

Graphical abstract
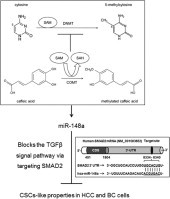
Abbreviations: CaA, 3,4-dihydroxycinnamic acid; CSCs, cancer stem cells; DNMT, DNA methyltransferases; HCC, hepatocellular carcinoma; miR-148a, microRNA-148a; qMSP, quantitative methylation-specific polymerase chain reaction; SAM, S-adenosylmethionine; TGFβ, transforming growth factor beta; TNBC, triple-negative breast cancer
Keywords: Cancer stem cells-like properties, Caffeic acid, Transforming growth factor beta-SMAD2 signal pathway, microRNA-148a, DNA methylation
Highlights
-
•
Caffeic acid (CaA) attenuates CSCs-like properties in human cancer cells.
-
•
CaA inhibits the activity/expression of SMAD2.
-
•
CaA elevates the expression of miR-148a by inducing DNA demethylation.
-
•
miR-148a targets SMAD2 in CaA-treated cells.
-
•
CaA attenuates CSCs-like properties via miR-148a.
Abstract
Current standard practices for treatment of cancers are less than satisfactory because of recurrence mediated by cancer stem cells (CSCs). Caffeic acid (CaA) is a novel anti-tumor agent that inhibits proliferation, migration, and invasion in human cancer cells. However, little is known about the functions of CaA in regulating CSCs-like properties and the potential molecular mechanisms. Here, we found that CaA attenuated the CSCs-like properties by the microRNA-148a (miR-148a)-mediated inhibition of transforming growth factor beta (TGFβ)-SMAD2 signaling pathway both in vitro and in vivo. CaA enhanced the expression of miR-148a by inducing DNA methylation. MiR-148a, which targeted the SMAD2-3′UTR, decreased the expression of SMAD2. Knockdown of miR-148a abolished the CaA-induced inhibition of TGFβ-SMAD2 signal pathway and the CSCs-like properties. Our study found a novel mechanism that CaA inhibits the CSCs-like properties via miR-148a-mediated inhibition of TGFβ-SMAD2 signaling pathway, which may help to identify a new approach for the treatment of human cancers.
1. Introduction
The cancer stem cells (CSCs) hypothesis states that the characteristic of neoplastic tissues is the existence of self-renewing, stem-like cells, which constitute a small portion of the neoplastic cells and exhibit the capacity to produce new tumors [1]. There is mounting evidence that CSCs occur in human solid tumors, including those found in breast, liver, colon and lung [1–3]. The elevation of CSCs-like properties is involved in the initiation, tumorigenesis, metastasis, angiogenesis, and recurrence of various human cancers [1,4]. The efficacy of standard chemotherapy treatments for cancer are decreased by the properties of CSCs, which include self-renewal, lack of differentiation, quiescence, distinct epigenetic state, deregulation of apoptotic/survival pathways, metabolism and interactions with the microenvironment [5,6]. For example, the cytotoxic chemotherapies such as taxanes and cisplatin are less than satisfactory partly because of the toxic/side-effects and induction the generation of CSCs [5,7]. So the continued search for novel naturally agents, which exhibit less cytotoxic and have repressive effects on CSCs or the cancer cells with CSCs-like properties are urgently needed.
Caffeic acid (3,4-dihydroxycinnamic acid, CaA), a naturally occurring hydroxycinnamic acid derivatives, is an active component in the phenolic propolis extract and also in a wide variety of plants [8]. Caffeic acid has several biological and pharmacological properties, such as antiviral, antioxidants, anti-inflammatory, and immunomodulatory activates [9]. In addition to these functions, CaA exerts anticancer effects. CaA decreases the viability of human hepatocellular carcinoma (HCC) HepG2 and Huh-7 cells in a time-dependent manner [10]. Meanwhile, it inhibits the DNA methylation in human breast cancer (BC) MCF-7 and MDA-MB-231 cells [11]. Moreover, CaA has been identified as a new matrix metalloproteinase-9 inhibitor, which may be involved in tumor cell invasion and metastasis [12]. Further, CaA and its derivative, caffeic acid phenethyl ester (CAPE), suppress the angiogenesis of human renal carcinoma cells by blocking STAT3-mediated VEGF expression [13]. Recent studies have demonstrated that, in breast CSCs isolated from MDA-MB-231 cells, CAPE causes pronounced changes in breast CSCs characteristics manifested by inhibition of self-renewal, progenitor formation, and clonal growth in soft agar [14]. However, the molecular mechanisms underlying in the CaA-induced repressive effects on CSCs or the cancer cells with CSCs-like properties remain largely uninvestigated.
We previously showed that blockage of TGFβ/SMADs signal pathway is involved in the glabridin-induced inhibition of CSCs-like properties in HCC cells [15]. Here we treated human triple-negative breast cancer (TNBC) cell line, MDA-MB-231, and human HCC cell line, MHCC97H, by CaA in vitro and in vivo to investigate the relationships among CaA, TGFβ/SMADs signal pathway, and CSCs-like properties, and to determine the underlying molecular mechanisms.
