonstrated therapeutic target (Bywater et al., 2012). Because there are numerous connections between nucleolar functions and synthesis of the antitumor protein p53, pre-rRNA processing was directly compared in colon carcinoma cells expressing p53 or not (Figure 4, lanes 9–12 vs. lanes 13–16). The pre-rRNA processing defects observed in the presence and absence of functional p53 were largely similar, indicating that p53 is not involved in the inhibition of ribosome biogenesis observed upon DIMT1L or WBSCR22-TRMT112 depletion (Figure 4, lanes 9–16; compare HCT116 p53+/+ and HCT116 p53−/−).
Finally, we examined the kinetics of the pre-rRNA processing defects in HeLa cells by in vivo metabolic labeling (Figure 5). Cells depleted of DIMT1L, WBSCR22, or TRMT112 for 3 d with a specific siRNA were pulse labeled with l-(methyl-3H)-methionine for 30 min, chased with an excess of cold methionine, and collected in a time-course experiment. Total RNA extracted at each time point was analyzed by denaturing agarose gel electrophoresis and the signal visualized by fluorography. In agreement with the Northern blot data (Figure 4 and Supplemental Figure S3), depletion of DIMT1L or WBSCR22 was found to have a strong effect on the kinetics of 18S rRNA synthesis. The effects were more moderate for TRMT112, but 18S rRNA formation was again delayed. In agreement with our steady-state analysis (Supplemental Figure S3), DIMT1L depletion, but not WBSCR22 depletion, led to accumulation of the 43S precursor (Figure 5, lanes 19–21). The other RNA precursors discussed earlier (e.g., 26S) are not detected in pulse-chase labeling experiments because they are either not abundant enough or too short-lived. Of interest, however, additional bands were detected below 41S after DIMT1L depletion (seen as a doublet in Figure 5, lanes 19–22). These are likely aberrant truncated intermediates, which, in the absence of faithful subunit assembly, are produced by activation of cryptic processing or increased degradation.
In conclusion, DIMT1L and WBSCR22-TRMT112 are involved in pre-rRNA processing steps leading to small-subunit rRNA production. DIMT1L is required for earlier processing steps than WBSCR22. This is consistent with the nucleolar subcellular location of DIMT1L and the nucleoplasmic distribution of WBSCR22. Furthermore, the processing defects do not depend on p53 and are essentially the same, with minor differences, in different cell types.
DIMT1L and WBSCR22-TRMT112 are required for 18S rRNA base methylation
Given their homology with the yeast proteins and the extreme conservation of the rRNA substrate at these positions, DIMT1L and WBSCR22-TRM112 are predicted to methylate, respectively, adenosines A1850/A1851 and the guanosine G1639 in human 18S rRNA (Figure 1A).
We tested this prediction by primer extension mapping in cells depleted of DIMT1L or WBSCR22 (Figure 6). The 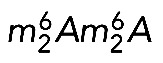 modification is readily detected by primer extension because it is a bulky modification blocking reverse transcriptase progression during cDNA synthesis, thus generating strong primer extension stops and a “stutter” (Lafontaine et al., 1995). In contrast, detection of m7G by primer extension ideally requires specific cleavage of the RNA at the site of modification. This is achieved by reduction with sodium borohydride followed by aniline-mediated strand scission at the reduced residue (Figaro et al., 2012; Létoquart et al., 2014).
modification is readily detected by primer extension because it is a bulky modification blocking reverse transcriptase progression during cDNA synthesis, thus generating strong primer extension stops and a “stutter” (Lafontaine et al., 1995). In contrast, detection of m7G by primer extension ideally requires specific cleavage of the RNA at the site of modification. This is achieved by reduction with sodium borohydride followed by aniline-mediated strand scission at the reduced residue (Figaro et al., 2012; Létoquart et al., 2014).
FIGURE 6:

DIMT1L and WBSCR22 are required for 18S rRNA 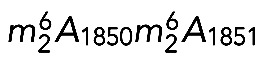 and m7G1639 methylation, respectively. (A–D) Primer extension mapping of the substrate nucleotides of DIMT1L and WBSCR222 on human 18S rRNA. Total RNA was extracted from HeLa cells depleted for 3 d with a DIMT1L-targeting (A) or WBSCR22-targeting (B) siRNA and analyzed by primer extension with oligonucleotide LD2120, LD2122, or LD2141. (C, D) The positions of the oligonucleotides and of the rRNA species. (E) PNO1 is required for efficient DIMT1L-mediated dimethylation. Total RNA was extracted from HeLa cells depleted for 3 d with a siRNA specific to DIMT1L or PNO1 and processed by primer extension with oligonucleotide LD2141. The level of residual dimethylation estimated with a Phosphorimager is indicated to the right. The results shown are means of three independent experiments. The sequences of the siRNAs used are listed in Supplemental Table S3 (WBSCR22#1, DIMT1L#2). (F) PNO1 is not required for the metabolic stability of DIMT1L. Total protein extract from HeLa cells depleted for 3 d with a siRNA specific to DIMT1L or PNO1 and tested by Western blotting with the antibodies indicated. Right, residual level of DIMT1L and PNO1 mRNAs assessed by qRT-PCR.
and m7G1639 methylation, respectively. (A–D) Primer extension mapping of the substrate nucleotides of DIMT1L and WBSCR222 on human 18S rRNA. Total RNA was extracted from HeLa cells depleted for 3 d with a DIMT1L-targeting (A) or WBSCR22-targeting (B) siRNA and analyzed by primer extension with oligonucleotide LD2120, LD2122, or LD2141. (C, D) The positions of the oligonucleotides and of the rRNA species. (E) PNO1 is required for efficient DIMT1L-mediated dimethylation. Total RNA was extracted from HeLa cells depleted for 3 d with a siRNA specific to DIMT1L or PNO1 and processed by primer extension with oligonucleotide LD2141. The level of residual dimethylation estimated with a Phosphorimager is indicated to the right. The results shown are means of three independent experiments. The sequences of the siRNAs used are listed in Supplemental Table S3 (WBSCR22#1, DIMT1L#2). (F) PNO1 is not required for the metabolic stability of DIMT1L. Total protein extract from HeLa cells depleted for 3 d with a siRNA specific to DIMT1L or PNO1 and tested by Western blotting with the antibodies indicated. Right, residual level of DIMT1L and PNO1 mRNAs assessed by qRT-PCR.
Primer extension mapping was performed on total RNA extracted from HeLa cells treated for 3 d with siRNAs against DIMT1L or WBSCR22 (Figure 6, A, B, and E). When primer LD2141, which specifically selects the nuclear 21S-C and its precursors (Figure 6C; Preti et al., 2013), was used to initiate cDNA synthesis on RNAs extracted from DIMT1L-depleted cells, we detected 57% reduction in dimethylation efficiency (Figure 6A). This observation is in agreement with the prediction that DIMT1L is the bona fide 18S rRNA dimethyltransferase. Because the 21S-C is nuclear, we further conclude that in human cells, the 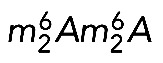 modification is synthesized in the nucleus, in contrast to the situation in yeast, in which this modification is reported to occur in the cytoplasm (Brand et al., 1977). RNA extracted from WBSCR22-depleted cells was specifically cleaved at N7G-modified residues and used as template for cDNA synthesis with primer LD2120 or LD2122 (Figure 6, B and C). As expected from the assumption that WBSCR22 is the bona fide 18S rRNA m7G methyltransferase, a substantial 62% reduction in methylation level was observed with both primers. Since m7G1639 is readily detected from primer LD2122, specific to a sequence 3' to the dimethylation site that would stop its extension, we further conclude that 18S rRNA N7G-methylation occurs prior to dimethylation and is thus also nuclear.
modification is synthesized in the nucleus, in contrast to the situation in yeast, in which this modification is reported to occur in the cytoplasm (Brand et al., 1977). RNA extracted from WBSCR22-depleted cells was specifically cleaved at N7G-modified residues and used as template for cDNA synthesis with primer LD2120 or LD2122 (Figure 6, B and C). As expected from the assumption that WBSCR22 is the bona fide 18S rRNA m7G methyltransferase, a substantial 62% reduction in methylation level was observed with both primers. Since m7G1639 is readily detected from primer LD2122, specific to a sequence 3' to the dimethylation site that would stop its extension, we further conclude that 18S rRNA N7G-methylation occurs prior to dimethylation and is thus also nuclear.
