

ABSTRACT
The human JC polyomavirus (JCPyV) establishes an asymptomatic, persistent infection in the kidneys of the majority of the population and is the causative agent of the fatal demyelinating disease progressive multifocal leukoencephalopathy (PML) in immunosuppressed individuals. The Mad-1 strain of JCPyV, a brain isolate, was shown earlier to require α2,6-linked sialic acid on the lactoseries tetrasaccharide c (LSTc) glycan for attachment to host cells. In contrast, a JCPyV kidney isolate type 3 strain, WT3, has been reported to interact with sialic acid-containing gangliosides, but the role of these glycans in JCPyV infection has remained unclear. To help rationalize these findings and probe the effects of strain-specific differences on receptor binding, we performed a comprehensive analysis of the glycan receptor specificities of these two representative JCPyV strains using high-resolution X-ray crystallography and nuclear magnetic resonance (NMR) spectroscopy, and correlated these data with the results of infectivity assays. We show here that capsid proteins of Mad-1 and WT3 JCPyV can both engage LSTc as well as multiple sialylated gangliosides. However, the binding affinities exhibit subtle differences, with the highest affinity observed for LSTc. Engagement of LSTc is a prerequisite for functional receptor engagement, while the more weakly binding gangliosides are not required for productive infection. Our findings highlight the complexity of virus-carbohydrate interactions and demonstrate that subtle differences in binding affinities, rather than the binding event alone, help determine tissue tropism and viral pathogenesis.
IMPORTANCE Viral infection is initiated by attachment to receptors on host cells, and this event plays an important role in viral disease. We investigated the receptor-binding properties of human JC polyomavirus (JCPyV), a virus that resides in the kidneys of the majority of the population and can cause the fatal demyelinating disease progressive multifocal leukoencephalopathy (PML) in the brains of immunosuppressed individuals. JCPyV has been reported to interact with multiple carbohydrate receptors, and we sought to clarify how the interactions between JCPyV and cellular carbohydrate receptors influenced infection. Here we demonstrate that JCPyV can engage numerous sialylated carbohydrate receptors. However, the virus displays preferential binding to LSTc, and only LSTc mediates a productive infection. Our findings demonstrate that subtle differences in binding affinity, rather than receptor engagement alone, are a key determinant of viral infection.
INTRODUCTION
JC polyomavirus (JCPyV) infects ∼50% of healthy individuals and causes an asymptomatic, lifelong persistent infection in the kidney (1, 2). The form of the virus that resides in the kidney is nonpathogenic and is excreted in the urine (3, 4). In immunosuppressed individuals, JCPyV can spread from the site of persistence to the central nervous system (CNS) (5, 6) and infect the glial cells astrocytes and oligodendroctyes (7, 8). Oligodendrocytes are myelin-producing cells, and astrocytes have been demonstrated to be critical to the process of CNS myelination (9–11). Infection of astrocytes and glial progenitor cells (GPCs), together with virus-induced cytolytic destruction of oligodendrocytes, causes the demyelinating disease progressive multifocal leukoencephalopathy (PML) (12–14). The disease, once considered fatal, is now managed with immune reconstitution therapy, but surviving patients remain severely debilitated (15). PML affects approximately 3 to 5% of HIV-1-positive individuals and patients receiving immunomodulatory therapies such as natalizumab and rituximab for immune-mediated diseases such as multiple sclerosis (MS), Crohn's disease, and rheumatoid arthritis (13, 16, 17). As of December 2014, there had been 517 cases of natalizumab-induced PML, and 23% of these PML patients have died, since the introduction of the drug in 2004 (Biogen Idec, 2014). Newly marketed therapies such as dimethyl fumarate and other fumaric acid ester-containing drugs prescribed for MS and psoriasis have also led to PML (18, 19), indicating an urgent need to better understand the disease pathogenesis of JCPyV.
Polyomaviruses have an icosahedral, nonenveloped protein capsid that is comprised of VP1, VP2, and VP3. VP1 is a pentameric protein present as 360 copies on the surface of the capsid, which are connected to neighboring pentamers through C-terminal extensions (20). Each VP1 pentamer also interacts with a VP2 or a VP3 molecule in the interior of the capsid (21), and together they encase the double-stranded DNA (dsDNA) genome. Surface loops connecting the β-strand core structure of VP1 mediate interactions with sialic acid receptors (22–27). The Mad-1 strain of JCPyV utilizes the sialic acid receptor motif α2,6-linked lactoseries tetrasaccharide c (LSTc) for attachment (24) and then enters by clathrin-dependent endocytosis supported by the 5-hydroxytryptamine 2 (5-HT2) family of serotonin receptors (28). Other members of the Polyomaviridae family, such as BK polyomavirus (BKPyV), Merkel cell polyomavirus (MCPyV), simian virus 40 (SV40), and mouse polyomavirus (MPyV), engage ganglioside receptors containing α2,3- and α2,8-linked sialic acids (23–25, 29, 30) and enter cells by caveola-dependent endocytosis (31). Gangliosides are a group of glycosphingolipids expressed on eukaryotic cells with a ceramide chain embedded in the membrane and an extracellular oligosaccharide (32).
JCPyV strains are classified into seven genotypes that possess amino acid differences at up to 13 positions within the VP1 capsid protein (33) (Fig. 1A). The JCPyV brain isolate Mad-1 strain, with a type 1 capsid, was found to specifically engage α2,6-linked sialic acid in the context of the LSTc pentasaccharide to mediate infection (24). However, other laboratories have demonstrated that virus-like particles (VLPs) of a strain referred to as WT3, with a type 3 capsid that differs from type 1 at 8 amino acid positions, are capable of engaging multiple sialic acid-containing ganglioside receptors (6). WT3 VLPs have been reported to bind to asialo-GM1, GD1a, GD1b, GD2, and GT1b (6) and GM1 and GM2 (Leonid Gorelik, Consortium for Functional Glycomics [CFG] [available at www.functionalglycomics.org]). Additionally, Mad-1 VLPs were demonstrated by virus overlay blotting assay to bind to a number of gangliosides, including GM3, GD2, GD3, GD1a, GD1b, GT1b, and GQ1b, but not GM1 or GM2 (34). However, the relevance of the interactions of JCPyV with multiple sialylated glycans in the context of JCPyV infection remained unclear.
FIG 1.

VP1 amino acid variations in JCPyV genotypes and crystal structure of WT3 VP1. (A) Classification of JCPyV strains into seven genotypes by phylogenetic analysis reveals VP1 amino acid variations (33). Colored spheres highlight residues differing between Mad-1 and WT3 VP1. Strain specific variations can be found within single genotypes. Bold letters indicate amino acids in Mad-1 VP1. N- and C-terminal residues (gray) are not included in the JCPyV Mad-1 VP1 crystal structure (24). (B) Crystal structure of WT3 VP1 pentamer with one VP1 monomer depicted in green. Spheres highlight residues (colored as in panel A) that differ between WT3 and Mad-1 VP1.
Cellular entry of the JCPyV strain Mad-1 has been shown to be mediated by 5-HT2 receptors (5-HT2Rs) (28), and infection of 5-HT2R-expressing HEK293A cells with Mad-1 and WT3 is equivalent (M. S. Maginnis and W. J. Atwood, unpublished results), suggesting that both strains utilize 5-HT2Rs for internalization. Further, the expression of 5-HT2Rs at the cell surface does not increase viral attachment to host cells (28). Therefore, JCPyV attachment and entry steps are likely related but distinct steps, and viral attachment to sialic acid receptors prior to entry plays a key regulatory role in infection and disease (24, 35). Viral isolates from the brains of individuals with PML contain polymorphisms in the capsid that are representative of both type 1 and type 3 strains (36), suggesting that if JCPyV utilizes multiple sialic acid-containing receptors, these polymorphisms might play a role in viral pathogenesis.
The goal of this study was to unravel the molecular details of carbohydrate receptor engagement by JCPyV in order to understand how recognition and specificity contribute to a pathogenesis that ultimately culminates in a devastating CNS disease. To clarify the role of sialic acid-containing receptors in JCPyV infection, we utilized a structure-function approach to define the role of these polymorphisms in JCPyV attachment to cellular receptors and infection of glial cells by generating WT3 VP1 in an infectious viral clone, in pseudoviruses, and in purified VP1 pentamers using the Mad-1 prototype PML strain as the backbone.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
SVGA cells (37) were grown in minimum essential medium (MEM) supplemented to contain 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (P-S) (Mediatech, Inc.) in a humidified incubator at 37°C with 5% CO2. HEK293A cells (ATTC) were grown in Dulbecco's modified Eagle medium (DMEM) supplemented to contain 10% FBS and 1% P-S. 293TT cells are derived from human embryonic kidney cells transformed with two copies of the SV40 large T antigen (a gift from Dan DiMaio's laboratory) and were grown in DMEM supplemented to contain 10% FBS, 0.1 mM nonessential amino acids (NEAA), and 250 μg/ml hygromycin B.
