

Abstract
Toll-like receptor 9 (TLR9) drives innate immune responses after recognition of foreign or endogenous DNA containing unmethylated CpG motifs. DNA-mediated TLR9 activation is highly implicated in the pathogenesis of several autoimmune skin diseases, yet its contribution to the inflammation seen in these diseases remains unclear. In this study, TLR9 ligand, CpGB DNA, was administered to mice via a subcutaneous osmotic pump with treatment lasting 1 or 4 weeks. Gene expression and immunofluorescence analyses were used to determine chemokine expression and cell recruitment in the skin surrounding the pump outlet. CpGB DNA skin treatment dramatically induced a marked influx of CD11b+ F4/80+ macrophages, increasing over 4 weeks of treatment, and induction of IFNγ and TNFα expression. Chemokines, CCL2, CCL4, CCL5, CXCL9 and CXCL10, were highly induced in CpGB DNA-treated skin, although abrogation of these signalling pathways individually did not alter macrophage accumulation. Flow cytometry analysis showed that TLR9 activation in the skin increased circulating CD11b+ CD115+ Ly6Chi inflammatory monocytes following 1 week of CpGB DNA treatment. Additionally, skin-resident CD11b+ cells were found to initially take up subcutaneous CpGB DNA and propagate the subsequent immune response. Using diphtheria toxin-induced monocyte depletion mouse model, gene expression analysis demonstrated that CD11b+ cells are responsible for the CpGB DNA-induced cytokine and chemokine response. Overall, these data demonstrate that chronic TLR9 activation induces a specific inflammatory response, ultimately leading to a striking and selective accumulation of macrophages in the skin.
Keywords: cell trafficking, chemokines, inflammation, monocytes/macrophages, skin
Introduction
Nucleic acids, including DNA, induce innate immune responses and are implicated in the pathogenesis of several autoimmune skin diseases. Overactivation of inflammatory pathways can lead to accumulation of immune cells and excessive production of inflammatory mediators that can cause severe tissue damage. Toll-like receptor 9 (TLR9), a pattern recognition receptor, recognizes DNA with unmethylated CpG motifs (1), and evidence suggests that it is highly involved in several autoimmune skin diseases, such as systemic lupus erythematosus (SLE), psoriasis and possibly systemic sclerosis (2,3). CpG DNA is thought to be released at high levels early in autoimmune disease, possibly due to increased or inadequately cleared cell debris, and might continually activate TLR9 in skin-resident cells over long periods of time (2,4,5). These observations emphasize the importance of understanding the immune response induced in the skin during chronic TLR9 activation.
In mouse models of CpG DNA-induced arthritis and septic shock, TLR9 activation has been shown to recruit macrophages and neutrophils and induce TNFα-mediated inflammation (6,7). Additionally, repeated intra-peritoneal injections of CpG DNA induce production of TNFα, IFNγ, IL-12 and IL-6, as well as increase the number of circulating monocytes (8–10). Few studies have examined the effect of CpG DNA exposure on the skin. Those studies have shown an influx of mononuclear cells and an increase in IL-1β, IL-6 and TNFα expression, after the administration of a single dose of CpG DNA (11–14). Thus, it remains unclear how chronic activation of TLR9 as expected in autoimmune disease would affect cytokine and chemokine production and subsequent immune cell recruitment.
Immune cells, including macrophages, dendritic cells and T cells, reside throughout the skin with macrophages representing about half of the cells in the dermis (3,11,15). In response to immune stimulus in the skin, dermal macrophages and dendritic cells have been suggested to be the major responders and initiators of inflammation (3,11). It remains unknown which skin-resident cells initially recognize CpG DNA and are responsible for initiating the immune response.
In this study, we examined the effects of chronic TLR9 activation on the skin using an osmotic pump mouse model. We observed specific monomorphic infiltration of CD11b+ F4/80+ macrophages. TLR9 activation induced expression of inflammatory mediators: TNFα, IFNγ and a discrete chemokine profile. Our data also show that dermal macrophage accumulation is associated with an increase in circulating inflammatory monocytes, suggesting this population is recruited into CpGB DNA-treated skin. Lastly, we demonstrate that dermal macrophages are the cells initially responding to subcutaneous CpGB DNA and driving the subsequent upregulation of inflammatory mediators and macrophage recruitment. Thus, chronic TLR9 activation leads to a surprisingly limited inflammatory cell response, which was associated with a particularly uniform recruitment of macrophages into the skin.
Materials and methods
Mice
C57BL/6 wild-type (WT), C57BL/6 TNFα−/−, C57BL/6 IFNγ−/−, C57BL/6 CXCR3−/−, C57BL/6 CCR5−/−, C57BL/6 CCL2−/− mice were obtained from Jackson Laboratories; C57BL/6 TLR9−/− mice were originally obtained from S. Akira (1) and provided by Ian Rifkin. All procedures were approved by the institutional animal care and use committee at Boston University Medical Campus.
Osmotic pump model
Briefly, mice were anesthetized by intra-peritoneal (i.p.) injections of ketamine (100 mg/kg) and xylazine (5 mg/kg). Osmotic pumps (Alzet, Cupertino, CA) designed to deliver CpGB DNA (ODN 1668; Integrate DNA Technologies, Inc. Coralville, Iowa; 1 μg/μl concentration) or phosphate-buffered saline (PBS) for either 7 or 28 days were sterilely implanted subcutaneously. At designated time points, mice were euthanized and the skin (approximately 1 cm2) surrounding the pump outlet was homogenized in TRIzol (Invitrogen, Grnad Island, NY) for RNA isolation or fixed in either formalin or O.C.T. compound for immunohistochemistry or immunofluorescence, respectively.