In yeast cells, depletion of the 40S subunit assembly factor Pno1 (also known as Dim2) strongly inhibits Dim1-mediated dimethylation (Vanrobays et al., 2004; Introduction). In pre-40S ribosomes, Dim1 and Pno1/Dim2 are located to either side of the incipient platform (Strunk et al., 2011). This physical proximity likely underlies the functional interaction between Dim1 and Dim2. The position of the platform (“Pt”) on mature 40S is shown in Figure 1B. Using the primer extension assay described, we tested whether efficient 18S rRNA dimethylation in human cells might likewise require PNO1, the human homologue of Dim2, and we found that it does (Figure 6E). It is not yet clear why PNO1 depletion consistently affected 18S rRNA dimethylation more than DIMT1L depletion. We hypothesized that this is because in cells depleted for PNO1, the substrate fails to adopt a suitable conformation to be modified. Finally, we were interested to know whether PNO1 is required for the metabolic stability of DIMT1L, and we found, in cells efficiently depleted for PNO1, that it is not (Figure 6F).
In conclusion, our primer extension mapping data demonstrate that DIMT1L and WBSCR22-TRMT112 are responsible, respectively, for 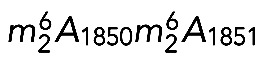 and m7G1639 in human 18S rRNA. We further show that DIMT1L-mediated dimethylation in human resembles dimethylation in yeast in that it requires hDIM2 (PNO1), but differs from yeast dimethylation in that it occurs in the nucleus before preribosome export to the cytoplasm.
and m7G1639 in human 18S rRNA. We further show that DIMT1L-mediated dimethylation in human resembles dimethylation in yeast in that it requires hDIM2 (PNO1), but differs from yeast dimethylation in that it occurs in the nucleus before preribosome export to the cytoplasm.
The methylation activities of DIMT1L and WBSCR22-TRMT112 are not necessary for pre-rRNA processing
In budding yeast, pre-rRNA processing requires the presence of the methyltransferases Dim1 and Bud23 within preribosomes but not their RNA-modifying catalytic activity (Lafontaine et al., 1995; White et al., 2008; Létoquart et al., 2014; Introduction).
To determine whether pre-rRNA processing in human cells requires the catalytic activity of DIMT1L and WBSCR22-TRMT112, we analyzed cell lines expressing constructs coding for catalytically deficient variants (Figures 7 and 8). The mutations introduced resulted in amino acid substitutions at key functional residues in the methyltransferases (Supplemental Figure S4). In DIMT1L, tyrosine 131 was replaced with glycine (Y131G), and in WBSCR22, mutations G63E and D82K were introduced, either individually or in combination (Figures 7 and 8 and Supplemental Figure S4). The selected residues are at, or directly adjacent to, the S-adenosyl-methionine–binding site. In budding yeast, mutations at the equivalent positions result in catalytically dead methyltransferases (White et al., 2008; Létoquart et al., 2014). In human cells, expression of the DIMT1L-Y131G allele led to only 15% residual rRNA dimethylation (Supplemental Figure S4A).
To allow a direct comparison of the effects of the various mutations, all variants were expressed in HEK293 cells as Flag-tagged proteins, under the control of a tetracycline-inducible promoter, from constructs inserted into the same genomic locus. This was achieved by using the Flp-In 293 T-REx system (see Materials and Methods). The presence of a Flag tag, resulting in a 3-kDa increase in protein size, made it easy to distinguish the endogenous proteins from the recombinant ones by Western blotting. The use of a Tet promoter allowed optimizing the level of recombinant protein produced so as to match that of the endogenous protein as closely as possible (see later discussion of Figures 7A and 8A). To analyze specifically the effects of the catalytic mutations, the endogenous mRNA transcripts of each methyltransferase were silenced with a siRNA. Expression of each mutant or control construct was made immune to this siRNA by an additional, silent mutation having no effect on the amino acid sequence of the protein but altering the siRNA-targeted sequence on the mRNA. As controls, a construct encoding the wild-type protein, with only the siRNA-resistant mutation, and an empty construct were integrated at the same locus.
Tetracycline at 0.2 μg/ml in the medium was found optimal for obtaining levels of mutant DIMT1L similar to that of endogenous protein (Figure 7A, lanes 6 and 10; the recombinant variants are 3 kDa larger). When cells were treated with a DIMT1L-targeting siRNA, the level of the endogenous protein dropped to below the detection level (Figure 7A, lanes 3/4, 7/8, and 11/12), whereas the mutant variants, encoded by siRNA-resistant mRNAs, were stably produced (Figure 7A, lanes 8 and 12).
When expression of a recombinant construct was not induced (lanes without tetracycline addition), cells depleted of endogenous DIMT1L accumulated the 26S pre-rRNA and/or the long truncated (asterisk) 18S precursor fragment (Figure 6B, lanes 3, 4, 7, and 11). This is consistent with our earlier results (Figure 4 and Supplemental Figure S3). When synthesis of the DIMT1L-Y131G variant was induced by addition of tetracycline to the medium, accumulation of these diagnostic RNAs was suppressed (Figure 6, compare lanes 11 and 12 and see quantitation). Hence the catalytic activity of DIMT1L is not required for pre-rRNA processing. As control, expression of the wild-type construct with the siRNA resistant mutation also suppressed the processing defect (disappearance of the 18S-E truncated form [asterisk] in Figure 6B, lane 8, as compared with lane 7), indicating efficient rescue.
Similarly, 0.2 μg/ml tetracycline proved optimal for production of the recombinant forms of WBSCR22, in most cases yielding levels similar to that of the endogenous protein (Figure 8A, lanes 6, 10, 14 and 18). Synthesis of endogenous WBSCR22 was efficiently suppressed by siRNAs (Figure 8A, lanes 3/4, 7/8, 11/12, 15/16, and 19/20). In contrast, the recombinant constructs were stably expressed, indicating that, as expected, the mRNAs encoding them were siRNA resistant (Figure 8A, lanes 8, 12, 16, and 20). In the absence of expression of a complementing construct, cells depleted of endogenous WBSCR22 accumulated the 18S-E pre-rRNA intermediate and the long and short truncated 18S precursor fragments (single and double asterisks; Figure 8B, lanes 3, 4, 7, 11, 15, and 19). This is consistent with our earlier results (Figures 4 and Supplemental Figure S3). Accumulation of these fragments was suppressed to a similar extent in cells producing the wild-type protein (Figure 8B, lane 8) or any of the three catalytically defective variants (Figure 8B, lanes 12, 16, and 20). This demonstrates that the catalytic activity of WBSCR22, like that of DIMT1L, is not required for pre-rRNA processing in human cells.
DISCUSSION
The ribosome is an extraordinary complex nanomachine whose blueprint has been extremely conserved throughout evolution across all three kingdoms of life (Melnikov et al., 2012). This is strikingly illustrated when one compares the ribosomes of simple and more complex eukaryotic cells, such as budding yeast and human cells (Ben-Shem et al., 2011; Anger et al., 2013). This resemblance has led to the assumption that ribosome biogenesis must be quite similar in yeast and humans. Recent research indicates that this is true, but only to some extent, as important specificities have begun to emerge (Sloan et al., 2013; Tafforeau et al., 2013). For example, the RNA exosome involved in ITS2 processing in yeast is additionally required for ITS1 maturation in humans, and the endoRNase MRP, which cleaves ITS1 in yeast, does not seem to play such a role in humans (Sloan et al., 2013; Tafforeau et al., 2013). Such specificities in pre-rRNA processing are making it absolutely imperative to conduct research directly on human cells. If ribosomopathies—that is, human syndromes associated with ribosome assembly dysfunctions (Narla and Ebert, 2010)—were to be modeled only in budding yeast, potential therapeutic targets might be selected for the wrong reasons and others left totally unnoticed.
