

Abstract
The complete mitochondrial genome (mitogenome) of the beet webworm, Spoladea recurvalis has been sequenced. The circular genome is 15,273 bp in size, encoding 13 protein-coding genes (PCGs), two rRNA genes, and 22 tRNA genes and containing a control region with gene order and orientation identical to that of other ditrysian lepidopteran mitogenomes. The nucleotide composition of the mitogenome shows a high A+T content of 80.9%, and the AT skewness is slightly negative (-0.023). All PCGs start with the typical ATN codons, except for COX1, which may start with the CGA codon. Nine of 13 PCGs have the common stop codon TAA; however, COX1, COX2 and ND5 utilize the T nucleotide and ND4 utilizes TA nucleotides as incomplete termination codons. All tRNAs genes are folded into the typical cloverleaf structure of mitochondrial tRNAs, except for the tRNASer(AGY) gene, in which the DHU arm fails to form a stable stem-loop structure. A total of 157 bp intergenic spacers are scattered in 17 regions. The overlapping sequences are 42 bp in total and found in eight different locations. The 329 bp AT-rich region is comprised of non-repetitive sequences, including the motif ATAG, which is followed by a 14 bp poly-T stretch, a (AT11 microsatellite-like repeat, which is adjacent to the motif ATTTA, and a 9 bp poly-A, which is immediately upstream from the tRNAMet gene. Phylogenetic analysis, based on 13 PCGs and 13 PCGs+2 rRNAs using Bayesian inference and Maximum likelihood methods, show that the classification position of Pyraloidea is inconsistent with the traditional classification. Hesperioidea is placed within the Papilionoidea rather than as a sister group to it. The Pyraloidea is placed within the Macrolepidoptera with other superfamilies instead of the Papilionoidea.
Introduction
The animal mitochondrial genome is a double-stranded circular DNA molecule, 14 to 20 kb in size, which encodes a conserved set of 37 genes, including 13 protein-coding genes (PCGs) plus the two ribosomal RNA (rRNA) genes and 22 transfer RNA (tRNA) genes [1,2]. Additionally, it also contains a control region, known as A+T-rich region in insects [3], including initiation sites of the transcription and replication of the mitogenome [1,4]. While the length of the A+T-rich region vary highly in that the presence of the indels and tandem duplicated elements [5]. The mitogenome is characterized by its small size, maternal inheritance, non-recombination, and rapid evolution [1,2,6]. Mitogenomes have been studied in a variety of fields, such as structural genomic [1,7], genetic resources [8], molecular evolution [9], population genetics [10], phylogeography [11], inter-ordinal and intra-ordinal relationships [12–14].
Lepidoptera (moths and butterflies) is the second largest order in the class Insecta with more than 160,000 species described around the world [15,16]. Despite the extreme taxonomic diversity in Lepidoptera, studies of lepidopteran mitogenomes have been very limited and confined to nine superfamilies: Tortricoidea, Bombycoidea, Noctuoidea, Pyraloidea, Geometroidea, Hesperioidea, Hepialoidea, Yponomeutoidea and Papilionoidea. As of 14 July 2014, 562 insect mitogenomes have been reported or deposited in GenBank. Of these mitogenomes, 151 complete or near complete mitogenomes are from the Lepidoptera, including 15 Pyraloidea species, such as Cnaphalocrocis medinalis [17], Chilo suppressalis [17], Diatraea saccharalis [18], Dichocrocis punctiferalis [19], Ostrinia furnacalis [20], Ostrinia nubilalis [20], Scirpophaga incertulas [21], Elophila interruptalis [22], Glyphodes quadrimaculalis [23], Ephestia kuehniella [24], Maruca testulalis [25], Paracymoriza distinctalis [26], Corcyra cephalonic (unpublished, NC_016866), Paracymoriza prodigalis (unpublished, NC_020094), and Maruca vitrata (unpublished, NC_024099). Characterization of our new mitogenome will help provide further insights into the understanding of phylogenetic evolutionary relationships in the Pyraloidea.
Taxonomically, S. recurvalis is a member of the family Crambidae, superfamily Pyraloidea. However, the number of reported mitogenome sequences in this superfamily is very limited. For the S. recurvalis mitogenome, only a partial of COX1 gene was reported [27,28]. In this study, we sequenced and described the complete mitogenome of S. recurvalis and compared its characteristics with other known lepidopteran mitogenomes. Then we reconstructed phylogenetic relationships within nine lepidopteran superfamilies using Bayesian inference (BI) and Maximum likelihood (ML) methods.
Materials and Methods
DNA sample extraction
Adult individuals of S. recurvalis were collected in Chengdu, China. The samples were preserved in 95% ethanol and stored at -20°C until used for DNA extraction. The whole genomic DNA was isolated from a single sample by applying phenol-chloroform protocol [18,29]. Product and quality of the DNA was assessed by electrophoresis in a 1.5% agarose gel and staining with ethidium bromide.
PCR amplification, cloning, and sequencing
The whole mitogenome of S. recurvalis was amplified in nine overlapping fragments. All primer sequences are shown in Table 1. Primers F1F, F4F, F4R, and F6R were from Cameron and Whiting [7], primers F3R and F5F were from Simon et al. [30], primer F8F was from Bybee et al. [31], and primer F8R was from Skerratt et al. [32]. The other specific primers were designed based on the conserved nucleotide sequences of the mitogenome sequences in homologous lepidopteran species, or the mitogenome fragments that we have previously sequenced.
Table 1. Primer sequences used in this study.
| Region | Primer pair | Primer sequence (5’→3’) | Size (kb) |
|---|---|---|---|
| s-rRNA→tRNAGln (Q) | AF1F | TTTAATAATAGGGTATCTAATCCTAGTTTAT | 0.86 |
| AF1R | GCACAATAGTTTTTGATACTTTTAGATATAGTTTG | ||
| s-rRNA →COX1 | F1F a | TCGTGGATTATCAATTAWTAAACAGATTCC | 3.0 |
| F1R | TATACTTCTGGATGTCCAAAGAAT | ||
| COX1 →tRNALys (K) | F2F | ACTCTACAAATCATAAAGATATTGG | 1.9 |
| F2R | GTTTAAGAGACCAGTACTTG | ||
| tRNALeu(UUR) →COX3 | F3F | TATGTAATGGATTTAAACC | 2.4 |
| F3R b | TCTACA AAATGTCAATATCA | ||
| COX3 →ND4 | F4F a | AGTAACYAAAGGRTTRCGATGAGG | 4.2 |
| F4R a | YCARCCTGAGCGAATTCARGCKGG | ||
| ND4 →CYTB | F5F b | GGAGCYTCAACATGAGCTTT | 2.5 |
| F5R | ATTACACCTCCAAGTTTATTTGGAAT | ||
| CYTB →rrnL | F6F | TATGTTCTTCCTTGAGGACAAATGTC | 2.1 |
| F6R a | AAATTACCTTAGGGATAACAGCG | ||
| ND1 →rrnL | F7F | ATCAAAAGGAGTTCGATTAGTTTC | 1.4 |
| F7R | CACTTGTTTATCAAAAACAT | ||
| rrnL →rrnS | F8F c | CACCGGTTTGAACTCAGATCA | 1.7 |
| F8R d | AAACTAGGATTAGATACCC |
PCR amplification conditions were as follows: an initial denaturation for 2 min at 95°C, followed by 35 cycles of denaturation for 40s at 92°C, annealing for 80 s at 53–57°C (depending on primer combinations), elongation for 1–4 min (depending on putative length of the fragments) at 62°C, and a final extension step of 72°C for 10 min. All PCR amplifications applied Takara LA Taq (Takara Co., Dalian, China) and performed on an Eppendorf Mastercycler and Mastercycler gradient. The PCR products were assessed by electrophoresis in a 1.5% agarose gel and staining with ethidium bromide. All PCR products were sequenced directly from both directions except for fragment F1. Since fragment F1 encompassed the A+T-rich region and some complex structures (e.g., poly-T and microsatellite-like repeat), giving rise to the failures of sequencing. So we utilized short PCR amplification with the primer pair AF1F and AF1R, and the PCR amplification conditions were as the long PCR amplifications. The PCR product of AF1 was purified with the E.Z.N.A. Gel Extraction Kit (Omega, USA) and ligated into the pMD19-T Vector (Takara Co., Dalian, China). Reconstructive plasmids were isolated from the transformed E. coli DH5α competent cells and sequenced with the primers M13-F and M13-R. All fragments were sequenced using ABI BigDye ver. 3.1 dye terminator sequencing technology and run on ABI PRISM 3730×1 capillary sequencers.
Sequence analysis and gene annotation
The whole mitogenome of S. recurvalis was assembled and completed by aligning the overlapping sequences of neighboring fragments using CLUSTAL X [33]. The 13 PCGs, two rRNA genes, and the A+T-rich region were identified by comparison with the homologous lepidopteran mitogenomes sequences (e.g., Cnaphalocrocis medinalis, NC_015985 and Maruca vitrata, NC_024099). The nucleotide sequences of 13 PCGs were translated into amino acid sequences on the basis of the invertebrate mitochondrial genetic code. The A+T content of nucleotide sequences and relative synonymous codon usage (RSCU) were calculated using the MEGA ver. 6.0 [34]. The AT skewness was calculated according to the formula: AT skew = [A-T] / [A+T] [35]. The secondary structure of rrnL and rrnS were drawn by XRNA 1.2.0 b (developed by B. Weiser and available at http://rna.ucsc.edu/rnacenter/xrna/xrna.html). The tRNA genes and secondary structures were identified using the tRNAscan-SE ver. 1.21 (http://selab.janelia.org/tRNAscan-SE/) [36]. The secondary structures of two tRNASer genes, which we were unable to predict by using the tRNAscan-SE, were analyzed by comparison with the nucleotide sequences of the tRNA genes in the Crambidae (e.g., Cnaphalocrocis medinalis, NC_015985 and Dichocrocis punctiferalis, JX448619). The tRNA genes secondary structures were drawn using DNA-SIS ver. 2.5 (Hitachi Engineering, Tokyo, Japan).