2. Materials and methods
2.1. Cell culture and reagents
Human TNBC cell line, MDA-MB-231, was obtained from American Type Culture Collection (ATCC, Rockville, MD, USA). Human HCC cell line, MHCC97H, was obtained from the Liver Cancer Institute, Zhongshan Hospital, Fudan University, Shanghai, China [16]. MDA-MB-231 cells were maintained at 37 °C in the absence of CO2 in L-15 medium (Life Technologies/Gibco, Grand Island, NY, USA), while the MHCC97H cells were maintained in 5% CO2 at 37 °C in Dulbecco’s Modified Eagle Medium (DMEM, Gibco). Both mediums were supplemented with 10% fetal bovine serum (FBS, Gibco), 100 U/ml penicillin, and 100 mg/ml streptomycin (Gibco). The caffeic acid (CaA, purity ⩾ 99%) and S-adenosylmethionine (SAM, a methyl donor) were purchased from Sigma Chemical Co. (St. Louis, MO, USA).
2.2. Animals
This study was performed according to a protocol approved by the Nanjing Medical University Institutional Animal Care and Use Committee (Permit Number: NJMU-IACUC-1403022). Briefly, the BALB/c nude mice were kept in a temperature controlled environment (20–22 °C) with a 12 h light dark cycle and with free access to drinking water and chow. For xenograft, 5 × 106 MHCC97H cells were injected subcutaneously into the right armpit of the mice. After the establishment of the tumors, CaA (0 or 10 mg/kg·BW) was administered intraperitoneally [i.p [13]] twice per week. Tumor volumes were measured weekly and tumor size was calculated using the formula: V = ½ (width2 × length) [8]. After 11 weeks, mice were sacrificed, and the tumor tissues were removed for further investigation.
2.3. Quantitative real-time polymerase chain reaction (qRT-PCR)
Primers used were listed in Table 1. The miRNA primers were synthesized by RiboBio Co. (Guangzhou, China). Total RNA was isolated using Trizol (Invitrogen, Carlsbad, USA) according to the manufacturer’s recommendations. For detection of mRNAs, total RNA (2 μg) was transcribed into cDNA using AMV Reverse Transcriptase (Promega, Madison, USA). For detection of miRNAs, 2 μg of total RNA, miRNA-specific stem-loop RT primers, and MMLV reverse transcriptase (Promega) were used in reverse transcription following the manufacturer’s protocol. qRT-PCR was performed using an ABI 7300 real-time PCR detection system (Applied Biosystems by Life Technologies, Grand Island, NY, USA). Fold changes in expression of each gene were calculated by a comparative threshold cycle (Ct) method using the formula 2−(ΔΔCt).
Table 1.
Primers used for qRT-PCR.
| Genes | Primers (5′–3′) |
|---|---|
| CD44 | TGAGCATCGGATTTGAGAC (F) |
| CATACTGGGAGGTGTTGGA (R) | |
| EpCAM | AAGGAGAAACAGGAAACCTC (F) |
| ACAGACACAGTCCAACTTCC (R) | |
| ALDH-1 | TGTCCAAGTCGGCATCAG (F) |
| GGCAGCCATTTCTTCTCA (R) | |
| 4-Oct | GCTTCCTCCACCCACTTCT (F) |
| GTATTCAGCCAAACGACCAT (R) | |
| BMI-1 | CTGATGACCCATTTACTGA (F) |
| CTCCACCTCTTCTTGTTTG (R) | |
| SMAD2 | GTTCCTGCCTTTGCTGAC (F) |
| TCTCTTTGCCAGGAATGCTT (R) | |
| Snail | TTCTCCCGAATGTCCCT (F) |
| TCAGCCTTTGTCCTGTAGC (R) | |
| CDH-1 | TGCTCACATTTCCCAACTC (F) |
| TCTGTCACCTTCAGCCATC (R) | |
| Lin-28B | CCTTGAGTCAATACGGGT (F) |
| ACGGTAATGACAGTCTCG (R) |
2.4. Western blots
Cell lysates were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (PVDF, Millipore, Billerica, USA); the immune complexes were detected by enhanced chemiluminescence (Cell Signaling Technology, Beverly, MA, USA). Antibodies used were those for total SMAD2, phospho-SMAD2 (Ser 465/467), SMAD4, DNA methyltransferases 1 (DNMT1, Cell Signaling Technology) and glyceraldehyde phosphate dehydrogenase (GAPDH, Sigma).
2.5. Analysis of side population (SP) cells
After treated MHCC97H cells were removed from the culture dish, washed, resuspended in DMEM/F-12 medium (Gibco) containing 5% FBS, and incubated in a 1.5 ml eppendorf tube at 37 °C for 10 m, they were labeled with 5 mg/ml Hoechst 33342 dye (Sigma) in the presence or absence of 50 mM verapamil (an inhibitor of ATP-binding cassette transporters, Sigma) at 37 °C for 90 m, followed by counterstaining with 1 mg/ml of propidium iodide (Sigma) to label dead cells. Then, 1 × 105 cells were passed through a FACSVantage fluorescence-activated cell sorter (Becton Dickinson, East Rutherford, USA).
2.6. Mammosphere assays
In non-adherent 24 well dishes (Costar, US), treated MDA-MB-231 cells (2 × 103) were suspended in defined, serum-free medium composed of DMEM-F12 (Gibco), 10 ng/ml of human recombinant basic fibroblast growth factor (bFGF, R&D Systems, Minneapolis, USA), and 10 ng/ml of epidermal growth factor (EGF, R&D Systems). Cells were grown for 10 days and fed every 48 h. Mammospheres were then counted under a microscope (Olympus, Tokyo, Japan).