Here we characterized two human methyltransferases with specificity toward the small ribosomal subunit rRNA. We show that their presence in cells, but not their RNA-methylating catalytic activity, is indispensable for pre-rRNA processing steps leading to the synthesis of the 18S rRNA (Figures 4, 5, 7, and 8 and Supplemental Figure S3). We demonstrate that DIMT1L is responsible for N6,N6 dimethylation at positions A1850 and A1851 and that WBSCR22 is required for N7 methylation at nucleotide G1639 (Figures 1 and 6; see also Haag et al., 2015). These nucleotide positions are equivalent to those modified in yeast by the homologous methyltransferases. We show that WBSCR22 interacts directly with TRMT112, forming a heterodimer, and that the latter protein is required for the metabolic stability of the former (Figure 3). Taken together, these findings indicate strong conservation of the functions of Dim1/DIMT1L and Bud23-Trm112/WBSCR22-TRMT112 in yeast and human cells (Lafontaine et al., 1994, 1995; White et al., 2008; Figaro et al., 2012; Sardana and Johnson, 2012; Létoquart et al., 2014).
In the late 1970s, fingerprint analysis revealed that both the m7G and 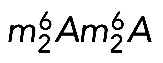 modifications are late maturation events in budding yeast (Brand et al., 1977). It was suggested that m7G is introduced into pre-18S rRNA precursors in the nucleus, and that
modifications are late maturation events in budding yeast (Brand et al., 1977). It was suggested that m7G is introduced into pre-18S rRNA precursors in the nucleus, and that 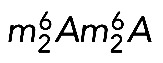 is synthesized after pre-18S rRNA export to the cytoplasm, before final processing (Brand et al., 1977). On this basis, Dim1-mediated dimethylation was used as a convenient marker of late cytoplasmic assembly events (e.g., Grandi et al., 2002; Schafer et al., 2003; Figaro et al., 2012). Primer extension mapping confirmed that, in budding yeast, both m7G1575 and
is synthesized after pre-18S rRNA export to the cytoplasm, before final processing (Brand et al., 1977). On this basis, Dim1-mediated dimethylation was used as a convenient marker of late cytoplasmic assembly events (e.g., Grandi et al., 2002; Schafer et al., 2003; Figaro et al., 2012). Primer extension mapping confirmed that, in budding yeast, both m7G1575 and 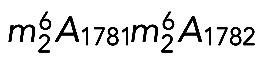 are introduced at the level of the 20S pre-rRNA, the direct precursor of the 18S rRNA (Lafontaine et al., 1995; Létoquart et al., 2014). Dimethylation in HeLa cells was reported long ago to be a late 18S rRNA modification (Salim and Maden, 1973), but whether it occurred in the nucleus or the cytoplasm was not determined in humans. Using primer extension mapping, we detected
are introduced at the level of the 20S pre-rRNA, the direct precursor of the 18S rRNA (Lafontaine et al., 1995; Létoquart et al., 2014). Dimethylation in HeLa cells was reported long ago to be a late 18S rRNA modification (Salim and Maden, 1973), but whether it occurred in the nucleus or the cytoplasm was not determined in humans. Using primer extension mapping, we detected 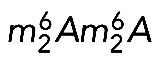 in HeLa cells with an oligonucleotide that selects the 21S pre-rRNA and its precursors (Figure 6). Because these pre-rRNA intermediates are nuclear, we conclude that dimethylation in human cells, in contrast to yeast cells (Brand et al., 1977), is a nuclear event.
in HeLa cells with an oligonucleotide that selects the 21S pre-rRNA and its precursors (Figure 6). Because these pre-rRNA intermediates are nuclear, we conclude that dimethylation in human cells, in contrast to yeast cells (Brand et al., 1977), is a nuclear event.
It is unclear at this stage whether specific individual RNA modifications, such as the rare base methylation, act as quality control checkpoints during subunit biogenesis. An attractive possibility is that they might contribute to signaling to the assembly machinery that key maturation steps have been completed. Preribosomes having passed this checkpoint would be committed to the next assembly step, and the others would be aborted (see later discussion on surveillance). Because 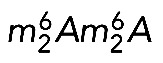 is a nuclear event in human cells, it could, for example, commit pre-40S subunits to export to the cytoplasm.
is a nuclear event in human cells, it could, for example, commit pre-40S subunits to export to the cytoplasm.
Several hundred ribosome assembly factors have been identified in budding yeast and, more recently, human cells (Wild et al., 2010; Tafforeau et al., 2013; Woolford and Baserga, 2013). The field is now facing the daunting task of assigning precise functions to them, identifying their binding sites or substrates on precursor ribosomes, and establishing the timing of their intervention—that is, a precise sequence of events. We show here that DIMT1L is involved in earlier processing steps than WBSCR22-TRM112 (Figures 4 and 5 and Supplemental Figure S3). This is consistent with the respective subcellular localizations of these proteins: the nucleolus and nucleoplasm. The nuclear localization of WBSCR22 is consistent with previous reports (Nakazawa et al., 2011; Ounap et al., 2013), but these studies failed to detect it in perinuclear vesicles (see later discussion).
Cells lacking DIMT1L accumulate the 43S, 26S, and 21S/21S-C pre-rRNAs. Hence the cleavages occurring at sites A0 and 1, which are normally concomitant, are disconnected in these cells, and cleavage at sites 1, C, and E is inhibited. Cells lacking WBSCR22 or TRMT112 accumulate the 18S-E precursor. This indicates inhibition of cleavage at site 3, a processing step reported to occur in the cytoplasm (Mullineux and Lafontaine, 2012). A likely explanation of this processing phenotype is that in the absence of WBSCR22-TRMT112, preribosomes are simply not exported to the cytoplasm. In agreement with this hypothesis, investigators have observed retention of fluorescently labeled pre-40S in the nucleus upon WBSCR22 depletion (Wild et al., 2010), and the 3′-extended forms of 18S-E that accumulates under these conditions are reported to be nuclear (Haag et al., 2015).
Because subtle variations in pre-rRNA processing are reported between cell lines (e.g., Sloan et al., 2014), we tested the effects of DIMT1L and WBSCR22-TRMT112 depletion on pre-rRNA processing in multiple cell types: cervix and colon carcinoma cell lines, embryonic kidney cells, and primary lung fibroblasts (Figures 4, 5, 7, and 8 and Supplemental Figure S3). The processing phenotypes observed were largely similar, but we did note a few differences. For example, DIMT1L depletion did not strongly affect mature rRNA accumulation in HeLa cells, but it did reduce the level of 18S rRNA in the other cell types tested.
Given the many connections between p53 and the synthesis of ribosomal components on one hand (Ruggero and Pandolfi, 2003) and between the functional integrity of the nucleolus and p53 metabolic stability on the other (James et al., 2014; Vlatkovic et al., 2014), we examined whether p53 somehow might contribute to the observed processing defects. Using a pair of isogenic colon carcinoma cell lines that only differ by the expression of p53 (Bunz et al., 1998), we found that p53 has no effect (Figure 4). It is noteworthy that our description of the requirements of WBSCR22 in pre-rRNA processing is consistent with and expands those of recent reports (Ounap et al., 2013; Haag et al., 2015).
In cells depleted of DIMT1L or WBSCR22-TRMT112, we consistently observed truncated 18S rRNA precursor fragments that appeared similar in length (Figures 4, 7, and 8 and Supplemental Figure S3). This suggests that in the absence of faithful subunit assembly, cryptic processing sites are activated, triggering increased turnover of precursor RNAs due to specific surveillance or increased sensitivity to cellular RNases. Such a specific surveillance may occur in a polarized perinuclear structure overlapping with the Golgi and lysosomes where WBSCR22-TRMT112 has been detected (Figure 2). Surveillance mechanisms are active at all steps of ribosome biogenesis, their purpose being to recognize misassembled ribosomes and target them for rapid clearance (Lafontaine, 2010). In principle, surveillance can occur anywhere in the cell. It can take place in dedicated degradation centers, such as those described in the nucleolus, the nucleoplasm, a perinuclear region, and the cytoplasm (discussed in Lafontaine, 2010). In yeast trm112Δ cells, pre-rRNAs undergo massive degradation, and this phenotype is suppressed upon inactivation of the nucleolar surveillance machinery by deletion of the gene encoding the poly(A)-polymerase Trf4 or Trf5 (Figaro et al., 2012). Both of these proteins are subunits of the TRAMP complexes acting as specific adaptors to the RNA Exosome (Schneider and Tollervey, 2013). We posit that WBSCR22-TRMT112 exerts functions in surveillance, in addition to being required for pre-rRNA processing and RNA methylation. We further speculate that the polarized perinuclear vesicles to which WBSCR22 and TRMT112 localize may be degradation centers. Of interest, a process termed piecemeal microautophagy of the nucleus (PMN) has been described in yeast during which the vacuole (the yeast lysosome) literally “pinches off” portions of the nucleus, including the nucleolus, in order to degrade them (Roberts et al., 2003; Kvam and Goldfarb, 2006; discussed in Lafontaine, 2010). Whether the hypothetical surveillance function of WBSCR22-TRMT112 in human cells might relate to a process similar to PMN in yeast remains to be investigated.
