

Abstract
Initiation of zygotic transcription in mammals is poorly understood. In mice, zygotic transcription is first detected shortly after pronucleus formation in 1-cell embryos, but the identity of the transcribed loci and mechanisms regulating their expression are not known. Using total RNA-Seq, we have found that transcription in 1-cell embryos is highly promiscuous, such that intergenic regions are extensively expressed and thousands of genes are transcribed at comparably low levels. Striking is that transcription can occur in the absence of defined core-promoter elements. Furthermore, accumulation of translatable zygotic mRNAs is minimal in 1-cell embryos because of inefficient splicing and 3′ processing of nascent transcripts. These findings provide novel insights into regulation of gene expression in 1-cell mouse embryos that may confer a protective mechanism against precocious gene expression that is the product of a relaxed chromatin structure present in 1-cell embryos. The results also suggest that the first zygotic transcription itself is an active component of chromatin remodeling in 1-cell embryos.
Keywords: gene expression, preimplantation mouse embryo, pre-mRNA splicing, RNA-Seq, transcription
Introduction
The oocyte-to-embryo transition (OET) entails a dramatic reprogramming of gene expression and conversion of a differentiated transcriptionally quiescent oocyte to totipotent blastomeres (De La Fuente & Eppig, 2001; Abe et al, 2010). The timing of zygotic genome activation (ZGA) is species dependent. Genome activation in mice is the earliest for mammals studied to date; the first wave of transcription (also referred to as the minor ZGA wave) starts at the mid-1-cell stage shortly after pronucleus formation, as evidenced by BrdU incorporation (Bouniol et al, 1995; Aoki et al, 1997) and expression of sperm-borne transgenes (Matsumoto et al, 1994). Although repetitive elements, for example, B2-containing sequences (Vasseur et al, 1985) and MuERV-L (Kigami et al, 2003), are expressed in 1-cell mouse embryos, transcription of single-copy genes is poorly understood, as is their function, which is not necessary for cleavage to the 2-cell stage (Warner & Versteegh, 1974).
Studies using plasmid-borne reporter genes have provided insights regarding mechanisms that govern transcription in 1-cell embryos. These studies demonstrated that reporter gene expression in 1-cell embryos does not require an enhancer for efficient expression, whereas an enhancer is required for efficient expression in 2-cell embryos (Wiekowski et al, 1991; Majumder et al, 1993). Thus, 1-cell embryos are transcriptionally permissive, but development to the 2-cell stage is accompanied by formation of a transcriptionally repressive state (DePamphilis, 1993) and genome-wide accumulation of repressive histone modification marks (Santos et al, 2005). This developmental change in transcriptional regulation involves DNA replication at the 2-cell stage because an enhancer is not required for efficient transcription when 2-cell embryos were treated with aphidicolin, which inhibits DNA replication (Wiekowski et al, 1991; Majumder et al, 1993; Henery et al, 1995; Forlani et al, 1998). The transcriptionally repressive state likely stems from chromatin structure because increasing histone acetylation in 2-cell embryos by treating the embryos with butyrate, an inhibitor of histone deacetylase, relieves the requirement for an enhancer for efficient expression of the reporter gene (Majumder et al, 1993; Wiekowski et al, 1993). Similar conclusions were reached using expression of the 2-cell transiently expressed endogenous Eif1a gene (Davis et al, 1996).
Microarray profiling identified zygotic mRNA expression at the 2-cell (major ZGA wave) but not the 1-cell stage (Hamatani et al, 2004; Wang et al, 2004; Zeng & Schultz, 2005). This finding suggests that mRNAs are either not expressed or microarray profiling is not sensitive enough to detect mRNAs produced during minor ZGA. More recent high-throughput sequencing (HTS) experiments identified hundreds of different mRNAs with increased abundance in 1-cell embryos (Park et al, 2013; Xue et al, 2013; Deng et al, 2014). However, due to increased RNA adenylation in 1-cell embryos (Piko & Clegg, 1982), poly(A) RNA sequencing results are difficult to interpret as they might reflect polyadenylation changes and not 1-cell transcription per se (Xue et al, 2013; Deng et al, 2014). The most comprehensive study regarding transcription in 1-cell embryos to date used SOLiD sequencing after ribosomal RNA depletion and identified ∽600 genes upregulated > 1.5-fold between the oocyte and 1-cell embryo (Park et al, 2013).
In this study, we employed total RNA sequencing to identify sequences transcribed in 1-cell embryos and to gain understanding about mechanisms that govern their expression. We show that pervasive transcription occurs in intergenic regions including many transposons whose transcription continues far into their genomic flanks; transcription can occur independently of defined core-promoter elements; over four thousand genes are transcribed in 1-cell embryos with ∽5% being transcribed transiently; the majority of genes transcribed in 1-cell embryos are also transcribed in 2-cell embryos when the major wave of genome activation occurs; and mRNAs transcribed at the 1-cell stage are mostly nonfunctional because their 3′ end processing and splicing are highly inefficient.
Results
Global analysis of the first wave of transcription
To explore the first wave of zygotic transcription, we characterized total RNA in metaphase II-arrested eggs (MII eggs) and embryos by HTS. We sequenced total RNA rather than selecting poly(A)-containing RNA (poly(A) RNA) because extensive RNA polyadenylation that occurs post-fertilization would be difficult to distinguish from bona fide zygotic mRNA synthesis (Piko & Clegg, 1982; Oh et al, 2000; Meijer et al, 2007). In addition, the RNA was not amplified to further minimize any deviation from the initial distribution of mRNAs. We isolated RNA from two sets of MII eggs and 1-cell embryos and one set of 2-cell and 4-cell embryos, morulae, and blastocysts. We also prepared RNA from 1-cell embryos in which transcription was inhibited by 5,6-dichlorobenzimidazole riboside (DRB), an inhibitor of RNA polymerase II activity (Sehgal et al, 1976), and 2-cell embryos treated with the DNA replication inhibitor aphidicolin (Bucknall et al, 1973) (Supplementary Table S1). Libraries from two sets of MII eggs and 1-cell embryos were subjected to 35-nt single-end sequencing (35SE); one set from each stage was subjected to 76-nt paired-end (76PE) sequencing with depths of 33–58 × 106 (Supplementary Table S1). Analysis of the sequencing results showed a high degree of reproducibility among the duplicate sets as well as for biological replicates (Supplementary Fig S1A). Comparison of our 76PE Illumina data and 50SE SOLiD data obtained by sequencing of total RNA depleted of ribosomal RNA from identical stages (Park et al, 2013) showed a high concordance of the signal distribution along chromosomes (r2 = ∽0.8, Supplementary Fig S1B) and a good similarity of transcriptome changes between different stages (r2 = ∽0.6, Supplementary Fig S1C), confirming the overall reproducibility of our data.
The relative abundance of rRNA, repetitive sequences, annotated mRNAs, and unique sequences in the individual libraries were consistent with previous measurements (Fig1A and Supplementary Fig S1D) (Piko & Clegg, 1982). For example, the fraction of mRNA in MII eggs and early cleavage-stage embryos is greater than that in somatic cells—for example, mRNA makes 2.5% of total RNA mass in HeLa cells (Jackson et al, 2000)—and the decline in the relative abundance of mRNA between the MII egg and 2-cell embryo correlates with the known degradation of maternal mRNA during this developmental period (Bachvarova & De Leon, 1980; Piko & Clegg, 1982). The fraction of mRNA was somewhat higher than previously reported (Piko & Clegg, 1982), which likely reflects that total RNA for HTS was size-selected for RNAs > 200 nt, causing a higher incidence of mRNAs in sequenced libraries. The small increase in the representation of rRNA and repeat-derived sequences at the expense of mRNA-derived reads in 1-cell embryos (Fig1A) could be a consequence of ongoing maternal mRNA degradation in 1-cell embryos and/or new transcription of rRNA and retrotransposons. The frequency of 5′ external transcribed spacer (ETS)-derived reads from the 45S rRNA precursor, an indirect proxy of rRNA transcription, did not suggest robust rRNA transcription in 1-cell embryos because the amount of 5′ ETS-derived reads in MII eggs was actually greater than that in 1-cell embryos (Supplementary Fig S2A). However, in agreement with rDNA transcription initiating during the 2-cell stage (Zatsepina et al, 2003), we observed a ∽fivefold increase in the number of 5′ ETS-derived reads between the 1-cell and 2-cell stages and a further more dramatic increase between 2-cell and 4-cell stages (Supplementary Fig S2B).
Figure 1.
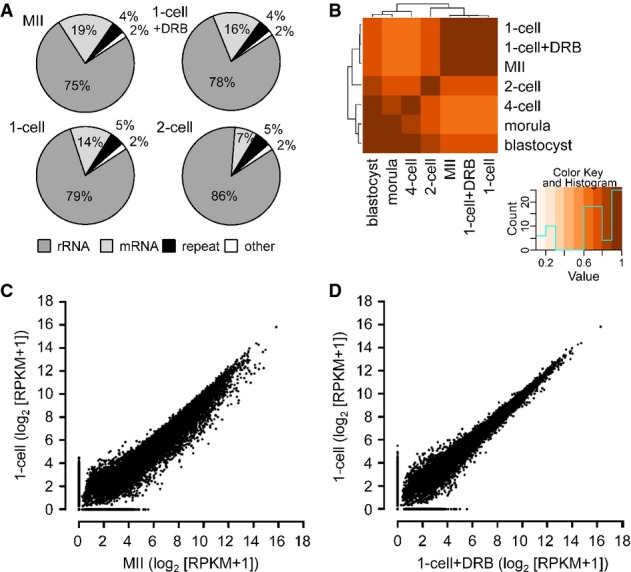
- A Composition of selected libraries produced from total RNA. Shown is the proportion of reads matching rRNA-derived transcripts (rRNA), transcripts produced from repeats identified by RepeatMasker (repeat), annotated mRNAs (mRNA), and other sequences (other).
- B Hierarchical clustering of 76-nt paired-end datasets from different stages of preimplantation development performed on Spearman correlations between log2 of RPKM values for transcripts.
- C, D Comparison of MII and 1-cell transcriptomes (C) and 1-cell and 1-cell + DRB transcriptomes (D). The graph was made by plotting expression values for mRNAs (log2[RPKM + 1]) from 76PE sequencing data. Each point represents one transcript whose position indicates its abundance value in indicated stages. RPKM values were increased by adding 1 in order to obtain log2 values above zero.
Unsupervised clustering of the samples based on reads mapping to exon sequences showed that the 1-cell embryo mRNA transcriptome was very similar to that of MII eggs and that DRB treatment had little apparent effect on the transcriptome (Fig1B). These results are consistent with previous transcriptome studies (Hamatani et al, 2004; Wang et al, 2004; Zeng & Schultz, 2005) and are further supported by quantification of reads mapping to exons in MII eggs, 1-cell, and 1-cell + DRB samples (Fig1C and 1D) lending confidence that new insights obtained from our HTS approach are warranted and not an experimental artifact.
