

Abstract
Experiments are described that probe the stability of N-substituted derivatives of the azadithiolate cofactor recently confirmed in the [FeFe] hydrogenases (Berggren, G., et al. Nature 2013, 499, 66). Acid-catalyzed hydrolysis of bis(thioester) BnN(CH2SAc)2 gives [BnNCH2SCH2]2 rather than azadithiol BnN(CH2SH)2. Treatment of BnN(CH2SAc)2 with NaOtBu generates BnN(CH2SNa)2, which was trapped with NiCl2(diphos) (diphos = 1,2-C2H4(PR2)2; R = Ph (dppe) and Cy (dcpe)) to give fully characterized complexes Ni[(SCH2)2NBn](diphos). The related N-aryl derivative Ni[(SCH2)2NC6H4Cl](diphos) was prepared analogously from 4-ClC6H4N(CH2SAc)2, NaOtBu, and NiCl2(dppe). Crystallographic analysis confirmed that these rare nonbridging [adtR]2− complexes feature distorted square planar Ni centers. The analogue Pd[(SCH2)2NBn](dppe) was also prepared. 31P NMR analysis indicates that Ni[(SCH2)2NBn](dppe) has basicity comparable to typical amines. As shown by cyclic voltammetry, the couple [M[(SCH2)2NBn](dppe)]+/0 is reversible near −2.0 V versus Fc+/0. The wave shifts to −1.78 V upon N-protonation. In the presence of CF3CO2H, Ni[(SCH2)2NBn](dppe) catalyzes hydrogen evolution at rate of 22 s−1 in the acid-independent regime, at room temperature in CH2Cl2 solution. In contrast to the instability of RN(CH2SH)2 (R = alkyl, aryl), the dithiol of tosylamide TsN(CH2SH)2 proved sufficiently stable to allow full characterization. This dithiol reacts with Fe3(CO)12 and, in the presence of base, NiCl2(dppe) to give Fe2[(SCH2)2NTs](CO)6 and Ni[(SCH2)2NTs](dppe), respectively.
INTRODUCTION
The identity of the dithiolate cofactor that supports the activity of the [FeFe] hydrogenases has been actively discussed since the original crystallographic descriptions of the enzymes1 from D. desulfuricans and C. pasteurianum. Model studies show that the amine in azadithiolates facilitates protonation of its Fe(I)Fe(I) derivatives (Figure 1).2,3 Furthermore, amine-containing dithiolates are required for oxidation of H2 by mildly electrophilic Fe(II)Fe(I) species.4 The idea5 of an amine poised close but not coordinated to a metal center has spawned the development of homogeneous catalysts featuring proton relays6 and is a foundation of our understanding of the second coordination sphere.7
Figure 1.
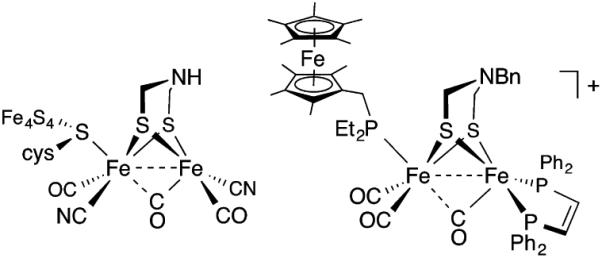
Active site of the H-cluster in [FeFe] hydrogenase enzyme (left) and typical model complex (right).
The first spectroscopic evidence for the azadithiolate cofactor versus, say, 1,3-propanedithiolate, came from electron–nuclear double resonance studies on [FeFe] hydrogenases that revealed coupling of Fe(I) to two 14N centers, proposed to be the CN− cofactor and the azadithiolate bridge.8 This assignment is supported by recent work on the C15N-enriched enzyme.9,10 The nature of the dithiolate has finally been settled, with experiments showing that synthetic [Fe2[(SCH2)2NH]-(CN)2(CO)4]2− reconstitutes apo-HydA1 from C. reinhardtii to give a highly active catalyst. Isostructural diiron complexes [Fe2(S2C3H6)(CN)2(CO)4]2 − and [Fe2[(SCH2)2O]-(CN)2(CO)4]2− are also accepted by the apoenzyme,11,12 but the resulting proteins exhibit very low catalytic activity.12
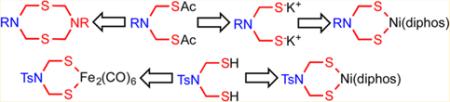
Now that the azadithiolate cofactor has been confirmed, it is of interest to examine the cofactor itself, free of the protein and, if possible, metals. First reported in 2001,13 complexes of [adtR]2− can be prepared by many routes, almost all of which involve installation of the cofactor on diiron centers (Scheme 1).13-16 Typical routes include condensation of CH2O and amines with Fe2(SH)2(CO)614,16 and alkylation of Fe2(μ-SLi)2(CO)6, the latter route only suited for tertiary amines of the type RN(CH2Cl)2.13 Related complexes have been developed with an azadiphosphide group (RN(CH2PR)22−)17 and a azadiselenide (RN(CH2Se)22−)18 bridging two iron centers.
Scheme 1.
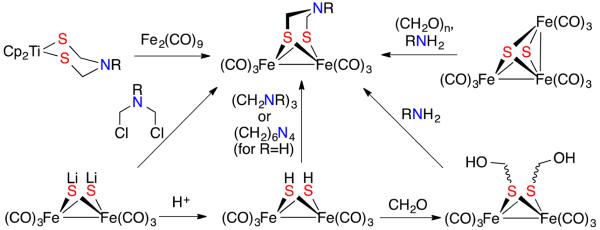
Synthetic Routes to Fe2[(SCH2)2NR](CO)6
In terms of obtaining the free cofactor, the diiron dithiolates represent obvious precursors to RN(CH2SH)2. Azadithiolato diiron complexes are, however, robust and exhibit no tendency to release the cofactor even in the presence of strong acids.
Free HN(CH2SH)2 and related derivatives might be anticipated to be unstable with respect to loss of hydrogen sulfide. This anticipated reactivity necessitates that the azadithiolates or their protonated derivatives be generated under mild reaction conditions. The bis(thioester) compounds of type RN(CH2SAc)2 emerged as attractive precursors to the azadithiolates. Indeed, hydrolysis of one such thioester has been claimed to afford [HOC2H4N(H)(CH2SH)2]Cl, the conjugate acid of an azadithiol.19
RESULTS AND DISCUSSION
Preparation and Hydrolysis of BnN(CH2SAc)2
Hydroxymethylation of primary amines in the presence of thioacetic acid is known to efficiently give bis(thioesters) RN-(CH2SAc)2.20 The relevant transformations are given in eqs 1 and 2.
| (1) |
| (2) |
This method appears general, and the initial phases of this study focused on benzyl derivatives. The acid-catalyzed hydrolysis of BnN(CH2SAc)2 was investigated as a route to the dithiol BnN(CH2SH)2. Upon treatment with aqueous HCl in EtOH, BnN(CH2SAc)2 was rapidly consumed with formation of a precipitate. 1H NMR analysis of the crude product with an internal standard showed that [BnNCH2SCH2]2 was present in ~60% yield. The reaction does not yield NMR-detectable amounts of trithiane (SCH2)3, a result consistent with the stoichiometry in eq 3.
| (3) |
Experiments were conducted testing the utility of [RNCH2SCH2]2 and the bis(thioesters) as precursors to diiron azadithiolate complexes. Reaction of [RNCH2SCH2]2 with Fe3(CO)12 yields Fe2(SCH2N(R)CH2)(CO)6 instead of the typical Fe2[(SCH2)2NBn](CO)6 derivatives featuring Fe2S2 tetrahedranes.21 Additionally, reaction of BnN(CH2SAc)2 with Fe3(CO)12 gave complex mixtures containing only traces of Fe2[(SCH2)2NBn](CO)6.