2.7. Cell transfection and luciferase reporter assay
Con-mimic and miR-148a-mimic were synthesized by RiBoBio. The pGL3-SMAD2-3′UTR (wild type, WT)-Luc construct and pGL3-SMAD2-3′UTR (mutant, MT, lacking the miR-148 binding sites)-Luc construct were synthesized by Shuntian Biology (Shanghai, China). The plasmid phRL-tk (used as an internal control for transfection efficiency and cytotoxicity of test chemicals) containing the Renilla luciferase gene was purchased from Promega. Briefly, cells were plated in 24-well culture dishes. When cells proliferated to 60 to 80% confluence after 24 h of culture, con-mimics or miR-148a-mimics was co-transfected with the respective reporter construct, by using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s protocol. The cells were lysed with passive lysis buffer (Promega), and the lysates were analyzed immediately with a 96-well plate luminometer (Berthold Detection System, Pforzheim, Germany). The amounts of luciferase and Renilla luciferase were measured with the Dual-Luciferase Reporter Assay System Kit (Promega) following the manufacturer’s instructions.
2.8. DNA methylation analysis
Cellular DNA was isolated using DNA purification kits (Qiagen, Germantown, MD, USA). The genomic DNA was modified with sodium bisulfite using the EpiTect Kit (Qiagen). DNA methylation was analyzed using a SYBR Green-based quantitative methylation-specific PCR (qMSP) as described previously [17]. Primers used were listed in Table 2. Briefly, 1 μl of bisulfite-treated DNA template was mixed with 10 μl of 2 × Power SYBR Green PCR Master Mix (Applied Biosystems) and a pair of primers in a final concentration of 400 nM. The PCR conditions included initial incubation at 50 °C for 2 min, denaturing at 95 °C for 10 min, and 40 cycles of denaturing at 95 °C for 15 s and annealing at 60 °C for 1 min.
Table 2.
Primers used for qMSP.
| Genes | Primers (5′–3′) |
|---|---|
| miR-148a | Methylated |
| TCGTCGGTAGTTAATAAGAGCG (F) | |
| ACGCCGCCCGCATGCTCCCCAAT (R) | |
| Unmethylated | |
| TTGTTTGTTGGTAGTTAATAAGAGTG (F) | |
| AAACACCACCCACATACTCCCCAAT (R) | |
| miR-155 | Methylated |
| TCGTTTAGGTCGGAGCGTAGCG (F) | |
| AATCGAAAATTTAAAACCG (R) | |
| Unmethylated | |
| TTGTTGTTTAGGTTGGAGTGTAGTG (F) | |
| AAAATCAAAAATTTAAAACCA (R) | |
2.9. Statistical analysis
Data were presented as the means ± SD. A Student’s t test, and a one-way analysis of variance (ANOVA) followed by Dunnett’s t test were used to assess significant differences between groups. P values <0.05 were considered statistically significant.
3. Results
3.1. CaA decreases the CSCs-like properties of human cancer cells
An increased exhibition of CSCs-like properties plays a key role in the initiation, development, and outcome of various human cancers [1]. CD44 and EpCAM are the cell-surface markers of HCC stem cells [18], while CD44 and ALDH-1 are the cell-surface markers of BC stem cells [5]. Here, CaA did not appreciably affect the viability of these cells at the concentration of 20 μM (Fig. S1A), but significantly decreased the expressions of CD44, EpCAM, and/or ALDH-1 (Fig. 1A and B). Oct-4, BMI-1, and Lin-28B are the key ‘stemness’ CSCs-associated genes in various cancers including HCC and TNBC [19–21]. Here, after exposure of these two cells to CaA, the expression of these mRNAs was significantly decreased (Fig. 1A and B). SP cells are enriched along with stem cells [5]. We then used flow cytometric analysis and found that the percentage of SP cells was decreased in the CaA-treated MHCC97H cells (Fig. 1C and D). Formation of mammospheroids demonstrates the capacity of cells for self-renewal and for the initiation/development of tumors, which are characteristics of bSCs and bCSCs [5]. As shown in Fig. 1E and F, CaA decreased the formation of mammospheroids effectively. Collectively, these results indicate that CaA attenuates the CSCs-like properties in MHCC97H and MDA-MB-231 cells.
Fig. 1.
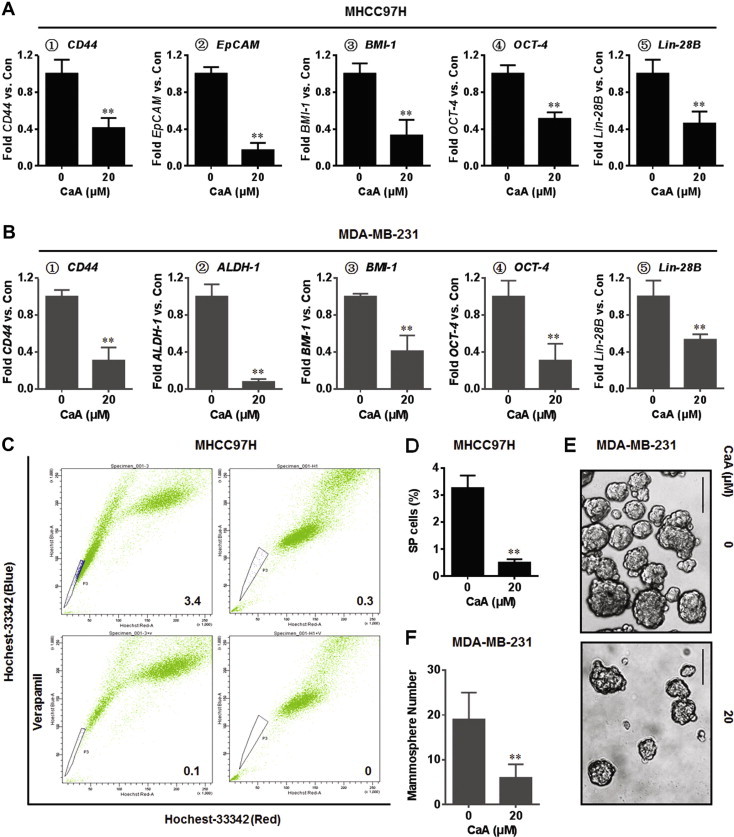
CaA decreases the CSCs-like properties of human cancer cells. MHCC97H and MDA-MB-231 cells were treated by 0 or 20 μM CaA for 72 h. (A) qRT-PCR analyses of CD44, EpCAM, BMI-1, Oct-4, and Lin-28B in MHCC97H cells (mean ± SD, n = 3). (B) qRT-PCR analyses of CD44, ALDH-1, BMI-1, Oct-4, and Lin-28B in MDA-MB-231 cells (mean ± SD, n = 3). (C and D) FACS analyses of the percentage of SP cells in MHCC97H cells (mean ± SD, n = 3). (E) Mammospheres formed by MDA-MB-231 cells (bar = 250 μm). (F) Mammospheres quantitation (mean ± SD, n = 3). ∗∗p < 0.01 compared with medium control cells.