Generation and propagation of the virus strain Mad-1/SVEΔ was previously described (38, 39). Mad-1/WT3C virus (PubMed accession code AAQ88264) was generated by introducing the VP1 mutations R74K, S116T, G133A, L157V, and K163T into the genomic JCPyV DNA of strain JC12 with a Mad-1 VP1 (40) subcloned into pUC19 (41). Mutations R74K, S116T, G133A, and K163T were introduced by site-directed mutagenesis using the Agilent QuikChange II site-directed mutagenesis kit (Qiagen) according to the manufacturer's instructions and the following mutagenesis primers (5′ to 3′): R74K, ACATTTGAAAAGTGACTCCCCAAATAAGGACATGCTTCCTT and AAGGAAGCATGTCCTTATTTGGGGAGTCACTTTCAAATGT; S116T, AGGTTATAGGGGTGACAACTTTGATGAATGTGCACTC and GAGTGCACATTCATCAAAGTTGTCACCCCTATAACCT; G133A, TGACAATGGTGCAGCGAAGCCAGTGCAGG and CCTGCACTGGCTTCGCTGCACCATTGTA; and K163T, GGGGTGCTTTTTAATTACAGAACAACGTACCCAGATGGAACAATTTTT and AAAAATTGTTCCATCTGGGTACGTTGTTCTGTAATTAAAAAGCACCC. L157V was introduced by Phusion mutagenesis using the primers (5′ to 3′) ATTACAGGGGGTGGTCTTTAATTACAGAAC and TCTAAAGCCTCCCCCCCAACAGA. Sequencing was performed at Genewiz Inc.
Mad-1 and Mad-1/WT3C viruses were propagated by transfecting 2 to 4 T150 flasks of SVGA cells with 32 μg of the infectious JCPyV clone with either the Mad-1 or Mad-1/WT3C VP1 that had been digested from the pUC19 plasmid using BamHI using Fugene. At 48 h posttransfection, cells were split into roller bottles (1,650 cm2) and incubated with complete MEM in a humidified roller cabinet at 37°C with 5% CO2. Infected cells were incubated for 2 weeks, with the medium changed at 1 week postinfection with complete MEM. Cells were harvested by scraping, and viruses were purified as described previously (39).
GM3 siRNA treatment.
siGENOME human ST3GAL5 small interfering RNAs (siRNAs) 1, 3, 4, and 17 were purchased from Thermo Scientific. The siRNA target sequences are as follows: 1, CAAUGGCGCUGUUAUUUGA; 3, GACCAUGCAUAAUGUGACA; 4, CGGAAGUUCUCCAGUAAAG; and 17, AGGAAUACUGCACGGAUUA. SignalSilence control siRNA (Cell Signaling Technology) was used as a control siRNA. SVGA cells were reverse transfected with siRNAs using RNAiMax transfection reagent (Invitrogen) by mixing transfection complexes in Opti-MEM in 24-well plates and incubating at room temperature for 20 min. Following incubation, 3 × 104 cells/well were added, and cells were incubated at 37°C for 72 h. Transfection efficiency was confirmed using BLOCK-iT Alexa Fluor Red control oligonucleotide (Life Technologies). At 72 h posttransfection, cells were either harvested for mRNA analysis or infected with JCPyV or SV40 as described below. For mRNA analysis, cells were harvested using CellStripper, washed in phosphate-buffered saline (PBS), and pelleted by centrifugation at 2,000 rpm at 4°C for 5 min. Cells were washed in PBS and pelleted, and RNA was extracted using the RNeasy minikit (Qiagen) with a DNase treatment (Qiagen). One microgram of RNA was reverse transcribed using Bio-Rad iScript cDNA synthesis according to the manufacturer's instructions, and 1 μl of the total reaction product was used for quantitative PCR (qPCR) analysis using Bio-Rad iQ SYBR green Supermix and Bio-Rad iQ5. Primers used for qPCR to detect ST3GAL5 (5′ to 3′) were CTGCCTTTGACATCCTTCAGT and CGATTGTGGGGACGTTCTTA, and those for the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) housekeeping gene were AGTCAGCCGCATCTTCTTTTGC and CAATACGACCAAATCCGTTGACT. Relative ST3GAL5 gene expression was determined by calculating the change in threshold cycle (ΔCT) relative to GAPDH (housekeeping) gene expression, and ΔΔCT was calculated relative to scrambled control siRNA.
JCPyV infection.
SVGA or HEK293A cells were washed following ganglioside treatment, infected with purified Mad-1 and Mad-1/WT3C JCPyV at a multiplicity of infection (MOI) of 5 focus-forming units (FFU)/cell in MEM containing 2% FBS in a volume of 100 to 200 μl, and incubated at 37°C for 1.5 h, and then complete MEM was added and cells were incubated at 37°C for 48 h (T antigen) or 72 h (VP1).
LSTc and GM1 pentasaccharide inhibition assay.
Purified Mad-1 and Mad-1/WT3C were pretreated with 5 mM LSTc (V Labs, Inc.) or GM1 (Enzo Life Sciences) pentasaccharides (diluted in sterile distilled water) in medium containing 2% fetal calf serum (FCS) on ice for 1 h. SVGA cells in 96-well plates were prechilled at 4°C for 30 min. Virus-pentasaccharide complexes were added to cells and incubated at 4°C for 1 h. Cells were washed with PBS twice, complete medium was added, and cells were incubated at 37°C for 72 h. Cells were fixed and stained by indirect immunofluorescence as described below.
Indirect immunofluorescence.
Cells were washed in PBS, fixed in cold methanol, and incubated at −20°C. Fixed cells were stained using PAB597, a hybridoma supernatant that produces a monoclonal antibody against JCPyV VP1 that was generously provided by Ed Harlow, as described previously (35) or using PAB962, a JCPyV-specific T antigen antibody that does not cross-react with SV40 T antigen and was provided by the Tevethia laboratory (Penn State University), as described previously (42). Cells were analyzed for nuclear VP1 or T antigen staining under a 10× or 20× objective using an Eclipse TE2000-U microscope (Nikon).
JCPyV pseudovirus production.
Pseudoviruses were produced by transfection of the VP1, VP2, VP3, and phGluc plasmids into 293TT cells using FuGENE 6 transfection reagent (Promega) in a 5:1:1:1 ratio. Mock pseudovirus controls were generated by transfecting 293TT cells with pUC19 control plasmid and the phGluc reporter plasmid in a 7:1 ratio. Cells were split 1:3 at 48 h posttransfection and harvested at 7 days posttransfection, and titers for properly encapsidated genomes were determined (43).
Pseudovirus luciferase infectivity assay.
Cells were plated in a 24 well or 96-well plate to 70% confluence and infected with equal particle equivalents (1 × 106 or 1 × 107 particles/well) of Mad-1 or Mad-1/WT3C pseudovirus in incomplete medium without phenol red. Infected cells were incubated at 37°C for 1.5 h and washed with PBS, and complete medium without phenol red was added and cells were incubated at 37°C for 72 h. Secreted luciferase was quantitated in 20 to 50 μl of cellular supernatants using the BioLux Gaussia luciferase assay (NE BioLabs) according to the manufacturer's instructions with an opaque 96-well microplate in a GloMax Multi detection system luminometer (Promega). Numbers of infected cells were also measured by quantifying green fluorescent protein (GFP)-positive cells by fluorescence microscopy using an Eclipse TE2000-U microscope (Nikon).
Protein expression, purification, and crystallization.
DNA coding for amino acids 22 to 289 of VP1 from the WT3 strain (UniProtKB accession number AAQ88264), obtained by gene synthesis (Eurofins MWG Operons, Germany), was subcloned into the pET15b vector (EMD Millipore) in frame with an N-terminal hexahistidine tag (His tag) and a thrombin cleavage site using the NcoI and BamHI restriction sites (24). Mad-1 and WT3 VP1 were expressed, purified, and crystallized as described earlier (24, 44). The C-terminal truncation of VP1 prevents assembly of VP1 pentamers to capsids and other incorrectly multimeric particles, whereas the N-terminal truncation was shown to help with the crystallization in the case of MPyV VP1 (22). Prior to crystallization, the His tag was cleaved off with thrombin (GE Healthcare) in solution, resulting in a short nonnative sequence at the (GSHM) N termini of Mad-1 and WT3 VP1.
Crystals of Mad-1 and WT3 VP1 pentamers, grown in space group C2 with one VP1 pentamer in the asymmetric unit, are highly isomorphous (see Tables S1 to S7 in the supplemental material). For comparative soaking experiments, crystals grown for 2 days with similar sizes (∼120 by 120 by 20 μm3) were soaked for 30 min in the reservoir solution complemented with oligosaccharide LSTc (5, 2.5, 1.25, 0.62, or 0.31 mM; Dextra, United Kingdom), GD1a, GD1b, or GM1 (10, 5, 2.5, or 1.25 mM; Elicityl SA, France), or GM2 (20, 10, 5, 2.5, or 1.25 mM; Elicityl SA, France). For cryoprotection, crystals were transferred for 2 s into a reservoir solution supplemented with 30% (vol/vol) glycerol and the respective ligand prior to freezing in liquid nitrogen.
Data collection and structure determination.