Trapping RN(CH2SNa)2 (R = Bn or 4-ClC6H4)
Given the instability of azadithiol RN(CH2SH)2 implicated by the previous experiments, the preparation of the corresponding dithiolate dianion was investigated. Treatment of BnN-(CH2SAc)2 with NaOMe in MeOH afforded mainly the known diether BnN(CH2OMe)2. A bulkier nucleophile might favor attack at the carbonyl centers instead of the methylene groups, and avoidance of protic solvents would preclude formation of any thiols, these being prone to the degradation pathway in eq 3. Indeed, experiments suggest that the dithiolate can be generated by treatment of RN(CH2SAc)2 (R = Bn, 4-ClC6H4) with NaOtBu in tetrahydrofuran (THF) at −78 °C. The putative RN(CH2SNa)2 was derivatized by treatment with NiCl2(diphos) (diphos = 1,2-C2H4(PR′2)2; R′ = Ph (dppe) and Cy (dcpe)). These trapping experiments afforded modest yields of orange microcrystalline solids identified as Ni[(SCH2)2NR]-(diphos) (eqs 4 and 5).
| (4) |
| (5) |
In the case of the electrophile NiCl2(dppe), the derivative Ni[(SCH2)2NR](dppe) was isolated and characterized according to 1H and 31P{1H} NMR and electrospray ionization mass spectrometry (ESI-MS) data. The complexes were stable as solids and in THF, CH2Cl2, and MeCN solution under an inert atmosphere. Single crystals of Ni[(SCH2)2NR](dppe) were grown from CH2Cl2/pentane. The solid-state structures of the two compounds are presented in Figures 2 and 3.
Figure 2.
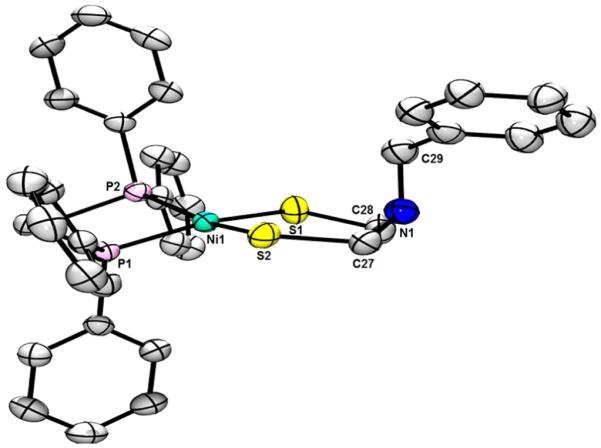
ORTEP of Ni[(SCH2)2NBn](dppe) with ellipsoids drawn at the 50% probability level. H atoms and disorder are omitted for clarity. Selected distances (Å): Ni1−S1, 2.1925(10); Ni1−S2, 2.1766(9); Ni1−P1, 2.1840(10); Ni1−P2, 2.1675(9). Selected angles (deg): S1−Ni1−S2, 100.46(3); P1−Ni1−P2, 85.84(3); P1−Ni1−S1, 165.05(3); P2−Ni1−S1, 88.93(3); P1−Ni1−S2, 88.20(3); P2−Ni1−S2, 163.16(3); C27−N1−C28, 112.8(2), C27−N1−C29, 114.6(3); C28−N1−C29, 113.7(2).
Figure 3.
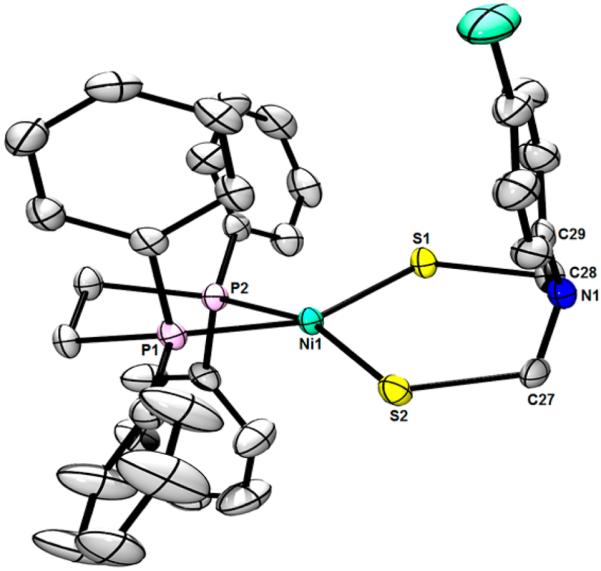
ORTEP of Ni[(SCH2)2NC6H4Cl](dppe) with ellipsoids drawn at the 75% probability level. H atoms and a solvent molecule are omitted for clarity. Selected distances (Å): Ni1−S1, 2.1737(9); Ni1−S2, 2.1792(10); Ni1−P1, 2.1651(10); Ni1−P2, 2.1663(10). Selected angles (deg): S1−Ni1−S2, 104.66(4); P1−Ni1−P2, 85.88(4); P1−Ni1−S1, 156.77(4); P2−Ni1−S1, 87.44(4); P1−Ni1−S2, 89.04(4); P2−Ni1−S2, 158.65(4); C27−N1−C28, 112.7(3), C27−N1−C29, 118.5 (3); C28−N1−C29, 112.00(2).
Crystallographic analysis revealed that Ni[(SCH2)2NBn]-(dppe) features a twisted NiS2P2 coordination. The average Ni−S (2.185 Å) and Ni−P bond lengths (2.176 Å) are similar to those in the propanedithiolate Ni(S2C3H6)(dppe) (2.203 and 2.160 Å, respectively).22 Substitution, however, of CH2 with NBn causes distortion of the ligand environment such that the NiS2 and NiP2 planes in Ni[(SCH2)2NBn](dppe) are twisted by 19.6° (this value being 7.6° for the propane-dithiolate). In the crystal, the Bn group participates in weak edge-to-face π−π stacking (4.3 Å) with the dppe ligand of a neighboring complex, orienting Bn toward Ni. In addition to stereoelectronic effects, this interaction causes the N lone pair to be directed away from the Ni site.
Crystallographic analysis of the complex Ni[(SCH2)2-NC6H4Cl](dppe) again reveals a twisted NiS2P2 core (Figure 3). The average Ni−S (2.179 Å) and Ni−P bond lengths (2.166 Å) are shortened relative to the benzyl derivative, highlighting a decrease in electron density at the metal center. The distortion of the ligand environments with R = 4-ClC6H4 is increased relative to the complex with R = Bn, with a twist of the NiS2 and NiP2 planes of 27.6° for the former derivative. The changes in bond lengths and coordination geometry likely reflect the weaker donicity of the [adtC6H4Cl]2− ligand. Containing a more basic diphosphine, NiCl2(dcpe) was converted to Ni[(SCH2)2NBn](dcpe), albeit in lower yield than in the dppe case. The 1H and 31P NMR (δ 73.6) and ESI-MS (m/z 676.3) data confirm formation of the target although the sample was not obtained in high purity. Nevertheless, the solid-state structure could be determined by diffraction (Figure 4).