3.2. CaA inhibits the TGFβ-SMADs signal pathway
TGFβ-SMADs signal pathway has been shown to increase the stem-like properties in human cancer cells, including HCC and TNBC [5,15]. We first examined the effects of CaA on the activation of SMAD2 and SMAD4 in TGFβ-treated MDA-MB-231 cells. As shown in Fig. 2A, CaA blocked the TGFβ-induced phosphorylation of SMAD2 in a dose-dependent manner. Interestingly, CaA attenuated the expression of total SMAD2 and SMAD4. So we next determined the effects of CaA on the expressions/activation of endogenous SMAD2 and SMAD4. As shown in Fig. 2B, with increased time of CaA exposure, there were greater decreased expressions of pSMAD2, total SMAD2, and SMAD4. Further, we examined the effects of CaA on the transcriptional activities of SMAD2 and its downstream factors (snail and CDH-1). As shown in Fig. 2C and D, there were decreased expressions of SMAD2 and snail, but increased expression of CDH-1 in CaA-treated cells. These results indicate that CaA inhibits the TGFβ-SMADs signaling pathway in MHCC97H and MDA-MB-231 cells. Further, the repressive effects of CaA on SMAD2 activity may not via the classical kinase signaling, because CaA decreased the expression of total SMAD2. MiRNAs are key regulators of gene expression, degrading mRNA or repressing translation in a post-transcriptional manner. Based on our current results, we hypothesized that miRNA might play a role in the CaA-induced attenuation of endogenous SMAD2.
Fig. 2.
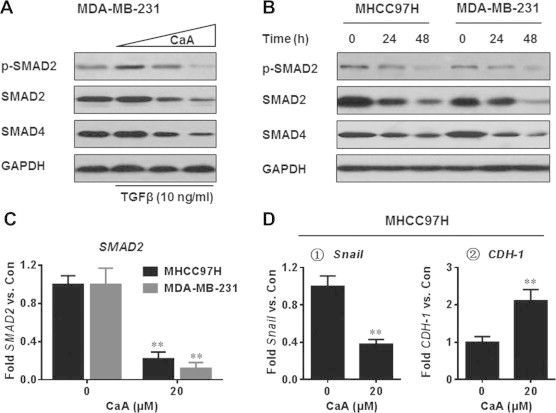
CaA inhibits the TGFβ-SMADs signal pathway. (A) After MDA-MB-231cells were pretreated by 0, 10, or 20 μM CaA for 48 h, they were exposed to 0 or 10 ng/ml TGFβ for 24 h. Western blot analyses of the expressions of SMAD2, p-SMAD2, and SMAD4. (B) MHCC97H and MDA-MB-231 cells were treated by 0 or 20 μM CaA for 24 or 48 h, respectively. Western blot analyses of the expressions of SMAD2, p-SMAD2, and SMAD4. (C) MHCC97H and MDA-MB-231 cells were treated by 0 or 20 μM CaA for 48 h, respectively. qRT-PCR analyses of SMAD2 (mean ± SD, n = 3). (D) MHCC97H cells were treated by 0 or 20 μM CaA for 48 h. qRT-PCR analyses of Snail and CDH-1 (mean ± SD, n = 3). ∗∗p < 0.01 compared with medium control cells.
3.3. MiR-148a is involved in the CaA-induced attenuation of SMAD2
We previously found that the expression of SMAD2 was repressed by miR-148a and miR-155 [15,22]. Meanwhile, it is predicted that SMAD2 might be regulated by other miRNAs, such as miR-206. So, we examined those potential miRNAs in CaA-treated MHCC97H cells. As shown in Fig. 3A, CaA elevated the expressions of these three miRNAs in MHCC97H cells. Interestingly, overexpression of miR-148a (Fig. 3B) elevated the expressions of miR-155 and miR-206 in MHCC97H and MDA-MB-231 cells (Fig. 3C). In addition, knockdown of miR-148a (Fig. 3D) partly attenuated the CaA-induced elevation of miR-155 and miR-206 in MHCC97H cells (Fig. 3E). So we chose miR-148a for further investigation.
Fig. 3.