Phylogenetic analysis
Phylogenetic analysis was performed based on the 13 PCGs of the complete mitogenome of S. recurvalis and 54 other lepidopteran mitogenomes downloaded from GenBank (Table 2). The mitogenomes of Anopheles gambiae (NC_002084) [37] and Drosophila melanogaster (NC_001709) [38] were used as outgroups. Nucleotide sequences of the 13 PCGs from the mitogenomes of the 54 lepidopteran, two outgroup species, and S. recurvalis were translated into amino acid sequences. They were aligned with CLUSTAL X using default settings, and then back-translated into nucleotide alignments, then the unaligned and unmatched regions were removed, and the remaining nucleotide alignments were concatenated together. Nucleotide sequences of two rRNA genes from the mitogenomes of the 57 species were aligned with CLUSTAL X using default settings, the unaligned and unmatched regions were removed, and then the concatenated nucleotide sequences were combined to the end of the aligned nucleotide of 13 PCGs respectively. The concatenated nucleotide alignments of 13 PCGs and 13 PCGs+2 rRNAs yielded a nucleotide matrix of 10,719 bp and 12072 bp in length, respectively, which were used for phylogenetic analysis with BI and ML methods.
Table 2. Comparisons characteristics of the Lepidoptera and two outgroup species mitogenomes.
| Superfamily | Species | Whole mitogenome | PCGs | lrRNA | srRNA | A+T-rich region | Acc.number | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Size (bp) | A+T (%) | A+T skew | Codon no. | A+T (%) | Size (bp) | A+T (%) | Size (bp) | A+T (%) | Size (bp) | A+T (%) | |||
| Bombycoidea | Actias selene | 15236 | 78.9 | -0.023 | 3725 | 77.3 | 1364 | 83.6 | 762 | 84.0 | 339 | 87.9 | NC_018133 |
| Antheraea pernyi | 15566 | 80.2 | -0.021 | 3729 | 78.5 | 1369 | 83.8 | 775 | 81.1 | 552 | 90.4 | NC_004622 | |
| Bombyx mandarina | 15928 | 81.7 | 0.055 | 3718 | 79.6 | 1377 | 84.8 | 783 | 86.0 | 747 | 95.2 | NC_003395 | |
| Bombyx mori | 15656 | 81.4 | 0.059 | 3716 | 79.5 | 1378 | 83.4 | 783 | 85.6 | 494 | 95.5 | AB070264 | |
| Eriogyna pyretorum | 15327 | 80.8 | -0.031 | 3727 | 79.3 | 1338 | 84.6 | 778 | 84.4 | 358 | 92.2 | NC_012727 | |
| Samia cynthia ricini | 15384 | 79.8 | -0.006 | 3728 | 78.2 | 1358 | 84.0 | 779 | 83.8 | 361 | 90.9 | NC_017869 | |
| Sphinx morio | 15299 | 81.2 | 0.001 | 3716 | 79.8 | 1379 | 84.6 | 773 | 85.2 | 316 | 92.7 | NC_020780 | |
| Geometroidea | Biston panterinaria | 15516 | 79.6 | 0.077 | 3726 | 77.3 | 1474 | 85.6 | 787 | 84.8 | 349 | 93.1 | NC_020004 |
| Phthonandria atrilineata | 15499 | 81.5 | 0.007 | 3722 | 79.0 | 1400 | 85.7 | 803 | 86.3 | 457 | 98.3 | NC_010522 | |
| Noctuoidea | Gynaephora menyuanensis | 15770 | 81.5 | 0.003 | 3728 | 79.7 | 1420 | 84.2 | 891 | 85.5 | 449 | 93.3 | NC_020342 |
| Helicoverpa armigera | 15347 | 81.0 | 0.001 | 3721 | 79.4 | 1395 | 84.7 | 794 | 85.9 | 328 | 95.1 | NC_014668 | |
| Hyphantria cunea | 15481 | 80.4 | 0.009 | 3721 | 77.5 | 1426 | 85.0 | 808 | 85.5 | 357 | 95.0 | NC_014058 | |
| Lymantria dispar | 15569 | 79.9 | 0.016 | 3732 | 77.8 | 1351 | 84.2 | 799 | 85.2 | 435 | 96.1 | NC_012893 | |
| Ctenoplusia agnata | 15261 | 81.1 | 0.012 | 3732 | 79.8 | 1328 | 84.3 | 784 | 85.5 | 334 | 93.4 | KC414791 | |
| Papilionoidea | Apatura metis | 15236 | 80.4 | -0.012 | 3702 | 78.9 | 1333 | 84.5 | 779 | 84.8 | 394 | 92.1 | NC_015537 |
| Aporia crataegi | 15140 | 81.2 | -0.024 | 3707 | 79.9 | 1326 | 85.4 | 779 | 85.5 | 354 | 95.2 | NC_018346 | |
| Argynnis hyperbius | 15156 | 80.8 | -0.025 | 3715 | 79.4 | 1330 | 84.4 | 778 | 85.2 | 349 | 95.4 | NC_015988 | |
| Artogeia melete | 15140 | 79.8 | 0.012 | 3711 | 81.4 | 1319 | 83.5 | 777 | 85.5 | 351 | 89.2 | EU597124 | |
| Athyma sulpitia | 15268 | 82.0 | -0.047 | 3730 | 80.6 | 1319 | 84.7 | 779 | 85.8 | 349 | 94.6 | JQ347260 | |
| Acraea issoria | 15245 | 79.8 | -0.023 | 3713 | 78.0 | 1331 | 83.8 | 788 | 83.8 | 430 | 96.0 | NC_013604 | |
| Calinaga davidis | 15267 | 80.4 | -0.044 | 3734 | 78.9 | 1337 | 83.8 | 773 | 85.4 | 389 | 92.0 | NC_015480 | |
| Hebomoia glaucippe | 15701 | 79.9 | -0.036 | 3718 | 78.0 | 1339 | 84.6 | 777 | 85.2 | 899 | 92.2 | NC_021123 | |
| Heliconius melpomene melpomene | 15325 | 81.7 | -0.037 | 3695 | 80.2 | 1364 | 85.6 | 779 | 85.1 | 268 | 95.9 | HE579083 | |
| Kallima inachus | 15183 | 80.3 | -0.013 | 3719 | 79.2 | 1335 | 82.8 | 774 | 85.1 | 376 | 92.0 | NC_016196 | |
| Melanitis leda | 15122 | 79.8 | -0.037 | 3719 | 82.4 | 1332 | 84.0 | 774 | 85.0 | 314 | 89.5 | JF905446 | |
| Papilio maackii | 15357 | 80.7 | -0.014 | 3716 | 79.2 | 1326 | 83.7 | 774 | 85.4 | 514 | 92.8 | KC433408 | |
| Papilio machaon | 15185 | 80.3 | -0.031 | 3716 | 79.0 | 1319 | 83.6 | 773 | 84.2 | 362 | 92.5 | NC_018047 | |
| Pieris rapae | 15157 | 79.7 | 0.013 | 3706 | 78.2 | 1320 | 84.0 | 764 | 85.0 | 393 | 91.6 | NC_015895 | |
| Protantigius superans | 15248 | 81.7 | -0.036 | 3709 | 80.3 | 1331 | 85.1 | 739 | 85.5 | 361 | 93.6 | NC_016016 | |
| Spindasis takanonis | 15349 | 82.4 | 0.004 | 3714 | 81.0 | 1333 | 85.6 | 777 | 84.7 | 371 | 94.6 | NC_016018 | |
| Coreana raphaelis | 15314 | 82.7 | -0.047 | 3707 | 81.5 | 1330 | 85.3 | 777 | 85.9 | 375 | 94.1 | NC_007976 | |
| Pyraloidea | Cnaphalocrocis medinalis | 15388 | 81.9 | -0.015 | 3724 | 80.5 | 1389 | 84.9 | 781 | 86.2 | 339 | 95.9 | NC_015985 |
| Chilo suppressalis | 15395 | 80.7 | 0.008 | 3729 | 78.8 | 1383 | 84.2 | 788 | 86.2 | 348 | 95.4 | NC_015612 | |
| Corcyra cephalonica | 15273 | 80.4 | -0.036 | 3713 | 78.9 | 1355 | 83.0 | 778 | 85.9 | 351 | 96.6 | NC_016866 | |
| Diatraea saccharalis | 15490 | 80.0 | 0.021 | 3721 | 77.8 | 1412 | 84.8 | 781 | 85.5 | 335 | 94.9 | NC_013274 | |
| Dichocrocis punctiferalis | 15355 | 80.6 | -0.025 | 3716 | 78.8 | 1360 | 84.5 | 793 | 86.0 | 338 | 96.5 | JX448619 | |
| ※ Ostrinia furnacalis | 14535 | 80.4 | 0.032 | 3714 | 79.4 | 1339 | 85.0 | 435 | 82.8 | - | - | NC_003368 | |
| ※ Ostrinia nubilalis | 14535 | 80.2 | 0.032 | 3714 | 79.1 | 1339 | 84.9 | 434 | 82.0 | - | - | NC_003367 | |
| Paracymoriza prodigalis | 15326 | 81.5 | 0.002 | 3714 | 80.0 | 1389 | 85.5 | 781 | 86.0 | 343 | 95.3 | NC_020094 | |
| Scirpophaga incertulas | 15223 | 77.1 | 0.031 | 3718 | 74.8 | 1314 | 82.0 | 768 | 84.1 | 403 | 92.8 | NC_021413 | |
| Elophila interruptalis | 15351 | 80.3 | -0.015 | 3712 | 78.6 | 1367 | 84.1 | 786 | 85.5 | 339 | 93.5 | NC_021756 | |
| Glyphodes quadrimaculalis | 15255 | 80.8 | -0.007 | 3714 | 79.2 | 1350 | 84.9 | 779 | 85.5 | 327 | 94.5 | NC_022699 | |
| Ephestia kuehniella | 15294 | 79.8 | -0.048 | 3714 | 78.1 | 1328 | 84.2 | 773 | 84.9 | 322 | 93.2 | NC_022476 | |
| Maruca vitrata | 15385 | 80.7 | -0.002 | 3772 | 79.3 | 1304 | 84.4 | 765 | 85.2 | 341 | 93.0 | NC_024099 | |
| Spoladea recurvalis | 15273 | 80.9 | -0.023 | 3719 | 79.3 | 1384 | 85.3 | 781 | 86.0 | 329 | 93.9 | KJ739310 | |
| Hesperioidea | Ctenoptilum vasava | 15468 | 80.5 | -0.029 | 3706 | 78.9 | 1343 | 84.1 | 774 | 85.7 | 429 | 88.1 | NC_016704 |