An evolutionarily conserved strategy to commit processed rRNAs to faithful modification?
Analysis of catalytically deficient Bud23 and Dim1 variants in yeast cells has revealed that neither m7G1575 nor 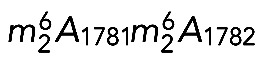 is required for growth, at least under optimal laboratory conditions (Lafontaine et al., 1998; White et al., 2008; Létoquart et al., 2014). On the other hand, both Dim1 and Bud23-Trm112 are either essential to, or important for, cell growth (Lafontaine et al., 1994; White et al., 2008; Figaro et al., 2012). Dim1 and Bud23-Trm112 associate with early precursor ribosomes (Schafer et al., 2003; Figaro et al., 2012) and are required for early nucleolar pre-rRNA processing steps (Lafontaine et al., 1995; White et al., 2008). The corresponding modifications occur later, on pre-18S rRNA precursors (Létoquart et al., 2014). In the case of
is required for growth, at least under optimal laboratory conditions (Lafontaine et al., 1998; White et al., 2008; Létoquart et al., 2014). On the other hand, both Dim1 and Bud23-Trm112 are either essential to, or important for, cell growth (Lafontaine et al., 1994; White et al., 2008; Figaro et al., 2012). Dim1 and Bud23-Trm112 associate with early precursor ribosomes (Schafer et al., 2003; Figaro et al., 2012) and are required for early nucleolar pre-rRNA processing steps (Lafontaine et al., 1995; White et al., 2008). The corresponding modifications occur later, on pre-18S rRNA precursors (Létoquart et al., 2014). In the case of 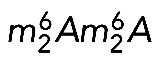 , modification takes place in the cytoplasm (Brand et al., 1977). These observations have two important implications. First, modification does not take place at the time of binding of the methyltransferase to the preribosomes. This implies that there is a regulated delay and a specific catalytic activation of the methyltransferase. Presumably, this simply involves the substrate acquiring competence for modification after sufficient maturation to adopt an appropriate (productive) conformation. Second, because only pre-RNAs having bound the methyltransferase are processed, all cleaved RNAs are destined to be modified. We interpret this as strong evidence of the existence of robust quality control (Lafontaine et al., 1995, 1998). Our results on human cells demonstrating that pre-rRNA cleavage requires the presence of the methyltransferase but not its catalytic activity now allow us to extend our model to humans. Remarkably, the quality control mechanism proposed to involve
, modification takes place in the cytoplasm (Brand et al., 1977). These observations have two important implications. First, modification does not take place at the time of binding of the methyltransferase to the preribosomes. This implies that there is a regulated delay and a specific catalytic activation of the methyltransferase. Presumably, this simply involves the substrate acquiring competence for modification after sufficient maturation to adopt an appropriate (productive) conformation. Second, because only pre-RNAs having bound the methyltransferase are processed, all cleaved RNAs are destined to be modified. We interpret this as strong evidence of the existence of robust quality control (Lafontaine et al., 1995, 1998). Our results on human cells demonstrating that pre-rRNA cleavage requires the presence of the methyltransferase but not its catalytic activity now allow us to extend our model to humans. Remarkably, the quality control mechanism proposed to involve 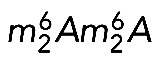 further appears primordial because in bacteria, KsgA is also required for efficient pre-rRNA processing and regulates the last steps of pre-30S subunit formation (Connolly et al., 2008; Xu et al., 2008; Introduction).
further appears primordial because in bacteria, KsgA is also required for efficient pre-rRNA processing and regulates the last steps of pre-30S subunit formation (Connolly et al., 2008; Xu et al., 2008; Introduction).
The exact role of the 18S rRNA m7G and 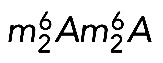 modifications in ribosome function is not fully understood. Clearly, however, both modifications are present at important, conserved ribosomal sites (the decoding site and a ridge between the P- and E-site tRNAs), and the absence of either is associated, in one way or another, with translational deficiencies. In budding yeast, dim1, bud23, and trm112 mutants are hypersensitive to the aminoglycoside antibiotic paromomycin (Lafontaine et al., 1998; Figaro et al., 2012; Létoquart et al., 2014), and a catalytically deficient dim1 mutant shows impaired translation in vitro (Lafontaine et al., 1998). In bacteria, ksgA mutants are resistant to the aminoglycoside antibiotic kasugamycin (Helser et al., 1971). In vitro, small subunits purified from ksgA-methyltransferase–deficient cells show decreased affinity for large subunits and the initiator tRNA, whereas in vivo, the absence of 16S rRNA dimethylation causes translational errors during both initiation and elongation, including increased readthrough of nonsense and frameshift mutations (van Buul et al., 1984; van Knippenberg, 1986; Rife, 2009). Finally, the crystal structure of Thermus thermophilus 30S subunits lacking
modifications in ribosome function is not fully understood. Clearly, however, both modifications are present at important, conserved ribosomal sites (the decoding site and a ridge between the P- and E-site tRNAs), and the absence of either is associated, in one way or another, with translational deficiencies. In budding yeast, dim1, bud23, and trm112 mutants are hypersensitive to the aminoglycoside antibiotic paromomycin (Lafontaine et al., 1998; Figaro et al., 2012; Létoquart et al., 2014), and a catalytically deficient dim1 mutant shows impaired translation in vitro (Lafontaine et al., 1998). In bacteria, ksgA mutants are resistant to the aminoglycoside antibiotic kasugamycin (Helser et al., 1971). In vitro, small subunits purified from ksgA-methyltransferase–deficient cells show decreased affinity for large subunits and the initiator tRNA, whereas in vivo, the absence of 16S rRNA dimethylation causes translational errors during both initiation and elongation, including increased readthrough of nonsense and frameshift mutations (van Buul et al., 1984; van Knippenberg, 1986; Rife, 2009). Finally, the crystal structure of Thermus thermophilus 30S subunits lacking 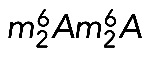 reveals that the modification facilitates a packing interaction near the decoding site, between helices 44 and 45 (Figure 1), through formation of an extensive hydrogen-bonding network (Demirci et al., 2010). Loss of this packing interaction perturbs rRNA structure in the neighboring A and P sites of the ribosome (Demirci et al., 2010), potentially explaining the translation defects of ksgA mutants.
reveals that the modification facilitates a packing interaction near the decoding site, between helices 44 and 45 (Figure 1), through formation of an extensive hydrogen-bonding network (Demirci et al., 2010). Loss of this packing interaction perturbs rRNA structure in the neighboring A and P sites of the ribosome (Demirci et al., 2010), potentially explaining the translation defects of ksgA mutants.
Given the extreme conservation of the mechanism and machineries involved in synthesizing the m7G and 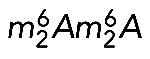 base modifications, it seems highly unlikely that they do not contribute any benefit to ribosome function, if only under specific conditions that remain to be determined (e.g., under stress, during development, etc.). High-resolution, high-throughput experimental strategies, such as ribosome profiling and quantitative mass spectrometry, should soon help us understand exactly what these modifications do in translation.
base modifications, it seems highly unlikely that they do not contribute any benefit to ribosome function, if only under specific conditions that remain to be determined (e.g., under stress, during development, etc.). High-resolution, high-throughput experimental strategies, such as ribosome profiling and quantitative mass spectrometry, should soon help us understand exactly what these modifications do in translation.
In conclusion, our work provides important novel insights into the function of two highly conserved human rRNA methyltransferases required in cell differentiation pathways and associated with severe diseases, including cancer. Despite overall conservation between yeast and humans, we highlight differences confirming that basic research on ribosome biogenesis must be conducted directly on human cells to allow selection of truly promising therapeutic targets before initiating costly drug development programs.