Diffraction data sets were collected at beamlines X06DA and X06SA at SLS (Villigen, Switzerland) and processed with XDS (45). The JCPyV Mad-1 VP1 pentamer structure (RCSB Protein Data Bank [PDB] accession number 3NXD) was used for molecular replacement with Phaser MR (46), included in the CCP4 program suite (47). Rigid-body and simulated annealing refinement was carried out with Phenix (48). Alternating rounds of model building in Coot (49) and refinement with Refmac5 (50), including 5-fold noncrystallographic symmetry restraints, the translation-libration-screw method (51), and CCP4 library- and user-defined restraints for the α2,3- and α2,6-glycosidic bond were performed. Coordinates and structure factor amplitudes have been deposited in PDB for the native and complex structures of WT3 VP1 pentamer with 5 mM LSTc, 10 mM GM1, 10 mM GD1a, 10 mM GD1b, and 20 mM GM2. Deposited Mad-1 VP1 pentamer complex structures were obtained with either 10 mM GM1, 10 mM GD1a, 10 mM GD1b, or 20 mM GM2. Structure figures were prepared with PyMOL (PyMOL Molecular Graphics System, version 1.3; Schrödinger, LLC).
Calculation of ligand electron density.
Unit cell parameters of all soaked crystals were treated as isomorphous. The unliganded WT3 VP1 data set was taken as a reference data set during data processing in XDS. CAD of CCP4 (47) was used to combine structure factor amplitudes and structure factor sigmas from all data sets of a concentration-dependent soaking experiment into one single file including free R flags and unit cell parameters from the reference data set (native WT3 VP1). Data sets were scaled (low- and high-resolution cutoff, 12.0 to 2.4 Å) to the reference data set with SCALEIT (52). Refinement with Refmac5 was done to obtain complex structure models in the referenced unit cell. Simulated annealing omit maps were calculated 5 Å around Neu5Ac or marker residues (Y80, Y160, and W200) with Phenix and FFT (CCP4 program suite) under each soaking condition. Masks 1 Å around the Neu5Ac moiety of the respective glycan ligand and around marker residues were generated with the CCP4 program NCSMASK (47). MAPMAN (53, 54) was used to estimate the Neu5Ac and marker residue electron densities by summation of the values for the grid points within the respective mask using the mstats command. Maps and masks were calculated with a grid spacing of ∼dmax/5, with maximum resolution dmax. Integrated electron densities were plotted against the ligand concentration. Data were analyzed and fitted assuming a simple equilibrium of the VP1-glycan interaction using GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA). Assuming a simple equilibrium, a dissociation constants Kd crystal for the interactions within the crystal can be estimated by applying a least-squares fit to the experimental data points and the equation electron densityobserved = [Cligand/(Cligand + Kd crystal)](electron densitymax − electron densitynat) + electron densitynat, where Cligand is the concentration of the ligand used for the soaking experiment. Electron densityobserved is the output from the MAPMAN electron density integration, whereas electron densitynat is the electron density at the respective position within the native crystal and electron densitymax is that observed for the glycan ligand in a fully occupancy binding site. Electron densitymax was not set to a defined value for the fit.
Saturation transfer difference (STD) NMR measurements.
All nuclear magnetic resonance (NMR) spectra were recorded using 3-mm tubes on a Bruker AVIII 600-MHz spectrometer fitted with a 5-mm cryogenic probe at 285 K and processed with TOPSPIN 3.0 (Bruker). Samples were prepared in D2O buffer containing 20 mM potassium phosphate (pH 7.4) and 150 mM NaCl. Protein concentrations were 20 μM VP1, and the GM1 glycan was added to 1 mM. A protein-free GM1 sample was used as a reference. The off- and on-resonance frequencies were set to −30 ppm and 6.9 ppm, respectively. The total relaxation delay was 3 s. A cascade of 40 Gaussian-shaped pulses with 50-ms duration each, corresponding to a strength of 65 Hz, and a saturation time of 2 s were used for selective excitation. A 10-ms continuous-wave spin lock filter with a strength of 3.7 kHz was employed. A total of 32,000 points were collected, and zero filling to 64,000 data points was employed. Spectra were multiplied with an exponential line broadening factor of 1 Hz prior to Fourier transformation.
Protein structure accession numbers.
Coordinates and structure factor amplitudes were deposited in PDB for the native (accession number 4X0Y) and the complex structures of WT3 VP1 pentamer with LSTc (4X13), GM1 (4X0Z), GD1a (4X11), GD1b (4X12), and GM2 (4X10). The Mad-1 VP1 pentamer complex structures obtained with GM1 (4X14), GD1a (4X16), GD1b (4X17), and GM2 (4X15) were deposited in PDB with the indicated accession numbers.
RESULTS
VP1 amino acid variations in JCPyV genotypes and glycan receptor specificity.
JCPyV strains are classified into seven genotypes that possess amino acid differences at up to 13 positions within VP1 (33) (Fig. 1A). Full-length VP1 from the prototype Mad-1 strain, a genotype 1 strain, differs at eight positions (residues 74, 116, 133, 157, 163, 320, 331, and 344) from VP1 of the urine isolate WT3 (genotype 3). Five of those residues are included in the crystal structure of the truncated, assembly-incompetent JCPyV Mad-1 VP1 pentamer comprising residues 22 to 289 (24). The remaining three residues are located in the C-terminal arms of VP1, where they help mediate assembly of the virion particle but are distant from the receptor-binding site (20). In order to assess strain- and genotype-specific glycan receptor engagement on the molecular level, we introduced WT3 polymorphisms into our VP1 construct and solved its X-ray structure (Fig. 1B; see Table S1 in the supplemental material). Structures of Mad-1 and WT3 VP1 are very similar and superimpose with root mean square deviation (RMSD) values of 0.2 Å and 0.5 Å for Cα atoms of monomers and pentamers, respectively. VP1 adopts the iconic jelly-roll fold of virus capsid proteins with two four-stranded β-sheets (strands B, I, D, and G and C, H, E and F) packed against each other, which assemble around a 5-fold axis and are linked on the outside of the virion by extended loops (24). Amino acid differences between Mad-1 and WT3 occur predominantly within or in close proximity to these surface loops (Fig. 1B).
WT3 VP1 engages sialic acids in diverse linkages.
In order to analyze glycan binding specificities of WT3, we solved structures of WT3 VP1 in complex with the glycan portions of a-series gangliosides GM1, GM2, and GD1a, b-series ganglioside GD1b, and the α2,6-linked Mad-1 receptor motif LSTc (Fig. 2; see Table S1 in the supplemental material). WT3 pentamers bound to all glycans, specifically interacting with terminal sialic acids in an almost identical manner independent of the presence of an α2,3-, α2,6-, or α2,8-linkage (Fig. 2; see Fig. S1 in the supplemental material). The conserved sialic acid-binding site is located in a groove at the junction of the extended BC1, BC2, DE, and HI loops of a VP1 monomer and the BC2 and DE loops of the counterclockwise (CCW) and clockwise (CW) neighboring VP1 monomers, respectively. The majority of interactions with the five different glycans involves functional groups of Neu5Ac (described in detail in reference 24). Glycan residues of LSTc, GM1, GM2, and GD1b contribute additional interactions. VP1 residue N123 forms a hydrogen bond with the carbonyl group of the GlcNAc N-acetyl group and fixes LSTc in a unique L-shaped conformation on top of the pentamer (Fig. 2A). The Glc and two Gal residues contribute to the buried surface area upon ligand engagement and are well ordered in the structure due to van der Waals interactions with VP1. In contrast, the Gal-(β1,3)-GalNAc arm of the GM1 glycan points in a different direction, resting in an elongated groove on the protein surface (Fig. 2B). The terminal Gal residue is recognized by hydrogen bonds to the side chains of N130 of the CCW VP1 monomer, S60, and the backbone of residue 61, as well as van der Waals interactions with N130CCW and S268 (Fig. 2B). However, this interaction is not seen in all binding sites, indicating structural flexibility of the terminal Gal. In contrast, the branched “ganglioside core” Neu5Ac-(α2,3)-(GalNAc-(β1,3))-Gal is conformationally more restrained through intramolecular interactions, and it is recognized by a hydrogen bond to K59 and van der Waals interactions with N130CW, S266, and S268. The Glc residue, which is linked to the membrane-anchoring ceramide in the context of the GM1 ganglioside, is pointing away from the protein and is not involved in contacts with VP1.
FIG 2.

JCPyV WT3 interacts with α2,3-, α2,6-linked Neu5Ac and α2,8-, α2,3-di-Neu5Ac in a highly plastic binding site. (A to E) Structures of WT3 VP1 bound to LSTc (A), GM1 (B), GM2 (C), GD1b (D), and GD1a (E) glycans. VP1 is shown in surface and cartoon representation. Residues involved in polar or van der Waals interactions in only one of the glycan complex structures are highlighted in pink or purple, respectively. Interactions are depicted using dashed lines, with direct and water-mediated contacts with the glycans in black, intramolecular interactions of glycans in blue, and interaction with either LSTc, GM1, or GM2 in pink. Glycans are shown in stick representation and colored according to atom type, with nitrogens in blue, oxygens in red, and carbons in orange for LSTc and in yellow for ganglioside-derived glycans, respectively. Water molecules are shown as red spheres. Black arrows indicate the part of LSTc that is linked to a lipid or protein (A), the direction of Glc that is further linked to the lipid in the context of the GM1 and GM2 gangliosides (B and C), and the O2 of the second Neu5Ac, to which the Gal residue within the branched GD1b ganglioside motif is attached (D). (F) Glycans used for comparative binding studies. Additionally, the GT1b ganglioside was used in cell supplementation assays. GT1b features, compared to GD1b, an additional α2,3-linked Neu5Ac at the Gal in the left arm of the branched motif.