| Erynnis montanus | 15530 | 81.8 | -0.001 | 3717 | 80.0 | 1408 | 85.1 | 780 | 85.8 | 389 | 95.1 | KC659955 | |
| Tortricoidea | Adoxophyes honmai | 15679 | 80.4 | -0.001 | 3733 | 78.4 | 1387 | 83.6 | 779 | 85.4 | 489 | 94.3 | NC_008141 |
| Adoxophyes orana | 15343 | 80.0 | -0.001 | 3723 | 78.3 | 1430 | 84.8 | 804 | 85.3 | 331 | 92.8 | JX872403 | |
| Cydia pomonella | 15253 | 80.1 | -0.004 | 3723 | 78.5 | 1361 | 83.8 | 813 | 85.5 | 327 | 96.0 | NC_020003 | |
| Grapholita molesta | 15716 | 80.9 | -0.004 | 3736 | 78.8 | 1377 | 84.8 | 772 | 85.4 | 770 | 96.0 | NC_014806 | |
| Spilonota lechriaspis | 15368 | 81.2 | -0.018 | 3737 | 80.6 | 1382 | 85.2 | 778 | 86.3 | 441 | 92.7 | NC_014294 | |
| Hepialoidea | Ahamus yunnanensis | 15816 | 82.3 | -0.006 | 3717 | 80.6 | 1329 | 86.0 | 770 | 86.1 | 978 | 89.4 | HM744695 |
| Thitarodes renzhiensis | 16173 | 81.3 | -0.001 | 3717 | 79.0 | 1335 | 85.4 | 779 | 85.4 | 1367 | 90.5 | HM744694 | |
| Yponomeutoidea | Plutella xylostella | 16079 | 81.4 | 0.005 | 3719 | 79.3 | 1379 | 85.0 | 783 | 86.1 | 981 | 91.6 | JF911819 |
| Drosophiloidea | Drosophila melanogaster | 19517 | 82.2 | 0.017 | 3716 | 77.2 | 1325 | 83.0 | 786 | 80.2 | 4601 | 95.6 | NC_001709 |
| Culicoidea | Anopheles gambiae | 15363 | 77.6 | 0.032 | 3733 | 75.9 | 1325 | 82.5 | 800 | 79.6 | 519 | 94.2 | NC_002084 |
※, Incomplete mitogenomes lack the partial rrnS gene, the entire A+T-rich region and partial tRNAMet gene. Termination codons were excluded in 13 PCGs.
By using Akaike Information Criterion (AIC) [39], the substitution model selection was calculated using the program Modeltest ver. 3.7 [40]. The TVM+I+G model was chosen as the best-fitting model for BI analysis with the dataset of 13 PCGs, and the second one was GTR+I+G. The GTR+I+G model was chosen as the best-fitting model for BI analysis with the dataset of 13 PCGs+2 rRNAs. The BI analysis was performed using MrBayes ver. 3.1 [41] under the following conditions: 10,000,000 generations, four independent chains (one cold chain and three hot chains) with tree sampling every 100 generations and a burn-in of 2500 trees. The confidence values of the BI tree were expressed as the Bayesian posterior probabilities. The posterior probabilities more than 0.9 were considered strongly-supported [42].
The ML analysis was performed using the PHYML online web server (http://www.atgc-montpellier.fr/phyml/) [43]. The setting conditions in the substitution model block were as follows: (a) Substitution model–“GTR”, (b) Equilibrium frequencies–“empirical”, (c) Transition/ transversion ratio–“fixed”, Proportion of invariable sites–“estimated”, Number of substitution rate categories–“4”, and (e) Gamma shape parameter–“estimated”. In the tree searching block, the setting conditions were (f) Starting tree(s)–“BIONJ”, (g) Type of tree improvement–“SPR”, (h) Number of random starting tree–“no”, (i) Optimise topology–“yes”, and (j) Optimise branch lengths–“yes”. Finally, in the branch support block, the settings conditions were (k) Fast likelihood-based method–“no”, (l) Perform bootstrap–“yes”, and (m) Perform bootstrap–“1000”. In ML analysis, nodes with bootstrap proportions over 70% were interpreted as well-supported [44]. The phylogenetic trees were viewed and edited using Figtree ver. 1.4 [45].
Results and Discussion
Genome structure and organization
The mitogenome of S. recurvalis was found to be a circular molecule with 15,273 bp in length, which is well within the range of other lepidopteran mitogenomes, with lengths ranging from 15,122 bp in M. leda to 16,173 bp in T. renzhiensis (Table 2). The mitogenome of S. recurvalis contained the typical set of 37 typical mitochondrial genes (13 PCGs, 22 tRNA genes, and two rRNA genes), and a major non-coding region known as the A+T-rich region, as has been found in other lepidopteran mitogenomes. Twenty-three genes were coded on the majority strand (J-strand) and the rest were coded on the minority strand (N-strand) (Table 3 and Fig 1). This mitogenome was submitted to GenBank under the accession number KJ739310.
Table 3. Annotation of the Spoladea recurvalis mitogenome.
| Gene | strand | Location | Size | Inc | Anticodon | Start codon | Stop codon |
|---|---|---|---|---|---|---|---|
| tRNAMet (M) | J | 1–69 | 69 | CAT | |||
| tRNAIle (I) | J | 70–134 | 65 | -3 | GAT | ||
| tRNAGln (Q) | N | 132–200 | 69 | 48 | TTG | ||
| ND2 | J | 249–1262 | 1014 | 13 | ATT | TAA | |
| tRNATrp (W) | J | 1276–1343 | 68 | -8 | TCA | ||
| tRNACys (C) | N | 1336–1404 | 69 | 7 | GCA | ||
| tRNATyr (Y) | N | 1412–1477 | 66 | 3 | GTA | ||
| COX1 | J | 1481–3011 | 1531 | CGA | T- | ||
| tRNALeu(UUR) | J | 3012–3078 | 67 | TAA | |||
| COX2 | J | 3079–3763 | 685 | -3 | ATG | T- | |
| tRNALys (K) | J | 3761–3831 | 71 | 6 | CTT | ||
| tRNAAsp (D) | J | 3838–3904 | 67 | GTC | |||
| ATP8 | J | 3905–4066 | 162 | -7 | ATT | TAA | |
| ATP6 | J | 4060–4734 | 675 | 3 | ATG | TAA | |
| COX3 | J | 4738–5526 | 789 | 2 | ATG | TAA | |
| tRNAGly (G) | J | 5529–5593 | 65 | TCC | |||
| ND3 | J | 5594–5947 | 354 | 6 | ATT | TAA | |
| tRNAAla (A) | J | 5954–6018 | 65 | 1 | TGC | ||
| tRNAArg (R) | J | 6018–6081 | 64 | TCG | |||
| tRNAAsn (N) | J | 6082–6150 | 69 | 11 | GTT | ||
| tRNASer(AGY) | J | 6162–6229 | 68 | 10 | GCT | ||
| tRNAGlu (E) | J | 6240–6305 | 66 | -2 | TTC | ||
| tRNAPhe (F) | N | 6304–6371 | 68 | GAA | |||
| ND5 | N | 6372–8094 | 1723 | 21 | ATT | T- | |
| tRNAHis (H) | N | 8116–8181 | 66 | GTG | |||
| ND4 | N | 8182–9521 | 1337 | -1 | ATG | TA- | |
| ND4L | N | 9521–9814 | 294 | 2 | ATG | TAA | |
| tRNAThr (T) | J | 9817–9883 | 67 | TGT | |||
| tRNAPro (P) | N | 9884–9949 | 66 | 2 | TGG | ||
| ND6 | J | 9952–10485 | 534 | 6 | ATC | TAA | |
| CYTB | J | 10492–11640 | 1149 | -2 | ATG | TAA | |
| tRNASer(UCN) | J | 11639–11704 | 66 | 15 | TGA | ||
| ND1 | N | 11720–12658 | 939 | 1 | ATG | TAA | |
| tRNALeu(CUN) | N | 12660–12728 | 69 | -16 | TAG | ||
| rrnL | N | 12713–14096 | 1384 | ||||
| tRNAVal (V) | N | 14097–14163 | 67 | TAC | |||
| rrnS | N | 14164–14944 | 781 | ||||
| A+T-rich Region | 14945–15273 | 329 |
Inc, intergenic nucleotides, negative values refer to overlapping nucleotides.
Fig 1. Map of the mitochondrial genome of Spoladea recurvalis.

Genes coded on the J strand (clockwise orientation) are blue- or green-colored, while the genes coded on the N strand (anti-clockwise orientation) are pink- or orange-colored. COX1, COX2 and COX3 refer to the cytochrome c oxidase subunits; CYTB refers to cytochrome B; ATP6 and ATP8 refer to ATP synthase subunits 6 and 8 genes; and ND1–ND6 and ND4L refer to the NADH dehydrogenase subunit 1–6 and 4L genes. tRNA genes are denoted as one-letter symbols according to the IUPAC-IUB single-letter amino acid codes: L1, L2, S1 and S2 refer to tRNALeu(CUN), tRNALeu(UUR), tRNASer(AGY), and tRNASer(UCN), respectively. CR refering to the A+T rich region and is brown-colored.