MATERIALS AND METHODS
Plasmid constructs
To overproduce WBSCR22 and TRMT112 in bacterial cells in order to test for direct interaction and complex formation, the WBSCR22 open reading frame was PCR amplified from plasmid pDL0737 with oligonucleotides H6wbNdeI and wbBglII introducing, respectively, at the 5′ end an NdeI site together with a sequence encoding a hexahistidine tag, and at the 3′ end, a BglII site (Supplemental Tables S1 and S2). This fragment was cloned into the pET11a vector between NdeI and BamHI to yield plasmid pVH475 (Supplemental Table S1). TRMT112 was overproduced from the previously described plasmid pFFh5 (Figaro et al., 2008). To generate cell lines stably expressing constructs encoding catalytically defective methyltransferase variants via siRNA-resistant mRNAs, DIMT1L and WBSCR22 were PCR amplified from pDL0846 and pDL0737, respectively, and cloned as BamHI-XhoI fragments into pDL0811 (pcDNA5/FRT/TO vector containing 2× FLAG tags under the control of a tetracycline-regulated promoter, a gift from N. Watkins [University of Newcastle, Newcastle upon Tyne, United Kingdom]). Site-directed mutagenesis (QuikChange II XL, 200521; Agilent) was used to introduce the siRNA-resistant and catalytic mutations. All constructs were sequenced. The resulting plasmids and the oligonucleotides used are listed in Supplemental Tables S1 and S2.
Cell culture
Cells were grown at 37°C in a humidified incubator under 5% CO2 in the following media: DMEM (D6429; Sigma-Aldrich)/10% fetal bovine serum (FBS; A&E Scientific) for HeLa (AIDS Ref. 153; National Institutes of Health), HEK293 (LifeTech), and FIB364 cells (HeLa cells stably expressing fibrillarin-GFP); MEM (30-2003; American Type Culture Collection [ATCC]/10% FBS for WI-38 cells (CCL-75; ATCC); and McCoy's 5a modified (Sigma-Aldrich)/10% FBS for HCT116 cells (CCL-247; ATCC). All media were supplemented with 50 U/ml penicillin and 50 μg/ml streptomycin (Life Technologies). The stable cell lines generated in this work were maintained in DMEM (Sigma-Aldrich) supplemented with FBS (10%), penicillin-streptomycin (Pen-Strep; 1%), hygromycin B (100 μg/ml; Invitrogen), and blasticidin (10 μg/ml; Life Technologies). Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich.
Creation of stable cell lines
Isogenic stable cell lines expressing alleles encoding methyltransferase-deficient variants were generated by transfecting FRT-HEK293 cells (Lifetech) with the corresponding pcDNA5-derived constructs (see Supplemental Table S1), using the Flp-In T-Rex system according to the manufacturer's protocol (LifeTech). The recipient cell line was cotransfected with the recombinant construct and a vector expressing the Flp recombinase. This resulted in targeted integration of all constructs at the same transcriptionally active genomic locus, giving rise to homogeneous expression levels. Individual clones were selected and analyzed.
siRNA inactivation and total RNA extraction
All siRNAs were purchased from Life Technologies (Silencer Select siRNA or Custom select; Supplemental Table S3). HeLa, WI-38, and HCT116 cells were reverse transfected as follows: 1.5 μl of 20 μM siRNA (10 nM final concentration) and 4 μl Lipofectamine RNAiMAX (Life Technologies) were mixed with 500 μl OPTI-MEM (Life Technologies) in each well of a six-well plate. After a 20-min incubation at room temperature, 1.5 × 105 cells, resuspended in 2.5 ml of medium without antibiotics, were seeded into each well. Inactivation was carried out for 72 h before total RNA extraction. Negative controls were transfected with a nontargeting scrambled (SCR) siRNA (ref. 4390844). Total RNA was extracted with the TRI Reagent (Life Technologies) according to the manufacturer's protocol and analyzed by northern blotting and/or quantitative qRT-PCR as described later.
For the experiments presented in Figures 7 and 8, HEK-293 Flp-In T-Rex cells lines (150,000 cells/ml) were transfected with siRNA targeting DIMT1L or WBSCR22 mRNA (LD070-s26097 or LD020-s41529, respectively) or with a nontargeting negative control siRNA (LD035_Scramble) as described previously (Tafforeau et al., 2013). Transfected cells were first cultured for 24 h in DMEM (Sigma-Aldrich) supplemented with FBS (10%) and Pen-Strep (1%) in the presence or absence of tetracycline at 0.2 μg/ml. After 24 h, the medium was replaced with DMEM (Sigma-Aldrich) supplemented with FBS (10%), Pen-Strep (1%), hygromycin B (100 μg/ml; Invitrogen), and blasticidin (10 μg/ml; Life Technologies) in the presence or absence of tetracycline at 0.2 μg/ml. Cells were cultured for an additional 24 h before total protein and total RNA extraction.
mRNA quantitation
The residual level of mRNAs was assessed by qRT-PCR. cDNA was synthesized (qSCript cDNA supermix; Quanta Biosciences) using as template total RNA extracted from HeLa cells transfected with a siRNA and incubated for 3 d. The qPCR was performed on a SetOne Plus system (Life Technologies) with the PerfeCta SYBR green supermix ROX (Quanta Biosciences) with the following specific primers: WBSCR22 (LD2476 × LD2477); TRMT112 (LD2472 × LD2473); DIMT1L (LD2637 × LD2638); PNO1 (LD2530 × LD2531); and glyceraldehyde-3-phosphate dehydrogenase (LD1818 × LD1819).
RNA electrophoresis and Northern blotting
For analysis of high–molecular weight RNA species, 3 μg of total RNA (for RKO and HCT116 cells) or 1.5 μg of total RNA (for WI-38 cells) was resolved on a denaturing agarose gel (6% formaldehyde/1.2% agarose in 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES]–EDTA buffer). For analysis of low–molecular weight species, 3 μg of total RNA was resolved on denaturing acrylamide gels (8% acrylamide-bisacrylamide 19:1/8 M urea in Tris-borate-EDTA buffer). Agarose gels were transferred overnight by capillarity (in 10× saline–sodium citrate) onto Hybond-N+ membranes. The membranes were prehybridized for 1 h at 65°C in 50% formamide, 5× SSPE, 5× Denhardt's solution, 1% (wt/vol) SDS, and 200 μg/ml fish sperm DNA solution (Roche). The 32P-labeled oligonucleotide probe was added and incubated for 1 h at 65°C and then overnight at 37°C. The probes are described in Supplemental Table S4. Northern blots were exposed to Fuji imaging plates (Fujifilm) and signals acquired with a Phosphorimager (FLA-7000; Fujifilm). The following rRNA fragments were quantified: the 47S pre-rRNA and the aberrant 34S (with the 5′-ETS probe, LD1844), the 47S/45S, 41S, 30S, 26S, 21S, and 18-E pre-rRNAs (ITS1 probe, LD1827), and the 32S (ITS2 probe, LD1828). Quantification was performed with native MultiGauge software, version 3.1. Each gel lane and RNA band was identified manually. The background was set automatically with the help of a polygonal lane (settings: H ratio = 10; V ratio = 75) and was then manually curated. The 28S/18S ratio was calculated with the Agilent RNA 6000 nano kit (5067-1511) on a BioAnalyzer 2100 (Agilent).
Pulse-chase analysis
We reverse transfected 75,000 HeLa cells with the appropriate siRNA as described under siRNA inactivation and total RNA extraction. After a 72-h incubation, the cells were washed and incubated for 30 min at 37°C in methionine-free DMEM (Invitrogen) supplemented with 10% dialyzed FBS (Sigma-Aldrich), labeled for 30 min with 50 μCi l-(methyl-3H)-methionine (50 mCi/ml; PerkinElmer), washed with complete growth medium supplemented with 0.3 mg/ml methionine, and incubated for 0, 15, 30, 60, 120, or 240 min in the same medium. The cells were then resuspended in 1 ml TRI Reagent (Life Technologies) for RNA extraction as described. Purified RNAs were separated on an agarose denaturing gel (6% formaldehyde/1.2% agarose in HEPES-EDTA buffer) for 16 h at 60 V and transferred to a GeneScreen membrane. The membrane was sprayed with tritium enhancer (PerkinElmer) and exposed and autoradiographed.