Diphtheria toxin-induced monocyte depletion mouse model
Itgam(CD11b)-DTR mice were obtained from Jackson Laboratories. CD11b-DTR and C57BL/6 WT mice were injected i.p. with 100 μl of diphtheria toxin (List Biological Laboratories, Inc. Campbell, CA) at a concentration of 0.02 μg per gram of mouse weight. Two days after PBS or CpGB DNA pump insertion, skin was collected for RNA isolation or immunofluorescence.
Subcutaneous injections of CpGB DNA
Mice were briefly anesthetized, and skin was shaved and cleaned. 50 μl of 1 μg/μl concentration of CpGB DNA (ODN 1668) conjugated to fluorescein isothiocyanate (FITC) (Invivogen) was injected subcutaneously. Skin samples were collected 4 h after the injection for immunofluorescence analysis. 250 μl of 1 μg/μl concentration of CpGB DNA or PBS was injected, and skin samples were collected 6 h after injection for qPCR and immunofluorescence analysis.
RNA isolation and gene expression analysis
RNA was isolated from skin homogenized in 2 ml of TRIzol (Invitrogen), using TissueLyser II (Qiagen, Manchester, UK), according to Invitrogen’s protocol for RNA isolation from TRIzol. Blood collected postmortem was mixed with 100 μl of 0.5 M EDTA pH8.0, and red blood cells were lysed using red blood cell lysing buffer (Sigma-Aldrich, St. Louis, MO). Remaining blood cells were resuspended in RLT lysis buffer, and RNA was isolated using the RNeasy Mini Kit (Qiagen). The concentration of total RNA isolated was measured (Nanodrop 1000; ThermoScientific, Tewksbury, MA), and 300 ng of RNA was used to make cDNA according to the Superscript II RT (Invitrogen) protocol using random primers. Gene expression assays (TaqMan; Applied Bio-systems, Foster City, CA) were used with the cDNA for quantitative real-time PCR analysis (qPCR) (StepOnePlus; Applied Biosystems). For chemokine array analysis, cDNAs were mixed with RT2SYBR green/ROX PCR Master Mix and used with RT2 Profiler PCR Array: Mouse Chemokines and Receptors according to the included protocol (SA Biosciences, Valencia, CA). Skin from mice treated with PBS and CpG was analysed using nanostring technology (16). Hundred nanograms of RNA per sample was used, and gene expression was normalized to the expression of eight housekeeping genes. The analysis was performed using GraphPad Software, Inc., La Jolla, CA.
Immunohistochemistry
Skin samples were fixed in formalin, embedded in paraffin and rehydrated. Sections were stained with haematoxylin and eosin. Additional sections were blocked with Fc Receptor Blocker and Background Buster (Innovex, Boston, MA), stained with rabbit polyclonal ki67 antibody (Novus Biologicals, Littleton, CO), then stained with secondary antibody against rabbit Ig (GE Healthcare Life Sciences, Pittsburgh, PA) and developed with Dako Chromogen System.
Immunofluorescence
Frozen skin sections were fixed in cold acetone and blocked with Fc Receptor Blocker and Background Buster (Innovex). Sections were stained with phycoerythrin (PE)-conjugated rat anti-mouse CD11b (BD Biosciences, San Jose, CA), allophycocyanin (APC)-conjugated rat anti-mouse Ly6G (eBioscience, San Diego, CA), Alexa Fluor 488- (Molecular Probes, Inc., Eugene, OR) conjugated rat anti-mouse F4/80 (BioLegend, London, UK) or Alexa Fluor 488- (Molecular Probes, Inc.) conjugated rat IgG2b κ isotype control (BioLegend). All sections were counterstained with Fluoroshield Mounting Medium with DAPI (Abcam, Cambridge, UK). Images were taken with Fluoview FV10i confocal microscope (Olympus, Center Valley, PA).
Flow cytometry
Isolated blood cells were labelled with APC-Cy7-conjugated rat anti-mouse CD11b (BioLegend), phycoerythrin (PE)-conjugated rat anti-mouse CD115 (eBioscience), Pacific Blue- (Molecular Probes, Inc.) conjugated rat anti-mouse Ly6C (BioLegend) and LIVE/DEAD Fixable Aqua Dead Cell Stain (Invitrogen). At least 1 000 000 events were acquired on a LSRII Flow Cytometer (BD Biosciences). Dead cells and doublets were excluded based on live/dead staining and side scatter properties. Data were analysed and graphed using FlowJo software (Tree Star, Ashland, OR).
Data analysis
qPCR data are normalized to mRNA expression of housekeeping gene (GAPDH) expression and expression of one control sample. All analyses graph the expression mean with the standard deviation (SD). P-values were calculated using 2-sided, unpaired t-test.
Results
One week of CpGB DNA treatment induced severe skin inflammation
Osmotic pumps containing CpGB DNA were sterilely inserted subcutaneously on the backs of wildtype C57BL/6 (B6) mice for 1-week treatments to determine the effects of chronic TLR9 activation on the skin. All 1-week CpGB DNA-treated mice had a small nodule in the skin at the pump outlet that was not seen in PBS-treated mice. A large accumulation of cells was seen in CpGB DNA-treated skin when examining the skin by haematoxylin and eosin staining (Fig. 1b). CpGB DNA treatment induced aggregation of these cells in the subcutaneous region, particularly, below the panniculus carnosus. These cells were a homogeneous population of mononuclear cells with large nuclei (Fig. 1c). Cell recruitment was not seen in the epidermal and dermal regions of CpGB-treated skin. PBS-treated mice showed neither cell accumulation nor noticeable structural changes in the skin (Fig. 1a).