Similar interactions hold GalNAc-(β1,4)-Gal in place in the GM2 complex structure, which shows interactions primarily with Neu5Ac (Fig. 2C). In the GD1b complex structure, the second Neu5Ac of the α2,8-α2,3-diNeu5Ac motif adds nonpolar interactions to the protein-glycan interaction network (Fig. 2D). The methyl group of the N-acetyl group is pointing against a hydrophobic platform formed by parts of the N130CW, S266, and S268 side chains. Interactions with the second arm of the branched motif GD1b are not observed. In the case of GD1a, defined electron density was observed only for a single Neu5Ac (Fig. 2E). We also solved equivalent complex structures of JCPyV Mad-1 VP1 (Fig. 3; see Table S2 and Fig. S1 in the supplemental material). Mad-1 VP1 engages GM1, GM2, GD1a, and GD1b glycans using the same contacts described above (Fig. 3 and 4).
FIG 3.

JCPyV Mad-1 engages α2,3-, α2,6-linked Neu5Ac and α2,8-, α2,3-di-Neu5Ac highly similarly to WT3. Complex structures of Mad-1 VP1 with GM1 (A), GM2 (B), GD1b (C), and GD1a (D) ganglioside glycans are shown. VP1 is shown in surface and cartoon representation. Interactions are depicted by dashed lines, with direct and water-mediated contacts with the glycans in black and intramolecular interactions of glycans in blue. VP1 amino acid differences between Mad-1 and WT3 are located predominantly within or close to the surface loops. Residue glycine 133 (green), which is an alanine in WT3, is located beneath N123, which undergoes an induced-fit movement to accommodate the terminal Neu5Ac during glycan engagement (21). Mad-1 residue serine 116, a threonine in WT3, is located below the hydrophobic cavity of the binding site that is formed by F67CW, F158CW, F57, and F262 and encloses the Neu5Ac N-acetyl group. The three additional mutations in WT3 VP1, R74K, L157V, and K163T, are distant from the binding site and are not shown in the close-up views.
FIG 4.

Strain-specific amino acid differences and engagement of GM1 and LSTc. Two VP1 monomers from JCPyV Mad-1 and WT3 are shown in cartoon representations with glycans depicted in orange (LSTc) or yellow (GM1). VP1 residues characteristic for either one of the two strains are colored in pink (Mad-1) or green (WT3). Other genotype-specific amino acid differences are shown in light blue and are labeled in black. Key residues of the glycan binding pocket, the hydrophobic cavity, and N123 are shown in gray sticks.
Binding affinities for LSTc and ganglioside glycans.
We hypothesized that binding affinities could modulate differences in glycan specificities between the two JCPyV strains. A comparative crystallographic approach, similar to strategies previously used for adenovirus fibers (55) and MPyV VP1 (56), was applied to obtain estimates for binding affinities. Isomorphous crystals of Mad-1 and WT3 VP1 were soaked in parallel in solutions supplemented with GM1, GM2, GD1a, GD1b, and LSTc glycans in a concentration-dependent manner, and structures were solved. With increasing glycan concentrations, simulated annealing Fobs − Fcalc omit electron density maps reveal better-defined features of the bound ligand until saturation is reached (Fig. 5A). The electron density within a 1.0-Å mask around the terminal Neu5Ac moieties was integrated and plotted against the respective glycan concentration (Fig. 5B and C). In the outcome of this, electron density binding curves could be fitted assuming a simple equilibrium of the VP1-glycan interaction. In the nonliganded VP1, water molecules are present in the binding site, contributing to the overall electron density at the position of Neu5Ac, when the occupancy is <1.0. Dissociation constants of about 0.5 mM and 6 mM for the strongest interaction with LSTc and the weakest interaction with GD1a, respectively, could be determined. Both strains engage LSTc with the highest affinity but possess similar, lower binding affinities for GD1a, GD1b, and GM2 (Fig. 5B and C). GD1a is engaged with the lowest affinity, consistent with the observation that only a single terminal Neu5Ac is engaged in all complexes (Fig. 2E). A reduced binding affinity is also observed for GD1b, in which only the second Neu5Ac contributes additional hydrophobic contacts with VP1, and for GM2 (Fig. 2D). Of the ganglioside glycans tested, GM1 binds best to JCPyV VP1, although the binding curves differ slightly between the two strains. While the estimated dissociation constant is about 1.2 mM for Mad-1 VP, WT3 VP1 engages the GM1 glycan with a binding affinity very similar to that seen for LSTc, with a dissociation constant of about 0.7 mM.
FIG 5.

Mad-1 and WT3 bind to LSTc with higher affinity than gangliosides. (A) Example for the concentration-dependent crystal soaking experiment. The simulated annealing omit map contoured at 3.0 σ around 2.0 Å of LSTc at 2-4 Å resolution for JCPyV WT3 shows better defined features of the ligand with higher concentration. LSTc (yellow) is shown as a reference in all panels. Data from soaked crystals were scaled to a reference data set, and density around the Neu5Ac moiety was used for electron density integration. (B and C) Concentration-dependent crystal soaking experiments and electron density integration of Fobs − Fcalc simulated annealing omit maps within a 1.0-Å mask around Neu5Ac moieties of LSTc, GM1, GM2, GD1a, or GD1b within the conserved binding site of Mad-1 and WT3 VP1. Values for the electron density obtained by MAPMAN (53, 54) are plotted against the ligand concentration. Water molecules in the binding site contribute to the overall electron density of Neu5Ac when the occupancy is <1.0.
In the crystal, only one binding site in the asymmetric unit is completely open toward the solvent, allowing engagement of all five glycans without steric hindrance or favorable interaction for a particular ligand. Therefore, a cross-validation between binding sites to estimate errors of the measurements is not possible. Electron densities for marker residues on the surface and in the hydrophobic core were instead integrated to test for the nonisomorphism of crystals and resulting differences of the electron density (see Fig. S2 in the supplemental material). The electron density of all marker atoms is clearly independent of the ligand concentrations. Therefore, measured differences in electron density values and binding affinities for the different ligands could be treated as significant.
WT3, Mad-1, and SV40 engage the GM1 glycan in a similar manner.
GM1 is the functional receptor of the closely related SV40 (23, 57). We therefore compared interactions of JCPyV and SV40 with GM1 in solution using saturation transfer difference (STD) NMR spectroscopy (58). This technique can be used to map protein-bound parts of the ligand in order to analyze and compare binding epitopes. STD NMR spectra clearly show that WT3 and Mad-1 VP1 interactions with GM1 are highly similar to the functional SV40 VP1-GM1 receptor interaction (Fig. 6A). Moreover, the superposition of VP1-GM1 complex structures illustrates a conserved binding mode, with distinct differences limited to the GalNAc-(β1,3)-Gal arm of GM1 (Fig. 6B).
FIG 6.

Interactions of JCPyV VP1 with the GM1 glycan reduces virus infection. (A) VP1 engages the GM1 glycan in solution in a manner similar to that for SV40 VP1. From top to bottom: NMR spectrum of the GM1 glycan and STD NMR difference spectra of WT3 VP1, Mad-1 VP1, and SV40 VP1 with GM1 glycan (50-fold molecular excess). (B) GM1 glycan engagement by SV40, Mad-1, and WT3 VP1. GM1 glycans are colored blue, pink, or green for SV40 (PDB 3BWR), Mad-1, and WT3, respectively. The Glc residue of GM1, which is not in contact with VP1, is not shown for clarity. (C) Pretreatment of Mad-1 or Mad-1/WT3C virus with LSTc or GM1 glycan reduces infection. Data represent the average number of infected cells per visual field (10×) for 4 fields of view of triplicate samples. Error bars indicate standard deviations (SD).
LSTc and GM1 glycans block JCPyV infection.
For functional characterization of the strain-related VP1 polymorphisms, we generated a Mad-1/WT3C virus by introducing WT3 sequences at positions 74, 116, 133, 157, and 163 into VP1 in the genomic Mad-1 DNA. To determine if the GM1 glycan could inhibit JCPyV infection, the purified JCPyV Mad-1 and Mad-1/WT3C viruses were pretreated with GM1 or LSTc pentasaccharide prior to addition to cells. Both glycans reduced JCPyV infection of SVGA cells (Fig. 6C). Interestingly, the infection was reduced to a greater extent with LSTc than with GM1, which likely reflects the increased affinity for LSTc. Furthermore, LSTc and GM1 binding sites are overlapping, and therefore, binding of either glycan to VP1 could block binding to either LSTc or GM1.
Exogenous addition of gangliosides does not enhance JCPyV infection.