Figure 4.
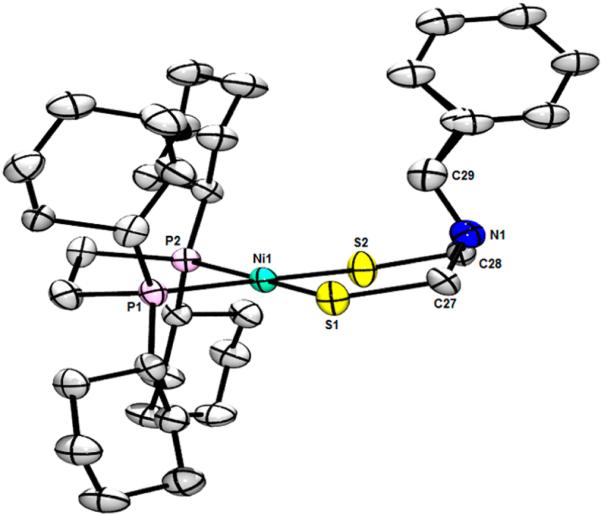
ORTEP of Ni[(SCH2)2NBn](dcpe)·CH2Cl2, showing one of the crystallographically independent complexes with ellipsoids drawn at the 50% probability level. H atoms, solvate, and disorder are omitted for clarity. Selected distances (Å): Ni1−S1, 2.1952(19); Ni1−S2, 2.1841(18); Ni1−P1, 2.1884(18); Ni1−P2, 2.1866(18); Ni2−S3, 2.1860(19); Ni2−S4, 2.176(2); Ni2−P3, 2.1774(19); Ni2−P4, 2.1797(18). Selected angles (deg): S1−Ni1−S2, 100.01(7); P1−Ni1−P2, 88.07(7); P1−Ni1−S1, 85.69(7); P2−Ni1−S1, 173.39(7); P1−Ni1−S2, 173.94(8); P2−Ni1−S2, 86.31(7); C27−N1−C28, 113.5(6), C27−N1−C29, 111.3(6); C28−N1−C29, 115.0(6). S4−Ni2−S3, 100.01(8); P3−Ni2−P4, 88.77(7); P3−Ni2−S3, 85.97(7); P4−Ni2−S3, 170.30(8); S4−Ni2−P3, 170.71(9); S4−Ni2−P4, 86.28(7); C62−N2−C63, 112.5(6); C62−N2−C64, 114.7(6); C63−N2−C64, 114.5(6).
The solid-state structure of Ni[(SCH2)2NBn](dcpe) mirrors that of the analogous dppe compound. While the Ni−P and Ni−S bond lengths in the two complexes are virtually identical, the ligand environment in the dcpe complex is less distorted, with the NiS2 and NiP2 planes being 2.9° and 10.5° apart in the two crystallographically independent complexes. This planarity might result from the greater size and σ-donicity of dcpe versus dppe. The anion in BnN(CH2SNa)2 was also trapped using PdCl2(dppe) as the electrophile, affording crystalline Pd-[(SCH2)2NBn](dppe), whose 1H NMR spectrum is similar to that for Ni[(SCH2)2NBn](dppe).
Protonation of Ni[(SCH2)2NBn](dppe)
Treatment of Ni[(SCH2)2NBn](dppe) with HOTf (1 equiv) in CD2Cl2 resulted in protonation, signaled by a change in color from orange to yellow-orange. The 31P NMR singlet only shifted by 3 ppm downfield upon protonation of the complex, but the 1H NMR spectrum drastically changed (see Supporting Information)—a new singlet was observed at δ 8.84, assigned to NH, and the symmetry of the Ni[(SCH2)2NBn](dppe) is lifted. While the SCH2N signals appeared as a broad multiplet for the neutral complex, they appear as doublets-of-triplets for the ammonium species. Consistent with a Cs-symmetric cation, the CH2Ph signal remains a doublet after protonation. The pKaMeCN of the amine was not determined since protonation labilizes the complex in this solvent. Nonetheless, addition of increasing amounts of BnNH3+ (pKaMeCN = 16.76) or Bu3NH+ (pKaMeCN = 18.03) to Ni[(SCH2)2NBn](dppe) shift the 31P NMR signals toward those of the fully protonated species (see Supporting Information). The complex Ni[(SCH2)2-NC6H4Cl)](dppe) did not survive protonation.
Electrochemical Behavior
Cyclic voltammograms (CVs) of Ni[(SCH2)2NBn](dppe) in CH2Cl2 feature irreversible oxidations at 0.11 V and a reduction at −2.02 V versus Fc0/+. The cathodic wave is shifted to −1.55 V upon addition of 1 equiv of CF3CO2H in CH2Cl2. The anodic shift is attributed to protonation of the amine and more facile reduction of the cation [Ni[(SCH2)2N(H)Bn](dppe)]+ relative to its conjugate base. Anodic shifts of this magnitude are observed for diiron azadithiolate catalysts.3,23 In the presence of CF3CO2H (1 equiv), no anodic peaks are observed when scanning in the positive direction. However, once [Ni[(SCH2)2NHBn]-(dppe)]+ has been reduced, the anodic peak corresponding to [Ni[(SCH2)2NBn](dppe)]0/+ reappears. These observations are consistent with the series of electron transfer and chemical steps in eqs 6–10.
| (6) |
| (7) |
| (8) |
| (9) |
| (10) |
Upon addition of increasing amounts of CF3CO2H to [Ni[(SCH2)2NHBn](dppe)]+, current increases are observed, which are attributed to catalytic reduction of the acid to give H2. The dependence of current on [CF3CO2H] plateaus at 500 mM of acid, which corresponds to a turnover frequency of 22 s−1 according to the usual analysis (see Supporting Information).24
The CV of Ni[(SCH2)2NC6H4Cl](dppe) in CH2Cl2 features an irreversible oxidation at 0.22 V and a quasi-reversible reduction at E1/2 = −1.88 V, both versus Fc0/+. The shifts to more positive potentials for the cathodic and anodic events for the ClC6H4 versus Bn derivatives further reflects the influence of the amine substituent on the metal center, despite its remoteness.
TsN(CH2SH)2 and Its Fe and Ni Derivatives
The lability of free azadithiols is proposed to be correlated to the basicity of the amine group. This hypothesis suggests that free azadithiols with electron-withdrawing groups on the nitrogen center could be stable. To address this question, the tosylamide (MeC6H4SO2NR2, abbreviated TsNR2) platform was selected since such sulfonamides are known to be nonbasic.25 The targeted TsN(CH2SH)2 was prepared from bis(thioester) TsN(CH SAc), which in turn was prepared from dichloride TsN(CH2Cl)2 (Scheme 2). Acid-catalyzed hydrolysis of the thioester afforded dithiol TsN(CH2SH)2 as analytically pure white crystals, the 1H NMR spectrum of which is readily assigned.
Scheme 2.