The most notable change in the 1-cell RNA population relative to that of MII eggs was a widespread occurrence of individual, rarely overlapping, DRB-sensitive reads whose density rarely exceeded a few reads per locus (∽0.1 counts per million (CPM), hence referred to as low CPM reads hereafter). In a display of a larger genomic region, low CPM reads appeared as an unevenly distributed ‘grass in a forest’ (Fig2 and Supplementary Fig S3). Low CPM reads appeared more frequently within gene-rich regions but were readily found in intergenic regions (Fig2A and Supplementary Fig S3A). Although low CPM reads were also observed at later embryonic stages, their appearance was not as uniform and striking as in 1-cell embryos.
Figure 2.
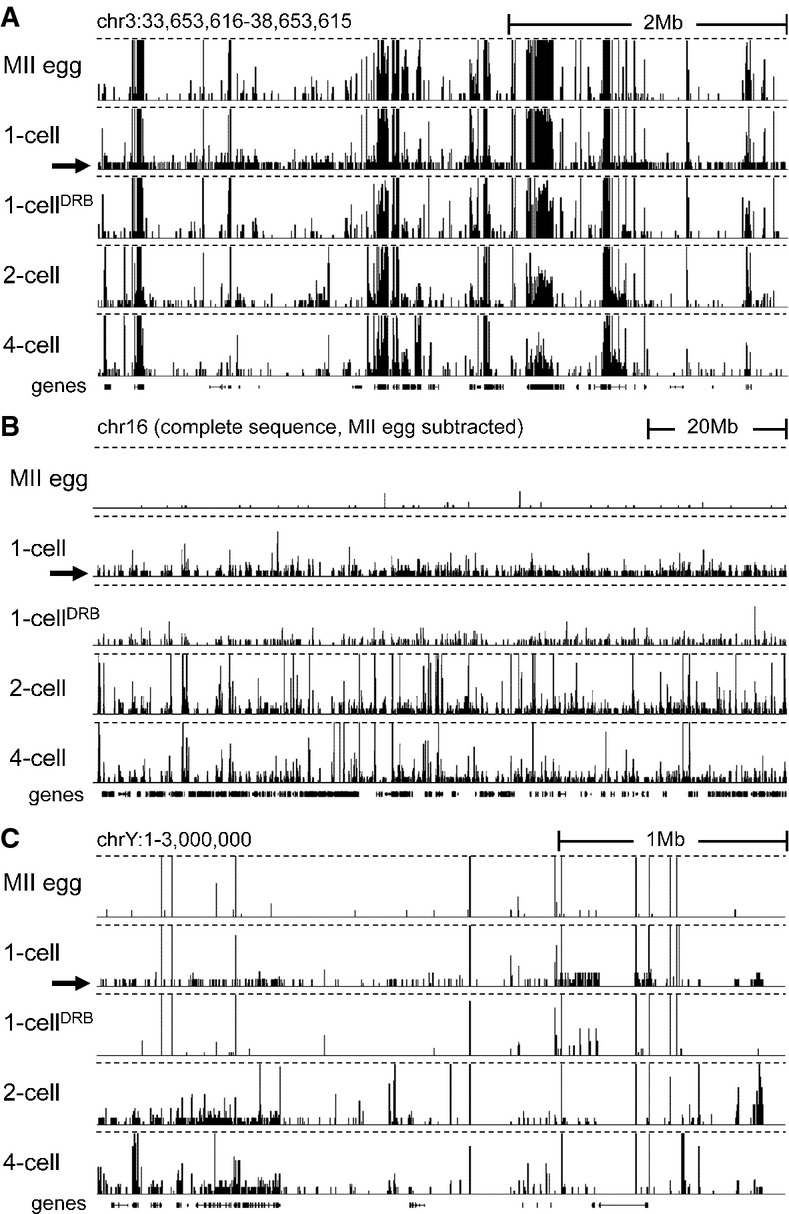
- Transcription at the 1-cell stage manifests as increased number of low CPM reads scattered across the genome. Shown are HTS data [a customized screenshot from the UCSC Genome browser (Karolchik et al, 2012)] from different stages mapped into a 5-Mb region of the genome with a variable gene density. The vertical scale was trimmed at 0.5 CPM; trimming is indicated by horizontal dashed lines. Note the appearance of low-density reads in 1-cell embryos (indicated by the black arrow), which are not observed in unfertilized oocytes and 1-cell embryos treated with DRB. Low CPM reads also appear in intergenic regions.
- Masking maternally transcribed regions reveals chromosome-wide presence of low CPM reads in 1-cell embryos. A UCSC browser screenshot of the chromosome 16 shows 76PE raw data mapped to the genome upon masking reads derived from oocytes. The vertical scale was trimmed at 20 reads; trimming is indicated by horizontal dashed lines. A small number of reads detected in MII eggs comes from repetitive sequences, which otherwise do not yield signal because of multi-mapping restrictions.
- Zygotic transcription from the Y chromosome initiates at the 1-cell stage. Shown are HTS data from different stages mapped into the ˜3-Mb annotated region of the chromosome Y. The vertical scale was trimmed at 0.5 CPM; trimming is indicated by horizontal dashed lines. The residual signal on the chromosome Y in MII eggs and 1-cell embryos treated with DRB is an artifact caused by common retrotransposon-derived sequences (mainly MT-derived).
Because the presence of low CPM reads became readily apparent when sequences present in MII eggs were masked (Fig2B), we examined the annotated proximal end of the Y chromosome (∽3 Mb) (Fig2C). Transcripts detected from this region must be of zygotic origin, and thus, low CPM reads would not be obscured by maternal mRNAs. Indeed, Y-chromosome-derived, DRB-sensitive low CPM reads were readily observed in the 76PE dataset (Fig2C), and low CPM reads were also found in 50SE data (Park et al, 2013) (Supplementary Fig S3B). Given that the low CPM reads originated from genes as well as intergenic regions, we explored these two transcript categories separately.
Widespread intergenic transcription in 1-cell embryos
The occurrence of low CPM reads in intergenic regions was first confirmed by quantitative analysis. We divided intergenic regions across the entire genome into 1-kb segments and determined the number of segments to which at least a single read was uniquely mapped. The number of 1-kb loci harboring uniquely mapped reads was threefold higher in 1-cell embryos than in MII eggs (Fig3A). This number decreased gradually with development and reached levels similar to those of MII eggs at the morula stage (Fig3A), while the number of loci harboring uniquely mapped reads in DRB-treated 1-cell embryos remained similar to that in MII eggs. Furthermore, aphidicolin treatment of 2-cell embryos to inhibit the DNA replication restored the number of loci to the level observed in 1-cell embryos. These results suggest the presence of a transcriptionally permissive state in 1-cell embryos might be governed by the same mechanisms that regulate expression of plasmid-borne reporter genes during this period of development and the 2-cell stage (Wiekowski et al, 1991; Majumder et al, 1993).
Figure 3.
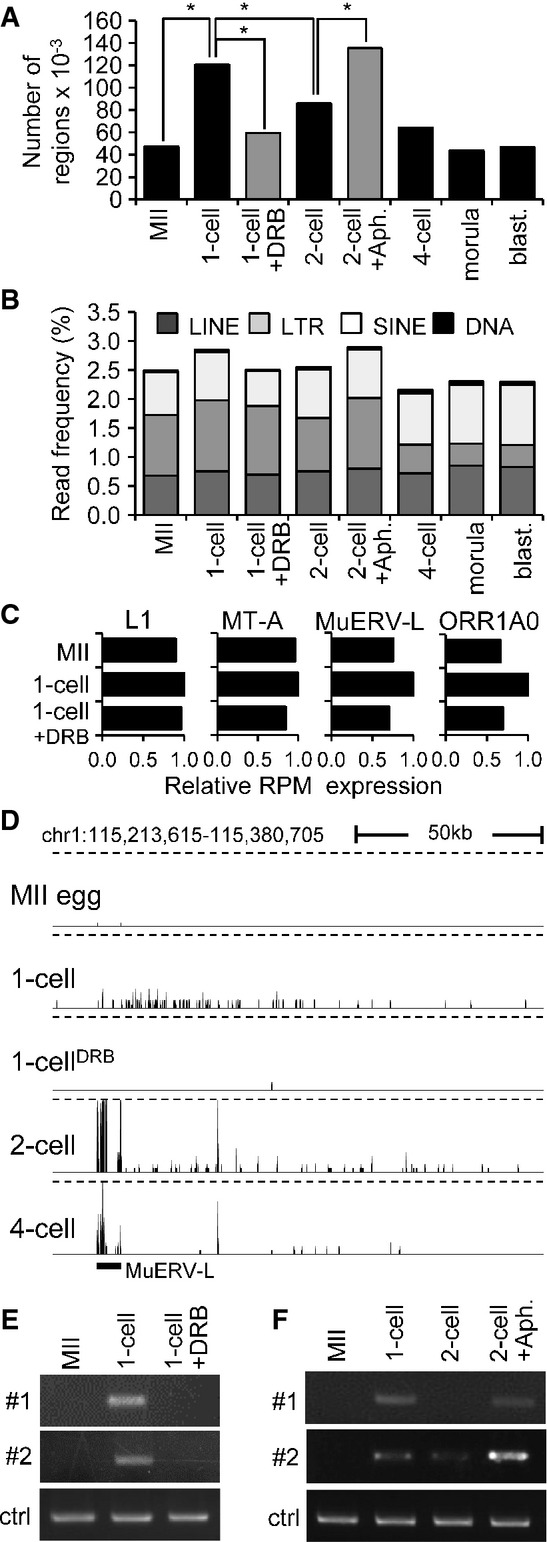
- A Quantitative analysis of transcription from intergenic regions in oocytes and preimplantation embryos. Intergenic regions were divided into 1-kb segments across the whole genome, and the number of segments to which at least a single read was uniquely mapped was determined. Results suggest that treatment of 1-cell embryos with DRB reduced the number of segments yielding intergenic transcripts. In contrast, treatment of 2-cell embryos with aphidicolin (Aph.) prevented reduction of the number of segments yielding intergenic transcripts. Bars representing samples treated with DRB and aphidicolin are gray to distinguish results from treated embryos and from the normal course of preimplantation development. An asterisk indicates a significant difference by chi-square test (P < 0.05).
- B Frequency of reads derived from mobile elements. Association of reads with a particular class of mobile DNA in each sample was determined by RepeatMasker and is displayed as a percentage of those reads in each library.
- C Relative changes of expression of four specific retrotransposons. Relative expression for each retrotransposon was calculated from RPKMs where expression in 1-cell embryos was set to one.
- D MuERV-L retrotransposons asymmetrically neighbor higher frequency of low CPM reads in large intergenic regions. Shown is an example of MuERV-L located in an intergenic region, which is not maternally expressed and becomes highly transcribed during zygotic genome activation at the 2-cell stage. A part of its transcription apparently invades almost 150 kb of its genomic flank, whereas the same region downstream of the MuERV-L yields higher frequency of low CPM reads in 1-cell stage in DRB-dependent manner. This pattern is most apparent for MuERV (see also Supplementary Fig S4B), but MT2 and ORR1AO insertions can also produce a similar asymmetric pattern (data not shown). The vertical scale was trimmed at 0.5 CPM; trimming is indicated by horizontal dashed lines.