The gene order and orientation of the S. recurvalis mitogenome were identical to that of other reported ditrysian lepidopteran mitogenomes, but differed from non-ditrysian groups with the ancestral arrangement of tRNAIle–tRNAGln–tRNAMet, such as the species Thitarodes renzhiensis and Ahamus yunnanensis in Hepialoidea [46]. All the ditrysian lineages of lepidopteran mitogenomes are characterized by the gene order tRNAMet–tRNAIle–tRNAGln, revealing a translocation of tRNAMet to a position 5'-upstream of tRNAIle, which differs from the hypothesized ancestral gene order of insects [47]. This suggests that the mitogenome arrangement of the lepidopteran insects may have evolved independently after splitting from a stem lineage of insects [48].
The nucleotide composition (A 39.5%, G 7.8%, T 41.4% and C 11.3%) of the S. recurvalis mitogenome indicated a high A+T content of 80.9%, which is well within the range of lepidopteran mitogenomes, which vary from 77.0% in S. incertulas to 82.7% in C. raphaelis, similar to that of C. medinalis (80.9%). The mitogenome A+T content was 79.3% in 13 PCGs, 85.3% in rrnL genes, 86.0% in rrnS genes, and 93.9% in the A+T-rich region, respectively. These values were consistent with the high values found in other lepidopteran mitogenomes (Table 2). The AT skewness of the mitogenome was -0.023, indicating the occurrence of more T nucleotides than A nucleotides, as has been found in other lepidopteran mitogenomes, with the values ranging from -0.048 in E. kuehniella to 0.059 in B. mori (Table 2).
Protein-coding genes
The PCGs regions of the S. recurvalis mitogenomes were consistent with those of other lepidopteran mitogenomes. Nine of the 13 PCGs (ND2, COX1, COX2, ATP8, ATP6, COX3, ND3, ND6, and CYTB) were coded on the majority strand (J-strand), and the remaining four PCGs (ND5, ND4, ND4L, and ND1) were coded on the minority strand (N-strand) (Table 3 and Fig 1). All PCGs initiated with a canonical start codon ATN with the exception of COX1, which may use an arginine CGA as the start codon. Specifically, seven PCGs (COX2, ATP6, COX3, ND4, ND4L, CYTB, and ND1) started with ATG, four PCGs (ND2, ATP8, ND3 and ND5) started with ATT, and one PCG (ND6) started with ATC. As for stop codon, nine PCGs (ND2, ATP8, ATP6, COX3, ND3, ND4L, ND6, CYTB and ND1) terminated with the standard stop codon TAA, whereas COX1, COX2 and ND5 used single T nucleotide, and ND4 used TA nucleotides as an incomplete stop codon. The non-canonical termination codons will be corrected by post-transcriptional modifications, such as polyadenylation, which occur during the mRNA maturation process [49,50]. The partial stop codons observed in most lepidopteran species minimize the intergenic spacers and gene overlaps may be one strategy for the selection of a stop codon [51].
The start codons for the COX1 gene of the lepidopteran insects have been the source of controversy in current studies. In other insects groups, some canonical codons were proposed as the COX1 start codon, such as TTA [52], TCG [53], TTG [54], and ACG [55]. In addition, some tetranucleotides, such as TTAG [48,56], ATAA [57–60], and some hexanucleotides, such as ATTTAA [37,61,62], TATCTA [63], and TATTAG [20,64,65], located immediately upstream of the CGA, have also been proposed as the start codons for the COX1 gene. However, a recent study, based on the transcript information of Anopheles funestus (Diptera), revealed that the translation initiation codon for the COX1 gene was TCG (Serine), rather than the atypical and longer codons which have been proposed for several other insects [66]. Data from the transcript map, with expressed sequence tags (ESTs) for the mitochondrial genome annotation of the legume pod borer Maruca vitrata (Lepidoptera: Crambidae), showed that the COX1 gene started with the CGA codon for arginine [67]. This start codon has been found previously well conserved in other lepidopteran species [68]; therefore, we tentatively designated CGA as the start codon of COX1 gene.
Codon usage
The relative synonymous codon usage (RSCU) value of the S. recurvalis mitogenome is summarized in Table 4. Excluding all initiation and termination codons, the 13 PCGs were 11,118 bp in total length, encoding 3,706 amino acid residues. The codons CCG, UGG, CGC, CGG, AGC, GGC, and AGG were not presented in these PCGs. The codons UUA (12.6%), AUU (12.0%), UUU (9.2%), AUA (7.0%), and AAU (6.7%) were the five most frequently used codons in the S. recurvalis mitogenome, accounting for 47.5%. These codons were all composed of A or U nucleotide, indicating the high biased usage of A and T nucleotides in the S. recurvalis PCGs. Likewise, the most frequent amino acids in the S. recurvalis mitochondrial proteins were Leu2 (13.0%), Ile (12.6%), Phe (10.1%), Met (7.0%), and Asn (6.6%), accounting for 49.3%. The least amino acid was Cys (0.8%). Codon usage of PCGs showed a significant bias of high A + T content, which played a major role in the A+T bias of the entire mitogenome.
Table 4. Codon usage in the PCGs of Spoladea recurvalis mitogenome.
| Codon | Count | % | RSCU | Codon | Count | % | RSCU | Codon | Count | % | RSCU | Codon | Count | % | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU(F) | 341 | 9.2 | 1.82 | UCU(S) | 109 | 2.9 | 2.63 | UAU(Y) | 181 | 4.9 | 1.88 | UGU(C) | 26 | 0.7 | 1.73 |
| UUC(F) | 33 | 0.9 | 0.18 | UCC(S) | 11 | 0.3 | 0.27 | UAC(Y) | 12 | 0.3 | 0.12 | UGC(C) | 4 | 0.1 | 0.27 |
| UUA(L) | 466 | 12.6 | 5.29 | UCA(S) | 87 | 2.3 | 2.10 | UAA(*) | - | - | - | UGA(W) | 95 | 2.6 | 2.00 |
| UUG(L) | 13 | 0.4 | 0.15 | UCG(S) | 5 | 0.1 | 0.12 | UAG(*) | - | - | - | UGG(W) | 0 | 0.0 | 0.00 |
| CUU(L) | 29 | 0.8 | 0.33 | CCU(P) | 66 | 1.8 | 2.10 | CAU(H) | 59 | 1.6 | 1.74 | CGU(R) | 17 | 0.5 | 1.31 |
| CUC(L) | 4 | 0.1 | 0.05 | CCC(P) | 14 | 0.4 | 0.44 | CAC(H) | 9 | 0.2 | 0.26 | CGC(R) | 0 | 0.0 | 0.00 |
| CUA(L) | 14 | 0.4 | 0.16 | CCA(P) | 46 | 1.2 | 1.46 | CAA(Q) | 58 | 1.6 | 1.87 | CGA(R) | 35 | 0.9 | 2.69 |
| CUG(L) | 3 | 0.1 | 0.03 | CCG(P) | 0 | 0.0 | 0.00 | CAG(Q) | 4 | 0.1 | 0.13 | CGG(R) | 0 | 0.0 | 0.00 |
| AUU(I) | 445 | 12.0 | 1.91 | ACU(T) | 81 | 2.2 | 2.13 | AAU(N) | 231 | 6.2 | 1.88 | AGU(S) | 33 | 0.9 | 0.80 |
| AUC(I) | 21 | 0.6 | 0.09 | ACC(T) | 5 | 0.1 | 0.13 | AAC(N) | 15 | 0.4 | 0.12 | AGC(S) | 0 | 0.0 | 0.00 |
| AUA(M) | 258 | 6.7 | 1.82 | ACA(T) | 65 | 1.8 | 1.71 | AAA(K) | 100 | 2.7 | 1.90 | AGA(S) | 87 | 2.3 | 2.10 |
| AUG(M) | 12 | 0.3 | 0.18 | ACG(T) | 1 | 0.0 | 0.03 | AAG(K) | 5 | 0.1 | 0.10 | AGG(S) | 0 | 0.0 | 0.00 |
| GUU(V) | 78 | 2.1 | 2.28 | GCU(A) | 81 | 2.2 | 2.45 | GAU(D) | 56 | 1.5 | 1.81 | GGU(G) | 50 | 1.3 | 1.02 |
| GUC(V) | 3 | 0.1 | 0.09 | GCC(A) | 4 | 0.1 | 0.12 | GAC(D) | 6 | 0.2 | 0.19 | GGC(G) | 0 | 0.0 | 0.00 |
| GUA(V) | 54 | 1.5 | 1.58 | GCA(A) | 43 | 1.2 | 1.30 | GAA(E) | 72 | 1.9 | 1.85 | GGA(G) | 127 | 3.4 | 2.58 |
| GUG(V) | 2 | 0.1 | 0.06 | GCG(A) | 4 | 0.1 | 0.12 | GAG(E) | 6 | 0.2 | 0.15 | GGG(G) | 20 | 0.5 | 0.41 |
A total of 3706 codons were analyzed excluding all initiation termination codons.
RSCU, relative sunonymous codon usage.
Transfer and ribosomal RNA genes
The mitogenome of S. recurvalis contained the typical set of 22 tRNAs genes as have been found in most lepidopteran mitogenomes. The tRNAs genes were scattered throughout the circular molecule and range from 64 bp (tRNAArg) to 71 bp (tRNALys) in size (Table 3). Fourteen tRNA genes were coded on the J-strand and the other eight on the N-strand, as with other lepidopteran mitogenomes (Table 3 and Fig 1). The putative secondary structure of the S. recurvalis tRNAs are shown in Fig 2. All tRNA genes were folded into the typical cloverleaf secondary structures, except for the tRNASer (AGY) gene, in which the dihydrouridine (DHU) arm was simplified as a loop, which has been observed in several other metazoan species, including insects [2]. The anticodons of the tRNA genes were identical to those most reported in insect mitogenomes.