Primer extension
Primer extension was carried out on 5 μg of total RNA and a molar excess of oligonucleotide (LD2141 for mapping the 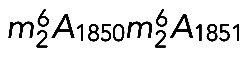 modifications, and LD2120 or LD2122 for m7G1639), using AMV Reverse Transcriptase (Promega). The RNA was then hydrolyzed and the cDNA precipitated and resolved on 6% sequencing gels. For mapping m7G1639, reverse transcription was performed on RNAs that had first been specifically cleaved at N7-methylated guanosine positions by borohydride reduction in 0.1 M NaBH4, followed by cleavage in a 1.0 M aniline/acetate, pH 4.5, buffer in the presence of 50 μg of m7GTP carrier (Létoquart et al., 2014). Primer extension stops were mapped with a DNA sequencing ladder generated with the same oligonucleotide as used for primer extension.
modifications, and LD2120 or LD2122 for m7G1639), using AMV Reverse Transcriptase (Promega). The RNA was then hydrolyzed and the cDNA precipitated and resolved on 6% sequencing gels. For mapping m7G1639, reverse transcription was performed on RNAs that had first been specifically cleaved at N7-methylated guanosine positions by borohydride reduction in 0.1 M NaBH4, followed by cleavage in a 1.0 M aniline/acetate, pH 4.5, buffer in the presence of 50 μg of m7GTP carrier (Létoquart et al., 2014). Primer extension stops were mapped with a DNA sequencing ladder generated with the same oligonucleotide as used for primer extension.
Protein overexpression and purification of the WBSCR22-TRMT112 complex
TRMT112 and polyhistidine-tagged (His6-tagged) WBSCR22 were produced in the Escherichia coli strain SoluBL21 DE3 cotransformed with suitable plasmids (see Supplemental Table S1) after induction overnight with isopropyl-β-d-thiogalactoside (IPTG; 1 mM) at 23°C at an optical density (600 nm) of 0.5 in Luria–Bertani medium containing ZnCl2 (100 μM final concentration), ampicillin (200 μg/ml), kanamycin (50 μg/ml), and chloramphenicol (15 μg/ml). The complexes obtained were purified as previously described for ScMtq2-Trm112 (Heurgue-Hamard et al., 2006), except for the use of His-Select Nickel affinity Gel (Sigma-Aldrich), for which we used the following resuspension/wash buffer (10 mM Tris-HCl, pH 7.5, 200 mM NaCl, 6 mM β-mercaptoethanol, 10 μM ZnCl2, 10 mM imidazole). Elution was done with the same buffer containing 50 mM imidazole.
Western blots
For total protein extraction, confluent cells from one well of a six-well plate were washed in 1 ml of cold phosphate-buffered saline (PBS), incubated on ice for 15 min in 300 μl of lysis buffer (20 mM Tris-HCl, pH 8.0, 0.5% NP-40, 150 mM NaCl, 1 mM EDTA, and protease inhibitor [Roche]), detached, and centrifuged at 20,000 × g for 10 min at 4°C. A 10-μg amount of protein (assayed by Bradford assay; Bio-Rad) was resolved on a 12% or a 4–12% polyacrylamide gel (Novex; Life Technologies) and transferred onto a nitrocellulose or polyvinylidene fluoride membrane. Membranes blocked in PBS supplemented with 5% bovine serum albumin (BSA) were incubated with primary antibody. For detection, we used the following primary antibodies: anti-DIMT1L (sc135130; Santa Cruz Biotechnology), anti-WBSCR22 (ab97911; Abcam), and anti-TRMT112 (H00051504-M09; Novus Biological) at 1:500 and anti-Flag (F3165; Sigma-Aldrich) at 1:1000. After washes in PBS/Tween-20, the membranes were incubated with horseradish peroxidase–tagged secondary antibodies (Santa Cruz Biotechnology). The signal was produced with the Supersignal WestPico Chemiluminescent Substrate (Thermo Scientific) or the Clarity Western ECL Substrate (Bio-Rad) and analyzed with the ChemiDoc imaging system fitted with an XRS camera (Bio-Rad). β-Actin (SC69879; Santa Cruz Biotechnology) was used as a loading control.
Microscopy
Immunofluorescence experiments were carried out on cells grown in 96-well plates. Cells were fixed in PBS/formaldehyde (4%) for 15 min at room temperature and rinsed three times for 5 min in PBS. The cells were then permeabilized by incubation in PBS supplemented with Triton X-100 (0.3%) and BSA (5%) for 60 min at room temperature. Primary antibodies targeting DIMT1L (ab69434; Abcam), WBSCR22 (HPA052185; Sigma-Aldrich), or TRMT112 (H00051504-M09; Novus Biologicals) were added at 1:500 in PBS supplemented with Triton X-100 (0.3%) and BSA (1%), and incubation was carried out for 16 h at 4°C. The cells were washed three times for 5 min in PBS before incubation for 1 h at room temperature with the appropriate secondary antibody (Alexa Fluor 488 or 594; Invitrogen) diluted at 1:1000 in PBS supplemented with Triton X-100 (0.3%) and BSA (1%). After washes in PBS, the cells were stained for 10 min at room temperature with 4′,6-diamidino-2-phenylindole (DAPI; 5 mg/ml, diluted 1:20,000; Sigma-Aldrich) and finally washed and stored in PBS before imaging. For lysosome or Golgi localization, cells were treated just before fixation, for 1 h at 37°C in the incubator, with Lyso- or Golgi-Tracker (LysoTracker Green DND-26 or CellLight Golgi-GFP BacMam 2.0; Life Technologies) diluted 1:20,000. The cells were washed once in PBS before being processed for immunofluorescence. Cells were observed with a Zeiss Axio Observer Z1 microscope driven by MetaMorph (MDS Analytical Technologies). High-resolution images were captured in the confocal mode with a Yokogawa spinning-disk head and the HQ2 camera with a laser illuminator from Roper (405-nm, 100-mW Vortran; 491-nm, 50-mW Cobolt Calypso; and 561-nm, 50-mW Cobolt Jive). To assess the specificity of the observed subcellular distribution of the relevant label, cells were incubated with secondary antibody alone and examined after the same exposure time. Images were analyzed with ImageJ (National Institutes of Health, Bethesda, MD).
Supplementary Material
Acknowledgments
We acknowledge N. Watkins for the gift of plasmid pcDNA5/FRT/TO. Christiane Zorbas was the recipient of a PhD fellowship from the “Fonds pour la formation à la Recherche dans l'Industrie et dans l'Agriculture” (FRIA) and Fonds Alice & David van Buuren. The laboratory of D.L.J.L. is supported by the Université Libre de Bruxelles, the Fonds National de la Recherche (FRS/FNRS), the Walloon Region (DGO6), and the European Research Development Fund. V.H.H. acknowledges financial support from the Centre National de la Recherche Scientifique, the Agence Nationale de la Recherche (ANR-14-CE09-0016-01), and the Initiative d'Excellence program of the French Government (Grant DYNAMO, ANR-11-LABX-0011).
Abbreviations used:
- Bd
body
- Bk
beak
- BSA
bovine serum albumin
- DAPI
4', 6'-diamidino-2-phenylindole
- DCS
decoding site
- EDTA
ethylenediaminetetraacetic acid
- FBS
fetal bovine serum
- FIB
fibrillarin
- GFP
green fluorescent protein
- H
head
- IPTG
isopropyl-β-d-thiogalactoside
- ITS
internal transcribed spacer
- Lf
left foot
- Mtase
methyltransferase
- Ni-NTA
nickel-nitrilotriacetic acid
- Nk
neck
- PBS
phosphate-buffered saline
- Pen-Strep
penicillin–streptomycin
- Pt
platform
- qRT-PCR
quantitative reverse transcription PCR
- Rf
right foot
- SCR
scramble
- Sh
shoulder
- siRNA
small interfering RNA
- Tet
tetracycline.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-02-0073) on April 7, 2015.
*These authors contributed equally.