Figure 1.
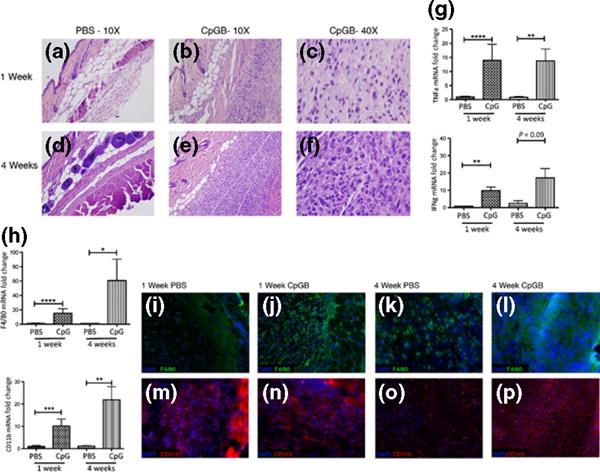
Chronic CpGB DNA skin treatment induces severe inflammation and an influx of CD11b+ F4/80+ cells. Haematoxylin and eosin staining of skin sections taken from mice treated with PBS or CpGB DNA for 1 or 4 weeks. (a) PBS 1 week treatment (b, c) CpGB DNA 1 week treatment (d) PBS 4 week treatment (e, f) CpGB DNA 4 week treatment (a, b, d, e) Images taken at 10× magnification. (c, f) Image taken at 40× magnification. (g) Gene expression analysis for TNFα and IFNγ. All genes are normalized to GAPDH and compared to the respective PBS-treated mouse. PBS 1 week: n = 8; CpGB 1 week: n = 11; PBS 4 week: n = 3; CpGB 4 week: n = 5. Data collected from five experiments. **P < 0.01; ****P < 0.0001. (h) Gene expression analysis for macrophage markers, F4/80 and CD11b. All genes are normalized to GAPDH and compared to the respective PBS-treated mouse. PBS 1 week: n = 8; CpGB 1 week: n = 11; PBS 4 week: n = 3; CpGB 4 week: n = 5. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (i–p) Immunofluorescence staining of skin treated with PBS or CpGB for 1 or 4 weeks; (i–l) F4/80 antibody conjugated to FITC; (m–p) CD11b antibody conjugated to PE; (i, m) PBS 1 week treatment; (j, n) 1 week CpGB DNA treatment; (k, o) 4 week PBS treatment; (l, p) 4 week CpGB DNA treatment. All images are taken at 20× magnification. Data collected from five experiments.
Mononuclear cell mass formed after 4 weeks of CpGB DNA treatment
CpGB DNA skin treatment for 4 weeks led to a remarkable accumulation of cells in the subcutaneous region of the skin (Fig. 1e). A large mass, approximately 1 cm in diameter, was observed at the pump outlet of these mice, an effect not seen in PBS-treated mice. Histological analysis of the 4-week CpGB DNA-treated skin showed a striking uniformity of these accumulated cells that were all large and mononuclear (Fig. 1f). Similar to 1-week CpGB DNA treatment, the effects of the 4-week treatment were localized to the subcutaneous region of the skin, with no changes seen in the epidermis or dermis.
Inflammatory mediators upregulated by chronic TLR9 activation
CpGB DNA skin treatment for 1 and 4 weeks upregulated expression of inflammatory cytokines, TNFα and IFNγ, compared to PBS-treated mice (Fig. 1g), in agreement with previously published studies that demonstrated this response occurs downstream of TLR9 after activation by CpGB DNA (1,6). For further confirmation, TLR9-deficient mice treated with CpGB DNA for 1 and 4 weeks did not show an induction of TNFα or IFNγ gene expression. (Figure S1a). CpGB DNA treatment did not increase expression of type I interferon-responsive genes after 1 week with only a minimal increase seen after 4 weeks (Figure S1b), distinguishing this response from TLR9 activation by CpGA DNA (data not shown).
CD11b+ F4/80+ macrophages accumulate in CpGB DNA-treated skin
To identify the accumulated cells, skin from CpGB DNA and PBS-treated mice was collected for gene expression analysis of immune cell markers. Expression of macrophages markers, F4/80 and CD11b, were significantly increased in the skin after 1 week of CpGB treatment in comparison with PBS-treated mice with an average increased expression of 15.3 ± 6.3- and 10.2 ± 3.1-fold (mean ± SD), respectively (Fig. 1h). These macrophage markers were further increased in 4-week CpGB DNA-treated mice with an average increased expression of 60 ± 29.6-fold for F4/80 and 22 ± 5.9-fold for CD11b compared to PBS-treated mice, correlating with the observed increase in the numbers of accumulated cells at this time point. Markers of other immune cells were also examined in the skin: CD11c (dendritic cells), Ly6G (neutrophils), CD19 (B cells) and CD3 (T cells). Expression of these cell markers was not increased in CpGB DNA-treated mice at either 1 or 4 weeks treatment time in comparison with the respective PBS-treated mice with the exception of CD11c that showed a trend towards increased expression in 4-week CpGB DNA-treated mice (Figure S1c). Average expression of Ly6G was significantly decreased in 1-week CpGB DNA-treated mice compared to PBS-treated mice. Further confirming that cell recruitment is specific to the activation of TLR9, TLR9-deficient mice treated with CpGB DNA did not show increased expression of these macrophage markers (Figure S1d).