JCPyV Mad-1 and WT3 demonstrate affinity for gangliosides GM1, GM2, GD1a, and GD1b as determined by structural analysis of the pentamers in complex with the respective glycans. To determine whether JCPyV is able to utilize gangliosides as functional receptors for infection, SVGA cells were supplemented with a panel of gangliosides prior to infection with JCPyV Mad-1, Mad-1/WT3C, BKPyV, and SV40. BKPyV utilizes the gangliosides GD1b and GT1b as functional receptors, while SV40 utilizes GM1, and exogenous ganglioside addition has been previously demonstrated to enhance infection (25, 59). Interestingly, supplementation of cells with gangliosides GM1, GM2, GD1a, GD1b, and GT1b did not alter JCPyV infection compared to that for the dimethyl sulfoxide (DMSO)-treated control. In contrast, addition of GD1b and GT1b enhanced BKPyV infection, and GM1 supplementation enhanced SV40 infection (Fig. 7A). Given the subtle differences in affinity of JCPyV Mad-1 and Mad-1/WT3C for ganglioside-derived glycans compared to LSTc, we hypothesized that these small differences might not be appreciable in a permissive cell type that has been demonstrated to require LSTc for infection (24). Therefore, we also tested whether ganglioside supplementation had an effect in a kidney cell line that is poorly permissive for JCPyV infection (28, 42). HEK293A cells were supplemented with gangliosides overnight and then infected with JCPyV Mad-1, Mad-1/WT3C, BKPyV, and SV40. While supplementation of cells with GD1b and GT1b enhanced BKPyV infection and exogenous GM1 addition resulted in increased SV40 infection, ganglioside supplementation had no effect on JCPyV infection (Fig. 7B). Additionally, supplementation of cells with gangliosides was not toxic to cells as measured by a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophe-nyl)-2H-tetrazolium (MTS) assay (data not shown).
FIG 7.
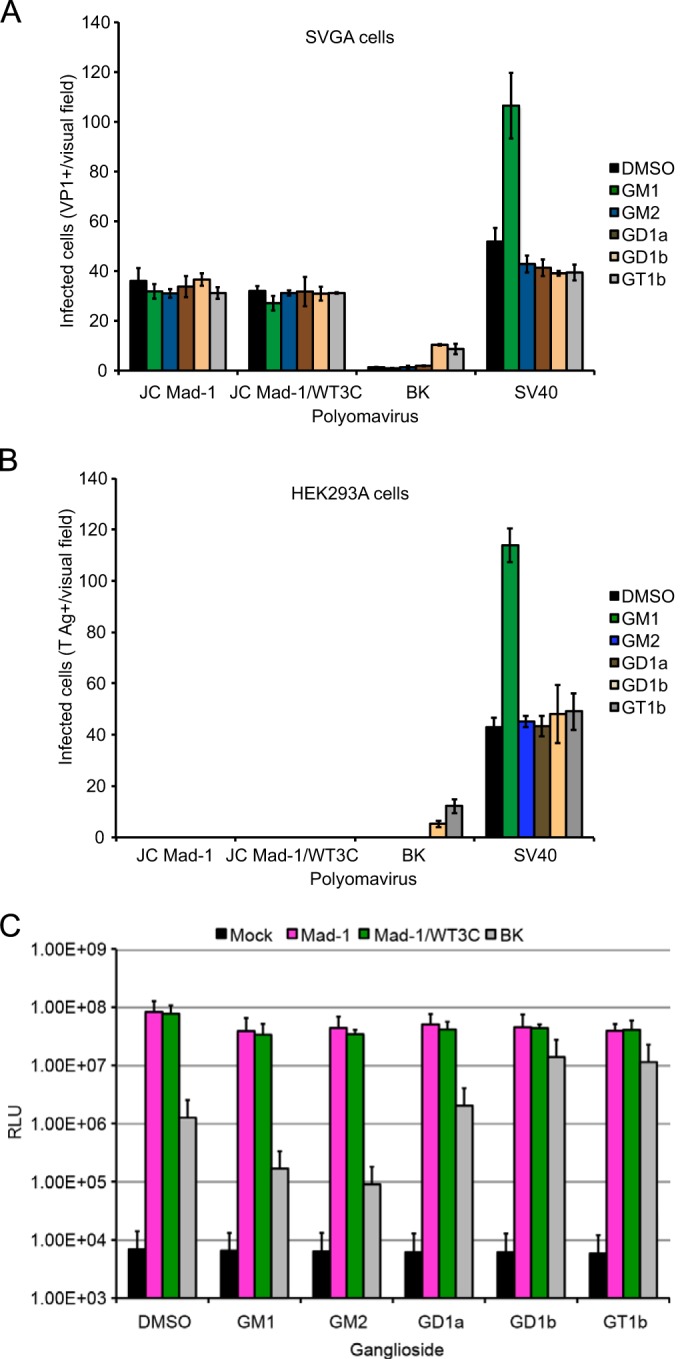
Exogenous addition of gangliosides does not alter JCPyV infection. (A) Permissive SVGA cells in a 24-well plate were supplemented with a 30 μM concentration of the indicated gangliosides or DMSO (vehicle control) overnight and then washed with medium and infected with JCPyV Mad-1, Mad-1/WT3C, or BKPyV and SV40 as controls. Infected cells were scored by indirect immunofluorescence for nuclear VP1 at 72 h. (B) Poorly permissive HEK293A cells were supplemented and infected as for panel A, and infected cells were scored by indirect immunofluorescence for nuclear T antigen at 48 h postinfection. Data represent the average number of infected cells per visual field for five 10× fields of view for triplicate samples. Error bars indicate SD. (C) SVGA cells were supplemented as for panel A and then infected with Mad-1, Mad-1/WT3C, or BK pseudovirus (control). Supernatant from infected cells was collected and analyzed for secreted luciferase. The average relative luciferase units (RLU) for triplicate samples are shown in log scale. Error bars represent SD. These data are representative of 3 experiments.
To extend these findings, we used the JCPyV pseudovirus system, which is more robust and has greater sensitivity. JCPyV pseudoviruses contain only the capsid components and a reporter genome, allowing for examination of only the initial steps in the virus life cycle from viral attachment through trafficking and transcription. We tested whether addition of gangliosides to SVGA (Fig. 7C) and HEK293A (data not shown) cells could enhance infection by JCPyV Mad-1, Mad-1/WT3C, and BKPyV pseudovirus infection. Addition of GD1b and GT1b enhanced BKPyV infection, while ganglioside supplementation had no effect on JCPyV infection (Fig. 7C).
Silencing of the GM3 synthase has no effect on JCPyV infection.
JCPyV Mad-1 and Mad-1/WT3C are both capable of binding to a panel of ganglioside structures. However, supplementation of cells with these gangliosides neither enhances nor reduces JCPyV infection. To test whether ganglioside expression contributes to JCPyV infection, GM3 synthase, the enzyme responsible for ganglioside biosynthesis, was silenced by siRNA. SVGA cells were transfected with RNA oligonucleotides specific for human ST3 β-galactoside α-2,3-sialyltransferase 5 (ST3GAL5), and the expression of GM3 synthase mRNA was determined by qPCR (Fig. 8A). The siRNA treatment of SVGA cells resulted in an ∼85% reduction in GM3 mRNA expression (Fig. 8A). SVGA cells treated with siRNA to GM3 synthase were infected with JCPyV Mad-1, Mad-1/WT3C, and SV40 (Fig. 8B). Only the GM3-17 siRNA resulted in a modest reduction in infection of Mad-1 and Mad-1/WT3C, while the other siRNAs did not reduce JCPyV infection. Treatment of SVGA cells with GM3 synthase siRNAs reduces SV40 infection by ∼75%, indicating that SV40 infection is dependent on ganglioside expression while JCPyV infection is not.
FIG 8.