Fig 2. Putative secondary structures for the 22 tRNA genes of Spoladea recurvalis mitogenome.
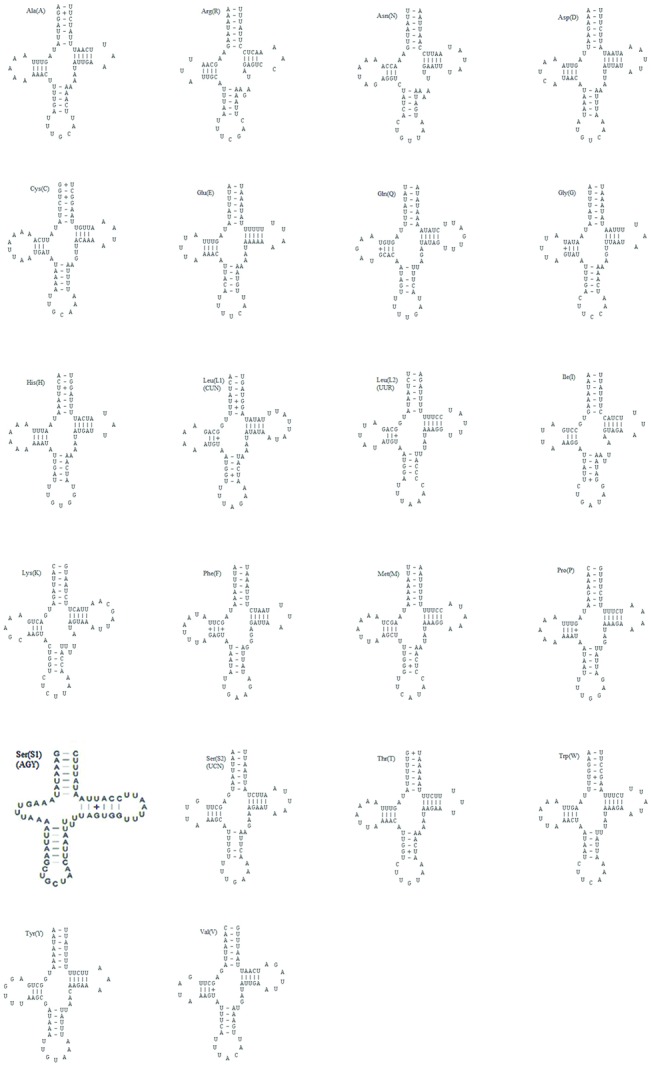
A total of 26 pairs of mismatched base pairs were found in 16 tRNA genes, including ten pairs in the amino acid acceptor stems, eight pairs in the DHU stems, seven pairs in the anticodon stems, and one pair in the pseudouridine (TΨC) stems. The 21 U–G mismatched bases may have formed weak bonds, while the other five U–U mismatches have not. The mismatched base pairs in tRNAs are modified via RNA-editing mechanisms that are well known in arthropod mitogenomes [69].
As seen in other insect mitogenomes, two rRNA genes (rrnL and rrnS) were present in the S. recurvalis mitogenome. The rrnL gene was located between the tRNALeu(CUN) and tRNAVal, and the rrnS gene was located between the tRNAVal and the A + T-rich region. The lengths of the rrnL gene and rrnS gene were 1384 bp and 781 bp, respectively, which are well within the lengths reported for these genes for other lepidopteran mitogenomes. The A + T contents of the rrnL gene and rrnS gene of S. recurvalis mitogenome were 85.3% and 86.0%, respectively. These values are also well within the range of other lepidopteran mitogenomes. The lengths of the rrnL genes varied from 1304 bp to 1474 bp and the A+T contents varied from 82.0% to 85.6%. The lengths of the rrnS genes varied from 739 bp to 891 bp and the A+T contents varied from 81.1% to 86.3% (Table 2).
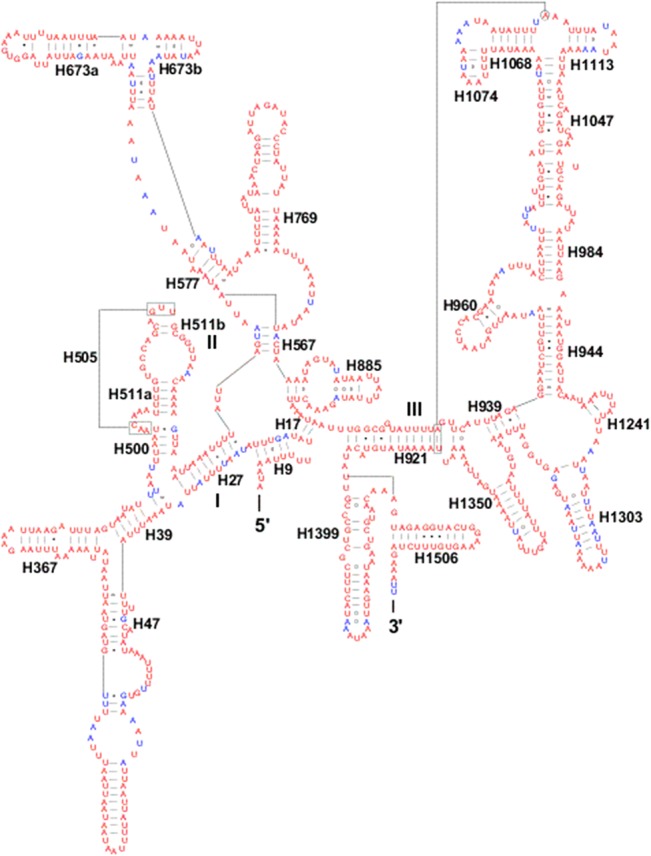
Both the secondary structure of rrnL and rrnS broadly conformed to the secondary structure models proposed for these genes from other insects [7,17,70–72]. In the mitogenome of S. recurvalis, six domains with 49 helices were present in the rrnL secondary structure (Fig 3A and 3B), as in M. sexta [7], C. medinalis [17], C. suppressalis [17], A. emma [70], L. malifoliella [71] and P. xylostella [72]. An internal 31bp large loop was located among the H991, H1057, and H1087, which is similar to that of C. medinalis, C. suppressalis, A. emma, L. malifoliella and P. xylostella but different from that of M. sexta. As in the rrnL secondary structure of M. sexta, C. medinalis, C. suppressalis, A. emma, and L. malifoliella, but differs from that of P. xylostella which contains a (TA)8 microsatellite-like repeat inserted into the loop region of H2347. This region is highly variable within Lepidoptera and a consistent secondary structure for it has not been found within the available lepidopteran mitogenomes [7].
Fig 3. Predicted rrnL secondary structure in the S. recurvalis mitogenome.

Tertiary interactions and base triples are connected by continuous lines. Base-pairing is indicated as follows: Watson-Crick pairs by lines, wobble GU pairs by dots and other non-canonical pairs by circles. A represents the 5’ half of rrnL; B represents the 3’ half of rrnL.
Three domains with 29 helices were present in the rrnS secondary structure of S. recurvalis mitogenome (Fig 4). A small loop was located in the H47 region of the rrnS gene compared to that of M. sexta and P. xylostella. This region has been found to be variable within lepidopteran species [7], which has been used to predict the phylogenetic relationships when it is combined with H39 and H367 [73].
Fig 4. Predicted rrnS secondary structure in the S. recurvalis mitogenome.
Tertiary interactions and base triples are connected by continuous lines. Base-pairing is indicated as follows: Watson-Crick pairs by lines, wobble GU pairs by dots and other non-canonical pairs by circles.
The H673, H1047, H1068, and H1074 present in the rrnS secondary structure of the S. recurvalis mitogenome were similar in length and secondary structures to those of C. medinalis and C. suppressalis, but different from those of M. sexta, indicating they are also variable regions in the secondary structure of rrnS gene within lepidopteran species [7]. The secondary structures of two rRNAs were predicted mainly based on sequence comparison and mathematical methods. The region in rrnS contains the H1047, H1068, and H1074 may yield several possible secondary structures, but it is not ascertained which one may be utilized among these structures [7].
Non-coding and overlapping region
The non-coding region of the S. recurvalis mitogenome was 157 bp in total, which consisted of 17 non-coding regions, ranging from 1 to 48 bp and including 5 major non-coding regions of more than 10 bp (Table 3). The longest intergenic spacer (Spacer1, 48bp) was located between the tRNAGln and ND2, with an extremely high richness in A + T nucleotides (95.8%). This spacer has been a feature common reported in the other lepidopteran mitogenomes, which has been sequenced to date, but which has not been found in non-lepidopteran insect species [7]. Spacer 2 (13 bp) was located between the ND2 and tRNATrp, which was only smaller than the 18 bp space of Hyphantria cunea in the similar location in lepidopteran mitogenomes [74]. Spacer 3 (11 bp) was located between the tRNAAsn and tRNASer (AGY), and Spacer 4 (21 bp) was located between the ND5 and tRNAHis. Spacer 5 (15 bp), which was located between the tRNASer(UCN) and ND1, contained the motif ‘‘ATACTAA”, which represents a conserved feature across lepidopteran insects [7]. The motif has been proposed as a possible mitochondrial transcription termination peptide-binding site (mtTERM protein) [4].
Eight overlapping sequences of the S. recurvalis mitogenome were found in eight different locations, ranging from 1 to 16 bp with a total of 42 bp. The longest overlapping sequence (16 bp) was located between the tRNALeu(CUN) and rrnL, and the second longest overlapping sequence (8 bp) was located between the tRNATrp and tRNACys. The third longest overlapping sequence was located between the ATP8 and ATP6 with a seven-nucleotide overlapping sequence (ATGATAA), which has been a common feature reported for many other lepidopteran mitogenomes, and also reported for many animal mtDNAs [1]. A similar-sized overlapping sequence in the same location has been reported in other lepidopteran mitogenomes [70]. The remaining overlapping sequences were all less than 3 bp.