Immunofluorescence staining of 1-week CpGB DNA-treated skin confirmed the gene expression analysis data that these cells are CD11b+ F4/80+ macrophages (Fig. 1j and n). Four-week CpGB DNA-treated skin showed remarkable widespread staining of these macrophage markers (Fig. 1l and p). CD11b+ and F4/80+ cells were also seen in PBS-treated skin (Fig. 1i, m and k, o) in agreement with previously published studies identifying a large number of skin-resident macrophages under normal, non-inflammatory conditions (11,15,17). This staining was performed in conjunction with an isotype control and an antibody against CD3 conjugated to PE that did not show positive staining, demonstrating the specificity of these cells in expressing only these macrophage markers (Figure S1e–h).
To further identify the accumulated cells, gene expression from skin of CpGB DNA and PBS-treated mice was collected for analysis by nanostring assay. Expression of M2 markers, ARG1 and CD163, were significantly downregulated while expression of M1 marker NOS was significantly upregulated (Figure S1i).
CpGB DNA treatment induces expression of macrophage chemoattractants in the skin
Chemokine, chemokine receptor and inflammatory cytokine expressions in 1-week CpGB DNA-treated skin were analysed using 84-gene chemokine arrays (SA Bioscience). Of all genes included in the array, only six chemokines and TNFα were found to have induced expression greater than threefold change in comparison with PBS-treated skin (Fig. 2a: CCL2-6.0 ± 3.1-fold change; CCL4-18.9 ± 7.8-fold change; CCL5-7.3 ± 6.3-fold change; CCL7-3.7 ± 2.2-fold change; CXCL10-4.1 ± 2.4-fold change; CXCL9-8.9 ± 5.1-fold change; TNF-16.0 ± 5.2-fold change). These observations made with chemokine arrays were confirmed using TaqMan gene expression assays for a larger group of mice. All chemokines identified in the array, with the exception of CCL7, were confirmed to have increased expression after 1-week CpGB DNA treatment (Fig. 2b: CCL2-5.3 ± 3.3-fold change; CCL4-29.3 ± 18.8-fold change; CCL5-5.9 ± 3.2-fold change; CXCL9-13.3 ± 7.7-fold change; CXCL10-4.4 ± 2.9-fold change). Expression of these chemokines remained induced after 4 weeks of CpGB DNA treatment compared to PBS-treated mice (CCL2-5.2 ± 1.5-fold change; CCL4-187.3 ± 81.9-fold change; CCL5-8.5 ± 2.4-fold change; CXCL9-17.5 ±7.9-fold change; CXCL10-5.8 ± 3.9-fold change). Complete data including expression from all genes analysed by the chemokine array can be found in Table S1. Chemokine gene expression analysis of TLR9-deficient mice treated with CpGB DNA for 1 and 4 weeks confirmed that induction of these chemokines is TLR9 dependent, except for CCL2, which showed induction in TLR9-deficient mice at 4 weeks (Figure S2a).
Figure 2.
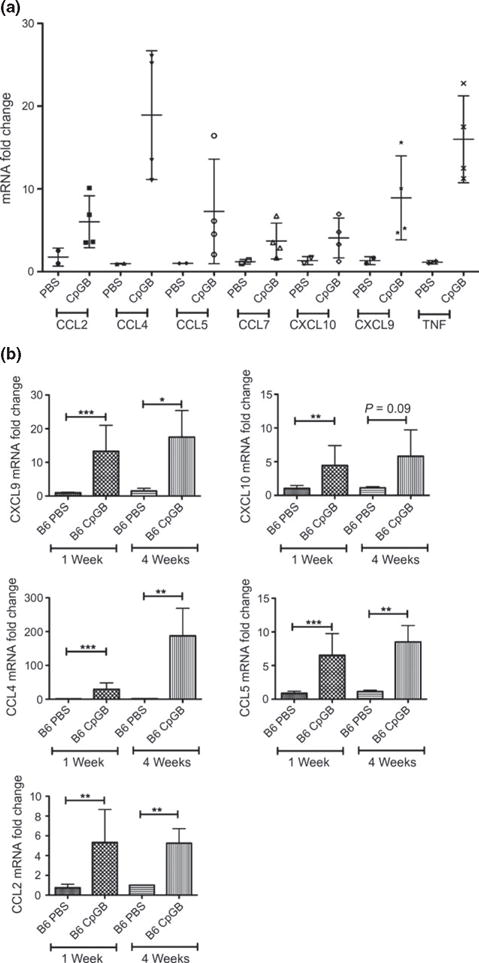
Chronic CpGB DNA skin treatment induces a specific chemokine response. (a) Chemokine gene expression array analysis. Only genes with an average fold change in CpGB DNA-treated skin greater than threefold compared to PBS-treated skin are shown. PBS: n = 2; CpGB: n = 4. Data collected from two experiments. (b) Gene expression analysis on the six induced chemokines identified in the array: CXCL9, CXCL10, CCL2, CCL4, CCL5, CCL7. PBS 1 week: n = 4; CpGB 1 week: n = 5; PBS 4 week: n = 3; CpGB 4 week: n = 5. Data collected from five experiments. For all genes, expression is normalized to housekeeping gene (a: HSP90 and b: GAPDH) and compared to the respective PBS-treated mouse. *P < 0.05; **P < 0.01; ***P < 0.001.