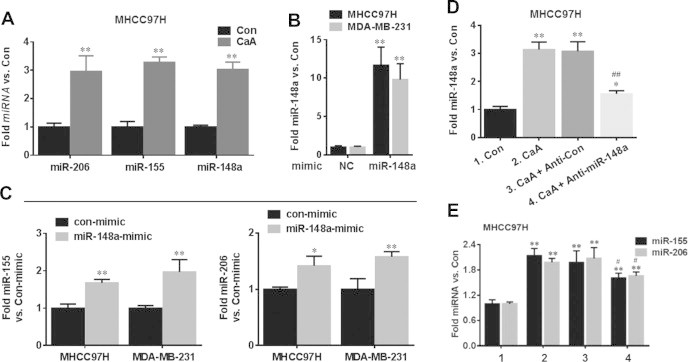
CaA enhances the expression of miR-148a, miR-155, and miR-206. MHCC97H cells were treated by 0 or 20 μM CaA for 48 h, respectively. (A) qRT-PCR analyses of miR-206, miR-155, and miR-148a (mean ± SD, n = 3). ∗∗p < 0.01 compared with medium control cells. (B and C) MHCC97H or MDA-MB-231 cells were transfected by con-mimic or miR-148a-mimic for 12 h, respectively. qRT-PCR analyses of miR-148a, miR-155 and miR-206 (mean ± SD, n = 3). ∗∗p < 0.01 compared with cells transfected by con-mimic. (D and E) After MHCC97H cells were transfected by anti-con or anti-miR-148a for 12 h, they were treated with 0 or 20 μM CaA for 48 h, respectively. qRT-PCR analyses of miR-148a, miR-155 and miR-206 (mean ± SD, n = 3). ∗∗p < 0.01 compared with medium control cells. #p < 0.05 and ##p < 0.01 compared with cells treated by 20 μM CaA alone or with anti-con-transfected cells treated by 20 μM CaA.
We then used luciferase reporter analysis to examine if SMAD2 was a direct target gene of miR-148a. The target sites and the wild type (WT)/or mutated (MT) SMAD2-3′UTR-Luc construct were exhibited in Fig. S2. Interestingly, co-transfected with pGL3-SMAD2-3′UTR-Luc construct (WT, but not MT) plus miR-148a-mimic led to a significant decrease of the luciferase activity in MHCC97H cells (Fig. 4A). Then we determined the effects of miR-148a on the expression of SMAD2. As shown in Fig. 4B–D, knockdown of miR-148a blocked the CaA-induced decreased expression of SMAD2. Further, overexpression of miR-148a by itself without CaA treatment decreased the SMAD2 level (Fig. 4E and F). These results suggest that CaA inhibits the SMAD2 by miR-148a in MHCC97H and MDA-MB-231 cells.
Fig. 4.
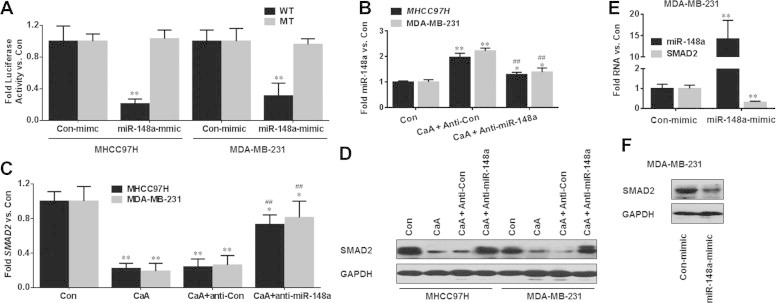
CaA decreased the expression of SMAD2 by miR-148a. (A) MHCC97H and MDA-MB-231 cells were co-transfected with pGL3-SMAD2-3′UTR (WT or MT)-Luc construct plus con-mimc or with pGL3-SMAD2-3′UTR (WT or MT)-Luc construct plus miR-148a-mimic for 12 h. Luciferase reporter assays of the effects of miR-148a on SMAD2-3′UTR. (B-D) After MHCC97H or MDA-MB-231 cells were transfected by anti-con or anti-miR-148a for 12 h, they were treated with 0 or 20 μM CaA for 48 h, respectively. qRT-PCR and/or Western blot analyses of miR-148a and SMAD2 (mean ± SD, n = 3). ∗∗p < 0.01 compared with medium control cells; ##p < 0.01 compared with cells treated by 20 μM CaA alone or with anti-con-transfected cells treated by 20 μM CaA. (E and F) MDA-MB-231 cells were transfected by con-mimic or miR-148a-mimic for 12 h, respectively. (E) qRT-PCR analyses of the expressions of miR-148a and SMAD2 (mean ± SD, n = 3). ∗∗p < 0.01 compared with cells transfected by con-mimic. (F) Western blots analyses of the expression of SMAD2.
3.4. CaA up-regulates the expression of miR-148a by demethylation
Studies indicate that there are many CpG-rich regions in miR-148a promoter, and that the silencing of miR-148a by DNA hypermethylation is an early event in human carcinogenesis [23,24]. Since biotransformation of CaA results in a deficiency of methyl donors, reducing DNA methylation (illustrated in Fig. 5), and since the catechol-O-methyltransferase (COMT), which is involved in the biotransformation of CaA, is rich in human HCC and BC cells [11,25], we hypothesized that CaA up-regulated the expression of miR-148a by demethylation.
Fig. 5.
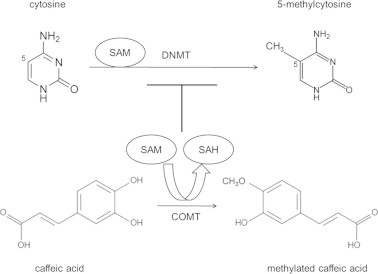
A pathway showing the demethylation effects induced by CaA. Schematic illustration of the enzymatic DNA methylation and its modulation by the COMT-mediated metabolic O-methylation of CaA. During the COMT-mediated metabolic O-methylation of CaA, there was a deficiency of methyl donors (SAM), however, the formation of SAH would be markedly increased. It is hypothesized that elevated intracellular levels of SAH due to metabolic O-methylation of CaA may exert a strong feedback inhibition of DNMT-mediated DNA methylation.