A + T-rich region
The A + T-rich region of the S. recurvalis mitogenome was located between the rrnS and tRNAMet with a length of 329 bp and with A+T nucleotides accounting for 93.9%, which are within the range of other lepidopteran mitogenomes, which vary from 88.1% in C. vasava to 98.3% in P. atrilineata (Table 2). The A + T-rich region has also been found to contain the origin sites for transcription and replication [4].
The A+T-rich of the S. recurvalis mitogenome region was comprised of non-repetitive sequences and had some features in common with other lepidopteran mitogenomes (Fig 5). In Bombyx, the ON (the origin of minority or light strand replication) has the motif ATAGA preceded by an 18 bp poly-T stretch and is located 21 bp upstream from the rrnS gene [75]. Though the length of the poly-T stretch varies between species, this motif ATAGA is conserved within Lepidoptera [7]. In S. recurvalis, the motif ATAG was similarly located 19 bp downstream from the rrnS gene and followed by a 14 bp poly-T stretch. The poly-T stretch has been postulated to be a transcription control and/or the initiation site of replication [76]. A (AT)11 microsatellite-like repeat was preceded by the motif ATTTA located in the 3' end of the A+T-rich region. A poly-A element was present upstream of tRNAMet as has been found in most lepidopteran mitogenomes.
Fig 5. The structure of the A+T-rich region of the Spoladea recurvalis mitogenome.

Phylogenetic relationships
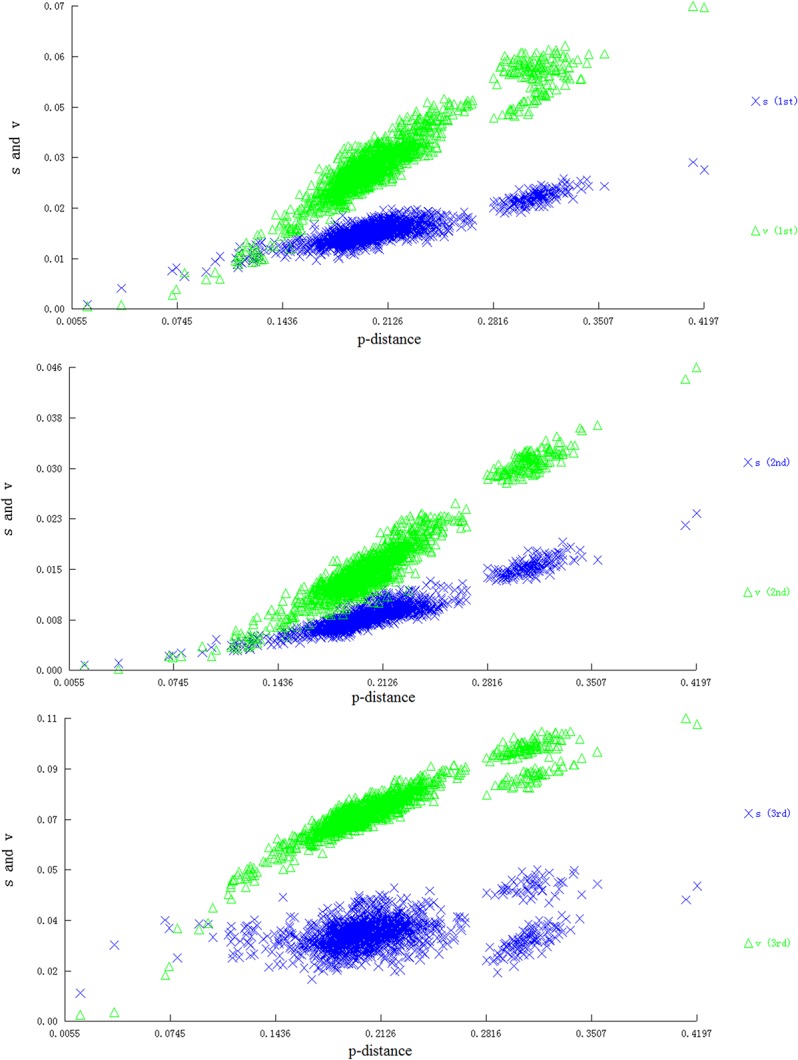
The estimated Transition/Transversion bias (R) of the first, second, and third codon positions of the 13 PCGs were 0.9, 0.6, and 3.7, respectively. The substitution rates were estimated under the Kimura 2-parameter model in MAGE ver. 6.0 [34]. The transversions and transitions in the first and the second codon position increased linearly with the extension of the phylogeny distance, while the transversions and transitions in the third codon position tended to reach the plateau state (Fig 6).
Fig 6. Substitution saturation analysis in the first, second, and third codon positions of 13 PCGs in Lepidoptera (s: transitions; v: transversions).
In our study, 54 lepidopteran mitogenomes were downloaded from Genebank to reconstruct phylogenetic relationships. The phylogenetic trees were inferred from the concatenated nucleotide sequences of 13 PCGs and 13 PCGs+2 rRNAs using BI and ML methods. The four tree topologies were almost identical to each other and indicated that S. recurvalis grouped with the other species within the Pyraloidea with strong posterior probabilities and bootstrap support. Only one node was weakly supported in the phylogenetic tree inferred from the dataset of 13 PCGs using ML method, two nodes were weakly supported in the phylogenetic tree inferred from the dataset of 13 PCGs+2 rRNAs using BI method and ML method, respectively (Fig 7B, 7C, 7D and 7E). In our analysis, the subfamilies relationships within the Pyraloidea are consistent with previous studies based on molecular and morphological characteristics [42,77,78].
Fig 7. Phylogeny of the Lepidoptera.

(A) The recent consensus view of lepidopteran relationships after Kristensen and Skalski (1999). (B) Phylogenetic tree inferred from nucleotide sequences of 13 PCGs using BI method (the numbers abutting branches refer to posterior probabilities). (C) Phylogenetic tree inferred from nucleotide sequences of 13 PCGs using ML method (the numbers abutting branches refer to bootstrap percentages). (D) Phylogenetic tree inferred from nucleotide sequences of 13 PCGs+2 rRNAs using BI method (the numbers abutting branches refer to posterior probabilities). (E) Phylogenetic tree inferred from nucleotide sequences of 13 PCGs+2 rRNAs using ML method (the numbers abutting branches refer to bootstrap percentages). Drosophila melanogaster (NC_001709) and Anopheles gambiae (NC_002084) were used as outgroups.
The 54 species represent nine Lepidoptera superfamilies: Tortricoidea, Bombycoidea, Noctuoidea, Pyraloidea, Geometroidea, Hesperioidea, Papilionoidea, Yponomeutoidea and Hepialoidea (a non-ditrysian superfamily). All of these superfamilies are shown to be monophyletic with the exception of the Papilionoidea. According to the recent consensus view of lepidopteran relationships by Kristensen and Skalski [16] (Fig 7A), the Bombycoidea, Geometroidea, Noctuoidea, and Papilionoidea are a group of the Macrolepidoptera; the Pyraloidea, together with the Macrolepidoptera are considered as the Obtectonera; the Tortricoidea together with the Obtectonera are considered as Apoditrysia. However, in our analysis, the superfamily Pyraloidea was placed within the Macrolepidoptera instead of the Papilionoidea. The phylogenetic relationships reconstructed in our study showed that Papilionoidea was a sister to the clade of (Pyraloidea+(Noctuoidea+(Bombycoidea+ Geometroidea))), which is congruent with previous studies [14,51,70–72, 74, 79–82], but differs from the morphological analysis of Kristensen and Skalski [16].
The phylogenetic relationships between Papilionoidea and Hesperioidea have been a subject of controversy in a long time [80]. Traditionally, Papilionoidea and Hesperioidea are considered as two different superfamilies. Hesperiidae (skippers) are usually placed in their own superfamily Hesperioidea while all other butterflies (Papilionidae, Pieridae, Lycaenidae and Nymphalidae) are placed in Papilionoidea [83]. Papilionoidea and Hesperioidea were proposed to be sister group based on a total-evidence analysis of both traditional morphological characters and new molecular characters from three gene regions (COXI, EF-1α and wingless) [83]. However, our results indicated that the superfamily Hesperioidea was placed within Papilionoidea and shared close relationships with (Pieridae+(Lycaenidae+Nymphalidae)), which is in accordance with previous studies based on morphological characters, nuclear genes, and mitogenome sequences [14,42,77,84].
Pyraloidea is not considered as being in the Macrolepidoptera, but grouped with the Macrolepidoptera, whereas the Papilionoidea is more distantly related from Macrolepidoptera [77]. According to the yet-to-be-tested hypothesis of Regier et al. [77], the position of the thoracic or abdominal ultrasound-detecting ‘‘ears”, which have never been previously theorized to have a common origin, are a candidate synapomorphy supporting Pyraloidea as a member of the Macrolepidoptera. Phylogenetic analysis by Regier et al. [77], based on five protein-coding nuclear genes (6.7 kb in total) of 123 species representing 55 families from 27 superfamilies of Ditrysia yielded the tree topologies were very similar to ours, but our tree topologies were even more strongly supported. Phylogenetic analysis in our study suggests that the complete mitogenome sequences are significant molecular markers for deep-level phylogenetic studies to verify morphological relationships and reconstruct phylogenetic relationships.
Acknowledgments
We sincerely thank Professor Timothy Moermond (University of Wisconsin-Madison) for help in revising the manuscript. We also thank three anonymous reviewers for their constructive comments and suggestions.
Data Availability
All relevant data are within the paper.
Funding Statement
This research was supported by the National Natural Science Foundation of China (grant No. 31272039).