Macrophage accumulation is not dependent on TNFα, IFNγ or individual chemokine signalling pathways
As TNFα and IFNγ are highly produced by CpGB DNA skin treatment (Fig. 1g) and are known to induce chemokine expression through activation of their receptors (18,19), we examined their role in the accumulation of macrophages and the induction of chemokines seen in CpGB DNA-treated skin. Surprisingly, TNFα-deficient mice treated with CpGB DNA for 1 week showed an induction of expression of macrophage markers, similar to the induction seen in wildtype (B6) CpGB DNA-treated mice, with even a modest increase of CD11b expression seen in TNFα-deficient mice (CD11b: P < 0.04, F4/80: P = ns; Figure S3a). In a similar manner, IFNγ-deficient mice also had an average expression of macrophage markers, CD11b and F4/80, comparable to the average expression in CpGB DNA-treated wildtype (B6) mice (CD11b: P < 0.02, F4/80: P = ns). Notably, expression of CpGB DNA-induced chemokines, CCL2, CCL5 and CXCL10, was actually increased in TNFα-deficient mice compared to wildtype (B6) mice, while CCL4 and CXCL9 expression remained the same (Figure S3b). In addition, chemokine expression was similar between wildtype (B6) an IFNγ-deficient CpGB DNA-treated mice, with the exception of CCL2 and CCL4, that showed increased expression in IFNγ-deficient mice.
As CpGB DNA treatment leads to a large accumulation of macrophages that is neither dependent on TNFα nor IFNγ signalling, we sought to determine which chemokines upregulated in CpGB DNA-treated mice were responsible for the recruitment of macrophages to the skin. Mice deficient of CCR5 (receptor for CCL4, CCL5), CXCR3 (receptor for CXCL9, CXCL10) or CCL2 were treated with CpGB DNA for 1 week. Expression of macrophage markers, CD11b and F4/80, did not show significant decreases in comparison with wildtype mice (Figure S2b–d).
CpGB DNA skin treatment leads to an increase in circulating CD11b+ CD115+ Ly6Chi inflammatory monocytes
As the recruitment of macrophages to the skin would likely derive from circulating monocytes, we analysed circulating leucocytes from mice treated with CpGB DNA for 1 week to determine whether blood monocyte populations were altered in comparison with PBS-treated mice. On average, 9.5 ± 2.1% (mean ± SD) of all live circulating leucocytes in PBS-treated mice were CD11b+ CD115+ monocytes (17,20,21). This population was significantly increased to an average of 21.1 ± 6.0% after 1 week of CpGB DNA treatment (P < 0.04; Fig. 3a and c). This increase of CD11b+ CD115+ monocytes after CpGB treatment was selective, as the population of CD11b+ CD115− cells, typically marking granulocytes (17,20,21), remained at 20.4 ± 4.5% on average in both PBS and CpGB DNA-treated mice (Fig. 3c, right panel). Analysis of Ly6C expression on circulating monocytes can subdivide this population into inflammatory monocytes (Ly6Chi) and patrolling monocytes (Ly6C−) (20–23). Granulocytes also express Ly6C but at intermediate levels compared to inflammatory monocytes. After gating on CD11b+ cells, Ly6Chi inflammatory monocytes were seen at an average of 19.5 ± 2.6% of circulating leucocytes in PBS-treated mice. The Ly6Chi inflammatory monocyte population significantly increased to 40.3 ± 12.1% of all CD11b+ cells after CpGB DNA skin treatment (P < 0.05; Fig. 3b and d). The increase of the inflammatory monocyte population was also selective, as the percentage of CD11b+ Ly6C− patrolling monocytes and CD11b+ Ly6Cint granulocytes remained constant between PBS and CpGB DNA-treated mice.
Figure 3.
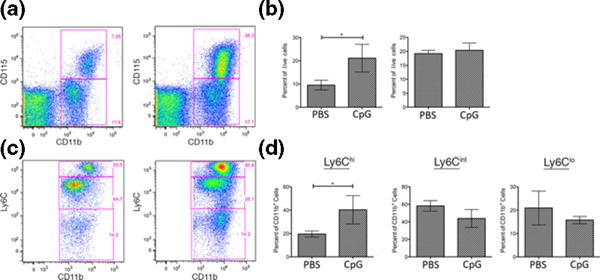
Inflammatory monocytes are increased in the blood following CpGB DNA skin treatment. Flow cytometry analysis of peripheral blood mononuclear cells from 1 week PBS or CpGB DNA-treated mice, using CD11b-APC_Cy7, CD115-PE, Ly6C-Pacific Blue, and AmCyan Live/Dead cell staining. Data shown is gated on live cells. Representative images are shown in dot plots. (a) CD11b and CD115 staining, showing selection of CD11b+ CD115+ cells (monocytes) and CD11b+ CD115− cells (neutrophils) (b) Gating on CD11b+ cells, Ly6C staining, showing selection of Ly6Chi cells (inflammatory monocytes), Ly6Cint cells (neutrophils), and Ly6C− cells (patrolling monocytes). (c, d) Quantification of per cent positive cells for each population, n = 3. Data collected from one experiment. *P < 0.05.
CpGB DNA-induced skin inflammation is dependent on dermal CD11b+ cells
To determine the cells initially recognizing CpGB DNA in the skin, wildtype (B6) mice were injected subcutaneously with CpGB DNA conjugated to fluorescein isothiocyanate (CpGB-FITC). Immunofluorescence of the skin 4 h following subcutaneous injection of CpGB-FITC identified CD11b+ cells as the major cell type positive for FITC staining (Fig. 4a). The FITC staining was perinuclear, in agreement with what has been described for localization of CpG DNA after binding to TLR9 (24). In order to address the possible role of granulocytes during the initial response to CpG, mice skin was collected 6 h following subcutaneous injection of CpG or PBS. There was no significant difference in Ly6G gene expression by qPCR between those mice injected with PBS and those injected with CpG (Figure S4a). Also, there was little expression of Ly6G by immunofluorescence (Figure S4b and c).