- E, F Validation of intergenic transcription at the 1-cell stage. Two intergenic regions harboring low CPM reads on chromosome 2 in 1-cell embryos were analyzed by RT–PCR. Both loci are annotated in Supplementary Fig S4C. Shown in (E) is RT–PCR analysis of intergenic transcription of two loci (shown in detail in Supplementary Fig S4C) in MII eggs and 1-cell embryos treated with and without DRB. ctrl = RT–PCR of a spiked α-rabbit globin mRNA demonstrating consistent RT–PCR efficiency across samples. Shown in (F) is the effect of inhibiting the second round of DNA replication in intergenic transcription at the 2-cell stage as examined by RT–PCR. Two-cell embryos were treated with aphidicolin (Aph.) 15 h after insemination to inhibit the second round of DNA replication. ctrl = RT–PCR of a spiked rabbit α-globin mRNA demonstrating consistent RT–PCR efficiency across samples. Experiments were performed three times with reproducible results; a representative result is shown (E, F).
Intergenic transcripts yielding low CPM reads could be either short (i.e., one read or pair of reads would represent one short transcription unit) or they could be fragments of rare long transcripts (i.e., one transcript would yield multiple sequenced fragments, most of which would be far from the actual transcription start site). Two observations suggest some intergenic low CPM reads were derived from fragmented long transcripts. First, when the sequenced libraries were combined to achieve greater depth, low CPM reads representing discrete short transcriptional units did not overlap but rather were more densely populated over a larger area (Supplementary Fig S3A). Second, numerous transcribed intergenic regions at the 1-cell stage correlated with downstream regions of several different retrotransposons whose transcription at the 2-cell stage apparently invaded their neighborhood.
Repetitive sequences represent a source of potential promoters for intergenic transcription. Accordingly, we analyzed repetitive element-derived reads and found that the 1-cell stage contained the highest frequency of retrotransposon-derived reads among all the samples (Fig3B). The increase in retrotransposon-derived read abundance between MII and 1-cell stage was DRB-sensitive and involved all major classes of retrotransposons (LINE, LTR, and SINE). Consistent with the development of a DNA replication-dependent transcriptionally repressive environment in 2-cell embryos, aphidicolin treatment resulted in higher frequency of retrotransposon-derived reads (Fig3B). When examined individually, different retrotransposons showed diverse patterns of expression and various levels of transcription at the 1-cell stage (Fig3C and Supplementary Fig S4A).
The best example of a retrotransposon-supplied promoter producing long intergenic transcripts was the type L mouse endogenous retrovirus (MuERV-L). Transcription of MuERV-L is detected in 1-cell embryos and is very high in 2-cell embryos (Kigami et al, 2003; Svoboda et al, 2004), and was confirmed by our HTS data (Supplementary Fig S4B). Strikingly, a genomic flank on one side of MuERV-L appeared transcribed up to 200 kb downstream of the element (Fig3D and Supplementary Fig S4B). In 1-cell embryos, we did not observe high read density over the retrotransposon but saw low CPM reads within the same area as in 2-cell embryos (Fig3D, Supplementary Fig S4B and C). This feature of MuERV-L elements seemed a general feature that yielded a prominent pattern that became apparent when sequencing data from larger genomic regions were displayed (Supplementary Fig S4B).
To confirm intergenic transcription in 1-cell embryos, we selected two intergenic regions for which reads were uniquely mapped in 1-cell embryos but not MII eggs and examined their expression by RT–PCR. One of these intergenic regions was not MuERV-associated (locus #1), whereas the other was downstream of an MuERV-L element (locus #2) (Supplementary Fig S4C). We first confirmed that transcription of these intergenic regions occurred only in 1-cell embryos but not in MII eggs or 1-cell embryos treated with DRB (Fig3E). We also observed that their expression decreased by the 2-cell stage but remained high when DNA replication was inhibited (Fig3F).
Taken together, these results suggest that a transcriptionally permissive state fosters promiscuous expression from intergenic regions in 1-cell embryos and includes retrotransposon transcription. Subsequently, promiscuous expression at the 2-cell stage is inhibited in DNA replication-dependent manner.
Core-promoter element-independent transcription in 1-cell embryos
To identify sequence features controlling 1-cell transcription, we constructed pGL3 luciferase vectors containing the promoter region of the Zp3 or Tktl1 gene (pGL3-Zp3 or pGL3-Tktl1, respectively), which are expressed in growing oocytes or embryos at the 1- and 2-cell stages, respectively (Hamamoto et al, 2014). These constructs, in principle, allow to test whether transcription in 1-cell embryos can initiate at ‘maternal’ (active only in the oocyte) and/or at ‘zygotic’ (active in preimplantation embryos) promoters. As a negative control, we used the original pGL3 luciferase vector lacking the gene promoter (pGL3-Basic). Growing oocytes microinjected with pGL3-Zp3 showed a significant level of luciferase activity as expected, whereas 1-cell embryos did not (Fig4A). Surprisingly, the pGL3-Basic vector supported luciferase activity in 1-cell embryos but not in oocytes at a level comparable to that of pGL3-Zp3 in oocytes (Fig4A). A corresponding observation was made when the transcriptional activity of pGL3-Tktl1 and pGL3-Basic was examined in 1- and 2-cell embryos (Fig4B).
Figure 4.
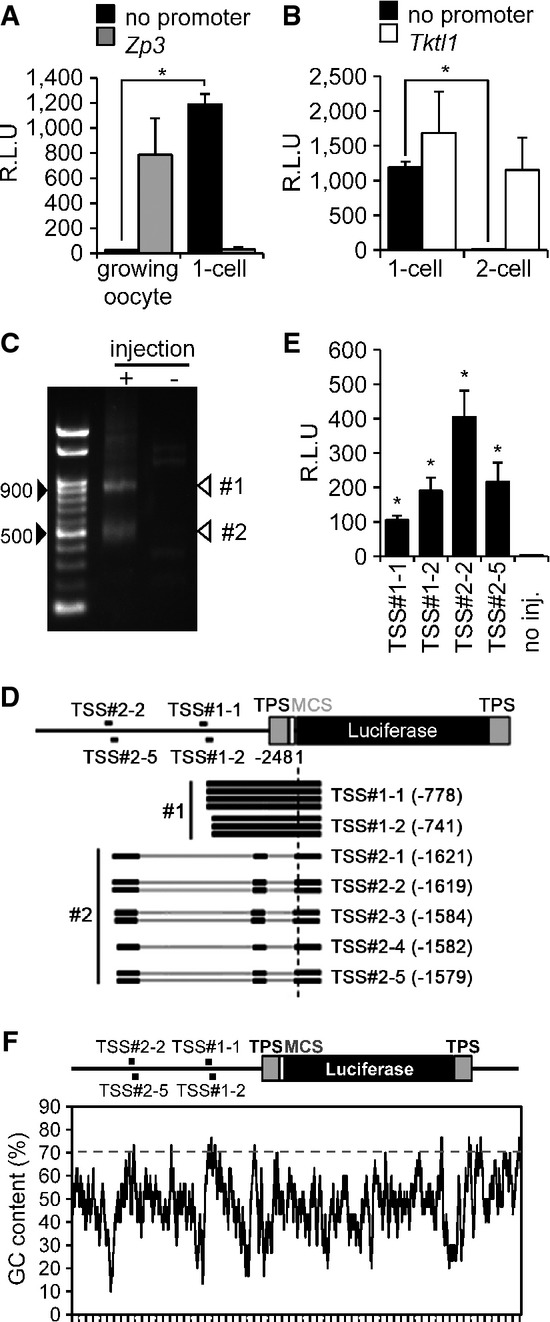
- Transcriptional activity in pGL3-Basic vector lacking a promoter in oocytes and 1-cell embryos. pGL3 vector with the Zp3 or no promoter sequence (pGL3-Zp3 or pGL3-Basic vector, respectively) was injected into the nuclei of growing oocytes or the male pronucleus of 1-cell embryos. After 8 h, luciferase activity was measured. Data are expressed as mean ± SEM (n = 3) and were analyzed using Student's t-test. Asterisks indicate significant differences (P < 0.05).
- Transcriptional activity in pGL3-Basic vector lacking a promoter in 1-cell and 2-cell embryos. The analysis was performed similarly as in (A), except a pGL3 vector with the Tktl1 promoter (pGL3-Tktl1) replaced pGL3-Zp3 and the experiment was performed in 1-cell and 2-cell embryos. Data are expressed as mean ± SEM (n = 3) and were analyzed using Student's t-test. Asterisks indicate significant differences (P < 0.05).
- Determination of the transcriptional start sites (TSSs) of the pGL3-Basic vector by 5′ RACE. The 5′ RACE technique generated two major DNA fragments of ˜900 and 500 bp. Black arrowheads indicate the 500- and 900-bp positions after gel electrophoresis; white arrowheads show the major products of 5′ RACE (TSS#1 and TSS#2).
- Schematic depiction of results from sequencing of seven and eight randomly selected RACE-derived cDNA clones of TSS#1 and TSS#2, respectively, which indicated two and five TSSs for TSS#1 (TSS#1-1 and TSS#1-2) and TSS#2 (TSS#2-1, TSS#2-2, TSS#2-3, TSS#2-4, and TSS#2-5), respectively, in the pGL3-Basic vector. Black lines represent the regions of pGL3-Basic vector corresponding to the results of DNA sequencing. Gray bars represent those of pGL3-Basic vector that were not found in the results of DNA sequencing. TPS represents the transcriptional pause site, a polyadenylation signal site. MCS represents the multi-cloning site.
- Analysis of promoter activity in the upstream regions of identified TSSs in the pGL3-Basic vector. The 76-bp regions upstream of the TSSs shown in (D) were inserted into the pEluc vector; virtually no luciferase activity is produced in 1-cell embryos microinjected with the pEluc vector (Hamamoto et al, 2014). The vectors were injected into the male pronucleus of 1-cell embryos, and luciferase activity was measured after 8 h. The experiments were performed three times. The data are presented as mean ± SEM and were analyzed using Student's t-test. Asterisks indicate significant differences between the sample without injection (no injection) and the other samples (P < 0.05).
- GC content in the pGL3-Basic vector. The percentage GC content was determined using a 30-bp sliding window analysis with a 1-bp shift.
These results suggested that the pGL3-Basic vector contains a cryptic promoter sequence upstream of the luciferase-coding region to support transcription in 1-cell embryos. To locate the corresponding transcriptional start site (TSS) in the pGL3-Basic vector, we performed 5′ RACE. We found two major amplicons ∽900 and 500 bp long [designated as products #1 and #2, respectively (Fig4C)]. These amplicons were cloned, and randomly selected clones were sequenced. cDNA fragments of several sizes were present in the two 5′ RACE products (Fig4D). Product #1 contained 935- and 898-bp cDNA fragments, indicating that the TSSs corresponding to these products were located 778 and 741 bp upstream of the Luc-coding region and were designated TSS#1-1 and TSS#1-2, respectively (Fig4D). Product #2 included 499-, 497-, 462-, 460-, and 457-bp cDNA fragments. Transcripts corresponding to these cDNAs were spliced, and their TSSs were located 1,621, 1,619, 1,584, 1,582, and 1,579 bp upstream of the Luc-coding region and designated TSS#2-1, TSS#2-2, TSS#2-3, TSS#2-4, and TSS#2-5, respectively. Interestingly, product #1 sequences retained an intron, which was spliced out in product #2 (Fig4D).