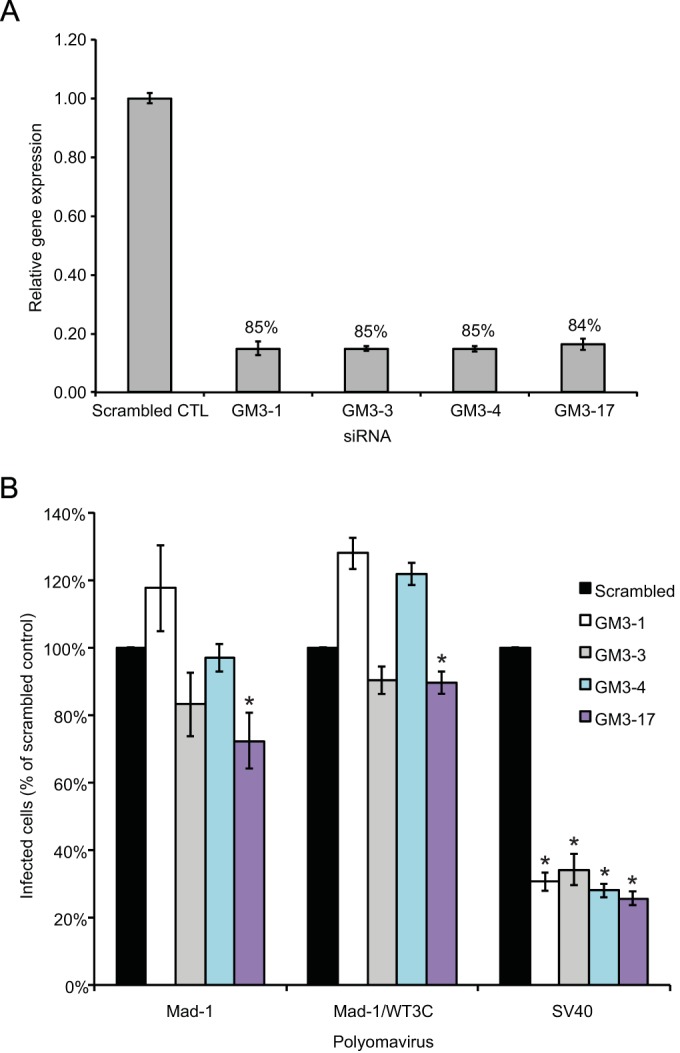
Gene silencing of GM3 synthase does not affect JCPyV infection in SVGA cells. SVGA cells were transfected with siRNAs targeted to the ST3GAL5 or control (scrambled) siRNA. At 72 h posttransfection, cells were either harvested for analysis of GM3 synthase mRNA expression (A) or infected (B). (A) RNA was extracted from 2 wells and reverse transcribed by reverse transcription-PCR (RT-PCR), and real-time PCR was performed with 1 μg cDNA using primers specific for GM3 synthase or GAPDH (housekeeping gene). Results are represented as the relative gene expression for triplicate real-time PCR samples relative to the GAPDH siRNA control. The percent reduction in gene expression relative to the scrambled control is expressed for each siRNA. (B) SVGA cells transfected with ST3GAL5 siRNAs or controls were infected with JCPyV or SV40. Infected cells were scored by indirect immunofluorescence. Data represent the average number of infected cells per visual field for four 10× fields of view for triplicate samples. Error bars indicate SD.
DISCUSSION
It was previously shown that JCPyV requires the α2,6-linked LSTc glycan for attachment and that engagement of LSTc is critical for infection and spread in culture (24). In addition to the linear LSTc motif, different sialylated glycans, as well as a number of a- and b-series ganglioside structures, have been implicated as receptor candidates for JCPyV (6, 34). However, it has remained unclear whether JCPyV can utilize all of these glycans to mount an infection, particularly as different JCPyV strains and genotypes were used in different studies. To help reconcile the existing findings about JCPyV glycan receptors and strain- or genotype-specific differences, we performed a comprehensive analysis of the binding specificities and affinities of the two representative JCPyV strains Mad-1 and WT3 for a range of glycan receptors using X-ray crystallography and NMR spectroscopy, and we correlated these data with infectivity assays. Our analysis unambiguously shows that both JCPyV strains are able to bind a range of sialylated receptors, including LSTc and several ganglioside motifs. The shallow, surface-exposed JCPyV VP1 binding site allows the engagement of various sialylated ligands without steric clashes, independent from the linkage of the terminal Neu5Ac residue. However, there are critical differences in binding affinities for the analyzed glycans, with the linear α2,6-linked LSTc glycan binding best to both strains. Distinct differences in affinities correlate with infectivity assays, which show that LSTc is utilized by both strains as a functional receptor whereas the other ganglioside-type receptors are not.
We estimate the dissociation constants (Kd crystal) for LSTc and ganglioside glycans to be in the millimolar range, i.e., 0.5 mM for the strongest interaction with LSTc and 6 mM for the weakest interaction with GD1a. Of the ganglioside glycans tested here, GM1 binds best to Mad-1 and WT3 VP1, with dissociation constants of about 1.2 and 0.7 mM, respectively. These estimated affinities are in line with affinities observed for other virus-glycan interactions (23, 55). Glycans with dissociation constants higher than 0.5 mM seem not to bind tightly enough to promote JCPyV infection. Thus, one could argue that a putative threshold for receptor binding might lie between 0.5 mM and 0.7 mM for the interaction of one glycan in one binding site. While differences in binding affinities are small, they will be amplified by a virus particle engaging a cell surface due to the availability of multiple low-affinity binding sites (60). For example, a relationship between the typically millimolar affinities for single protein-glycan interactions and femtomolar avidities observed for virus binding has been established for influenza virus, explaining how a relatively modest decrease in the affinity of one binding site can abolish virus binding (61).
As the SV40 GM1 and BKPyV GD1b binding is still conserved in JCPyV, we aimed to retarget JCPyV toward engagement of GM1 gangliosides rather than LSTc via site-directed mutagenesis at residue N123 of JCPyV. This amino acid, which is a glycine in SV40 and BKPyV (25), is the key residue that makes contacts with both arms of the L-shaped LSTc, yet potential Neu5Ac contacts would be preserved. Evaluation by crystal structure analysis revealed abolished binding to LSTc in the L-shape conformation and conserved engagement of GM1, but supplementation of cells with GM1 ganglioside did not alter cell binding or infection (data not shown). Thus, it seems that there are additional evolutionary constraints on JCPyV toward utilizing LSTc, leading JCPyV VP1 to favor this interaction over GM1 (or GD1b) ganglioside engagement. In contrast to the case for SV40 and BKPyV, JCPyV entry is achieved by clathrin-mediated rather than cholesterol-dependent endocytosis (28, 62, 63). It is tempting to speculate that the increased affinity for LSTc has coevolved with changes in the viral entry pathway of JCPyV. Only a few amino acid changes in the VP1 binding site are sufficient for switching glycan binding specificity, but the adaption of alternative attachment and entry mechanisms may require additional genetic changes. JCPyV infects only a narrow range of cell types and entry, and subsequent infection is supported by 5-HT2 family of serotonin receptors (28). In fact, infection of 5-HT2R-expressing HEK293A cells with viruses containing either a Mad-1 or WT3 capsid is equivalent (28; Maginnis and Atwood, unpublished results), yet exogenous addition of gangliosides in HEK293A cells, which express low levels of 5-HT2Rs, does not lead to a productive infection (Fig. 7B), indicating that both strains of JCPyV require 5-HT2Rs for productive infection. In contrast, BKPyV and SV40 can still switch their glycan receptor specificity through a single VP1 point mutation, but these viruses both utilize lipid-linked ganglioside receptors and share a cholesterol-dependent entry mechanism (25, 59). Binding of JCPyV to LSTc may induce organizational and/or structural changes at the plasma membrane that favor interactions with the entry receptor 5-HT2R.
Sialic acid receptors commonly govern outcomes in viral infection and pathogenesis due to evolution and switches of receptor specificity (64). For instance, avian influenza A virus hemagglutinin (HA) attachment protein engages α2,3-linked sialic acids, which are abundant in the gastrointestinal tracts of birds, but it cannot infect humans due to the scarcity of these receptors on cells of the human respiratory tract (65). However, when a switch in sialic acid utilization from α2,3-Neu5Ac to α2,6-Neu5Ac occurs in avian or porcine reservoirs, influenza viruses from nonhuman species can be transmitted to human hosts, as α2,6-linked sialic acids are highly expressed on cells of the human respiratory tract. Influenza A virus strains with HA preference for human α2,6-linked receptors can lead to further human-to-human transmissibility and cause widespread infection in humans and viral pandemics (65). It was recently shown that certain BKPyV serotypes engage a distinct spectrum of cell surface attachment receptors, resulting in different cellular attachment mechanisms and cell tropism (66). Consequently, these findings raise questions whether different JCPyV genotypes use distinct glycan receptors besides LSTc and therefore exhibit altered tissue tropism resulting in different levels of virulence in vivo and are a possible risk factor for PML as proposed earlier (67). The VP1 amino acid differences in the two strains investigated here comprise the majority of genotype-specific variability (Fig. 1A). Due to the location of remaining VP1 amino acid changes not investigated in this study (Fig. 4), it is unlikely that other genotypes possess altered LSTc specificities not detected here. Consequently, genotype- and strain-related variations differ from PML-associated VP1 mutations, which are located within the central Neu5Ac binding site and render JCPyV variants noninfectious in cell culture (35). PML-associated mutations are commonly found in patients with PML in the background of different genotypes, but mechanisms by which they arise and their effects on cellular tropism remain undefined.
Our findings help explain conflicting reports on JCPyV glycan specificity. Biochemical methods such as glycan microarray screening, virus overlay blotting, and enzyme-linked immunosorbent assays to investigate glycan binding in vitro have variable detection limits and drawbacks. Binding alone does not indicate whether a glycan is a receptor or a pseudoreceptor. Due to the relatively low affinity of virus-glycan interactions and avidity effects, the virus and the presented glycan have a significant impact on the experimental outcome and its meaning for the physiological tissue tropism (68). Critical differences in binding affinities, rather than the binding events alone, can therefore determine receptor usage. Although a varying susceptibility of certain genotypes for PML development cannot be excluded, our study shows that it is unlikely that genotype-specific VP1 variations alter JCPyV attachment mechanisms or influence the risk for PML development. Our findings resolve a long-standing question in the field and also provide a proper framework for interpreting how interactions with viral receptors must be analyzed for functionality in infectious assays to define the outcomes on viral pathogenicity.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Stehle, Atwood, and DiMaio laboratories for critical discussion. We also thank the staff at beamlines X06SA and X06DA of the Swiss Light Source (Villigen, Switzerland) for assistance with X-ray data collection and Remco Sprangers, MPI for Developmental Biology in Tübingen, Germany, for assistance with NMR data collection.