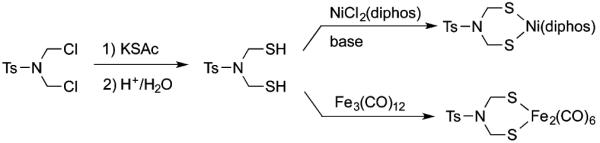
Synthesis of TsN(CH2SH)2 and Its Complexes
The robustness of TsN(CH2SH)2 is further indicated by its efficient conversion to dithiolato complexes. Thus, treatment of the dithiol with Fe3 (CO)12 in hot toluene gave Fe2[(SCH2)2NTs](CO)6, isolated as red crystals. This synthetic method is analogous to the use of alkyl- and arylthiols.26 In addition to its characterization by IR and NMR spectroscopy, Fe2[(SCH2)2NTs](CO)6 was examined by single-crystal X-ray diffraction (Figure 5). Hexacarbonyl Fe2[(SCH2)2NTs](CO)6 is structurally similar to many diiron(I) dithiolates. The Fe centers are within the sum of their covalent radii (2.64 Å for low-spin Fe) with Fe−Fe distance (2.5145(4) Å) being typical for such compounds, for example, 2.5047(6) Å for Fe2[(SCH2)2NPh](CO)6.14 Like this phenyl analogue, the N center in the Ts derivative is roughly trigonal planar, the sum of its bond angles being 357°. Reflecting the nonbasic nature of this tosylamido center, a solution of the diiron complex was found to be unaffected by HBF4. The nonbasicity of Fe2[(SCH2)2NTs](CO)6 is in contrast to the easy protonation of other azadithiolato complexes with R = alkyl or aryl.3
Figure 5.
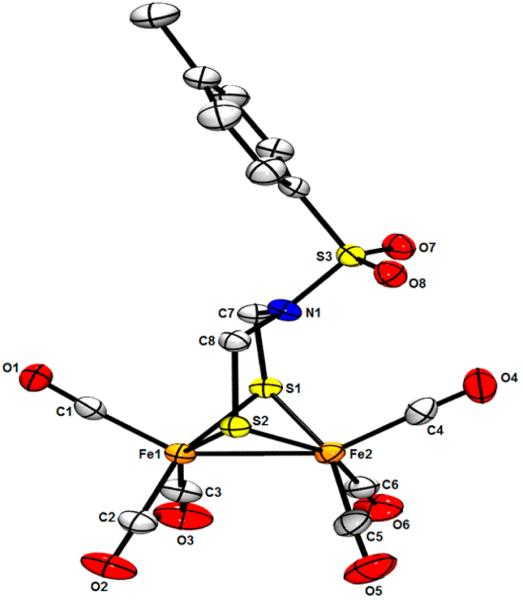
ORTEP of Fe2[(SCH2)2NTs](CO)6 with ellipsoids drawn at the 50% probability level and H atoms omitted for clarity. Selected distances (Å): Fe1−Fe2, 2.5145(4); Fe1−S1, 2.2537(5); Fe2−S2, 2.2547(5); Fe2−S1, 2.2619(5); Fe2−S2, 2.2547(5). Selected angles (deg): Fe1−S1−Fe2, 67.675(15); Fe1−S2−Fe2, 67.718(15); S1−Fe1−S2, 85.735(17); S1−Fe1−Fe2, 56.318(14); C7−N1−S3, 116.45(14); C7−N1−S3, 119.70(11); C8−N1−S3, 120.63(11).
In the presence of base, TsN(CH2SH)2 was also found to react with NiCl2(dppe) to give Ni[(SCH2)2NTs](dppe), isolated as air-stable red crystals. The structure of this Ni complex was verified by X-ray diffraction (Figure 6).
Figure 6.
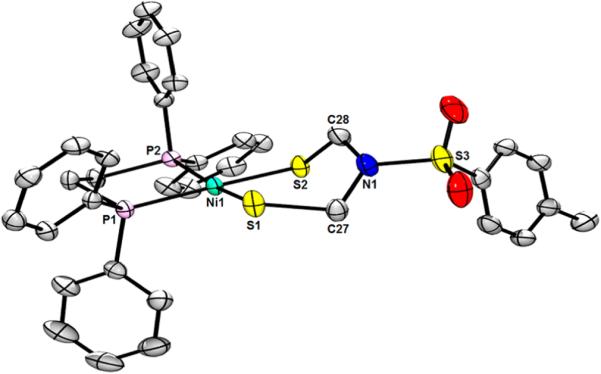
ORTEP of Ni[(SCH2)2NTs](dppe) with ellipsoids drawn at the 50% probability level. H atoms and disorder are omitted for clarity. Selected distances (Å): Ni1−S1, 2.1943(5); N1−S2, 2.1963(5); Ni1−P1, 2.1804(5); Ni1−P2, 2.1873(5). Selected angles (deg): S1−Ni1−S2, 94.568(19); P1−Ni1−P2, 87.143(18); P1−Ni1−S1, 90.505(18); P2−Ni1−S1, 168.68(2); P1−Ni1−S2, 173.82(2); P2−Ni1−S2, 88.549(18); C27−N1−C28, 116.95(15), C27−N1−S3, 119.69(13); C28−N1−S3, 117.26(13).
The complex Ni[(SCH2)2NTs](dppe) was found to be virtually isostructural to its NBn congener, however, with less distortion of the Ni center (the NiS2 and NiP2 interplanar angle being 11.7°). As with the diiron complex, the N atom is approximately planar (angle sum = 354°), but the Ni complex differs in that the sp2-hybridization causes puckering of the chelate ring, in contrast to the idealized chair/boat conformation in Fe2[(SCH2)2NTs](CO)6.
CONCLUSIONS
The present work was motivated by the recent confirmation of the azadithiolate ([adtH]2−) cofactor in the [FeFe] hydrogenases.11 Our studies were intended to probe the stability of the free azadithiol and its salts. A simplifying aspect of our endeavor was the use of the N-substituted analogues of [adtH]2− rather than the secondary amine itself. This compromise was necessary because the precursor HN-(CH2SAc)2 is unknown, and a synthesis of this compound could not be devised. The work described here led us to two instructive conclusions.
1. Azadithiolate Salts and Azadithiols Are Labile
The instability is associated with the proximity of the basic nitrogen centers to the thiol groups, this labile motif also being present in the yet-to-be-isolated thioaminals (R2NCH2SH). In contrast, related compounds are stable when SH is replaced by S-alkyl27,28 or when the spacer between the thiol and the amine is extended, as is the case for cysteamine derivatives (R2NCH2CH2SH).27 When the N-substituent is electron-withdrawing, the corresponding dithiolates appear more stable. This effect is indicated by the improved yields of Ni[(SCH2)2-NC6H4Cl](dppe) relative to the N-benzyl derivative. More dramatically, N-substitution with p-toluenesulfonyl (Ts) allowed full characterization of the dithiol TsN(CH2SH)2. This dithiol was shown to react efficiently with iron carbonyls to give the corresponding azadithiolatodiiron(I) complex. In this complex, however, the nitrogen center is insufficiently basic to sustain protonation. In terms of their ease of synthesis and low basicity, the [adtTs]2− derivatives resemble analogous N-acetylated ligands [adtAc]2−.29
2. Azadithiolate is Stabilized by Coordination, Even to One Metal
Although hundreds of [adtR]2− complexes have been reported, few are not components of a diiron complex. In this paper, the N-substituted [adtR]2− ligands were stabilized through formation of complexes of the familiar Ni(dithiolate)-(dppe) motif.30 Aside from the nickel complexes reported in this paper, the titanocene derivatives (C5H4R)2Ti(adtR) (prepared from (C5H4R′)2Ti(SH)2 and (CH2NR)3; R′ = H, Me; R = Ph, Me) feature nonbridging [adtR]2− complexes.15
Finally, these results have some bearing on the biosynthesis of [FeFe] hydrogenase. Containing three unusual cofactors, CO, CN−, and [adtH]2−, as well as the attached 4Fe−4S cluster, the active site is assembled following a multistep maturation pathway.10,31 The source of CO and CN− has been elucidated, but the origin of [adtH]2− remains unsolved. As demonstrated here, the cofactor in its various protonated forms [HnadtH](2−n)− is expected to be too unstable to persist in the absence of metal ions. Indeed, the biochemical evidence32 and some literature precedents33 suggest that the azadithiolate is biosynthesized on an Fe-containing scaffold.