To confirm this hypothesis, qMSP was conducted. As shown in Fig. 6A and B, the average methylation level of miR-148a promoter was high in medium control MHCC97H cells, but low in CaA-treated MHCC97H cells. Then, we determined the effects of CaA on the methylation status for miR-155 [a SMAD2 associated miRNA, which is epigenetically silenced in human cancers [26]]. As shown in Fig. S3A and B, CaA also decreased the average methylation level in miR-155 promoter. We then tested the functional relevance of CaA-induced DNA demethylation and increased expression of miR-148a and miR-155. As shown in Fig. 6C and S3C, hypermethylation treatment by SAM dramatically blocked the CaA-induced elevation of miR-148a and miR-155.
Fig. 6.
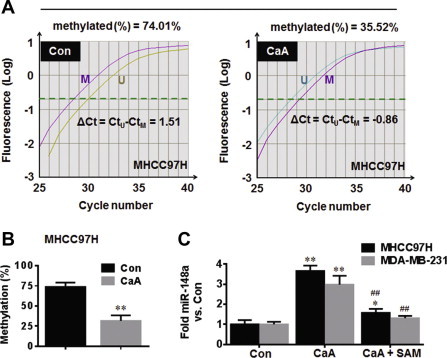
CaA up-regulates the expression of miR-148a by demethylation. MHCC97H cells were treated by 0 or 20 μM CaA for 48 h. (A) qMSP analyses and (B) average methylation level of miR-148a promoter. (C) MHCC97H or MDA-MB-231 cells were treated by 0 or 20 μM CaA in the presence or absence of 100 μM SAM for 48 h, respectively. qRT-PCR analyses of miR-148a (mean ± SD, n = 3). ∗∗p < 0.01 compared with medium control cells, ##p < 0.01 compared with cells treated by 20 μM CaA alone.
More importantly, miR-148a has been identified to target DNMT1, which down-regulates the expression of DNMT1 in breast cancer cells [23]. Here in our present study, overexpression of miR-148a (Fig. 7A) decreased the expression of DNMT1 (Fig. 7B); moreover, CaA decreased the expression of DNMT1 in MDA-MB-231 cells (Fig. 7C); further, knockdown of miR-148a (Fig. 7D) blocked this phenomenon (Fig. 7E). These results suggest that CaA up-regulates the expression of miR-148a by demethylation in MHCC97H and MDA-MB-231 cells.
Fig. 7.
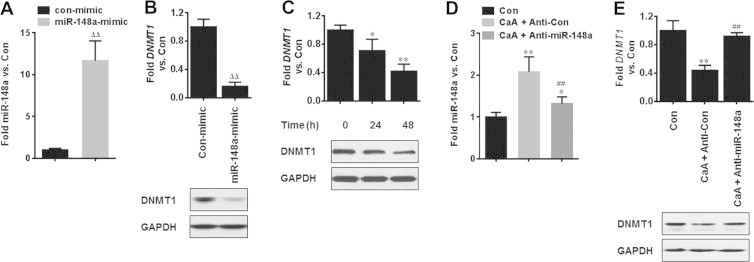
Relationships between CaA, miR-148a, and DNMT1. (A and B) MDA-MB-231 cells were transfected by con-mimic or miR-148a-mimic for 12 h, respectively. qRT-PCR and/or Western blot analyses of the expression of miR-148a and DNMT1. (C) MDA-MB-231 cells were treated by 0 or 20 μM CaA for 24 or 48 h, respectively. qRT-PCR (top) and Western blot (bottom) analyses of the expression of DNMT1. (D and E) After MDA-MB-231 cells were transfected by anti-con or anti-miR-148a for 12 h, they were treated with 0 or 20 μM CaA for 48 h, respectively. qRT-PCR and/or Western blot analyses of the expression of miR-148a and DNMT1. ∗p < 0.05 and ∗∗p < 0.01 compared with medium control cells, ##p < 0.01 compared with cells treated by 20 μM CaA plus anti-con, ΔΔp < 0.01 compared with cells transfected by con-mimic.
3.5. CaA decreases the CSCs-like properties of by miR-148a
Since the activation of TGFβ-SMADs signal enhances the CSCs-like properties [15], and since miR-148a targeted SMAD2, we hypothesized that CaA attenuated the CSCs-like properties by miR-148a. Here, knockdown of miR-148a blocked the CaA-induced decreased expressions of CD44, EpCAM, and ALDH-1 (Fig. 8A and B). Moreover, knockdown of miR-148a blocked the CaA-induced inhibition of the percentage of SP cells in MHCC97H cells (Fig. 8C and D). Further, knockdown of miR-148a blocked the CaA-induced decreased formation of mammospheroids in MDA-MB-231 cells (Fig. 8E and F). These results indicate that CaA decreases the CSCs-like properties of by miR-148a in MHCC97H and MDA-MB-231 cells.
Fig. 8.