This work was funded by NIH grant PPG P01NS065719-16 (W.J.A., D.D., and T.S.) and Ruth L. Kirchstein National Research Service Awards F32NS064870 (M.S.M.) and F32NS070687 (C.D.S.N.) from the National Institute of Neurological Disorders and Stroke. Research reported in this project was also supported by Institutional Development Award (IDeA) NIH P20GM103423 (M.S.M.) from the National Institute of General Medical Sciences and P01CA16038 (D.D.) from the National Cancer Institute. Core facilities that support research in the Atwood laboratory are funded by P30GM103410 (W.J.A.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00489-15.
REFERENCES
- 1.Kean JM, Rao S, Wang M, Garcea RL. 2009. Seroepidemiology of human polyomaviruses. PLoS Pathog 5:e1000363. doi: 10.1371/journal.ppat.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Egli A, Infanti L, Dumoulin A, Buser A, Samaridis J, Stebler C, Gosert R, Hirsch HH. 2009. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J Infect Dis 199:837–846. doi: 10.1086/597126. [DOI] [PubMed] [Google Scholar]
- 3.Yogo Y, Kitamura T, Sugimoto C, Ueki T, Aso Y, Hara K, Taguchi F. 1990. Isolation of a possible archetypal JC virus DNA sequence from nonimmunocompromised individuals. J Virol 64:3139–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daniel AM, Swenson JJ, Mayreddy RP, Khalili K, Frisque RJ. 1996. Sequences within the early and late promoters of archetype JC virus restrict viral DNA replication and infectivity. Virology 216:90–101. doi: 10.1006/viro.1996.0037. [DOI] [PubMed] [Google Scholar]
- 5.Dubois V, Dutronc H, Lafon ME, Poinsot V, Pellegrin JL, Ragnaud JM, Ferrer AM, Fleury HJ. 1997. Latency and reactivation of JC virus in peripheral blood of human immunodeficiency virus type 1-infected patients. J Clin Microbiol 35:2288–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorelik L, Reid C, Testa M, Brickelmaier M, Bossolasco S, Pazzi A, Bestetti A, Carmillo P, Wilson E, McAuliffe M, Tonkin C, Carulli JP, Lugovskoy A, Lazzarin A, Sunyaev S, Simon K, Cinque P. 2011. Progressive multifocal leukoencephalopathy (PML) development is associated with mutations in JC virus capsid protein VP1 that change its receptor specificity. J Infect Dis 204:103–114. doi: 10.1093/infdis/jir198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silverman L, Rubinstein LJ. 1965. Electron microscopic observations on a case of progressive multifocal leukoencephalopathy. Acta Neuropathol 5:215–224. doi: 10.1007/BF00686519. [DOI] [PubMed] [Google Scholar]
- 8.Zurhein G, Chou SM. 1965. Particles resembling papova viruses in human cerebral demyelinating disease. Science 148:1477–1479. doi: 10.1126/science.148.3676.1477. [DOI] [PubMed] [Google Scholar]
- 9.Liedtke W, Edelmann W, Bieri PL, Chiu FC, Cowan NJ, Kucherlapati R, Raine CS. 1996. GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelination. Neuron 17:607–615. doi: 10.1016/S0896-6273(00)80194-4. [DOI] [PubMed] [Google Scholar]
- 10.Spiegel I, Peles E. 2006. A new player in CNS myelination. Neuron 49:777–778. doi: 10.1016/j.neuron.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 11.Bradl M, Lassmann H. 2010. Oligodendrocytes: biology and pathology. Acta Neuropathol 119:37–53. doi: 10.1007/s00401-009-0601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Astrom KE, Mancall EL, Richardson EP Jr. 1958. Progressive multifocal leuko-encephalopathy; a hitherto unrecognized complication of chronic lymphatic leukaemia and Hodgkin's disease. Brain 81:93–111. doi: 10.1093/brain/81.1.93. [DOI] [PubMed] [Google Scholar]
- 13.Ferenczy MW, Marshall LJ, Nelson CD, Atwood WJ, Nath A, Khalili K, Major EO. 2012. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev 25:471–506. doi: 10.1128/CMR.05031-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kondo Y, Windrem MS, Zou L, Chandler-Militello D, Schanz SJ, Auvergne RM, Betstadt SJ, Harrington AR, Johnson M, Kazarov A, Gorelik L, Goldman SA. 2014. Human glial chimeric mice reveal astrocytic dependence of JC virus infection. J Clin Invest 124:5323–5336. doi: 10.1172/JCI76629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirsch HH, Kardas P, Kranz D, Leboeuf C. 2013. The human JC polyomavirus (JCPyV): virological background and clinical implications. APMIS 121:685–727. doi: 10.1111/apm.12128. [DOI] [PubMed] [Google Scholar]
- 16.Carson KR, Focosi D, Major EO, Petrini M, Richey EA, West DP, Bennett CL. 2009. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol 10:816–824. doi: 10.1016/S1470-2045(09)70161-5. [DOI] [PubMed] [Google Scholar]
- 17.Bloomgren G, Richman S, Hotermans C, Subramanyam M, Goelz S, Natarajan A, Lee S, Plavina T, Scanlon JV, Sandrock A, Bozic C. 2012. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med 366:1870–1880. doi: 10.1056/NEJMoa1107829. [DOI] [PubMed] [Google Scholar]
- 18.Ermis U, Weis J, Schulz JB. 2013. Case reports of PML in patients treated for psoriasis. N Engl J Med 369:1081. doi: 10.1056/NEJMc1307680. [DOI] [PubMed] [Google Scholar]
- 19.Ermis U, Weis J, Schulz JB. 2013. PML in a patient treated with fumaric acid. N Engl J Med 368:1657–1658. doi: 10.1056/NEJMc1211805. [DOI] [PubMed] [Google Scholar]
- 20.Liddington RC, Yan Y, Moulai J, Sahli R, Benjamin TL, Harrison SC. 1991. Structure of simian virus 40 at 3.8-A resolution. Nature 354:278–284. doi: 10.1038/354278a0. [DOI] [PubMed] [Google Scholar]
- 21.Chen XS, Stehle T, Harrison SC. 1998. Interaction of polyomavirus internal protein VP2 with the major capsid protein VP1 and implications for participation of VP2 in viral entry. EMBO J 17:3233–3240. doi: 10.1093/emboj/17.12.3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stehle T, Harrison SC. 1997. High-resolution structure of a polyomavirus VP1-oligosaccharide complex: implications for assembly and receptor binding. EMBO J 16:5139–5148. doi: 10.1093/emboj/16.16.5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neu U, Woellner K, Gauglitz G, Stehle T. 2008. Structural basis of GM1 ganglioside recognition by simian virus 40. Proc Natl Acad Sci U S A 105:5219–5224. doi: 10.1073/pnas.0710301105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neu U, Maginnis MS, Palma AS, Stroh LJ, Nelson CD, Feizi T, Atwood WJ, Stehle T. 2010. Structure-function analysis of the human JC polyomavirus establishes the LSTc pentasaccharide as a functional receptor motif. Cell Host Microbe 8:309–319. doi: 10.1016/j.chom.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neu U, Allen SA, Blaum BS, Liu Y, Frank M, Palma AS, Stroh LJ, Feizi T, Peters T, Atwood WJ, Stehle T. 2013. A structure-guided mutation in the major capsid protein retargets BK polyomavirus. PLoS Pathog 9:e1003688. doi: 10.1371/journal.ppat.1003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neu U, Khan ZM, Schuch B, Palma AS, Liu Y, Pawlita M, Feizi T, Stehle T. 2013. Structures of B-lymphotropic polyomavirus VP1 in complex with oligosaccharide ligands. PLoS Pathog 9:e1003714. doi: 10.1371/journal.ppat.1003714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khan ZM, Liu Y, Neu U, Gilbert M, Ehlers B, Feizi T, Stehle T. 2014. Crystallographic and glycan microarray analysis of human polyomavirus 9 VP1 identifies N-glycolyl neuraminic acid as a receptor candidate. J Virol 88:6100–6111. doi: 10.1128/JVI.03455-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Assetta B, Maginnis MS, Gracia Ahufinger I, Haley SA, Gee GV, Nelson CD, O'Hara BA, Allen Ramdial SA, Atwood WJ. 2013. 5-Ht2 receptors facilitate Jc polyomavirus entry. J Virol 87:13490–13498. doi: 10.1128/JVI.02252-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erickson KD, Garcea RL, Tsai B. 2009. Ganglioside GT1b is a putative host cell receptor for the Merkel cell polyomavirus. J Virol 83:10275–10279. doi: 10.1128/JVI.00949-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qian M, Tsai B. 2010. Lipids and proteins act in opposing manners to regulate polyomavirus infection. J Virol 84:9840–9852. doi: 10.1128/JVI.01093-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neu U, Stehle T, Atwood WJ. 2009. The Polyomaviridae: contributions of virus structure to our understanding of virus receptors and infectious entry. Virology 384:389–399. doi: 10.1016/j.virol.2008.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maccioni HJ, Quiroga R, Ferrari ML. 2011. Cellular and molecular biology of glycosphingolipid glycosylation. J Neurochem 117:589–602. doi: 10.1111/j.1471-4159.2011.07232.x. [DOI] [PubMed] [Google Scholar]