Figure 4.
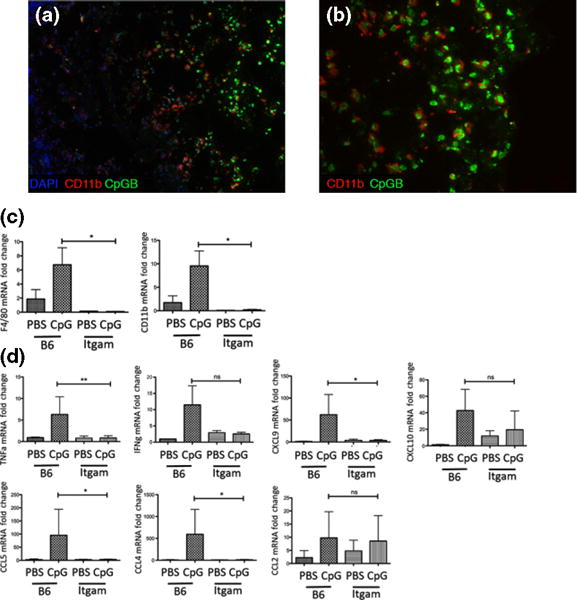
CpGB DNA-induced skin inflammation is dependent on dermal CD11b+ cells. Immunofluorescence staining for CD11b in B6 skin 4 h following subcutaneous injection with CpGB-FITC (Subpanels a, b). CpGB-FITC – green; CD11b-PE – red; Nuclei-DAPI – blue. (a) 10× magnification (b) 20× magnification. Representative image from two experiments. Gene expression analysis from diphtheria toxin depletion model CD11b-DTR (Subpanels c, d) (c) Gene expression analysis of macrophage markers: CD11b and F4/80. (d) Gene expression analysis of cytokines: TNFα and IFNγ and chemokines: CCL2, CCL4, CCL5, CXCL9 and CXCL10. For all treated mice, PBS n = 4; CpGB n = 6. Data collected from two experiments. Gene expression is normalized GAPDH and compared to the respective PBS-treated mouse. *P < 0.05; **P < 0.01.
To determine whether CD11b+ cells drive CpGB DNA-induced skin inflammation, we treated monocyte-depleted mice with CpGB DNA for 2 days. CD11b+ cells were depleted using a diphtheria toxin (DT) inducible model, where the CD11b promoter drives the expression of the human diphtheria toxin receptor (DTR) deleting only CD11b+ cells. Wildtype (B6) and CD11b-DTR mice were injected i.p. with DT one day prior to treatment with PBS or CpGB DNA for 2 days by osmotic pump. CD11b-DTR mice showed a complete loss of CD11b+ and F4/80+ cells in the skin, along with a loss of circulating monocytes (Figure S5). Skin in these mice did not show macrophage recruitment after CpGB DNA treatment (Fig. 4c). Cytokine and chemokine expression analysis in the skin demonstrated that CD11b-depleted mice (CD11b-DTR) were able to induce neither TNFα and IFNγ expression nor expression of chemokines: CCL2, CCL4, CCL5, CXCL9 and CXCL10 after CpGB DNA treatment, whereas these chemokines and CD11b+ F4/80+ macrophages were highly induced in DT-treated wildtype mice (Fig. 4d).
Discussion
The study of chronic TLR9 activation in the skin showed a specific inflammatory response characterized by increased expression of a select group of cytokines and chemokines, along with the immense accumulation of CD11b+ F4/80+ macrophages in the skin. Dermal macrophages recognized and induced the initial immune response to TLR9 ligand, CpGB DNA, leading to an increase in circulating inflammatory monocytes that likely supply the influx of macrophages into the skin. Overall, these data are supported by the current paradigm that macrophages are recruited from the blood during proinflammatory immune responses (25,26) and demonstrates that local immune stimuli can induce a systemic inflammatory response.
As important protectors of barrier immunity, resident macrophages are found in high numbers in both human and mouse skin during normal, non-inflammatory conditions (3,11,15,27). Thus, as efficient phagocytic cells, it is not surprising that they are the first responders to subcutaneous CpGB DNA. Interestingly, CD11b depletion of macrophages demonstrated that dermal DCs and structural skin cells did not play an important role in the cytokine and chemokine response after CpGB DNA treatment. This effect may be facilitated by the subdermal location of the osmotic pump, an area that typically contains many macrophages. Even so, it is unexpected that other skin-resident cells did not induce an immune response in the absence of dermal macrophages. These data point to skin-resident macrophages as crucial mediators of inflammation following CpGB DNA exposure.
Surprisingly, TLR9 activation of dermal macrophages led to a specific accumulation of inflammatory macrophages. Upon histological examination, it was clear that all of the recruited cells were morphologically similar and expressed the same macrophage cell markers, CD11b and F4/80. They showed upregulated M1 marker, NOS2, and decreased M2 markers, Arg1 and CD163. This M1 phenotype is different from what is seen when mice are pretreated with CpG systemically 6 days before wound healing, where a marked increase in M2 macrophage marker RELM-a is seen in association with accelerated wound healing (28). In another study in which CpG was administered topically, increased IL-12 was seen more typical of an M1 response (29). So differences in kinetics, dose and/or route of administration appear to be key to the effect of TLR9 stimulation on dermal macrophages.