REFERENCES
- Anger AM, Armache JP, Berninghausen O, Habeck M, Subklewe M, Wilson DN, Beckmann R. Structures of the human and Drosophila 80S ribosome. Nature. 2013;497:80–85. doi: 10.1038/nature12104. [DOI] [PubMed] [Google Scholar]
- Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. The structure of the eukaryotic ribosome at 3.0 A resolution. Science. 2011;334:1524–1529. doi: 10.1126/science.1212642. [DOI] [PubMed] [Google Scholar]
- Boehringer D, O'Farrell HC, Rife JP, Ban N. Structural insights into methyltransferase KsgA function in 30S ribosomal subunit biogenesis. J Biol Chem. 2012;287:10453–10459. doi: 10.1074/jbc.M111.318121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand RC, Klootwijk J, Van Steenbergen TJ, De Kok AJ, Planta RJ. Secondary methylation of yeast ribosomal precursor RNA. Eur J Biochem. 1977;75:311–318. doi: 10.1111/j.1432-1033.1977.tb11531.x. [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Bywater MJ, Poortinga G, Sanij E, Hein N, Peck A, Cullinane C, Wall M, Cluse L, Drygin D, Anderes K, et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell. 2012;22:51–65. doi: 10.1016/j.ccr.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly K, Rife JP, Culver G. Mechanistic insight into the ribosome biogenesis functions of the ancient protein KsgA. Mol Microbiol. 2008;70:1062–1075. doi: 10.1111/j.1365-2958.2008.06485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirci H, Murphy Ft, Belardinelli R, Kelley AC, Ramakrishnan V, Gregory ST, Dahlberg AE, Jogl G. Modification of 16S ribosomal RNA by the KsgA methyltransferase restructures the 30S subunit to optimize ribosome function. RNA. 2010;16:2319–2324. doi: 10.1261/rna.2357210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll A, Grzeschik KH. Characterization of two novel genes, WBSCR20 and WBSCR22, deleted in Williams-Beuren syndrome. Cytogenet Cell Genet. 2001;95:20–27. doi: 10.1159/000057012. [DOI] [PubMed] [Google Scholar]
- Fernandez-Pevida A, Kressler D, de la Cruz J. Processing of preribosomal RNA in Saccharomyces cerevisiae. Wiley Interdiscip Rev RNA. 2015;6:191–209. doi: 10.1002/wrna.1267. [DOI] [PubMed] [Google Scholar]
- Figaro S, Scrima N, Buckingham RH, Heurgue-Hamard V. HemK2 protein, encoded on human chromosome 21, methylates translation termination factor eRF1. FEBS Lett. 2008;582:2352–2356. doi: 10.1016/j.febslet.2008.05.045. [DOI] [PubMed] [Google Scholar]
- Figaro S, Wacheul L, Schillewaert S, Graille M, Huvelle E, Mongeard R, Zorbas C, Lafontaine DLJ, Heurgue-Hamard V. Trm112 is required for Bud23-mediated methylation of the 18S rRNA at position G1575. Mol Cell Biol. 2012;32:2254–2267. doi: 10.1128/MCB.06623-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu D, Brophy JA, Chan CT, Atmore KA, Begley U, Paules RS, Dedon PC, Begley TJ, Samson LD. Human AlkB homolog ABH8 Is a tRNA methyltransferase required for wobble uridine modification and DNA damage survival. Mol Cell Biol. 2010;30:2449–2459. doi: 10.1128/MCB.01604-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandi P, Rybin V, Bassler J, Petfalski E, Strauss D, Marzioch M, Schafer T, Kuster B, Tschochner H, Tollervey D, et al. 90S pre-ribosomes include the 35S pre-rRNA, the U3 snoRNP, and 40S subunit processing factors but predominantly lack 60S synthesis factors. Mol Cell. 2002;10:105–115. doi: 10.1016/s1097-2765(02)00579-8. [DOI] [PubMed] [Google Scholar]
- Granneman S, Petfalski E, Swiatkowska A, Tollervey D. Cracking pre-40S ribosomal subunit structure by systematic analyses of RNA-protein cross-linking. EMBO J. 2010;29:2026–2036. doi: 10.1038/emboj.2010.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean H, Breton M, Sirand-Pugnet P, Tardy F, Thiaucourt F, Citti C, Barre A, Yoshizawa S, Fourmy D, de Crecy-Lagard V, Blanchard A. Predicting the minimal translation apparatus: lessons from the reductive evolution of mollicutes. PLoS Genet. 2014;10:e1004363. doi: 10.1371/journal.pgen.1004363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag S, Kretschmer J, Bohnsack MT. WBSCR22/Merm1 is required for late nuclear pre-ribosomal RNA processing and mediates N7-methylation of G1639 in human 18S rRNA. RNA. 2015;21:180–187. doi: 10.1261/rna.047910.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helser TL, Davies JE, Dahlberg JE. Change in methylation of 16S ribosomal RNA associated with mutation to kasugamycin resistance in Escherichia coli. Nat New Biol. 1971;233:12–14. doi: 10.1038/newbio233012a0. [DOI] [PubMed] [Google Scholar]
- Henras A, Plisson-Chastang C, O'Donohue M-F, Chakraborthy A, Gleizes P-E. An overview of pre-rRNA processing in eukaryotes. Wiley Interdiscip Rev RNA. 2015;6:225–242. doi: 10.1002/wrna.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurgue-Hamard V, Graille M, Scrima N, Ulryck N, Champ S, van Tilbeurgh H, Buckingham RH. The zinc finger protein Ynr046w is plurifunctional and a component of the eRF1 methyltransferase in yeast. J Biol Chem. 2006;281:36140–36148. doi: 10.1074/jbc.M608571200. [DOI] [PubMed] [Google Scholar]
- James A, Wang Y, Raje H, Rosby R, DiMario P. Nucleolar stress with and without p53. Nucleus. 2014;5:402–426. doi: 10.4161/nucl.32235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jangani M, Poolman TM, Matthews L, Yang N, Farrow SN, Berry A, Hanley N, Williamson AJ, Whetton AD, Donn R, Ray DW. The methyltransferase WBSCR22/Merm1 enhances glucocorticoid receptor function and is regulated in lung inflammation and cancer. J Biol Chem. 2014;289:8931–8946. doi: 10.1074/jbc.M113.540906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvam E, Goldfarb DS. Nucleus-vacuole junctions in yeast: anatomy of a membrane contact site. Biochem Soc Trans. 2006;34:340–342. doi: 10.1042/BST0340340. [DOI] [PubMed] [Google Scholar]
- Lafontaine DLJ. A “garbage can” for ribosomes: how eukaryotes degrade their ribosomes. Trends Biochem Sci. 2010;35:267–277. doi: 10.1016/j.tibs.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Lafontaine DLJ. Noncoding RNAs in eukaryotic ribosome synthesis and function. Nat Struct Mol Biol. 2015;22:11–19. doi: 10.1038/nsmb.2939. [DOI] [PubMed] [Google Scholar]
- Lafontaine DLJ, Delcour J, Glasser AL, Desgres J, Vandenhaute J. The DIM1 gene responsible for the conserved m6(2)Am6(2)A dimethylation in the 3'-terminal loop of 18 S rRNA is essential in yeast. J Mol Biol. 1994;241:492–497. doi: 10.1006/jmbi.1994.1525. [DOI] [PubMed] [Google Scholar]
- Lafontaine DLJ, Preiss T, Tollervey D. Yeast 18S rRNA dimethylase Dim1p: a quality control mechanism in ribosome synthesis. Mol Cell Biol. 1998;18:2360–2370. doi: 10.1128/mcb.18.4.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontaine DLJ, Vandenhaute J, Tollervey D. The 18S rRNA dimethylase Dim1p is required for pre-ribosomal RNA processing in yeast. Genes Dev. 1995;9:2470–2481. doi: 10.1101/gad.9.20.2470. [DOI] [PubMed] [Google Scholar]
- Létoquart J, Huvelle E, Wacheul L, Bourgeois G, Zorbas C, Graille M, Heurgué-Hamard V, Lafontaine DLJ. Structural and functional studies of Bud23-Trm112 reveal 18S rRNA N7-G1575 methylation occurs on late 40S precursor ribosomes. Proc Natl Acad Sci USA. 2014;111:E5518–E5526. doi: 10.1073/pnas.1413089111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikov S, Ben-Shem A, Garreau de Loubresse N, Jenner L, Yusupova G, Yusupov M. One core, two shells: bacterial and eukaryotic ribosomes. Nat Struct Mol Biol. 2012;19:560–567. doi: 10.1038/nsmb.2313. [DOI] [PubMed] [Google Scholar]