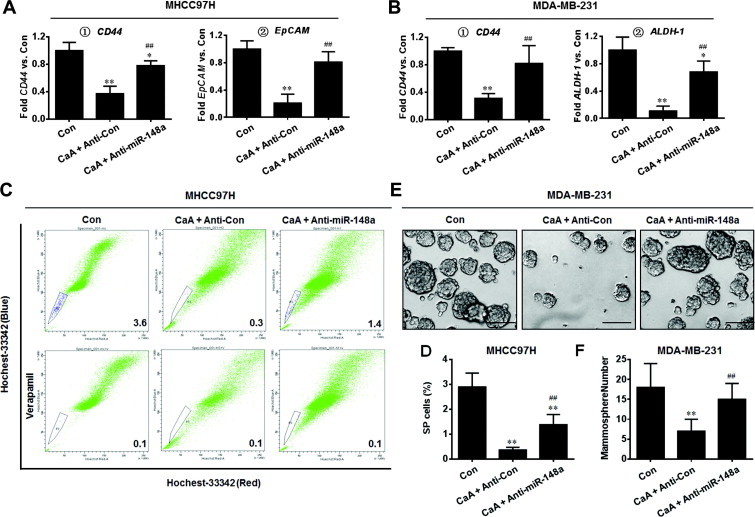
CaA decreases the CSCs-like properties of by miR-148a. After MHCC97H or MDA-MB-231 cells were transfected by anti-con or anti-miR-148a for 12 h, they were treated with 0 or 20 μM CaA for 72 h, respectively. (A) qRT-PCR analyses of CD44 and EpCAM in MHCC97H cells (mean ± SD, n = 3). (B) qRT-PCR analyses of CD44 and ALDH-1 in MDA-MB-231 cells (mean ± SD, n = 3). (C and D) FACS analyses of the percentage of SP cells in MHCC97H cells (mean ± SD, n = 3). (E) Free-floating, viable mammospheres formed by MDA-MB-231 cells (bar = 250 μm). (F) Mammospheres quantitation (mean ± SD, n = 3); ∗p < 0.05 and ∗∗p < 0.01 compared with medium control cells, and ##p < 0.01 compared with anti-con-transfected cells treated by 20 μM CaA.
3.6. Effects of CaA on miR-148a/SMAD2 and CSCs-like properties in vivo
Finally, we investigated the effects of CaA on miR-148a/SMAD2 signaling and on the CSCs-like properties in vivo. Firstly, we conducted a mice xenograft model, 5 × 106 MHCC97H cells were injected subcutaneously into the right armpit of the mice. After the establishment of the tumors, CaA (0 or 10 mg/kg·BW) was administered intraperitoneally twice per week for 11 weeks. As shown in Fig. 9A, CaA decreased the tumor growth. However, the body weight and liver weight were similar in both vehicle and CaA-treated mice (Table. 3). Then we further determined the effects of CaA on miR-148a, SMAD2, and CSCs-like properties. For each group, total RNA isolated from tumors (n = 6) were mixed, and qRT-PCR were performed. As shown in Figs. 9B–G, in CaA-treated group, there were decreased expressions of CD44, EpCAM, Oct-4, Lin-28B, and SMAD2, but increased expression of miR-148a. These results indicate that CaA improves the expression of miR-148a but inhibits the expression of SMAD2 and CSCs-like properties in vivo.
Fig. 9.
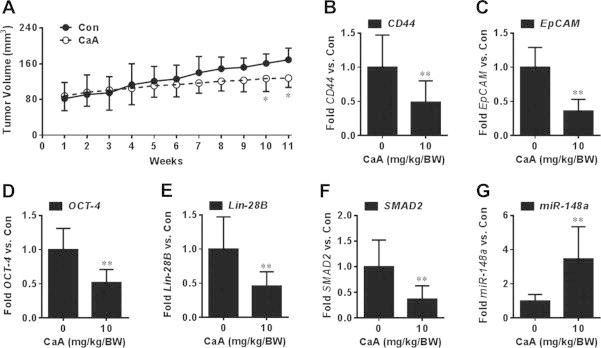
Effects of CaA on miR-148a/SMAD2 and CSCs-like properties in vivo. After MHCC97H cells were injected subcutaneously into the right armpit of the mice for 3 weeks, CaA (0 or 10 mg/kg·BW) was administered (i.p) twice per week. After 11 weeks, the mice were sacrificed, and the tumor tissues were removed for further investigation. (A) Tumor volumes were measured weekly and tumor size was calculated using the formula: V = ½ (width2 × length); (B–G) Total RNA isolated from tumors (n = 6) were mixed, qRT-PCR analyses of the expression of (B) CD44, (C) EpCAM, (D) Oct-4, (E) Lin-28B, (F) SMAD2, and (G) miR-148a (mean ± SD, n = 3). ∗∗p < 0.01 compared tumors treated with no CaA.
Table 3.
Liver weight and body weight in nude mice treated with vehicle (0.1% DMSO) or CaA (10 mg/kg·BW).
| Treatment | Liver weight | Body weight |
|---|---|---|
| Vehicle (0.1% DMSO) | 2.62 ± 0.19 | 27.79 ± 3.55 |
| CaA (10 mg/kg·BW) | 2.37 ± 0.27 | 30.22 ± 1.61 |
4. Discussion
Hydroxycinnamic acid derivatives are reported to have anticancer, anti-inflammatory, and antioxidant properties. Their natural origin and ubiquitous occurrence have prompted strong interest in their use as anticancer agents [27]. CaA is the major representative of hydroxycinnamic acids [27]. Previous studies have indicated that the daily intake of coffee was associated with a reduced incidence of colon and rectal cancer. Michels et al. reported that participants who regularly consumed two or more cups of decaffeinated coffee per day had a 52% lower incidence of rectal cancer than those who never consumed decaffeinated coffee [28]. A similar conclusion has also been reported by Tavani et al., who found that, compared with coffee non-drinkers, the risk of colon cancer was reduced in drinkers of 4 or more cups/day [29]. In the human diet, daily intake of CaA in coffee drinkers is about 0.5–1 g (approximately 0.5–1 mM), and the absorption ratio of CaA is about 95%. However, part of CaA from foods will enter into the blood circulation, but most will reach the colon [30], so we used a relative lower concentration of CaA (20 μM) to investigate the functions of CaA on the progression of cancers. Here, CaA did not appreciably affect the viability of MHCC97H and MDA-MB-231 cells (Fig. S1A), moreover, there was also no detectable cytotoxicity of CaA on L-02 (human liver epithelial cell line) and MCF-10A (human breast epithelial cell line) cells (Fig. S1B). However, CaA attenuates the CSCs-like properties in MHCC97H and MDA-MB-231 cells, which may be associated with the inhibition of TGFβ/SMAD2 signaling.