EXPERIMENTAL SECTION
General Considerations
Unless otherwise stated, reactions were conducted using standard Schlenk techniques, and all reagents were purchased from Sigma-Aldrich or Fisher. Unless otherwise noted, all solvents were HPLC grade and purified using an alumina filtration system (Glass Contour, Irvine, CA). ESI-MS data were acquired using a Waters Micromass Quattro II or ZMD spectrometer with analytes in dilute CH2Cl2 solution. Analytical data were acquired using an Exeter Analytical CE-440 elemental analyzer. NMR data were acquired using Varian U400, U500, and VXR500 spectrometers. Chemical shifts (in parts per million) were referenced to residual solvent peaks (for 1H, 13C) or external 85% H3PO4 (for 31P). Solution IR spectra were recorded on a PerkinElmer Spectrum 100 FTIR spectrometer. Crystallographic data were collected using a Siemens SMART diffractometer equipped with a Mo Kα source (λ = 0.710 73 Å) and an Apex II detector.
BnN(CH2SAc)2
In a modification of the method of Izawa,20 a solution of benzylamine (10.72 g, 100 mmol) in 100 mL of 95% EtOH was treated with formaldehyde (37% in MeOH/H2O, 24 mL). The mixture was heated at 60 °C for 30 min and then treated with AcSH (14.1 mL, 200 mmol). After 2 h, the reaction mixture was cooled to −17 °C, when colorless crystals formed. The crystals were isolated by filtration, washed with cold 95% EtOH, and dried briefly under vacuum. Yield: 22.52 g (79%). 1H NMR (400 MHz, deuterated dimethyl sulfoxide (DMSO-d6), 298 K): δ 7.34−7.23 (m, 5H, C6H5), 4.51 (s, 4H, C(O)CH2), 3.61 (s, 2H, PhCH2), 2.35 (s, 6H, CH3). 13C NMR (75 MHz, DMSO-d6, 298 K): δ 196 (C=O), 137 (Ph−C1), 129 (PhC2, PhC2′), 128 (PhC2, PhC2′), 128 (Ph−C1), 56.4 (C(O)−CH2), 53.6 (PhCH2), 31.5 (CH3). Anal. Calcd for C13H17NO2S2: C, 55.09; H, 6.05; N, 4.94. Found: C, 55.24; H, 6.02; N, 4.98%.
Hydrolysis of BnN(CH2SAc)2
A solution of BnN(CH2SAc)2 (0.58 g, 2.00 mmol) in 10 mL of EtOH was treated with a 2 M solution of HCl in Et2O (4.00 mL, 8.00 mmol) under Ar. After the mixture was stirred for 15 h, the white precipitate was isolated by filtration in air and washed with 2 × 5 mL of EtOH, yielding 0.370 g of crude product. 1H NMR analysis (1,3,5-C6H3(OMe)3 integration standard) of the precipitate showed 63% [BnNCH2SCH2]2, indicating a yield of 0.23 g of [BnNCH2SCH2]2 (theoretical yield: 330 mg). The reaction of BnN(CH2SAc)2 with NaOMe afforded the known compound BnN(CH2OMe)2.34
[BnNCH2SCH2]2
The following is an adaptation of a method for the α-methylbenzylamine derivative.35 A solution of benzylamine (4.9 g, 0.045 mol) in 90 mL of MeOH was added to cold 37% aqueous formaldehyde (27 mL, 0.352 mol) at 0 °C. After it was stirred for 5 min at 0 °C, the solution was treated with a solution of NaSH·xH2O (13.53 g, 0.12 mol) in 115 mL of MeOH. After it was stirred for 5 min at 0 °C for 24 h, the cloudy solution was concentrated to half its volume under vacuum causing an oil to precipitate. Approximately 30 mL of the oil was separated from the reaction mixture. The oil was mixed with 30 mL of acetone and diluted with 60 mL of EtOH. The solution was concentrated under vacuum until white crystals formed. The crystals were isolated by filtration and washed with 2 × 20 mL of EtOH. Yield: (0.560 g, 18%). 1H NMR (500 MHz, DMSO-d6) δ 7.24−7.34 (m, J = 15.8, 7.4 Hz, 10H), 4.54/4.51/4.38/4.35 (ABq, 8H), 3.87 (s, 4H). These data match those from samples prepared according to literature methods.23,24 Anal. Calcd for C18H22N2S2: C, 65.41; H, 6.71; N, 8.48. Found: C, 65.18; H, 6.66; N, 8.43%. ESI-MS (m/z): 331 (MH+).
Ni[(SCH2)2NBn](dppe)
A slurry was prepared of BnN(CH2SAc)2 (283.4 mg, 1.00 mmol), NaOtBu (192.2 mg, 2.00 mmol), and NiCl2(dppe) (528.0 mg, 1.00 mmol) in THF (45 mL) at −78 °C with stirring. After 4 h, the purple-red mixture was allowed to warm to room temperature and filtered, with the filtrate being carefully layered with pentane. Red-orange crystals of the product were carefully isolated using a pipet, the remainder of the material (an orange powder) being recrystallized from CH2Cl2/pentane. The combined crops were washed with pentane (2 × 10 mL) and dried to afford the title compound as red-orange crystals. Yield: 210 mg (32%). 1H NMR (500 MHz, CD2Cl2, 298 K): δ 7.89 (m, 8H, H2), 7.53 (m, 4H, H4), 7.49 (m, 8H, H3), 7.33 (d, 3JHH = 7.5 Hz, 2H, H2-CH2Ph), 7.24 (dd, 3JHH = 7.5 Hz, 3JHH = 7.5 Hz, 2H, H3-CH2Ph), 7.19 (d, 3JHH = 7.5 Hz, 1H, H4-CH2Ph), 3.99 (s, 2H, CH2Ph), 3.90 (d, 4JPH = 4.3 Hz, 4H, SCH2), 2.18 (d, 3JPH = 16.9 Hz, 4H, PCH2). 31P{1H} NMR (202 MHz, CD2Cl2, 298 K): δ 57.3. ESI-MS: m/z 654.1 (MH+). Red prisms of Ni[(SCH2)2NBn](dppe) formed upon slow diffusion of pentane layered onto a concentrated CH2Cl2 solution of the title compound at −28 °C.