References
- 1. Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999; 27: 1767–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wolstenholme DR. Animal mitochondrial DNA: structure and evolution. Int Rev Cytol. 1992; 141: 173–216. [DOI] [PubMed] [Google Scholar]
- 3. Shadel GS, Clayton DA. Mitochondrial transcription initiation. Variation and conservation. J Biol Chem. 1993; 268: 16083–16086. [PubMed] [Google Scholar]
- 4. Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. BBA-Bioenergetics. 1999; 1410: 103–123. [DOI] [PubMed] [Google Scholar]
- 5. Wei SJ, Shi M, He JH, Sharkey M, Chen XX. The complete mitochondrial genome of Diadegma semiclausum (Hymenoptera: Ichneumonidae) indicates extensive independent evolutionary events. Genome. 2009; 52: 308–319. 10.1139/g09-008 [DOI] [PubMed] [Google Scholar]
- 6. Scheffler I. Mitochondria Wiley-Liss, New York; 1999. [Google Scholar]
- 7. Cameron SL, Whiting MF. The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene. 2008; 408: 112–123. [DOI] [PubMed] [Google Scholar]
- 8. Kim I, Cha SY, Yoon MH, Hwang JS, Lee SM, Sohn HD, et al. The complete nucleotide sequence and gene organization of the mitochondrial genome of the oriental mole cricket, Gryllotalpa orientalis (Orthoptera: Gryllotalpidae). Gene. 2005; 353: 155–168. [DOI] [PubMed] [Google Scholar]
- 9. Zakharov EV, Caterino MS, Sperling FAH. Molecular phylogeny, historical biogeography, and divergence time estimates for swallowtail butterflies of the genus Papilio (Lepidoptera: Papilionidae). Syst Biol. 2004; 53: 193–215. [DOI] [PubMed] [Google Scholar]
- 10. Hurst GDD, Jiggins FM. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. P Roy Soc B-Biol Sci. 2005; 272: 1525–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Avise JC, Nelson WS, Bowen BW, Walker D. Phylogeography of colonially nesting seabirds, with special reference to global matrilineal patterns in the sooty tern (Sterna fuscata). Mol Ecol. 2000; 9: 1783–1792. [DOI] [PubMed] [Google Scholar]
- 12. Cameron SL, Barker SC, Whiting MF. Mitochondrial genomics and the new insect order Mantophasmatodea. Mol Phylogenet Evol 2006; 38: 274–279. [DOI] [PubMed] [Google Scholar]
- 13. Cameron SL, Lambkin CL, Barker SC, Whiting MF. A mitochondrial genome phylogeny of Diptera: whole genome sequence data accurately resolve relationships over broad timescales with high precision. Syst Entomol. 2007; 32: 40–59. [Google Scholar]
- 14. Kim MJ, Kang AR, Jeong HC, Kim KG, Kim I. Reconstructing intraordinal relationships in Lepidoptera using mitochondrial genome data with the description of two newly sequenced lycaenids, Spindasis takanonis and Protantigius superans (Lepidoptera: Lycaenidae). Mol Phylogenet Evol. 2011; 61: 436–445. 10.1016/j.ympev.2011.07.013 [DOI] [PubMed] [Google Scholar]
- 15. Kristensen NP, Scoble MJ, Karsholt O. Lepidoptera phylogeny and systematics: the state of inventorying moth and butterfly diversity. Zootaxa. 2007; 1668: 699–747. [Google Scholar]
- 16. Kristensen NP, Skalski AW. Phylogeny and paleontology In: Kristensen NP, editor. Lepidoptera: Moths and Butterflies. Evolution, Systematics, and Biogeography, Handbook of Zoology Vol I, Part 35. Berlin and New York: De Gruyter; 1999. pp. 7–25. [Google Scholar]
- 17. Chai HN, Du YZ, Zhai BP. Characterization of the complete mitochondrial genomes of Cnaphalocrocis medinalis and Chilo suppressalis (Lepidoptera: Pyralidae). Int J Biol Sci. 2012; 8: 561–579. 10.7150/ijbs.3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li W, Zhang X, Fan Z, Yue B, Huang F, King E, et al. Structural characteristics and phylogenetic analysis of the mitochondrial genome of the sugarcane borer, Diatraea saccharalis (Lepidoptera: Crambidae). DNA Cell Biol. 2011; 30: 3–8. 10.1089/dna.2010.1058 [DOI] [PubMed] [Google Scholar]
- 19. Wu QL, Gong YJ, Shi BC, Gu Y, Wei SJ. The complete mitochondrial genome of the yellow peach moth Dichocrocis punctiferalis (Lepidoptera: Pyralidae). Mitochondr DNA. 2013; 24: 105–107. [DOI] [PubMed] [Google Scholar]
- 20. Coates BS, Sumerford DV, Hellmich RL, Lewis LC. Partial mitochondrial genome sequences of Ostrinia nubilalis and Ostrinia furnicalis . Int J Biol Sci. 2005; 1: 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cao SS, Yu WW, Sun M, Du YZ. Characterization of the complete mitochondrial genome of Tryporyza incertulas, in comparison with seven other Pyralid moths. Gene. 2014; 533: 356–365. 10.1016/j.gene.2013.07.072 [DOI] [PubMed] [Google Scholar]
- 22.Park JS, Kim MJ, Kim SS, Kim I. Complete mitochondrial genome of an aquatic moth, Elophila interruptalis (Lepidoptera: Crambidae). Mitochondr DNA. 2013; 1–3. [DOI] [PubMed]
- 23.Park J S, Kim M J, Ahn S J, Kim I. Complete mitochondrial genome of the grass moth Glyphodes quadrimaculalis (Lepidoptera: Crambidae). Mitochondr DNA, 2013: 1–3. [DOI] [PubMed]
- 24. Traut W, Vogel H, Glöckner G, Hartmann E, Heckel DG. High-throughput sequencing of a single chromosome: a moth W chromosome. Chromosome Res. 2013; 21: 491–505. 10.1007/s10577-013-9376-6 [DOI] [PubMed] [Google Scholar]
- 25.Zou Y, Ma W, Zhang L, He S, Zhang X, Zeng T. The complete mitochondrial genome of the bean pod borer, Maruca testulalis (Lepidoptera: Crambidae: Spilomelinae). Mitochondr DNA. 2014; 1–2. [DOI] [PubMed]
- 26.Ye F, You P. The complete mitochondrial genome of Paracymoriza distinctalis (Lepidoptera: Crambidae). Mitochondr DNA. 2014; 1–2. [DOI] [PubMed]
- 27. Haines WP, Rubinoff D. Molecular phylogenetics of the moth genus Omiodes Guenee (Crambidae: Spilomelinae), and the origins of the Hawaiian lineage. Mol Phylogenet Evol. 2012; 65: 305–316. 10.1016/j.ympev.2012.06.021 [DOI] [PubMed] [Google Scholar]
- 28. Hebert PDN, de Waard JR, Zakharov EV, Prosser SWJ, Sones JE, Mantle B, et al. A DNA 'Barcode Blitz': Rapid Digitization and Sequencing of a Natural History Collection. PLoS One. 2013; 8: e68535 10.1371/journal.pone.0068535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sambrook J, Russell D. Molecular cloning: a laboratory manual 3rd ed., New York, Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 30. Simon C, Buckley TR, Frati F, Stewart JB, Beckenbach AT. Incorporating molecular evolution into phylogenetic analysis, and a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu Rev Ecol Evol S. 2006; 37: 545–579. [Google Scholar]
- 31. Bybee SM, Taylor SD, Riley Nelson C, Whiting MF. A phylogeny of robber flies (Diptera: Asilidae) at the subfamilial level: molecular evidence. Mol Phylogenet Evol. 2004; 30: 789–797. [DOI] [PubMed] [Google Scholar]
- 32. Skerratt LF, Campbell NJH, Murrell A, Walton S, Kemp D, Barker S. The mitochondrial 12S gene is a suitable marker of populations of Sarcoptes scabiei from wombats, dogs and humans in Australia. Parasitol Res. 2002; 88: 376–379. [DOI] [PubMed] [Google Scholar]
- 33. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTALX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997; 25: 4876–4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol Biol Evol. 2013; 30: 2725–2729. 10.1093/molbev/mst197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perna NT, Kocher TD. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 1995; 41: 353–358. [DOI] [PubMed] [Google Scholar]
- 36. Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997; 25: 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beard C, Hamm D, Collins F. The mitochondrial genome of the mosquito Anopheles gambiae: DNA sequence, genome organization, and comparisons with mitochondrial sequences of other insects. Insect Mol Biol. 1993; 2: 103–124. [DOI] [PubMed] [Google Scholar]
- 38. de Bruijn MH. Drosophila melanogaster mitochondrial DNA, a novel organization and genetic code. Nature. 1983; 304: 234–241. [DOI] [PubMed] [Google Scholar]
- 39. Akaike H. A new look at the statistical model identification. IEEE T Automat Contr. 1974; 19: 716–723. [Google Scholar]
- 40. Posada D, Crandall KA. Modeltest: testing the model of DNA substitution. Bioinformatics. 1998; 14: 817–818. [DOI] [PubMed] [Google Scholar]
- 41. Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001; 17: 754–755. [DOI] [PubMed] [Google Scholar]
- 42. Mutanen M, Wahlberg N, Kaila L. Comprehensive gene and taxon coverage elucidates radiation patterns in moths and butterflies. Proc R Soc B. 2010; 277: 2839–2848. 10.1098/rspb.2010.0392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guindon S, Lethiec F, Duroux P, Gascuel O. PHYML Online—a web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 2005; 33: W557–W559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hillis DM, Bull JJ. An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst Biol. 1993; 42: 182–192. [Google Scholar]
- 45.Rambaut A. FigTree, a graphical viewer of phylogenetic trees. 2007. Available: http://treebioed.ac.uk/software/figtree.