- Mullineux ST, Lafontaine DLJ. Mapping the cleavage sites on mammalian pre-rRNAs: where do we stand. Biochimie. 2012;94:1521–1532. doi: 10.1016/j.biochi.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Nakazawa Y, Arai H, Fujita N. The novel metastasis promoter Merm1/Wbscr22 enhances tumor cell survival in the vasculature by suppressing Zac1/p53-dependent apoptosis. Cancer Res. 2011;71:1146–1155. doi: 10.1158/0008-5472.CAN-10-2695. [DOI] [PubMed] [Google Scholar]
- Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115:3196–3205. doi: 10.1182/blood-2009-10-178129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohbayashi I, Konishi M, Ebine K, Sugiyama M. Genetic identification of Arabidopsis RID2 as an essential factor involved in pre-rRNA processing. Plant J. 2011;67:49–60. doi: 10.1111/j.1365-313X.2011.04574.x. [DOI] [PubMed] [Google Scholar]
- Ounap K, Kasper L, Kurg A, Kurg R. The human WBSCR22 protein is involved in the biogenesis of the 40S ribosomal subunits in mammalian cells. PLoS One. 2013;8:e75686. doi: 10.1371/journal.pone.0075686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preti M, O'Donohue MF, Montel-Lehry N, Bortolin-Cavaille ML, Choesmel V, Gleizes PE. Gradual processing of the ITS1 from the nucleolus to the cytoplasm during synthesis of the human 18S rRNA. Nucleic Acids Res. 2013;41:4709–4723. doi: 10.1093/nar/gkt160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rife JP. Roles of the ultra-conserved ribosomal RNA methyltransferase KsgA in ribosome biogenesis. In: Grosjean H, editor. DNA and RNA Modification Enzymes: Structure, Mechanism, Function and Evolution. Austin, TX: Landes Bioscience; 2009. pp. 512–526. [Google Scholar]
- Roberts P, Moshitch-Moshkovitz S, Kvam E, O'Toole E, Winey M, Goldfarb DS. Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae. Mol Biol Cell. 2003;14:129–141. doi: 10.1091/mbc.E02-08-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggero D, Pandolfi PP. Does the ribosome translate cancer. Nat Rev Cancer. 2003;3:179–192. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- Salim M, Maden BE. Early and late methylations in HeLa cell ribosome maturation. Nature. 1973;244:334–336. doi: 10.1038/244334a0. [DOI] [PubMed] [Google Scholar]
- Sardana R, Johnson AW. The methyltransferase adaptor protein Trm112 is involved in biogenesis of both ribosomal subunits. Mol Biol Cell. 2012;23:4313–4322. doi: 10.1091/mbc.E12-05-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer T, Strauss D, Petfalski E, Tollervey D, Hurt E. The path from nucleolar 90S to cytoplasmic 40S pre-ribosomes. EMBO J. 2003;22:1370–1380. doi: 10.1093/emboj/cdg121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, Tollervey D. Threading the barrel of the RNA exosome. Trends Biochem Sci. 2013;38:485–493. doi: 10.1016/j.tibs.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Lafontaine DLJ. A new perspective on eukaryotic ribosomal RNA base modification. Trends Biochem. 2015 doi: 10.1016/j.tibs.2015.07.008. (in press) [DOI] [PubMed] [Google Scholar]
- Sloan KE, Bohnsack MT, Schneider C, Watkins NJ. The roles of SSU processome components and surveillance factors in the initial processing of human ribosomal RNA. RNA. 2014;20:540–550. doi: 10.1261/rna.043471.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan KE, Mattijssen S, Lebaron S, Tollervey D, Pruijn GJ, Watkins NJ. Both endonucleolytic and exonucleolytic cleavage mediate ITS1 removal during human ribosomal RNA processing. J Cell Biol. 2013;200:577–588. doi: 10.1083/jcb.201207131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songe-Moller L, van den Born E, Leihne V, Vagbo CB, Kristoffersen T, Krokan HE, Kirpekar F, Falnes PO, Klungland A. Mammalian ALKBH8 possesses tRNA methyltransferase activity required for the biogenesis of multiple wobble uridine modifications implicated in translational decoding. Mol Cell Biol. 2010;30:1814–1827. doi: 10.1128/MCB.01602-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strunk BS, Loucks CR, Su M, Vashisth H, Cheng S, Schilling J, Brooks CL, 3rd, Karbstein K, Skiniotis G. Ribosome assembly factors prevent premature translation initiation by 40S assembly intermediates. Science. 2011;333:1449–1453. doi: 10.1126/science.1208245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafforeau L, Zorbas C, Langhendries JL, Mullineux ST, Stamatopoulou V, Mullier R, Wacheul L, Lafontaine DLJ. The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of Pre-rRNA processing factors. Mol Cell. 2013;51:539–551. doi: 10.1016/j.molcel.2013.08.011. [DOI] [PubMed] [Google Scholar]
- Tiedemann RE, Zhu YX, Schmidt J, Shi CX, Sereduk C, Yin H, Mousses S, Stewart AK. Identification of molecular vulnerabilities in human multiple myeloma cells by RNA interference lethality screening of the druggable genome. Cancer Res. 2012;72:757–768. doi: 10.1158/0008-5472.CAN-11-2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuhisa JG, Vijayan P, Feldmann KA, Browse JA. Chloroplast development at low temperatures requires a homolog of DIM1, a yeast gene encoding the 18S rRNA dimethylase. Plant Cell. 1998;10:699–711. doi: 10.1105/tpc.10.5.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Buul CP, Visser W, van Knippenberg PH. Increased translational fidelity caused by the antibiotic kasugamycin and ribosomal ambiguity in mutants harbouring the ksgA gene. FEBS Lett. 1984;177:119–124. doi: 10.1016/0014-5793(84)80994-1. [DOI] [PubMed] [Google Scholar]
- van Knippenberg PH. Structural and functional aspects of the N6, N6 dimethyladenosines in 16S ribosomal RNA. In: Hardesty B, Kramer G, editors. Structure, Function, and Genetics of Ribosomes. Berlin: Springer-Verlag; 1986. pp. 412–424. [Google Scholar]
- Vanrobays E, Gelugne JP, Caizergues-Ferrer M, Lafontaine DLJ. Dim2p, a KH-domain protein required for small ribosomal subunit synthesis. RNA. 2004;10:645–656. doi: 10.1261/rna.5162204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlatkovic N, Boyd MT, Rubbi CP. Nucleolar control of p53: a cellular Achilles' heel and a target for cancer therapy. Cell Mol Life Sci. 2014;71:771–791. doi: 10.1007/s00018-013-1361-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins NJ, Bohnsack MT. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip Rev RNA. 2012;3:397–414. doi: 10.1002/wrna.117. [DOI] [PubMed] [Google Scholar]
- White J, Li Z, Sardana R, Bujnicki JM, Marcotte EM, Johnson AW. Bud23 methylates G1575 of 18S rRNA and is required for efficient nuclear export of pre-40S subunits. Mol Cell Biol. 2008;28:3151–3161. doi: 10.1128/MCB.01674-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieckowski Y, Schiefelbein J. Nuclear ribosome biogenesis mediated by the DIM1A rRNA dimethylase is required for organized root growth and epidermal patterning in Arabidopsis. Plant Cell. 2012;24:2839–2856. doi: 10.1105/tpc.112.101022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild T, Horvath P, Wyler E, Widmann B, Badertscher L, Zemp I, Kozak K, Csucs G, Lund E, Kutay U. A protein inventory of human ribosome biogenesis reveals an essential function of exportin 5 in 60S subunit export. PLoS Biol. 2010;8:e1000522. doi: 10.1371/journal.pbio.1000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolford JL, Jr, Baserga SJ. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics. 2013;195:643–681. doi: 10.1534/genetics.113.153197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, O'Farrell HC, Rife JP, Culver GM. A conserved rRNA methyltransferase regulates ribosome biogenesis. Nat Struct Mol Biol. 2008;15:534–536. doi: 10.1038/nsmb.1408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.