Protonation of Ni[(SCH2)2NBn](dppe)
A solution of Ni-[(SCH2)2NBn](dppe) (5.4 mg, 8.25 μmol) in 0.5 mL of CD2Cl2 in a J-young tube was treated with HOTf (8.4 μL, 1 M) resulting in a color from orange to yellow-orange. 1H NMR (500 MHz, CD2Cl2): δ 8.84 (br. s, 1H, N(H)Bn), 7.80−7.36 (m, 20 H, PPh2), 4.19 (dt, 3JHH = 12.4, 4.0 Hz, 2H, SCH2N), 3.79 (dt, 3JHH = 12.4, 4.2 Hz, 2H, SCH2N). (ddt, 3JPH = 14.3 Hz, 3JHH = 7.9, 3.7 Hz, 4H, PCH2). 31P{1H} NMR (202 MHz, CD2Cl2, 298 K): δ 61.03.
Ni[(SCH2)2NBn](dcpe)
This compound was prepared as yellow-orange crystals analogously to Ni[(SCH2)2NBn](dppe), using NiCl2(dcpe) as the precursor. Yield: 28%. 1H NMR (500 MHz, CD2Cl2, 298 K): δ 7.42−7.18 (m, 5H, Ph), 3.95 (s, 2H, CH2Ph), 3.89 (d, 4JPH = 3.0 Hz, 4H, SCH2), 2.58 (d, 3JPH = 12.7 Hz, 2H, PCH), 2.37 (d, 3JPH = 12.7 Hz, 4H, PCH2), 2.28 (d, 3JPH = 13.8 Hz, 2H, PCH), 2.20−1.46 (m, 40H, (CH2)5). 31P NMR (202 MHz, CD2Cl2, 298 K): δ 73.6. ESI-MS: m/z 676.3 (MH+). Orange blocks of Ni[(SCH2)2-NBn](dcpe)·CH2Cl2 formed upon slow diffusion of pentane layered onto a concentrated CH2Cl2 solution of the title compound at −28 °C.
Pd[(SCH2)2NBn](dppe)
This compound was prepared as orange crystals analogously to Ni[(SCH2)2NBn](dppe), using PdCl2(dppe) as the precursor. Yield: 24%. 1H NMR (500 MHz, CD2Cl2, 298 K): δ 7.82 (m, 8H, H2), 7.52 (m, 4H, H4), 7.48 (m, 8H, H3), 7.35 (d, 3JHH = 7.6 Hz, 2H, H2-CH2Ph), 7.24 (dd, 3JHH = 7.9 Hz, 3JHH = 7.9 Hz, 2H, H3-CH2Ph), 7.18 (m, 1H, H4-CH2Ph), 4.19 (s, 2H, CH2S), 4.19 (s, 2H, CH2S), 4.17 (s, 2H, CH2Ph), 2.41 (s, 2H, PCH2), 2.37 (s, 2H, PCH2). 31P{1H} NMR (202 MHz, CD2Cl2, 298 K): δ 50.3. ESI-MS: m/z 702.1 (MH+). Anal. Calcd for C35H35NP2Pd2S2: C, 59.87; H, 5.02; N, 1.99. Found: C, 59.55; H, 5.12; N, 1.64%.
ClC6H4N(CH2SAc)2
A solution of 4-chloroaniline (12.76 g, 100 mmol) in 100 mL of 95% EtOH was treated with formaldehyde (37% in methanol/H2O, 24 mL). The mixture was heated at 60 °C for 30 min and then treated with AcSH (14.1 mL, 200 mmol). After 2 h, the reaction mixture was poured into ~100 mL of ice−water, when yellow crystals formed. The crystals were collected by filtration and washed with 100 mL of cold EtOH. Yield: 24.6 g (81%). 1H NMR (500 MHz, DMSO-d6, 298 K): δ 7.29 (d, 3JHH = 7.69 Hz, 2H, C6H4), 6.80 (d, 3JHH = 7.69 Hz, 2H, C6H4), 5.10 (s, 4H, C(O)CH2), 2.35 (s, 6H, CH3). 13C NMR (75.47 MHz, DMSO-d6, 298 K): δ 196 (C=O), 143 (C6H4), 129 (C6H4), 124 (C6H4), 116 (C6H4), 52.1 (C(O)CH2), 31.2 (CH3). Anal. Calcd for C12H14ClNO2S2: C, 47.44; H, 4.64; N, 4.61. Found: C, 47.13; H, 4.5; N, 4.61%. Also prepared analogously was 4-MeC6H4N(CH2SAc)2. Anal. Calcd for C13H17NO2S2: C, 55.09; H, 6.05; N, 4.94. Found: C, 55.24; H, 6.02; N, 4.98%.
Ni[(SCH2)2NC6H4Cl](dppe)
A slurry was prepared of ClC6H4N-(CH2SAc)2 (304.8 mg, 1.00 mmol), NaOtBu (192.2 mg, 2.00 mmol), and NiCl2(dppe) (528.0 mg, 1.00 mmol) in THF (45 mL) at −78 °C with stirring. After 4 h, the purple-red mixture was allowed to warm to room temperature, resulting in a red solution and some green solid precipitate. The solids were removed by filtration and carefully layered with pentane, affording red-orange crystals. Yield: 505 mg (75%). 1H NMR (500 MHz, CD2Cl2, 298 K): δ 7.89 (m, 8H, H2), 7.53 (m, 4H, H4), 7.49 (m, 8H, H3), 7.33 (d, 3JHH = 7.5 Hz, 2H, H2-CH Ph), 7.24 (dd, 3JHH = 7.5 Hz, 3JHH = 7.5 Hz, 2H, H3-CH2Ph), 7.19 (d, 3JHH = 7.5 Hz, 1H, H4-CH2Ph), 3.99 (s, 2H, CH2Ph), 3.90 (d, 4JPH = 4.3 Hz, 4H, SCH2), 2.18 (d, 3JPH = 16.9 Hz, 4H, PCH2) (NMR analysis of the green solid was the same, except for the presence of THF). 31P{1H} NMR (202 MHz, CD2Cl2, 298 K): δ 57.3. ESI-MS: m/z 654.1 (MH+). Red prisms of Ni[(SCH2)2NBn](dppe) formed upon slow diffusion of pentane layered onto a concentrated CH2Cl2 solution of the title compound at °C. Anal. Calcd for C34H32ClNNiP2S2·CH2Cl2: C, 55.33; H, 4.51; N, 1.84. Found: C, 55.72; H, 4.35; N, 2.08%.
TsN(CH2SAc)2
A suspension of KSAc (2.73 g, 23.9 mmol) and TsN(CH2Cl)236 (3.06 g, 11.4 mmol) in THF (60 mL) was stirred for 27 h, after which the reaction mixture was filtered to remove NaCl and the colorless filtrate was evaporated to afford a yellow oily residue. A CH2Cl2 extract of the crude product was chromatographed on silica gel eluting with 4:1 hexane/EtOAc. The second band was collected, and the solvent was removed to yield the product TsN(CH2SAc)2 as colorless crystals. Yield: 3.04 g (77%). 1H NMR (400 MHz, CDCl3, 293 K): δ 7.69 (d, 2H, H2), 7.32 (d, 2H, H3), 4.89 (s, 4H, NCH2S), 2.45 (s, 3H, CH3), 2.31 (s, 6H, C(O)CH3). Anal. Calcd for C13H17NO4S3: C, 44.94; H, 4.93; N, 4.03. Found: C, 45.17; H, 4.76, N, 4.32%.