We also noted that these TSSs were located upstream of the transcriptional pause site (TPS) (Enriquez-Harris et al, 1991), which harbors the consensus sequence of the polyadenylation signal located upstream of the Luc-coding region that reduces luciferase activity derived from background transcription (Fig4D), suggesting that transcription starting from these TSSs was not efficiently terminated by the TPS. Therefore, it was formally possible that transcription initiated at TSS#1 or TSS#2 in the growing oocytes and 2-cell embryos, as well as in 1-cell embryos, but was terminated at the TPS in the oocytes and 2-cell embryos, which resulted in the absence of luciferase activity in these cells. To test this hypothesis, we conducted RT–PCR to detect transcription initiated from TSS#1 and TSS#2. DNA fragments were amplified from 1-cell embryos but not oocytes or 2-cell embryos (Supplementary Fig S5A), indicating that transcription starting from these TSSs occurred only in 1-cell embryos.
To identify the promoter sequences employed by 1-cell embryos, we inserted the 76-bp regions upstream of four TSSs TSS#1-1, TSS#1-2, TSS#2-2, and TSS#2-5, into the pEluc vector (Toyobo, Tokyo), which contains tandem polyadenylation signals and completely terminated transcription upstream of the Luc-coding region; there is virtually no luciferase activity in 1-cell embryos microinjected with pEluc vector (Hamamoto et al, 2014). Interestingly, reporter gene assays of all 76-bp regions upstream of TSS#1-1, TSS#1-2, TSS#2-2, and TSS#2-5 yielded significant luciferase activities (Fig4E) despite no known proximal or core-promoter elements, that is, upstream promoter elements (GC-box) and core-promoter elements (TATA-box, B recognition element (BRE), initiator (Inr), and downstream promoter element (DPE)), were common to all four 76-bp regions upstream or downstream of these TSSs (Supplementary Fig S5B).
In mammals, the promoter regions of many genes have a high G/C content and lack a classical TATA-box (Sandelin et al, 2007; Fenouil et al, 2012). Thus, we searched for G/C-rich regions 76 bp upstream of the aforementioned four TSSs. Using a 30-bp sliding window analysis with a 1-bp shift, we found that all four 76-bp upstream regions contained 30-bp sequences whose G/C contents were > 70% (Fig4F). This finding suggests that these regions are involved in transcriptional regulation independent of known core-promoter elements in 1-cell embryos. In addition, the presence of several closely located TSSs suggests that cryptic initiation of transcription can occur without a specific promoter element in 1-cell embryos. Taken together, the plasmid-borne reporter gene analysis suggested that transcription in 1-cell embryos can initiate from sites lacking well-characterized core-promoter elements.
Analysis of genes transcribed in 1-cell embryos
Genes transcribed in 1-cell embryos fall into two categories: those that have transcripts also detected in MII eggs and those that are not detected in MII eggs (or their abundance is very low). For the former class, it was difficult to determine whether the gene was transcribed in 1-cell embryos because the maternally derived transcript would represent the bulk of the transcripts present in the 1-cell embryo. Thus, we first focused on the second category. To select expressed mRNAs, we used empirically determined criteria for reads per kilobase per million (RPKM) ≤ 0.04 and RPKM ≥ 0.12 in MII eggs and 1-cell embryos, respectively, combined with a minimal fourfold increase between MII and 1-cell and a fourfold reduction following DRB treatment. In addition, to minimize false positives stemming from processed pseudogenes and reads mapping to short transcripts, we selected only spliced mRNAs > 500 nt long. Using these criteria, we selected 96 candidate genes transcribed in 1-cell embryos (Supplementary Table S2). Illustrative examples are found in Fig5A, and RT–PCR confirmation of their expression is shown in Fig5B.
Figure 5.
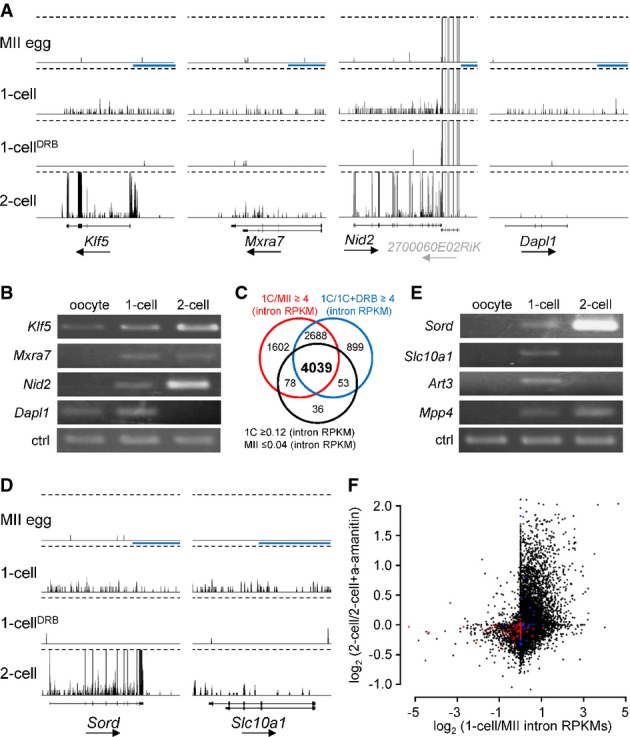
- Examples of four genes exhibiting DRB-dependent 1-cell transcription, which were selected using mRNA (exon) RPKM-based filtering. Shown are 76PE HTS data from four different stages visualized in a customized UCSC browser. All sets are shown at the same scale 0–0.5CPM. Dashed lines indicate trimming at 0.5CPM. The horizontal blue scale bar = 10 kb. The black arrow under each gene name indicates transcription orientation. A simplified exon–intron structure of each gene is shown above the gene name.
- RT–PCR analysis of the four genes from (A). ctrl = RT–PCR of a spiked α-rabbit globin mRNA demonstrating consistent RT–PCR efficiency across samples.
- Intron RPKM-based filtering conditions for selecting genes transcribed in 1-cell embryos. The Venn diagram shows an overlap between gene populations where (i) RPKMs of introns in 1-cell embryos were more than fourfold greater than those in MII eggs oocytes (red circle), (ii) RPKMs of introns in 1-cell embryos were more than fourfold greater than those in 1-cell embryos treated with DRB, and (iii) the numbers of reads mapped to introns for a gene were ≥ 0.12 RPKM in 1-cell embryos and ≤ 0.04 in MII eggs.
- Examples of two genes identified among the 4,039 genes depicted in (C). Figure design is the same as in (A).
- RT–PCR analysis of expression of four genes found among the 4,039 genes identified by filtering conditions described in (C). Ctrl amplicon is the same as in (B).
- Relationship between the minor and major wave of ZGA. Each point represents one transcript (gene). The y-axis depicts α-amanitin sensitivity in 2-cell embryos [log2-fold change inferred from microarray profiling (Zeng & Schultz, 2005)]. The x-axis depicts 1-cell/MII intron RPKM ratio. Red points represent the 100 most expressed oocyte-specific genes, and blue points represent the 100 most expressed testis-specific genes [tissue specificity and level of expression were inferred from BioGPS source data (Su et al, 2004)].
Interestingly, inspection of each of the 96 candidate genes revealed a frequent occurrence of intron-derived reads in 1-cell embryos. In fact, their density was virtually the same as that observed over exons (Fig5A) suggesting that our HTS primarily detects nascent transcripts. We decided therefore to use intron-derived reads to estimate the number of genes transcribed in 1-cell embryos for which the maternal transcript was still present and thereby prevented detection of exon-derived reads. To select these genes, we used the same criteria for intron-based selection as for mRNA selection. Using these criteria, we identified 4,039 genes transcribed in 1-cell embryos in DRB-dependent manner (Fig5C and Supplementary Table S3). Inspection of individual genes confirmed the presence of widespread synthesis of nascent transcripts and showed that exon-based filtering (Supplementary Table S2) also missed many genes whose maternal transcripts were very low but whose expression values for exons fell below the cutoff (Fig5D). RT–PCR analysis of several selected genes confirmed their transcription in 1-cell embryos (Fig5E).
Microarray data from a panel of ∽40 mouse tissues (Su et al, 2004) revealed variable expression patterns of genes transcribed in 1-cell embryos (Supplementary Fig S6A). Expression of more than a half of the genes was ubiquitous, whereas a minority exhibited high tissue specificity (Supplementary Fig S6A). The chromosomal distribution of the 4,039 coding genes suggested that no chromosome was preferentially transcribed (data not shown). Of note is that at least 268 of the 4,039 genes appeared transiently expressed at the 1-cell stage because transcripts were not detected in 4-cell embryos, morulae, and blastocysts, and their RPKM intron values were higher in 1-cell than in 2-cell embryos (Supplementary Table S3). Transcription of these genes could reflect that within the promiscuous first wave of transcription, there is embedded transcription of a subset of genes constituting a part of a 1-cell-specific transcription program (Supplementary Fig S6B).
To examine whether transcription in 1-cell embryos represents expression of genes that are expressed during the major wave of zygotic transcription in 2-cell embryos, we superimposed intronic RPKM results onto relative changes observed in 2-cell embryos treated with α-amanitin (Zeng & Schultz, 2005). This analysis unmasked a remarkably strong relationship between 1-cell transcribed genes and genes whose expression is α-amanitin sensitive in 2-cell embryos (Fig5F). Interestingly, inspection of HTS profiles of genes highly sensitive to α-amanitin showed that Psat1, Rps19, and many others, which did not pass our selection criteria and were not included in the list of 4,039 genes, nevertheless appeared transcribed in 1-cell embryos (Supplementary Fig S6C). This finding suggested that our filtering was conservative and that transcription in 1-cell embryos occurs in more than 4,000 genes; that is, transcription is broad-based in 1-cell embryos and occurs across the entire genome. Nevertheless, a certain degree of selectivity exists for transcription in 1-cell embryos because analysis of 100 genes highly expressed on MII eggs indicated that most were not transcribed in 1-cell embryos (Fig5F). A similar result was found for testis-specific genes (Fig5F).
Deficient mRNA processing in 1-cell embryos
As described above, a substantial fraction of RNA sequence reads of genes transcribed in 1-cell embryos contained introns and sequences well beyond where transcription should have terminated. For example, the reads for Klf5 were evenly mapped to introns and exons in 1-cell embryos and beyond the polyA signal (Fig6A), although the reads in MII eggs and 1-cell embryos were mapped only to exons of Zp3, an oocyte-specific gene (Philpott et al, 1987) (Fig6A). Similar mapping patterns of the reads to introns and beyond polyA signals were identified for many genes (Fig5A and D). These results suggest transcripts from genes transcribed in 1-cell embryos are not processed properly, that is, neither spliced nor terminated correctly.
Figure 6.