TLR9 expression by various cell types in the skin has been difficult to detect using available reagents. However, our data not surprisingly clearly show dermal macrophages take up and respond to CpG indicating they have these receptors. Keratinocytes also appear to express TLR9 though perhaps at lower levels (28,30). Subcutaneous osmotic pump delivery clearly favours delivery to dermal and subdermal tissues, and CpG may not reach the epidermis in adequate concentration to have an effect on this cell layer in our model.
Chemokine expression was analysed to determine the set of chemokines that recruited such a specific cell type. Several chemokines were found to be upregulated in CpGB-treated skin: CCL2, CCL4, CCL5, CXCL9 and CXCL10. The receptors for these chemokines, CCR2, CCR5 and CXCR3, are also expressed on dendritic cells and T cells in addition to monocytes/macrophages (31). Although the highly upregulated expression of these chemokines led to a homogenous influx of macrophages, abrogation of these chemokine pathways individually did not alter macrophage recruitment after CpGB treatment. Thus, this specific combination of chemokines does not appear to drive the specificity of this recruitment. Furthermore, these data suggest that these chemokines are redundant in their function, as all of them are chemotactic for macrophages. Overall, these data indicate that blocking one single chemokine pathways will not likely provide a strong therapeutic for blocking macrophage recruitment in autoimmune skin disease.
Another interesting aspect of the CpGB DNA osmotic pump model is that these macrophages drastically increase in number over time. While the continuous recruitment of inflammatory monocytes likely drives the accumulation, it appears that these cells are remaining at the site of inflammation as we did not observe histological evidence of apoptosis or necrosis. These inflammatory macrophages do not upregulate chemokine receptor, CCR7, which would induce migration out of the skin and into the skin draining lymph nodes, supporting the current hypothesis that inflammatory macrophages remain in the periphery (32). Despite seeing a discrete increase in circulating inflammatory monocytes, we surprisingly did not see a change in monocyte/macrophage accumulation in CpG-treated mice deleted of CCL2, the chemokine most closely associated with monocyte mobilization from the bone marrow through CCR2 (33). Despite this, we feel most likely CpG is recruiting inflammatory monocytes from the bone marrow through a combination of chemokines. Another alternative is that the CpG is entering the circulation and reaching the bone marrow to affect CCR2/CCL2, CXCL12 or other chemokine regulating monocyte egress from the bone marrow. Similar effects have been already shown with systemic administration of a variety of TLR ligands including CpG (34). Our data would support the notion that multiple chemokines can serve this function and not just CCL2.
There has been some evidence that TLR9 activation can promote the survival of macrophages, and as there is a continuous source of TLR9 ligand in this model, the macrophages do not die nor induce resolution of the inflammation (35). In autoimmune skin disease, macrophages can be found in increased numbers in affected skin, which is likely an important aspect of pathogenesis as macrophages induce widespread tissue damage (21). Thus, interfering with TLR9-mediated survival could be a promising therapeutic target, as death of these cells could lead to resolution of the local immune response.
Overall, this study of TLR9 activation by CpGB DNA has identified a signature for chronic TLR9 activation in the skin. In addition to increased CCL2, CCL4, CCL5, CXCL9 and CXCL10 expression, local TLR9 activation led to an increase in circulating inflammatory monocytes that likely drive subcutaneous macrophage accumulation. In autoimmune skin diseases where TLR9 activation is implicated in pathogenesis, it is likely one of multiple immune pathways activated in the disease. This signature provides a valuable tool for identifying the specific role of TLR9 activation in disease. Taken together, these data further emphasize the importance of macrophages in propagating TLR9-mediated autoimmune skin disease and how selective TLR activation can result in selective cell recruitment and survival.
Supplementary Material
Table S1. Chemokine array for CpGB DNA-treated skin.
Figure S1. TLR9-dependent TNFα and IFNγ induction, no type I interferon response to CpGB DNA treatment, and specific recruitment of macrophages to CpGB DNA treated skin.
Figure S2. Chemokine expression after CpGB DNA treatment is TLR9-dependent, and cell recruitment is not dependent on CXCR3, CCR5, or CCL2 signaling.
Figure S3. CpGB DNA-induced chemokine expression and cell recruitment are not dependent on IFNγ or TNFα signaling.
Figure S4. Initial CpGB DNA-induced skin inflammation is not dependent on granulocytes.
Figure S5. CD11b depletion by diphtheria toxin inducible mouse model.
Acknowledgments
This work was supported by National Institutes of Health Research Training in Immunology Grant 5T32 AI007309 to Allison Mathes and National Institutes of Health Grants 1P50AR060780, 5P30AR061271 and 2R01AR051089 to Robert Lafyatis.
Footnotes
Conflict of interests
The authors have declared no conflicting interests.
Supporting Information
Additional supporting data may be found in the supplementary information of this article.