TsN(CH2SH)2
A solution of HCl (12 M aqueous, 4.88 mL, 5.76 mmol) in MeOH (100 mL) was sparged with N2 and then added to a solution of TsN(CH2SAc)2 (0.993 g, 2.86 mmol) in MeOH (20 mL). The resulting homogeneous solution was stirred for 21 h at 50 °C, afforded a white residue. A CH2Cl2 after which solvent evaporation a extract of this solid was chromatographed (SiO2, CH2Cl2 eluent). The first band was collected and evaporated to dryness to give the dithiol as a white solid. Yield: 310 mg (41%). 1H NMR (400 MHz, CDCl3, 293 K): δ 7.72 (d, 2H, H2), 7.34 (d, 2H, H3), 4.66 (d, 4H, NCH2S), 2.44 (s, 3H, CH3), 1.79 (t, 2H, SH). Anal. Calcd for C9H13NO2S3: C, 41.04; H, 4.97; N, 5.32. Found: C, 41.31; H, 4.99; N, 5.19%. mp 51−53 °C.
Fe2[(SCH2)2NTs](CO)6
A solution of TsN(CH2SH)2 (263.4 mg, 1.00 mmol) and Fe3(CO)12 (514.1 mg, 1.02 mmol) in toluene (30 mL) was stirred at 90 °C for 3 h, during which time the solution assumed a red color. The reaction mixture was evaporated to yield 472 mg of brick-red solid, which crystallized from CH2Cl2 (5 mL) upon the addition of hexanes followed by cooling. Yield: 189 mg (35%). 1H NMR (400 MHz, CDCl3, 293 K): δ 7.50 (d, 2H), 7.30 (d, 2H), 3.65 (s, 4H), 2.43 (s, 3H). FTIR (CH2Cl2): νCO 2080, 2043, 2005, 1985 cm−1. Anal. Calcd for C15H11Fe2NO8S3: C, 33.29; H, 2.05; N, 2.59. Found: C, 33.10; H, 1.85; N, 2.63%. Red prisms of Fe2[(SCH2)2NTs]-(CO)6 formed upon slow diffusion of hexane layered onto a concentrated CH2Cl2 solution of the title compound.
Ni[(SCH2)2NTs](dppe)
A solution of TsN(CH2SH)2 (1.57 g, 6.98 mmol) in 100 mL of CH2Cl2 was added to a slurry of NiCl2(dppe) (2.63 g, 5.00 mmol) and 150 mL of CH2Cl2. A solution of NaOEt (0.68 g, 10.0 mmol) in 45 mL of EtOH was added into the mixture. The mixture darkened in color, the NiCl2(dppe) dissolved, and NaCl precipitated. After the reaction mixture was stirred for 3 h, solvent was removed under vacuum. The product was extracted into CH2Cl2 (90 mL). The extract was concentrated to a volume of 10 mL. Brick-red powder precipitated from the concentrate upon the addition of 90 mL of hexane. Yield: 3.15 g (88%). 1H NMR (500 MHz, CD2Cl2, 293 K): δ 7.74 (dd, 8H, Ph), 7.63 (d, 2H, Ts), 7.52 (t, 4H, Ph), 7.46 (t, 8H, Ph), 7.21 (d, 2H, Ts), 4.21 (d, 4H, NCH2S), 2.38 (s, 3H, CH3), 2.19 (d, 4H, PCH2). 13C{1H} NMR (125 MHz, CD2Cl2, 293 K): δ 143.2, 134, 132, 130, 129, 128, 125, 121, 43.7, 27.5, 21.6. 31P{1H} NMR (202 MHz, CD2Cl2, 293 K): δ 57.6. Anal. Calcd for C35H35NNiO2P2S3: C, 58.51; H, 4.91; N, 1.95. Found: C, 57.88; H, 4.74; N, 2.06%. Red prisms of Ni[(SCH2)2NTs](dppe) formed upon slow diffusion of hexane layered onto a concentrated CH2Cl2 solution of the title compound.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by the National Institutes of Health (No. GM061153). R.A. is grateful for the Rubicon grant from The Netherlands Organisation for Scientific Research (NWO). We thank Drs. A. Fuller and C. Richers for advice and assistance.
Footnotes
Supporting Information
NMR, ESI-MS, and IR spectra, crystallographic data and structures in CIFs, CV data, and plot of ipcat/ip of [Ni[(SCH2)2NHBn](dppe)]+ (1 mM) versus [CF3CO2H] added, where ip is the current with 1 mM acid. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem.5b00290.
Notes
The authors declare no competing financial interest.
REFERENCES
- 1.Nicolet Y, Lemon BJ, Fontecilla-Camps JC, Peters JW. Trends Biochem. Sci. 2000;25:138–143. doi: 10.1016/s0968-0004(99)01536-4. [DOI] [PubMed] [Google Scholar]
- 2.Barton BE, Olsen MT, Rauchfuss TB. J. Am. Chem. Soc. 2008;130:16834–16835. doi: 10.1021/ja8057666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carroll ME, Barton BE, Rauchfuss TB, Carroll PJ. J. Am. Chem. Soc. 2012;134:18843–18852. doi: 10.1021/ja309216v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Camara JM, Rauchfuss TB. J. Am. Chem. Soc. 2011;133:8098–8101. doi: 10.1021/ja201731q. [DOI] [PMC free article] [PubMed] [Google Scholar]; Camara JM, Rauchfuss TB. Nat. Chem. 2012;4:26–30. doi: 10.1038/nchem.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicolet Y, de Lacey AL, Vernede X, Fernandez VM, Hatchikian EC, Fontecilla-Camps JC. J. Am. Chem. Soc. 2001;123:1596–1601. doi: 10.1021/ja0020963. [DOI] [PubMed] [Google Scholar]
- 6.Bullock RM, Appel AM, Helm ML. Chem. Commun. 2014;50:3125–3143. doi: 10.1039/c3cc46135a. [DOI] [PubMed] [Google Scholar]; DuBois DL. Inorg. Chem. 2014;53:3935–3960. doi: 10.1021/ic4026969. [DOI] [PubMed] [Google Scholar]
- 7.Shook RL, Borovik AS. Inorg. Chem. 2010;49:3646–3660. doi: 10.1021/ic901550k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Silakov A, Wenk B, Reijerse E, Lubitz W. Phys. Chem. Chem. Phys. 2009;11:6592–6599. doi: 10.1039/b905841a. [DOI] [PubMed] [Google Scholar]; Lubitz W, Ogata H, Rüdiger O, Reijerse E. Chem. Rev. 2014;114:4081–4148. doi: 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- 9.Stich TA, Myers WK, Britt RD. Acc. Chem. Res. 2014;47:2235–2243. doi: 10.1021/ar400235n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Myers WK, Stich TA, Suess DLM, Kuchenreuther JM, Swartz JR, Britt RD. J. Am. Chem. Soc. 2014;136:12237–12240. doi: 10.1021/ja507046w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Berggren G, Adamska A, Lambertz C, Simmons TR, Esselborn J, Atta M, Gambarelli S, Mouesca J-M, Reijerse E, Lubitz W, Happe T, Artero V, Fontecave M. Nature. 2013;499:66–69. doi: 10.1038/nature12239. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Esselborn J, Lambertz C, Adamska-Venkatesh A, Simmons T, Berggren G, Noth J, Siebel J, Hemschemeier A, Artero V, Reijerse E, Fontecave M, Lubitz W, Happe T. Nat. Chem. Biol. 2013;9:607–609. doi: 10.1038/nchembio.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siebel JF, Adamska-Venkatesh A, Weber K, Rumpel S, Reijerse E, Lubitz W. Biochem. 2015;54:1474–1483. doi: 10.1021/bi501391d. [DOI] [PubMed] [Google Scholar]