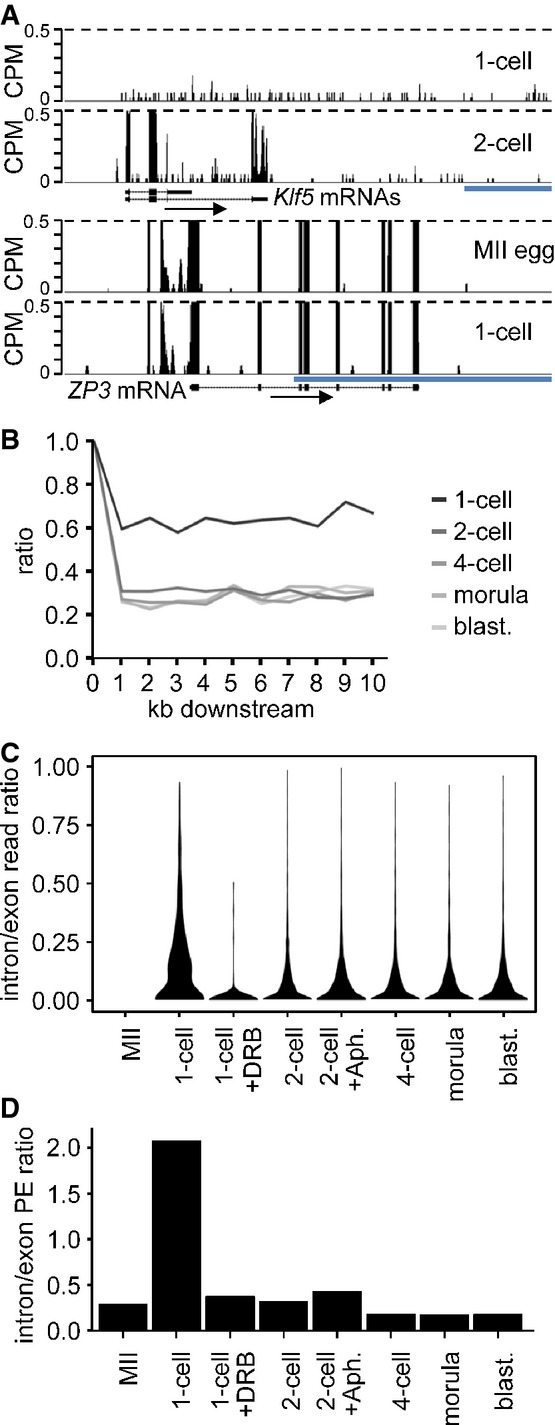
- Example of a gene transcribed at 1-cell stage. Shown are reads from 1-cell- and 2-cell-derived paired-end-sequenced libraries mapping to the Klf5 gene region. Their distribution indicates inefficient splicing and 3′ end processing in 1-cell embryos. In contrast to the 2-cell embryo, the 1-cell embryo does not show any enrichment of exon-derived reads (exons are depicted as black rectangles) and no apparent transcriptional termination at the 3′ end. A detailed analysis of all reads mapping to the Klf5 gene identified a single read derived from a spliced Klf5 transcript. Below is shown profile of Zp3, an abundantly expressed oocyte-specific gene with very well defined exon–intron boundaries, which are retained also at the 1-cell stage. The vertical scale was trimmed at 0.5 CPM; trimming is indicated by horizontal dashed lines. The blue scale bars represent 10 kb.
- Transcription termination analysis. Lines represent median ratios of read counts per kb (RPK) of reads downstream of transcription termination site to exons for gene sets transcribed in 1-cell embryos and subsequent stages but not in MII eggs. Downstream regions for genes with at least one RPK in exons are divided into 1-kb slices, and reads in each slice are counted and divided by the RPK value of the respective exon (point 1, 100%). The 1-cell stage shows higher downstream to exon read ratio indicating the extension of transcription past the polyA site.
- Violin plot distributions of intron/exon read count ratios per cell stage for genes not transcribed in MII eggs. Intron and exon read counts were normalized to 1 kb length (RPK) and divided to obtain the read ratio for each region transcribed at the 1-cell stage or later. The 1-cell stage shows a shift toward higher intron/exon ratios indicating that a larger proportion of transcripts contain unspliced intronic regions, compared to the later stages. The MII stage is displayed as control and contains no values.
- Comparison of unspliced/spliced read pair ratios per cell stage. Read counts where one end maps to intron/exon junction or entirely in intron and the other end maps to the adjacent exon were labeled as ‘unspliced’. The ‘spliced’ pairs were selected so that the one end maps either to the splice site and covers two adjacent exons or with each end mapping to separate, adjacent exons. The ratios of unspliced/spliced pair counts normalized to 1-kb length were calculated for all transcribed regions in 1,786 transcripts not expressed at MII and expressed at later stages. The 1-cell stage shows a clear dominance of unspliced reads over the spliced ones, indicating the increased incidence of intron retention in sequenced transcripts.
To estimate the efficiency of 3′ end processing, we compared read frequencies upstream and downstream of polyA sites where the nearest exon was > 10 kb further downstream from the polyA site. Results of this analysis suggested a high incidence of read-through the termination signal in 1-cell embryos that decreased with development (Fig6B). To estimate the efficiency of splicing, we analyzed the relative ratio of exon and intron-derived frequencies among genes transcribed in 1-cell embryos (Fig6C) and exon–exon/exon–intron localization of paired-end reads (Fig6D). Compared to embryos at the 2-cell stage and beyond, 1-cell embryos showed in a DRB-dependent manner a high proportion of intron-derived reads as evidenced by the markedly wider violin plot above 0.10 in Fig6C and a higher ratio of unspliced RNA fragments relatively to spliced ones (Fig6D). These latter results strongly suggest that nascent transcripts in 1-cell embryos are very poorly spliced.
To confirm the presence of unspliced transcripts in 1-cell embryos, we conducted RT–PCR using primer sets for the Klf5, Nid2, and Mxra7 genes. The primer sets were designed within a single exon (primer set PCR A) or across the splicing junction (primer set PCR B) (Fig7A). All three PCR B products were detected in the 1-cell embryos. Although the amount of product derived from PCR A was higher in 2-cell embryos than in 1-cell embryos, the amount of product obtained from PCR B was markedly higher in 1-cell embryos, suggesting a deficiency in mRNA splicing (Fig7A). To ascertain whether the deficiency in splicing was restricted to the 1-cell stage, we microinjected a ftz pre-mRNA, which was composed of two exons sandwiching an intron, into the nucleus of growing oocytes, and 1- and 2-cell embryos and examined whether the pre-mRNA was spliced into mature mRNA (Fig7B). The mature mRNA was readily detected in oocytes and 2-cell embryos but not in 1-cell embryos.
Figure 7.
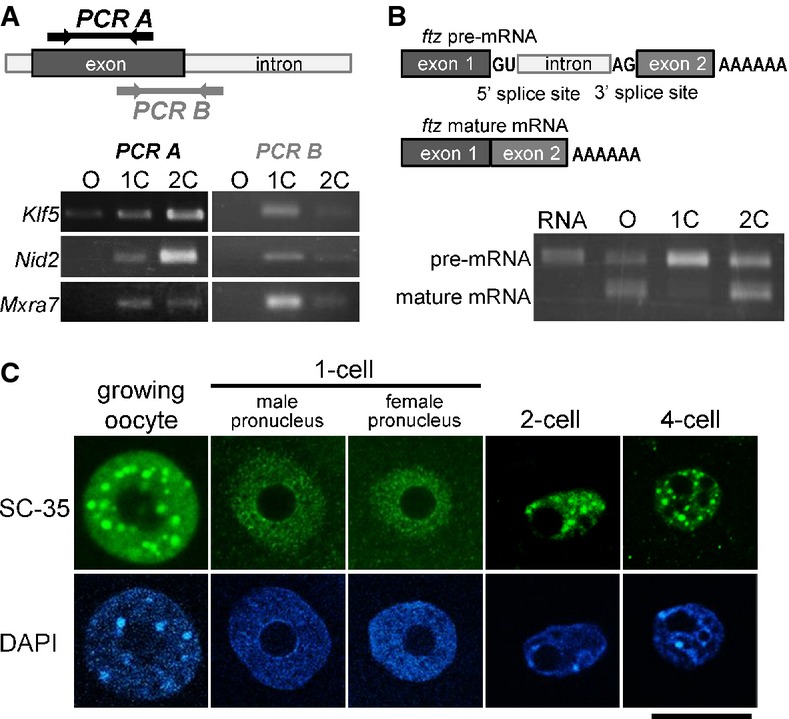
- RT–PCR analysis of unspliced introns. Upper portion shows the locations for which primer pairs were designed. Lower portion depicts results of an RT–PCR analysis using the primer pairs described in the upper diagram.
- Upper diagrams show the structures of pre- and mature ftz mRNAs. Synthesized ftz pre-mRNA was microinjected into growing oocytes, and 1- and 2-cell-stage embryos. Lower portion shows the results of RT–PCR using primer pairs for exons 1 (forward) and 2 (reverse).
- Immunofluorescence with an anti-SC-35 antibody of the nuclei of growing oocytes, and 1-, 2-, and 4-cell-stage embryos. The position of the nucleus, detected with DAPI, is shown in the lower portion of the figure. Scale bar, 20 μm.
The male pronucleus (PN) supports a higher level of transcription than the female PN (Henery et al, 1995; Aoki et al, 1997). Thus, it was formally possible that splicing occurs in the female PN but was not detected. To address this issue, we generated parthenogenetic embryos and examined the occurrence of unspliced Klf5, Nid2, Mxra7, and Sord transcripts. We readily detected unspliced transcripts in the parthenogenotes (Supplementary Fig S7A), suggesting that inefficient splicing was not specific to male pronuclei. Importantly, 50SE data (Park et al, 2013) also detected unspliced Klf5, Nid2, Mxra7, and Sord transcripts (Supplementary Fig S7B), corroborating reduced efficiency of posttranscriptional processing (Supplementary Fig S8). Of note is that 50SE data from 1-cell parthenogenotes also supported reduced posttranscriptional processing in female PN (Supplementary Fig S8B and C).
Finally, we examined the distribution of SC-35 in oocytes, 1-cell embryos, and later embryonic stages. SC-35 is a component of nuclear speckles, which are associated with storage of splicing factors required for pre-mRNA splicing (Huang & Spector, 1992; Kim et al, 2011). Interestingly, whereas male and female pronuclei yielded comparable signal, they both lacked nuclear speckles. In contrast, nuclear speckles were clearly observed in growing oocytes, and 2-cell and 4-cell embryos (Fig7C). These results suggest that the splicing machinery might not be fully formed or functions inefficiently in 1-cell embryos.
Discussion
Despite variable timing, ZGA in vertebrates occurs in a similar manner in which an initial minor ZGA wave is followed by a major second wave. Although the major burst of zygotic expression is well characterized in mice (Schultz, 2002; Zeng & Schultz, 2005), Xenopus (Paranjpe et al, 2013), and zebrafish (Haberle et al, 2014), little is known about the identity of the first transcripts expressed during the initial wave, except for zebrafish (Heyn et al, 2014).