- 46. Cao YQ, Ma CA, Chen JY, Yang DR. The complete mitochondrial genomes of two ghost moths, Thitarodes renzhiensis and Thitarodes yunnanensis: the ancestral gene arrangement in Lepidoptera. BMC Genomics. 2012; 13: 276 10.1186/1471-2164-13-276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Boore JL, Lavrov DV, Brown WM. Gene translocation links insects and crustaceans. Nature. 1998; 392: 667–668. [DOI] [PubMed] [Google Scholar]
- 48. Kim I, Lee EM, Seol KY, Yun EY, Lee YB, Hwang JS, et al. The mitochondrial genome of the Korean hairstreak, Coreana raphaelis (Lepidoptera: Lycaenidae). Insect Mol Biol. 2006; 15: 217–225. [DOI] [PubMed] [Google Scholar]
- 49. Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981; 290: 470–474. [DOI] [PubMed] [Google Scholar]
- 50. Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981; 290: 457–465. [DOI] [PubMed] [Google Scholar]
- 51. Hao J, Sun Q, Zhao H, Sun X, Gai Y, Yang Q. The Complete Mitochondrial Genome of Ctenoptilum vasava (Lepidoptera: Hesperiidae: Pyrginae) and Its Phylogenetic Implication. Comp Funct Genom. 2012; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yamauchi MM, Miya MU, Nishida M. Complete mitochondrial DNA sequence of the Japanese spiny lobster, Panulirus japonicus (Crustacea: Decapoda). Gene. 2002; 295: 89–96. [DOI] [PubMed] [Google Scholar]
- 53. Nardi F, Carapelli A, Dallai R, Frati F. The mitochondrial genome of the olive fly Bactrocera oleae: two haplotypes from distant geographical locations. Insect Mol Biol. 2003; 12: 605–611. [DOI] [PubMed] [Google Scholar]
- 54. Lutz-Bonengel S, Sänger T, Pollak S, Szibor R. Different methods to determine length heteroplasmy within the mitochondrial control region. Int J Legal Med. 2004; 118: 274–281. [DOI] [PubMed] [Google Scholar]
- 55. Ogoh K, Ohmiya Y. Complete mitochondrial DNA sequence of the sea-firefly, Vargula hilgendorfii (Crustacea, Ostracoda) with duplicate control regions. Gene. 2004; 327: 131–139. [DOI] [PubMed] [Google Scholar]
- 56. Yukuhiro K, Sezutsu H, Itoh M, Shimizu K, Banno Y. Significant levels of sequence divergence and gene Rearrangements have occurred between the mitochondrial Genomes of the wild mulberry silkmoth, Bombyx mandarina, and its close relative, the domesticated silkmoth, Bombyx mori . Mol Biol Evol. 2002; 19: 1385–1389. [DOI] [PubMed] [Google Scholar]
- 57. Clary DO, Wolstenholme DR. The mitochondrial DNA molecular of Drosophila yakuba: nucleotide sequence, gene organization, and genetic code. J Mol Evol. 1985; 22: 252–271. [DOI] [PubMed] [Google Scholar]
- 58. Flook PK, Rowell CHF, Gellissen G. The sequence, organization, and evolution of the Locusta migratoria mitochondrial genome. J Mol Evol. 1995; 41: 928–941. [DOI] [PubMed] [Google Scholar]
- 59. Crease TJ. The complete sequence of the mitochondrial genome of Daphnia pulex (Cladocera: Crustacea). Gene. 1999; 233: 89–99. [DOI] [PubMed] [Google Scholar]
- 60. Ballard JWO. Comparative genomics of mitochondrial DNA in members of the Drosophila melanogaster subgroup. J Mol Evol. 2000; 51: 48–63. [DOI] [PubMed] [Google Scholar]
- 61. Mitchell SE, Cockburn AF, Seawright JA. The mitochondrial genome of Anopheles quadrimaculatus species A: complete nucleotide sequence and gene organization. Genome. 1993; 36: 1058–1073. [DOI] [PubMed] [Google Scholar]
- 62. Spanos L, Koutroumbas G, Kotsyfakis M, Louis C. The mitochondrial genome of the Mediterranean fruit fly, Ceratitis capitata . Insect Mol Biol. 2000; 9: 139–144. [DOI] [PubMed] [Google Scholar]
- 63. Wilson K, Cahill V, Ballment E, Benzie J. The complete sequence of the mitochondrial genome of the crustacean Penaeus monodon: are malacostracan crustaceans more closely related to insects than to branchiopods? Mol Biol Evol. 2000; 17: 863–874. [DOI] [PubMed] [Google Scholar]
- 64. Liu Y, Li Y, Pan M, Dai F, Zhu X, Lu C, et al. The complete mitochondrial genome of the Chinese oak silkmoth, Antheraea pernyi (Lepidoptera: Saturniidae). Acta Bioch Bioph Sin. 2008; 40: 693–703. [PubMed] [Google Scholar]
- 65. Feng X, Liu DF, Wang NX, Zhu CD, Jiang GF. The mitochondrial genome of the butterfly Papilio xuthus (Lepidoptera: Papilionidae) and related phylogenetic analyses. Mol Biol Rep. 2010; 37: 3877–3888. 10.1007/s11033-010-0044-z [DOI] [PubMed] [Google Scholar]
- 66. Krzywinski J, Grushko OG, Besansky NJ. Analysis of the complete mitochondrial DNA from Anopheles funestus: An improved dipteran mitochondrial genome annotation and a temporal dimension of mosquito evolution. Mol Phylogenet Evol. 2006; 39: 417–423. [DOI] [PubMed] [Google Scholar]
- 67. Margam VM, Coates BS, Hellmich RL, Agunbiade T, Seufferheld MJ, Sun W, et al. Mitochondrial Genome Sequence and Expression Profiling for the Legume Pod Borer Maruca vitrata (Lepidoptera: Crambidae). PLoS One. 2011; 6, e16444 10.1371/journal.pone.0016444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kim MI, Baek JY, Kim MJ, Jeong HC, Kim KG, Bae CH, et al. Complete nucleotide sequence and organization of the mitogenome of the red-spotted apollo butterfly, Parnassius bremeri (Lepidoptera: Papilionidae) and comparison with other lepidopteran insects. Mol Cells. 2009; 28: 347–363. 10.1007/s10059-009-0129-5 [DOI] [PubMed] [Google Scholar]
- 69. Lavrov DV, Brown WM, Boore JL. A novel type of RNA editing occurs in the mitochondrial tRNAs of the centipede Lithobius forficatus . Proc Natl Acad Sci USA. 2000; 97: 13738–13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lu HF, Su TJ, Luo AR, Zhu CD, Wu CS. Characterization of the complete mitochondrion genome of Diurnal Moth Amata emma (Butler) (Lepidoptera: Erebidae) and its phylogenetic implications. PLoS One. 2013; 8: e72410 10.1371/journal.pone.0072410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Wu YP, Zhao JL, Su TJ, Li J, Yu F, Chesters D, et al. The complete mitochondrial genome of Leucoptera malifoliella Costa (Lepidoptera: Lyonetiidae). DNA Cell Biol. 2012; 31: 1508–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wei SJ, Shi BC, Gong YJ, Li Q, Chen XX. Characterization of the mitochondrial genome of the diamondback moth Plutella xylostella (Lepidoptera: Plutellidae) and phylogenetic analysis of advanced moths and butterflies. DNA Cell Biol. 2013; 32: 173–187. 10.1089/dna.2012.1942 [DOI] [PubMed] [Google Scholar]
- 73. Wei SJ, Min S, Sharkey MJ, Achterberg C, Chen XX. Comparative mitogenomics of Braconidae (Insecta: Hymenoptera) and the phylogenetic utility of mitochondrial genomes with special reference to Holometabolous insects. BMC Genomics. 2010;11: 371 10.1186/1471-2164-11-371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Liao F, Wang L, Wu S, Li YP, Zhao L, Huang GM, et al. The complete mitochondrial genome of the fall webworm, Hyphantria cunea (Lepidoptera: Arctiidae). Int J Biol Sci. 2010; 6: 172–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Saito S, Tamura K, Aotsuka T. Replication origin of mitochondrial DNA in insects. Genetics. 2005; 171: 1695–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhang DX, Hewitt GM. Insect mitochondrial control region: A review of its structure, evolution and usefulness in evolutionary studies. Biochem Syst Ecol. 1997; 25: 99–120. [Google Scholar]
- 77. Regier JC, Zwick A, Cummings MP, Kawahara AY, Cho S, Weller S, et al. Toward reconstructing the evolution of advanced moths and butterflies (Lepidoptera: Ditrysia): an initial molecular study. BMC Evol Biol. 2009; 9: 280 10.1186/1471-2148-9-280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nuss M. Global information system of Pyraloidea (GlobIZ). 2006. Available: http://www.pyraloidea.org.
- 79. van Nieukerken EJ, Kaila L, Kitching IJ, Kristensen P, Lees DC, Minet J, et al. In: Zhang ZQ, editor. Order Lepidoptera. Animal Biodiversity: An Outline of Higher-Level Classification and Survey of Taxonomic Richness New Zealand, Auckland: Magnolia Press; 2011. pp. 212–221. [Google Scholar]
- 80. Minet J. Tentative reconstruction of the ditrysian phylogeny (Lepidoptera: Glossata). Insect Syst Evol. 1991; 22: 69–95. [Google Scholar]
- 81. Jiang ST, Hong GY, Yu M, Li N, Yang Y, Liu YQ, et al. Characterization of the complete mitochondrial genome of the giant silkworm moth, Eriogyna pyretorum (Lepidoptera: Saturniidae). Int J Biol Sci. 2009; 5: 351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yang X, Xue D, Han H. The complete mitochondrial genome of Biston panterinaria (Lepidoptera: Geometridae), with phylogenetic utility of mitochondrial genome in the Lepidoptera. Gene. 2013; 515: 349–358. 10.1016/j.gene.2012.11.031 [DOI] [PubMed] [Google Scholar]
- 83. Wahlberg N, Braby MF, Brower AV, de Jong R, Lee MM, Nylin S, et al. Synergistic effects of combining morphological and molecular data in resolving the phylogeny of butterflies and skippers. Proc R Soc B. 2005; 272: 1577–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Heikkilä M, Kaila L, Mutanen M, Peña C, Wahlberg N. Cretaceous origin and repeated tertiary diversification of the redefined butterflies. Proc R Soc B. 2012; 279: 1093–1099. 10.1098/rspb.2011.1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.