References
- 1.Hemmi H, Takeuchi O, Kawai T, et al. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 2.Lin JH, Dutz JP, Sontheimer RD, et al. Clin Rev Allergy Immunol. 2007;33:85–106. doi: 10.1007/s12016-007-0031-x. [DOI] [PubMed] [Google Scholar]
- 3.Nestle FO, Di Meglio P, Qin JZ, et al. Nat Rev Immunol. 2009;9:679–691. doi: 10.1038/nri2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marshak-Rothstein A, Rifkin IR. Annu Rev Immunol. 2007;25:419–441. doi: 10.1146/annurev.immunol.22.012703.104514. [DOI] [PubMed] [Google Scholar]
- 5.Rifkin IR, Leadbetter EA, Busconi L, et al. Immunol Rev. 2005;204:27–42. doi: 10.1111/j.0105-2896.2005.00239.x. [DOI] [PubMed] [Google Scholar]
- 6.Campbell JD, Cho Y, Foster ML, et al. J Clin Invest. 2009;119:2564–2576. doi: 10.1172/JCI38294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deng GM, Verdrengh M, Liu ZQ, et al. Arthritis Rheum. 2000;43:2283–2289. doi: 10.1002/1529-0131(200010)43:10<2283::AID-ANR16>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 8.Nardini E, Morelli D, Aiello P, et al. J Cell Physiol. 2005;204:889–895. doi: 10.1002/jcp.20360. [DOI] [PubMed] [Google Scholar]
- 9.Santiago-Raber ML, Baudino L, Alvarez M, et al. J Autoimmun. 2011;37:171–179. doi: 10.1016/j.jaut.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 10.Velayudham A, Hritz I, Dolganiuc A, et al. J Hepatol. 2006;45:813–824. doi: 10.1016/j.jhep.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 11.Kupper TS, Fuhlbrigge RC. Nat Rev Immunol. 2004;4:211–222. doi: 10.1038/nri1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu L, Zhou X, Shi J, et al. Immunology. 2003;110:341–347. doi: 10.1046/j.1365-2567.2003.01739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Molne L, Collins LV, Tarkowski A. J Invest Dermatol. 2003;121:294–299. doi: 10.1046/j.1523-1747.2003.12352.x. [DOI] [PubMed] [Google Scholar]
- 14.Zhao Q, Temsamani J, Zhou RZ, et al. Antisense Nucleic Acid Drug Dev. 1997;7:495–502. doi: 10.1089/oli.1.1997.7.495. [DOI] [PubMed] [Google Scholar]
- 15.Dupasquier M, Stoitzner P, van Oudenaren A, et al. J Invest Dermatol. 2004;123:876–879. doi: 10.1111/j.0022-202X.2004.23427.x. [DOI] [PubMed] [Google Scholar]
- 16.Northcott PA, Shih DJ, Remke M, et al. Acta Neuropathol. 2012;123:615–626. doi: 10.1007/s00401-011-0899-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lucas T, Waisman A, Ranjan R, et al. J Immunol. 2010;184:3964–3977. doi: 10.4049/jimmunol.0903356. [DOI] [PubMed] [Google Scholar]
- 18.Hu X, Ivashkiv LB. Immunity. 2009;31:539–550. doi: 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roach DR, Bean AG, Demangel C, et al. J Immunol. 2002;168:4620–4627. doi: 10.4049/jimmunol.168.9.4620. [DOI] [PubMed] [Google Scholar]
- 20.Auffray C, Sieweke MH, Geissmann F. Annu Rev Immunol. 2009;27:669–692. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- 21.Sunderkotter C, Nikolic T, Dillon MJ, et al. J Immunol. 2004;172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 22.Geissmann F, Manz MG, Jung S, et al. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ziegler-Heitbrock L, Ancuta P, Crowe S, et al. Blood. 2010;116:e74–e80. doi: 10.1182/blood-2010-02-258558. [DOI] [PubMed] [Google Scholar]
- 24.Latz E, Schoenemeyer A, Visintin A, et al. Nat Immunol. 2004;5:190–198. doi: 10.1038/ni1028. [DOI] [PubMed] [Google Scholar]
- 25.Helming L. Curr Biol. 2011;21:R548–R550. doi: 10.1016/j.cub.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 26.Jenkins SJ, Ruckerl D, Cook PC, et al. Science. 2011;332:1284–1288. doi: 10.1126/science.1204351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zaba LC, Fuentes-Duculan J, Steinman RM, et al. J Clin Invest. 2007;117:2517–2525. doi: 10.1172/JCI32282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hergert B, Grambow E, Butschkau A, et al. Wound Repair Regen. 2013;21:723–729. doi: 10.1111/wrr.12084. [DOI] [PubMed] [Google Scholar]
- 29.Kim YS, Kim Y, Lee KJ, et al. Int Arch Allergy Immunol. 2007;144:315–324. doi: 10.1159/000106458. [DOI] [PubMed] [Google Scholar]
- 30.Curry JL, Qin JZ, Bonish B, et al. Arch Pathol Lab Med. 2003;127:178–186. doi: 10.5858/2003-127-178-IIRRIN. [DOI] [PubMed] [Google Scholar]
- 31.Charo IF, Ransohoff RM. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 32.Randolph GJ, Inaba K, Robbiani DF, et al. Immunity. 1999;11:753–761. doi: 10.1016/s1074-7613(00)80149-1. [DOI] [PubMed] [Google Scholar]
- 33.Shi C, Pamer EG. Nat Rev Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi C, Jia T, Mendez-Ferrer S, et al. Immunity. 2011;34:590–601. doi: 10.1016/j.immuni.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sester DP, Brion K, Trieu A, et al. J Immunol. 2006;177:4473–4480. doi: 10.4049/jimmunol.177.7.4473. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Chemokine array for CpGB DNA-treated skin.
Figure S1. TLR9-dependent TNFα and IFNγ induction, no type I interferon response to CpGB DNA treatment, and specific recruitment of macrophages to CpGB DNA treated skin.
Figure S2. Chemokine expression after CpGB DNA treatment is TLR9-dependent, and cell recruitment is not dependent on CXCR3, CCR5, or CCL2 signaling.
Figure S3. CpGB DNA-induced chemokine expression and cell recruitment are not dependent on IFNγ or TNFα signaling.
Figure S4. Initial CpGB DNA-induced skin inflammation is not dependent on granulocytes.
Figure S5. CD11b depletion by diphtheria toxin inducible mouse model.