In mice, the first cleavage separates the earliest transcription from the major wave of transcription and reprogramming of gene expression, which takes place in 2-cell embryos. The transcription in 1-cell embryos, however, still remains poorly understood two decades since its discovery (Matsumoto et al, 1994). Here, we provide genome-wide characterization of the initial transcription wave during mammalian ZGA. We used HTS to explore the dynamics of total RNA composition during oocyte-to-embryo transition, focusing primarily on the 1-cell transcriptome. By using total RNA, we obtained a comprehensive and well-mapped dataset that provides information beyond the poly(A) RNA fraction, whose analysis is prone to artifacts. For example, 14 minor ZGA genes identified in mouse and human embryos using poly(A) RNA HTS (Xue et al, 2013) are maternally expressed and not upregulated in either Park et al (2013) or in our dataset (data not shown).
We find that transcription in 1-cell embryos has unique features consistent with genome-wide, promiscuous, and low-level transcriptional activation uncoupled from efficient production of functional mRNAs. This transcription manifests as low CPM reads, whose analysis is limited by their amount. The following lines of evidence argue that these low-abundance reads are not an experimental artifact but originate from transcripts synthesized in 1-cell embryos and reflect genome-wide transcription during the minor transcription wave: (i) The low CPM reads are reproducible and their presence is strongly reduced following DRB treatment. (ii) The distribution of these reads is not random; they are partially associated with genes and specific repetitive elements. (iii) By increasing depth of the data (by combining HTS data from the same stage), gaps were filled between clustered low CPM reads rather than generating distinct peaks or equalizing low CPM read distribution across the genome. (iv) The presence of newly synthesized RNAs from intergenic and gene regions was detected by RT–PCR. (v) Comparable results were obtained when examining a previously published 50SE SOLiD HTS MII, 1-cell and 2-cell data, which provide better sequencing depth (∽1 × 108 total mapped reads after rRNA subtraction from 1-cell embryos vs. ∽2 × 107 total non-rRNA reads from 1-cell embryos in our dataset) (Park et al, 2013). Remarkably, the 50SE dataset offers only a partially improved coverage of novel transcripts in 1-cell samples and does not permit either reliable annotation of transcription start sites in 1-cell embryos or for assembly of minor ZGA transcript sequences. Thus, reliable annotation and assembly of intergenic transcripts appearing during minor ZGA will likely require a sequencing depth larger than in current datasets and paired-end sequencing.
Three types of promoters seem to function in 1-cell embryos: promoters of protein coding genes, promoters of various retrotransposons, and cryptic promoters lacking a defined promoter structure. Although HTS data do not allow reliable determination of TSS positions and minimal promoter features required for transcription in 1-cell embryos, transcription factors available in the zygote likely provide some degree of selectivity. These transcription factors would explain why many genes expressed during the major ZGA wave are transcribed in 1-cell embryos whereas highly expressed maternal and testis-specific genes are poorly transcribed (Fig5F). We speculate that spurious transcription from oocyte-specific promoters is absent in 1-cell embryos because oocyte-specific transcription factors are no longer present and their absence would facilitate reprogramming of gene expression during OET. According to this model, spurious transcription is sufficient for luciferase expression from a promoterless vector in 1-cell embryos, whereas insertion of the Zp3 promoter would have a suppressive effect.
The dynamic chromatin structure at 1-cell embryos is likely a key factor underlying the transcriptionally permissive state. A more relaxed chromatin structure in 1-cell embryos than at later developmental stages offers an explanation that transcription in 1-cell embryos does not require defined core-promoter elements as well as the genome-wide opportunistic transcription occurring in genes, retrotransposons, and intergenic regions. Such a developmental change likely underlies the lack of a requirement for an enhancer for expression in 1-cell embryos and why enhancers stimulate transcription starting at the 2-cell stage, a requirement that is relieved by histone hyperacetylation (Wiekowski et al, 1991, 1993; Majumder et al, 1993; Schultz, 1993); transcription factors require enhancers to access promoters in repressive chromatin and core-promoter elements are essential for stable transcription in eukaryotic cells (Smale & Kadonaga, 2003). Consistent with these findings is that genomic DNA is more sensitive to DNase I at the 1-cell than the 2-cell stage (Cho et al, 2002), suggesting that chromatin in 1-cell embryos is less compact, that is, less mature, than at later developmental stages. Also, consistent with such a developmental change in chromatin structure is that histone mobility is much higher in 1-cell embryos than in 2-cell embryos (Aoki, unpublished results). Such promiscuous transcription in 1-cell embryos, however, also presents a threat to genome integrity and an obstacle to establish a specific gene expression pattern required for continued development. Inefficient posttranscriptional processing in 1-cell embryos may therefore confer a protective mechanism against a promiscuous expression. Noteworthy is that a recent report suggests that the earliest transcribed zygotic genes in the zebrafish are intron poor (Heyn et al, 2014). In light of our result, it is possible to speculate that functionally relevant intron-poor genes might be actually selected for because they would have a higher chance to produce functional transcript than genes with multiple introns.
That 1-cell transcription may not produce translated/translatable transcripts has been previously suggested, but processing of nascent endogenous transcripts from 1-cell embryos was never examined in depth. For example, protein synthesis from a paternally provided gene is observed only at the 2-cell stage (Matsumoto et al, 1994), and mRNAs from endogenous genes were first detected by microarrays at the 2-cell stage (Zeng & Schultz, 2005). Transcripts with intronic sequences have also been detected in bovine preimplantation embryos during the course of genome activation (Graf et al, 2014). We find that the 1-cell HTS library contains an unusually high proportion of sequences derived from intronic sequences as well as a high occurrence of unspliced RNAs. Inefficient splicing is not restricted to the male PN as parthogenetic zygotes also fail to splice efficiently nascent transcripts.
One-cell embryos do splice nascent transcripts, albeit poorly, as evidenced by occasional reads mapping across exon–exon junctions in genes present in MII eggs. In addition, the spliced transcripts are produced from microinjected plasmid reporters (Fig4D and Zeng & Schultz, 2005). Although the splice sites in Fig4D are, in fact, cryptic (which would still support the notion of aberrant posttranscriptional processing), the physiological relevance of splicing observed upon microinjection of thousands of copies of extrachromosomal DNA is unclear. Furthermore, splicing was not the only posttranscriptional process that was impaired in 1-cell embryos. We also find that transcription readily passes through polyadenylation sites without accumulation of properly terminated transcripts. A combination of reduced efficiency of splicing and polyadenylation (as well as activation of cryptic splice sites) could provide a robust barrier that minimizes the risk of retrotransposition and aberrant gene expression at a time when the embryo is most susceptible to such risk.
ftz pre-mRNA microinjection into oocytes and 1-cell and 2-cell embryos (Fig7B) provides strong evidence that splicing of nascent transcripts in 1-cell embryos is inefficient, but the molecular basis for inefficient splicing is unclear. The observed absence of SC-35-containing nuclear speckles, which retain pre-mRNA splicing factors (Spector & Lamond, 2011), could either be a cause or consequence of dysfunctional splicing. Inefficient splicing could also be a consequence of the chromatin composition in 1-cell embryos. Exons contain biased chromatin signatures (Spies et al, 2009). As our understanding of the relationship between splicing and chromatin structure evolves (reviewed in Bentley, 2014; de Almeida & Carmo-Fonseca, 2014), genome-wide acquisition of splicing linked to chromatin remodeling in early embryos offers an interesting model for further testing.
Finally, our results provide novel insights into the question of importance of 1-cell transcription. Inhibiting transcription starting at the 1-cell stage with α-amanitin does not prevent cleavage to the 2-cell stage but does inhibit cleavage to the 4-cell stage (Warner & Versteegh, 1974). Unfortunately, the irreversible nature of α-amanitin inhibition does not allow to demonstrate importance of transcription in 1-cell embryos per se. Interestingly, when 1-cell embryos are cultured in the presence of DRB until the early 2-cell stage and then transferred to DRB-free medium, development arrests at the 2-cell stage (Aoki, unpublished observations). These findings imply that transcription in 1-cell embryos is essential for the development. Thus, it is possible that, despite inefficient posttranscriptional processing, some 1-cell transcripts are functional—either producing proteins or functioning as untranslated long noncoding RNAs. Noncoding RNAs, some of which do not require splicing (Hutchinson et al, 2007), can play essential roles in various cellular processes, including regulation of chromatin structure in early embryos (Casanova et al, 2013). Finally, it is important to consider that transcription per se (and not its products) in 1-cell embryos is important for proper maturation of chromatin. Numerous examples have been shown where the polymerase II complex changes chromatin structure, from nucleosome repositioning, replacement histone deposition, to changes of histone modifications, which include acetylation (reviewed in Butler & Dent, 2012; Das & Tyler, 2013; Weber & Henikoff, 2014). In this model, transcription and chromatin during the minor ZGA form a feedback loop where open chromatin promotes genome-wide pioneering transcription uncoupled from posttranscriptional processing. Pioneering transcription in turn facilitates chromatin remodeling that leads to a properly established chromatin structure and functional posttranscriptional processing during the major ZGA in 2-cell embryos.
Materials and Methods
Collection and culture of oocytes and embryos
Growing oocytes were obtained from 12- to 14-day-old B6D2F1 mice (SLC Japan). The ovaries were transferred to HEPES-buffered KSOM and punctured with a 30-gauge needle. Liberated oocytes with attached follicle cells were harvested, and the follicle cells were gently removed using a narrow-bore glass pipette. The oocytes were then transferred into α-minimal essential medium (α-MEM; Life Technologies, Inc., Grand Island, NY, USA) containing 5% fetal bovine serum and 10 ng/ml epidermal growth factor (both from Sigma-Aldrich).
Metaphase II-arrested eggs (MII eggs) were obtained from superovulated 3-week-old C57BL6/J and B6D2F1 female mice (SLC Japan, Shizuoka) that were first injected intraperitoneally with 5 IU of equine chorionic gonadotropin (eCG; ASKA Pharmaceutical Co., Tokyo, Japan) followed 48 h later with 5 IU of human chorionic gonadotropin (hCG; ASKA Pharmaceutical). MII eggs were collected from the ampullae of oviducts 15 h post-hCG injection and transferred to human tubal fluid medium (Lawitts & Biggers, 1993) supplemented with 10 mg/ml BSA (Sigma-Aldrich, Saint Louis, MO, USA). The eggs derived from C57BL6/J and B6D2F1 mice were inseminated with spermatozoa obtained from the caudal epididymis of adult C57BL6/J and ICR mice (SLC Japan), respectively. The spermatozoa were incubated for 2 h in TYH medium (Toyoda et al, 1971) and human tubal fluid medium supplemented with 10 mg/ml BSA for the eggs from C57BL6/J and B6D2F1 female mice, respectively, in an atmosphere of 5% CO2/95% air at 38°C prior to use for insemination. Four to six hours after insemination, the eggs were washed and cultured in potassium simplex optimized medium (Quinn & Begley, 1984). The 1-, 2-, and 4-cell-, and morula- and blastocyst-stage embryos were collected at 13, 32, 48, 72, and 96 h after insemination, respectively.
For DRB and aphidicolin treatment, 1-cell embryos were transferred into KSOM containing 120 μM DRB (Sigma-Aldrich) or 3 μg/ml aphidicolin (Sigma-Aldrich) at 4 (before initiation of transcription (Aoki et al, 1997)) or 16 h post-insemination (embryos enter M phase at 16 h after insemination), respectively.