- 13.Lawrence JD, Li H, Rauchfuss TB, Bénard M, Rohmer M-M. Angew. Chem., Int. Ed. 2001;40:1768–1771. doi: 10.1002/1521-3773(20010504)40:9<1768::aid-anie17680>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 14.Li H, Rauchfuss TB. J. Am. Chem. Soc. 2002;124:726–727. doi: 10.1021/ja016964n. [DOI] [PubMed] [Google Scholar]
- 15.Angamuthu R, Carroll ME, Ramesh M, Rauchfuss TB. Eur. J. Inorg. Chem. 2011;2011:1029–1032. doi: 10.1002/ejic.201001208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Z, Liu J-H, He C-J, Jiang S, Åkermark B, Sun L-C. J. Organomet. Chem. 2007;692:5501–5507. [Google Scholar]
- 17.Das P, Capon J-F, Gloaguen F, Pétillon FY, Schollhammer P, Talarmin J, Muir KW. Inorg. Chem. 2004;43:8203–8205. doi: 10.1021/ic048772+. [DOI] [PubMed] [Google Scholar]
- 18.Gao S, Fan J, Sun S, Peng X, Zhao X, Hou J. Dalton Trans. 2008:2128–2135. doi: 10.1039/b717497g. [DOI] [PubMed] [Google Scholar]
- 19.Lysenko NM. Zh. Org. Khim. 1974;10:2049. [Google Scholar]
- 20.Izawa T, Terao Y, Suzuki K. Tetrahedron: Asymmetry. 1997;8:2645–2648. [Google Scholar]
- 21.Lin T, Ulloa OA, Rauchfuss TB, Gray DL. Eur. J. Inorg. Chem. 2014;2014:4109–4114. doi: 10.1002/ejic.201402413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu W, Marr AC, Wang Q, Neese F, Spencer DJE, Blake AJ, Cooke PA, Wilson C, Schröder M. Proc. Natl. Acad. Sci. U. S. A. 2005;102:18280–18285. doi: 10.1073/pnas.0505779102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bourrez M, Steinmetz R, Gloaguen F. Inorg. Chem. 2014;53:10667–10673. doi: 10.1021/ic501815m. [DOI] [PubMed] [Google Scholar]
- 24.Fraze K, Wilson AD, Appel AM, Rakowski DuBois M, DuBois DL. Organometallics. 2007;26:3918–3924. [Google Scholar]
- 25.(a) Green TW, Wuts PGM. Protective Groups in Organic Synthesis. Wiley-Interscience; New York: 1999. [Google Scholar]; (b) Hartung R, Golz G, Schlaf S, Silvennoinen G, Polborn K, Mayer P, Pfaendler HR. Synthesis. 2009:495–501. [Google Scholar]
- 26.Tard C, Pickett C. J. Chem. Rev. 2009;109:2245–2274. doi: 10.1021/cr800542q. [DOI] [PubMed] [Google Scholar]
- 27.Böhme H, Hartke K. Chem. Ber. 1963;96:604–606. [Google Scholar]
- 28.(a) Böhme H, Kietz K, Leidreiter KD. Arch. Pharm. 1954;287:198–209. doi: 10.1002/ardp.19542870406. [DOI] [PubMed] [Google Scholar]; (b) Wellmar U. J. Heterocycl. Chem. 1998;35:1531–1532. [Google Scholar]; (c) Miyazawa S, Ikeda K, Achiwa K, Sekiya M. Chem. Lett. 1984:785–788. [Google Scholar]; (d) Rakhimova EB, Vasil’yeva IV, Khalilov LM, Ibragimov AG, Dzhemilev UM. Chem. Heterocycl. Compd. 2012;48:1050–1057. [Google Scholar]; (e) Murzakova NN, Prokof’ev KI, Tyumkina TV, Ibragimov AG. Russ. J. Org. Chem. 2012;48:588–593. [Google Scholar]
- 29.(a) Stanley JL, Rauchfuss TB, Wilson SR. Organometallics. 2007;26:1907–1911. doi: 10.1021/om0611150. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Song L-C, Wang L-X, Yin B-S, Li Y-L, Zhang X-G, Zhang Y-W, Luo X, Hu QM. Eur. J. Inorg. Chem. 2008;2008:291–297. [Google Scholar]
- 30.(a) Rauchfuss TB, Roundhill DM. J. Am. Chem. Soc. 1975;97:3386–3392. [Google Scholar]; (b) Kochem A, Weyhermüller T, Neese F, van Gastel M. Organometallics. 2015;34:995–1000. [Google Scholar]; (c) Gan L, Groy TL, Tarakeshwar P, Mazinani SKS, Shearer J, Mujica V, Jones AK. J. Am. Chem. Soc. 2015;137:1109–1115. doi: 10.1021/ja509779q. [DOI] [PubMed] [Google Scholar]
- 31.(a) Swanson KD, Duffus BR, Beard TE, Peters JW, Broderick JB. Eur. J. Inorg. Chem. 2011;2011:935–947. [Google Scholar]; (b) Dinis P, Suess DLM, Fox SJ, Harmer JE, Driesener RC, De La Paz L, Swartz JR, Essex JW, Britt RD, Roach PL. Proc. Nat. Acad. Sci. U.S.A. 2015 00, 0000. [Google Scholar]
- 32.Peters JW, Broderick JB. Annu. Rev. Biochem. 2012;81:429–450. doi: 10.1146/annurev-biochem-052610-094911. [DOI] [PubMed] [Google Scholar]
- 33.(a) Fugate CJ, Stich TA, Kim EG, Myers WK, Britt RD, Jarrett JT. J. Am. Chem. Soc. 2012;134:9042–9045. doi: 10.1021/ja3012963. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jarrett JT. In: Iron-Sulfur Clusters in Chemistry and Biology. Rouault TA, editor. Walter de Gruyter; Berlin, Germany: 2014. pp. 107–131. [Google Scholar]
- 34.Mityuk AP, Denisenko AV, Dacenko OP, Grygorenko OO, Mykhailiuk PK, Volochnyuk DM, Shishkin OV, Tolmachev AA. Synthesis. 2010;2010:493–497. doi: 10.1021/ol101866x. [DOI] [PubMed] [Google Scholar]
- 35.Cadenas-Pliego G, Jésus Rosales-Hoz M. d., Contreras R, Flores-Parra A. Tetrahedron: Asym. 1994;5:633–640. [Google Scholar]
- 36.Beermann C, Ramloch H. Ger. Offen. DE1915898. 1970 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.