- 33.Cubitt CL, Cui X, Agostini HT, Nerurkar VR, Scheirich I, Yanagihara R, Ryschkewitsch CF, Stoner GL. 2001. Predicted amino acid sequences for 100 JCV strains. J Neurovirol 7:339–344. doi: 10.1080/13550280152537201. [DOI] [PubMed] [Google Scholar]
- 34.Komagome R, Sawa H, Suzuki T, Suzuki Y, Tanaka S, Atwood WJ, Nagashima K. 2002. Oligosaccharides as receptors for JC virus. J Virol 76:12992–13000. doi: 10.1128/JVI.76.24.12992-13000.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maginnis MS, Stroh LJ, Gee GV, O'Hara BA, Derdowski A, Stehle T, Atwood WJ. 2013. Progressive multifocal leukoencephalopathy-associated mutations in the JC polyomavirus capsid disrupt lactoseries tetrasaccharide c binding. mBio 4(3):e00247–00213. doi: 10.1128/mBio.00247-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sunyaev SR, Lugovskoy A, Simon K, Gorelik L. 2009. Adaptive mutations in the JC virus protein capsid are associated with progressive multifocal leukoencephalopathy (PML). PLoS Genet 5:e1000368. doi: 10.1371/journal.pgen.1000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Major EO, Miller AE, Mourrain P, Traub RG, de Widt E, Sever J. 1985. Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc Natl Acad Sci U S A 82:1257–1261. doi: 10.1073/pnas.82.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vacante DA, Traub R, Major EO. 1989. Extension of JC virus host range to monkey cells by insertion of a simian virus 40 enhancer into the JC virus regulatory region. Virology 170:353–361. doi: 10.1016/0042-6822(89)90425-X. [DOI] [PubMed] [Google Scholar]
- 39.Nelson C, Carney D, Derdowski A, Lipovsky A, Gee G, O'Hara B, Williard P, DiMaio D, Sello J, Atwood W. 2013. A retrograde trafficking inhibitor of ricin and Shiga-like toxins inhibits infection of cells by human and monkey polyomaviruses. mBio 4(6):00729–13. doi: 10.1128/mBio.00729-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen BJ, Atwood WJ. 2002. Construction of a novel JCV/SV40 hybrid virus (JCSV) reveals a role for the JCV capsid in viral tropism. Virology 300:282–290. doi: 10.1006/viro.2002.1522. [DOI] [PubMed] [Google Scholar]
- 41.Gee GV, Tsomaia N, Mierke DF, Atwood WJ. 2004. Modeling a sialic acid binding pocket in the external loops of JC virus VP1. J Biol Chem 279:49172–49176. doi: 10.1074/jbc.M409326200. [DOI] [PubMed] [Google Scholar]
- 42.Maginnis MS, Haley SA, Gee GV, Atwood WJ. 2010. Role of N-linked glycosylation of the 5-HT2A receptor in JC virus infection. J Virol 84:9677–9684. doi: 10.1128/JVI.00978-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gee GV, O'Hara BA, Derdowski A, Atwood WJ. 2013. Pseudovirus mimics cell entry and trafficking of the human polyomavirus JCPyV. Virus Res 178:281–286. doi: 10.1016/j.virusres.2013.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nelson CD, Derdowski A, Maginnis MS, O'Hara BA, Atwood WJ. 2012. The VP1 subunit of JC polyomavirus recapitulates early events in viral trafficking and is a novel tool to study polyomavirus entry. Virology 428:30–40. doi: 10.1016/j.virol.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kabsch W. 2010. Xds. Acta Crystallogr D Biol Crystallogr 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCoy AJ. 2007. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr 63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Afonine PV, Grosse-Kunstleve RW, Adams PD. 2005. A robust bulk-solvent correction and anisotropic scaling procedure. Acta Crystallogr D Biol Crystallogr 61:850–855. doi: 10.1107/S0907444905007894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murshudov GN, Vagin AA, Dodson EJ. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 51.Painter J, Merritt EA. 2006. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr D Biol Crystallogr 62:439–450. doi: 10.1107/S0907444906005270. [DOI] [PubMed] [Google Scholar]
- 52.Howell PL, Smith GD. 1992. Identification of heavy-atom derivatives by normal probability methods. J Appl Crystallogr 25:81–86. doi: 10.1107/S0021889891010385. [DOI] [Google Scholar]
- 53.Kleywegt GJ, Jones TA. 1996. xdlMAPMAN and xdlDATAMAN—programs for reformatting, analysis and manipulation of biomacromolecular electron-density maps and reflection data sets. Acta Crystallogr D Biol Crystallogr 52:826–828. doi: 10.1107/S0907444995014983. [DOI] [PubMed] [Google Scholar]
- 54.Kleywegt GJ. 1999. Experimental assessment of differences between related protein crystal structures. Acta Crystallogr D Biol Crystallogr 55:1878–1884. doi: 10.1107/S0907444999010495. [DOI] [PubMed] [Google Scholar]
- 55.Burmeister WP, Guilligay D, Cusack S, Wadell G, Arnberg N. 2004. Crystal structure of species D adenovirus fiber knobs and their sialic acid binding sites. J Virol 78:7727–7736. doi: 10.1128/JVI.78.14.7727-7736.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stehle T, Harrison SC. 1996. Crystal structures of murine polyomavirus in complex with straight-chain and branched-chain sialyloligosaccharide receptor fragments. Structure 4:183–194. doi: 10.1016/S0969-2126(96)00021-4. [DOI] [PubMed] [Google Scholar]
- 57.Tsai B, Gilbert JM, Stehle T, Lencer W, Benjamin TL, Rapoport TA. 2003. Gangliosides are receptors for murine polyoma virus and SV40. EMBO J 22:4346–4355. doi: 10.1093/emboj/cdg439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mayer M, Meyer B. 1999. Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew Chem Int Ed Engl 38:1784–1788. [DOI] [PubMed] [Google Scholar]
- 59.Magaldi TG, Buch MH, Murata H, Erickson KD, Neu U, Garcea RL, Peden K, Stehle T, DiMaio D. 2012. Mutations in the GM1 binding site of simian virus 40 VP1 alter receptor usage and cell tropism. J Virol 86:7028–7042. doi: 10.1128/JVI.00371-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szklarczyk OM, Gonzalez-Segredo N, Kukura P, Oppenheim A, Choquet D, Sandoghdar V, Helenius A, Sbalzarini IF, Ewers H. 2013. Receptor concentration and diffusivity control multivalent binding of Sv40 to membrane bilayers. PLoS Comput Biol 9:e1003310. doi: 10.1371/journal.pcbi.1003310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiong X, Coombs PJ, Martin SR, Liu J, Xiao H, McCauley JW, Locher K, Walker PA, Collins PJ, Kawaoka Y, Skehel JJ, Gamblin SJ. 2013. Receptor binding by a ferret-transmissible H5 avian influenza virus. Nature 497:392–396. doi: 10.1038/nature12144. [DOI] [PubMed] [Google Scholar]
- 62.Anderson HA, Chen Y, Norkin LC. 1996. Bound simian virus 40 translocates to caveolin-enriched membrane domains, and its entry is inhibited by drugs that selectively disrupt caveolae. Mol Biol Cell 7:1825–1834. doi: 10.1091/mbc.7.11.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eash S, Querbes W, Atwood WJ. 2004. Infection of vero cells by BK virus is dependent on caveolae. J Virol 78:11583–11590. doi: 10.1128/JVI.78.21.11583-11590.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stroh LJ, Stehle T. 2014. Glycan Engagement by viruses: receptor switches and specificity. Annu Rev Virol 1:285–306. doi: 10.1146/annurev-virology-031413-085417. [DOI] [PubMed] [Google Scholar]
- 65.Imai M, Kawaoka Y. 2012. The role of receptor binding specificity in interspecies transmission of influenza viruses. Curr Opin Virol 2:160–167. doi: 10.1016/j.coviro.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pastrana DV, Ray U, Magaldi TG, Schowalter RM, Cuburu N, Buck CB. 2013. BK polyomavirus genotypes represent distinct serotypes with distinct entry tropism. J Virol 87:10105–10113. doi: 10.1128/JVI.01189-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Agostini HT, Ryschkewitsch CF, Mory R, Singer EJ, Stoner GL. 1997. JC virus (JCV) genotypes in brain tissue from patients with progressive multifocal leukoencephalopathy (PML) and in urine from controls without PML: increased frequency of JCV type 2 in PML. J Infect Dis 176:1–8. doi: 10.1086/514010. [DOI] [PubMed] [Google Scholar]
- 68.Demarco ML, Woods RJ, Prestegard JH, Tian F. 2010. Presentation of membrane-anchored glycosphingolipids determined from molecular dynamics simulations and NMR paramagnetic relaxation rate enhancement. J Am Chem Soc 132:1334–1338. doi: 10.1021/ja907518x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.