RNA extraction, preparation of the RNA-Seq library, and HTS
Total RNA was extracted from 3,000, 3,000, 4,500, 2,800, 1,400, and 700 MII eggs, 1-, 2-, and 4-cell embryos, morulae, and blastocysts obtained from C57BL6/J mice, respectively, using Isogen (Nippon Gene, Tokyo, Japan), according to the manufacturer's instructions. In the second trials for MII eggs and 1-cell embryos, similar numbers of cells used for the first trials were used for RNA extraction. RNA quality analysis and size-selection (> 200 nt) were performed on the Bioanalyzer RNA Pico Chip (Agilent Technologies, Santa Clara, CA, USA). RNA-Seq libraries were constructed using the mRNA-Seq Sample Preparation Kit (Illumina, San Diego, CA, USA) without selection of polyadenylated RNA. Briefly, size-selected total RNA was fragmented to 40–900-nt fragments in fragmentation buffer at 94°C for 5 min, reverse-transcribed with random primers, and ligated with adaptors. cDNA templates were amplified by PCR in the conditions as follows: the initial heat treatment at 98°C for 30 s followed by 15 cycles of 98°C for 10 s, 65°C for 30 s, 72°C for 30 s, and final treatment of 72°C for 5 min. RNA-Seq libraries were subjected to sequencing using Genome Analyzer IIx (Illumina), and 35-nt single-end and 76-nt paired-end sequencing reads were mapped on the mouse genome. The data were deposited into the ArrayExpress database under reference #E-MTAB-2950. Additional details concerning read mapping and bioinformatics analyses can be found in Supplementary Experimental Procedures.
Reverse transcription and polymerase chain reaction
Total RNA was isolated from 100 MII eggs and embryos using Isogen (Nippon Gene) and treated with RQ1 RNase-Free DNase (Promega, Madison, WI, USA) according to the manufacturer's instructions. As an external control, 50 pg rabbit globin mRNA was added prior to total RNA isolation. The isolated total RNA was subjected to reverse transcription using a PrimeScript RT–PCR kit (Takara Bio Inc., Otsu, Japan) according to the manufacturer's instructions. PCR was performed in a thermal cycler (iCycler; Bio-Rad, Berkeley, CA, Japan) using Ex Taq DNA polymerase (Takara) with 35–38 cycles of 95°C for 30 s, 57–60°C for 30 s, and 72°C for 60 s. After the electrophoresis on an agarose gel, the PCR products were stained with ethidium bromide. The primers and PCR conditions are shown in Supplementary Table S4.
Synthesis and microinjection of ftz pre-mRNA
To prepare truncated ftz pre-mRNAs, pGEM pre-ftz (donated by Mutsuhito Ohno in Kyoto University) was amplified by PCR using a forward primer containing the T7 promoter sequence and a reverse primer containing polyA (40 nt) (Supplementary Table S4). Pre-mRNAs were synthesized using in vitro transcription with the mMASSAGE mMACHINE T7 kit (Life Technologies) according to the manufacturer's instructions.
Microinjection was performed on an inverted microscope (ECLIPSE TE300; Nicon Corporation, Tokyo, Japan) using a microinjector (IM300; Narishige Co., Ltd, Tokyo, Japan). Synthesized RNA diluted in nuclease-free water was placed on ice until microinjection. Growing oocytes and embryos obtained from B6D2F1 mice were transferred to HEPES-buffered KSOM, and then, the synthesized RNA was injected into the nuclei in growing oocytes, the nuclei in single blastomeres of 2-cell-stage embryos 29 h after insemination and the male PN in 1-cell-stage embryos 10 h after insemination using narrow glass capillaries (GC100 TF-10; Harvard Apparatus Ltd., Kent). The concentration and volume of injected RNA are 200 ng/μl and 5 pl, respectively. After 1 h of culture in α-MEM (oocytes) and KSOM (embryos), the oocytes and embryos were collected in Isogen for reverse transcription and polymerase chain reaction.
Luciferase assay
A total of 10 pl of 200 ng/μl plasmid DNA was injected into the nuclei of growing oocytes, male PN in 1-cell embryos from B6D2F1 mice 7–9 h after insemination, and the nuclei in single blastomeres of 2-cell-stage embryos were injected 25–27 h after insemination. After 8 h of culture in α-MEM (oocytes) and KSOM (embryos), the oocytes and embryos were collected in 25 μl of phosphate-buffered saline (PBS; Takara Bio Inc.) containing 1 mg/ml BSA (0.1% BSA/PBS) for luciferase assays. Luciferase activity was measured using the ONE-Glo Luciferase Assay System (Promega) and Emerald Luc Luciferase Assay Reagent (Toyobo) for the pGL3-vector and pEluc-test vector, respectively, according to the manufacturer's instructions. A total of 30 oocytes or embryos were used in each assay.
Immunostaining
Growing oocytes and embryos were fixed with 4.0% paraformaldehyde (Wako, Osaka, Japan) in PBS for 15 min. After washing three times with PBS/0.1% BSA, the cells were permeabilized with 0.5% Triton X-100 (Wako) in PBS for 15 min and then incubated overnight at 4°C with an anti-SC 35 antibody (Cat#S4045, Sigma-Aldrich) diluted 1/100 in PBS/0.1% BSA. The cells were washed and incubated with Alexa488-conjugated anti-mouse IgG secondary antibody (Invitrogen, Carlsbad, CA, USA) for 1 h at room temperature. After washing, they were mounted in Vectashield (Vector Laboratories, Burlingame, CA, USA) containing 4′,6-diamidino-2-phenylindole (DAPI; Dojindo Laboratories, Kumamoto) for DNA staining. Confocal digital images were collected using a confocal laser scanning microscope (LSM 5 EXCITER; Carl Zeiss MicroImaging GmbH, Oberkochen).
Acknowledgments
We thank M. Ohno for providing pGEM pre-ftz. Computation time was provided by the Supercomputer System at the Human Genome Center, Institute of Medical Science, University of Tokyo. This work was supported in part by Grants-in-Aid (to F. A.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (#20062002, #25252054). RMS was supported by a grant from NIH (HD022681). VF and KV were supported through the European Commission Seventh Framework Program (Integra-Life; grant 315997 to KV), EMBO Young Investigator Program (Installation grant 1431/2006 to KV), and Croatian Ministry of Science, Education and Sports grant 119-0982913-1211. PS was supported by a Czech Science Foundation grant P305/12/G034. Collaborations were supported by an Academy of Sciences of the Czech Republic project M200521202 (PS and KV) and a Czech Ministry of Education grant KONTAKT II LH13084 (PS and RMS).
Author contributions
FA designed the experiments. KA, RY, MC, and YS conducted the experiments. KA, RY, VF, MGS, KV, PS, RMS, and FA analyzed the data. VF and KV contributed to the statistical analysis. KA, PS, RMS, and FA wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figures
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Supplementary Information
References
- Abe K-I, Inoue A, Suzuki MG, Aoki F. Global gene silencing is caused by the dissociation of RNA polymerase II from DNA in mouse oocytes. J Reprod Dev. 2010;56:502–507. doi: 10.1262/jrd.10-068a. [DOI] [PubMed] [Google Scholar]
- de Almeida SF, Carmo-Fonseca M. Reciprocal regulatory links between cotranscriptional splicing and chromatin. Semin Cell Dev Biol. 2014;32:2–10. doi: 10.1016/j.semcdb.2014.03.010. [DOI] [PubMed] [Google Scholar]
- Aoki F, Worrad DM, Schultz RM. Regulation of transcriptional activity during the first and second cell cycles in the preimplantation mouse embryo. Dev Biol. 1997;181:296–307. doi: 10.1006/dbio.1996.8466. [DOI] [PubMed] [Google Scholar]
- Bachvarova R, De Leon V. Polyadenylated RNA of mouse ova and loss of maternal RNA in early development. Dev Biol. 1980;74:1–8. doi: 10.1016/0012-1606(80)90048-2. [DOI] [PubMed] [Google Scholar]
- Bentley DL. Coupling mRNA processing with transcription in time and space. Nat Rev Genet. 2014;15:163–175. doi: 10.1038/nrg3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouniol C, Nguyen E, Debey P. Endogenous transcription occurs at the 1-cell stage in the mouse embryo. Exp Cell Res. 1995;218:57–62. doi: 10.1006/excr.1995.1130. [DOI] [PubMed] [Google Scholar]
- Bucknall RA, Moores H, Simms R, Hesp B. Antiviral effects of aphidicolin, a new antibiotic produced by Cephalosporium aphidicola. Antimicrob Agents Chemother. 1973;4:294–298. doi: 10.1128/aac.4.3.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler JS, Dent SY. Chromatin ‘resetting’ during transcription elongation: a central role for methylated H3K36. Nat Struct Mol Biol. 2012;19:863–864. doi: 10.1038/nsmb.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova M, Pasternak M, El Marjou F, Le Baccon P, Probst AV, Almouzni G. Heterochromatin reorganization during early mouse development requires a single-stranded noncoding transcript. Cell Rep. 2013;4:1156–1167. doi: 10.1016/j.celrep.2013.08.015. [DOI] [PubMed] [Google Scholar]
- Cho T, Sakai S, Nagata M, Aoki F. Involvement of chromatin structure in the regulation of mouse zygotic gene activation. Anim Sci J. 2002;73:113–122. [Google Scholar]
- Das C, Tyler JK. Histone exchange and histone modifications during transcription and aging. Biochim Biophys Acta. 2013;1819:332–342. doi: 10.1016/j.bbagrm.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis W, De Sousa PA, Schultz RM. Transient expression of translation initiation factor eIF-4C during the 2-cell stage of the preimplantation mouse embryo: identification by mRNA differential display and the role of DNA replication in zygotic gene activation. Dev Biol. 1996;174:190–201. doi: 10.1006/dbio.1996.0065. [DOI] [PubMed] [Google Scholar]
- De La Fuente R, Eppig JJ. Transcriptional activity of the mouse oocyte genome: companion granulosa cells modulate transcription and chromatin remodeling. Dev Biol. 2001;229:224–236. doi: 10.1006/dbio.2000.9947. [DOI] [PubMed] [Google Scholar]
- Deng Q, Ramskold D, Reinius B, Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science. 2014;343:193–196. doi: 10.1126/science.1245316. [DOI] [PubMed] [Google Scholar]
- DePamphilis ML. Origins of DNA replication in metazoan chromosomes. J Biol Chem. 1993;268:1–4. [PubMed] [Google Scholar]
- Enriquez-Harris P, Levitt N, Briggs D, Proudfoot NJ. A pause site for RNA polymerase II is associated with termination of transcription. EMBO J. 1991;10:1833–1842. doi: 10.1002/j.1460-2075.1991.tb07709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenouil R, Cauchy P, Koch F, Descostes N, Cabeza JZ, Innocenti C, Ferrier P, Spicuglia S, Gut M, Gut I, Andrau J-C. CpG islands and GC content dictate nucleosome depletion in a transcription-independent manner at mammalian promoters. Genome Res. 2012;22:2399–2408. doi: 10.1101/gr.138776.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forlani S, Bonnerot C, Capgras S, Nicolas JF. Relief of a repressed gene expression state in the mouse 1-cell embryo requires DNA replication. Development. 1998;125:3153–3166. doi: 10.1242/dev.125.16.3153. [DOI] [PubMed] [Google Scholar]
- Graf A, Krebs S, Zakhartchenko V, Schwalb B, Blum H, Wolf E. Fine mapping of genome activation in bovine embryos by RNA sequencing. Proc Natl Acad Sci U S A. 2014;111:4139–4144. doi: 10.1073/pnas.1321569111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberle V, Li N, Hadzhiev Y, Plessy C, Previti C, Nepal C, Gehrig J, Dong X, Akalin A, Suzuki AM, van IJcken WF, Armant O, Ferg M, Strahle U, Carninci P, Muller F, Lenhard B. Two independent transcription initiation codes overlap on vertebrate core promoters. Nature. 2014;507:381–385. doi: 10.1038/nature12974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamamoto G, Suzuki T, Suzuki MG, Aoki F. Regulation of transketolase like 1 gene expression in the murine one-cell stage embryos. PLoS ONE. 2014;9:e82087. doi: 10.1371/journal.pone.0082087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamatani T, Carter MG, Sharov AA, Ko MSH. Dynamics of global gene expression changes during mouse preimplantation development. Dev Cell. 2004;6:117–131. doi: 10.1016/s1534-5807(03)00373-3. [DOI] [PubMed] [Google Scholar]
- Henery CC, Miranda M, Wiekowski M, Wilmut I, DePamphilis ML. Repression of gene expression at the beginning of mouse development. Dev Biol. 1995;169:448–460. doi: 10.1006/dbio.1995.1160. [DOI] [PubMed] [Google Scholar]
- Heyn P, Kircher M, Dahl A, Kelso J, Tomancak P, Kalinka AT, Neugebauer KM. The earliest transcribed zygotic genes are short, newly evolved, and different across species. Cell Rep. 2014;6:285–292. doi: 10.1016/j.celrep.2013.12.030. [DOI] [PubMed] [Google Scholar]
- Huang S, Spector DL. U1 and U2 small nuclear RNAs are present in nuclear speckles. Proc Natl Acad Sci U S A. 1992;89:305–308. doi: 10.1073/pnas.89.1.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson JN, Ensminger AW, Clemson CM, Lynch CR, Lawrence JB, Chess A. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genom. 2007;8:39. doi: 10.1186/1471-2164-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DA, Pombo A, Iborra F. The balance sheet for transcription: an analysis of nuclear RNA metabolism in mammalian cells. FASEB J. 2000;14:242–254. [PubMed] [Google Scholar]
- Karolchik D, Hinrichs AS, Kent WJ. The UCSC Genome Browser. Curr Protoc Bioinformatics. 2012;Chapter 1 doi: 10.1002/0471250953.bi0104s40. Unit 1 4. [DOI] [PubMed] [Google Scholar]
- Kigami D, Minami N, Takayama H, Imai H. MuERV-L is one of the earliest transcribed genes in mouse one-cell embryos. Biol Reprod. 2003;68:651–654. doi: 10.1095/biolreprod.102.007906. [DOI] [PubMed] [Google Scholar]
- Kim Y-D, Lee J-Y, Oh K-M, Araki M, Araki K, Yamamura K-I, Jun C-D. NSrp70 is a novel nuclear speckle-related protein that modulates alternative pre-mRNA splicing in vivo. Nucleic Acids Res. 2011;39:4300–4314. doi: 10.1093/nar/gkq1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawitts JA, Biggers JD. Culture of preimplantation embryos. Methods Enzymol. 1993;225:153–164. doi: 10.1016/0076-6879(93)25012-q. [DOI] [PubMed] [Google Scholar]
- Majumder S, Miranda M, DePamphilis ML. Analysis of gene expression in mouse preimplantation embryos demonstrates that the primary role of enhancers is to relieve repression of promoters. EMBO J. 1993;12:1131–1140. doi: 10.1002/j.1460-2075.1993.tb05754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K, Anzai M, Nakagata N, Takahashi A, Takahashi Y, Miyata K. Onset of paternal gene activation in early mouse embryos fertilized with transgenic mouse sperm. Mol Reprod Dev. 1994;39:136–140. doi: 10.1002/mrd.1080390203. [DOI] [PubMed] [Google Scholar]
- Meijer HA, Bushell M, Hill K, Gant TW, Willis AE, Jones P, de Moor CH. A novel method for poly(A) fractionation reveals a large population of mRNAs with a short poly(A) tail in mammalian cells. Nucleic Acids Res. 2007;35:e132. doi: 10.1093/nar/gkm830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh B, Hwang S, McLaughlin J, Solter D, Knowles BB. Timely translation during the mouse oocyte-to-embryo transition. Development. 2000;127:3795–3803. doi: 10.1242/dev.127.17.3795. [DOI] [PubMed] [Google Scholar]
- Paranjpe SS, Jacobi UG, van Heeringen SJ, Veenstra GJ. A genome-wide survey of maternal and embryonic transcripts during Xenopus tropicalis development. BMC Genom. 2013;14:762. doi: 10.1186/1471-2164-14-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-J, Komata M, Inoue F, Yamada K, Nakai K, Ohsugi M, Shirahige K. Inferring the choreography of parental genomes during fertilization from ultralarge-scale whole-transcriptome analysis. Genes Dev. 2013;27:2736–2748. doi: 10.1101/gad.227926.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpott CC, Ringuette MJ, Dean J. Oocyte-specific expression and developmental regulation of ZP3, the sperm receptor of the mouse zona pellucida. Dev Biol. 1987;121:568–575. doi: 10.1016/0012-1606(87)90192-8. [DOI] [PubMed] [Google Scholar]
- Piko L, Clegg KB. Quantitative changes in total RNA, total poly(A), and ribosomes in early mouse embryos. Dev Biol. 1982;89:362–378. doi: 10.1016/0012-1606(82)90325-6. [DOI] [PubMed] [Google Scholar]
- Quinn P, Begley AJ. Effect of human seminal plasma and mouse accessory gland extracts on mouse fertilization in vitro. Aust J Biol Sci. 1984;37:147–152. doi: 10.1071/bi9840147. [DOI] [PubMed] [Google Scholar]
- Sandelin A, Carninci P, Lenhard B, Ponjavic J, Hayashizaki Y, Hume DA. Mammalian RNA polymerase II core promoters: insights from genome-wide studies. Nat Rev Genet. 2007;8:424–436. doi: 10.1038/nrg2026. [DOI] [PubMed] [Google Scholar]
- Santos F, Peters AH, Otte AP, Reik W, Dean W. Dynamic chromatin modifications characterise the first cell cycle in mouse embryos. Dev Biol. 2005;280:225–236. doi: 10.1016/j.ydbio.2005.01.025. [DOI] [PubMed] [Google Scholar]
- Schultz RM. Regulation of zygotic gene activation in the mouse. BioEssays. 1993;15:531–538. doi: 10.1002/bies.950150806. [DOI] [PubMed] [Google Scholar]
- Schultz RM. The molecular foundations of the maternal to zygotic transition in the preimplantation embryo. Hum Reprod Update. 2002;8:323–331. doi: 10.1093/humupd/8.4.323. [DOI] [PubMed] [Google Scholar]
- Sehgal PB, Darnell JE, Jr, Tamm I. The inhibition by DRB (5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole) of hnRNA and mRNA production in HeLa cells. Cell. 1976;9:473–480. doi: 10.1016/0092-8674(76)90092-1. [DOI] [PubMed] [Google Scholar]
- Smale ST, Kadonaga JT. The RNA polymerase II core promoter. Annu Rev Biochem. 2003;72:449–479. doi: 10.1146/annurev.biochem.72.121801.161520. [DOI] [PubMed] [Google Scholar]
- Spector DL, Lamond AI. Nuclear speckles. Cold Spring Harb Perspect Biol. 2011;3:a000646. doi: 10.1101/cshperspect.a000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies N, Nielsen CB, Padgett RA, Burge CB. Biased chromatin signatures around polyadenylation sites and exons. Mol Cell. 2009;36:245–254. doi: 10.1016/j.molcel.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda P, Stein P, Anger M, Bernstein E, Hannon GJ, Schultz RM. RNAi and expression of retrotransposons MuERV-L and IAP in preimplantation mouse embryos. Dev Biol. 2004;269:276–285. doi: 10.1016/j.ydbio.2004.01.028. [DOI] [PubMed] [Google Scholar]
- Toyoda Y, Yokoyama M, Hosi T. Studies on the fertilization of mouse eggs in vitro: I. in vitro fertilization of eggs by fresh epididymal sperm. Jpn J Anim Reprod. 1971;16:147–151. [Google Scholar]
- Vasseur M, Condamine H, Duprey P. RNAs containing B2 repeated sequences are transcribed in the early stages of mouse embryogenesis. EMBO J. 1985;4:1749–1753. doi: 10.1002/j.1460-2075.1985.tb03846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QT, Piotrowska K, Ciemerych MA, Milenkovic L, Scott MP, Davis RW, Zernicka-Goetz M. A genome-wide study of gene activity reveals developmental signaling pathways in the preimplantation mouse embryo. Dev Cell. 2004;6:133–144. doi: 10.1016/s1534-5807(03)00404-0. [DOI] [PubMed] [Google Scholar]
- Warner CM, Versteegh LR. In vivo and in vitro effect of alpha-amanitin on preimplantation mouse embryo RNA polymerase. Nature. 1974;248:678–680. doi: 10.1038/248678a0. [DOI] [PubMed] [Google Scholar]
- Weber CM, Henikoff S. Histone variants: dynamic punctuation in transcription. Genes Dev. 2014;28:672–682. doi: 10.1101/gad.238873.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiekowski M, Miranda M, DePamphilis ML. Regulation of gene expression in preimplantation mouse embryos: effects of the zygotic clock and the first mitosis on promoter and enhancer activities. Dev Biol. 1991;147:403–414. doi: 10.1016/0012-1606(91)90298-h. [DOI] [PubMed] [Google Scholar]
- Wiekowski M, Miranda M, DePamphilis ML. Requirements for promoter activity in mouse oocytes and embryos distinguish paternal pronuclei from maternal and zygotic nuclei. Dev Biol. 1993;159:366–378. doi: 10.1006/dbio.1993.1248. [DOI] [PubMed] [Google Scholar]
- Xue Z, Huang K, Cai C, Cai L, Jiang C-Y, Feng Y, Liu Z, Zeng Q, Cheng L, Sun YE, Liu J-Y, Horvath S, Fan G. Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature. 2013;500:593–597. doi: 10.1038/nature12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatsepina O, Baly C, Chebrout M, Debey P. The step-wise assembly of a functional nucleolus in preimplantation mouse embryos involves the cajal (coiled) body. Dev Biol. 2003;253:66–83. doi: 10.1006/dbio.2002.0865. [DOI] [PubMed] [Google Scholar]
- Zeng F, Schultz RM. RNA transcript profiling during zygotic gene activation in the preimplantation mouse embryo. Dev Biol. 2005;283:40–57. doi: 10.1016/j.ydbio.2005.03.038. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Supplementary Information