In general, the TGFβ signaling is initiated with ligand-induced oligomerization of serine/threonine receptor kinases and phosphorylation of the cytoplasmic signaling molecules, Smad2 and Smad3 [31]. Although the switch of TGFβ from a tumor suppressor to a tumor promoter is not well defined, there is evidence that TGFβ plays an important role as an activator of tumor growth and metastasis. In the liver, TGFβ is the most potent hepatic pro-fibrogenic cytokine predominantly produced by activated mesenchymal cells upon chronic liver damage [32,33]. Further, TGFβ is an important link between chronic injury, cirrhosis, and HCC, and may serve as a key target for HCC therapy [32,33]. In BC, activation of TGFβ attributes to the initiation of EMT, which increases tumor cell invasion. Meanwhile, the EMT process induced by TGFβ generates cells with a stable bCSCs phenotype, which is demonstrated by increased self-renewing and tumorigenicity [34,35]. Currently, TGFβ inhibitors have been proposed and are being developed as anti-metastatic agents [33,36]. One proposal is that inhibition of TGFβ blocks the generation of CSCs, which enhances the chemotherapy action against TNBC [5]. In our present study, CaA decreased the TGFβ-induced phosphorylation of SMAD2. Importantly, CaA also attenuated the expression of total SMAD2. Based on these results, we hypothesized that the repressive effects of CaA on the endogenous SMAD2 may be via miRNAs, resulting in the degradation of mRNAs.
MiRNAs are small non-coding RNAs that regulate gene expression by binding to the 3′-UTR of the target mRNA and inhibiting translation or targeting the mRNA for degradation [37]. Currently, the miRNAs networks and their regulation of mRNA translation and protein expression in the CSCs-like properties remain to be elucidated. Here, we picked out three SMAD2 associated miRNAs, miR-148a, miR-155, and miR-206. Interestingly, miR-148a elevated the expressions of miR-155 and miR-206. There are two possible reasons for this. First, miR-155 is reported to be epigenetically silenced in human cancers [26]. Meanwhile, miR-148a has been identified as a DNMT1 target miRNA in breast cancer cells [23]. So the up-regulation of miR-155 by miR-148a might be via the DNA demethylation; Second, Winbanks et al. reported that TGFβ reduced the endogenous expression of the primary and mature forms of miR-206 [38]. Since miR-148a targeted SMAD2, we hypothesized that the up-regulation of miR-206 by miR-148a might be via the inhibition of TGFβ/SMADs pathway. Based on these results, we chose miR-148a for further investigation.
MiR-148a was first shown to modulate the levels of cytochrome P450 3A4 via post-transcriptionally regulating the 3′UTR of the Pregnane X Receptor (PXR) mRNA [39]. Meanwhile, miR-148a is also a pro-apoptotic miRNA that targets Bcl-2 [40]. In addition, inhibition of miR-148a by hyper-methylation is associated with metastasis in many tumor types and with up-regulation of metastasis-associated genes [41]. Further miR-148a is involved in the anti-metastasis of HCC cells by the inhibitions of Wnt1-mediated EMT and acquirement of CSCs-like properties [37]. In breast cancer MCF-7 cells, miR-148a inhibits angiogenesis by targeting erythroblastic leukemia viral oncogene homolog 3 (ERBB3), which blocks the downstream pathway activation including protein kinase B (PKB), extracellular signal regulated kinases (ERKs), and hypoxia-inducible factor 1 alpha (HIF-1α) [42]. We previously found that, miR-148a down-regulated SMAD2 in glabridin-treated HepG2 cells [15]. Here, by use of luciferase reporter assay, qRT-PCR, and western blot, we confirmed the direct relationship between miR-148a and SMAD2 in CaA-treated MHCC97H cells. Further, we found that the up-regulation of miR-148a by CaA might via the DNA demethylation in miR-148a promoter.
Methylation of CaA requires SAM as a cofactor, which causes the demethylation of DNA [11]. A previous study indicates that there are many CpG-rich regions in miR-148a promoter [23]. So we hypothesized that CaA up-regulated the expression of miR-148a by demethylation. Our data showed that the average methylation level of miR-148a promoter in CaA-treated MHCC97H cells was lower than that in medium control cells. Further, hypermethylation treatment by SAM dramatically blocked the CaA-induced elevation of miR-148a. Moreover, as shown in Fig. S3A–C, CaA also decreased the average methylation level in miR-155 promoter. DNMTs are responsible for DNA methylation [11]. Interestingly, miR-148a has been identified to target DNMT1, which down-regulates the expression of DNMT1 in breast cancer cells [23]. In our study, overexpression of miR-148a decreased the expression of DNMT1 (Fig. 7A and B). Further, CaA decreased the expression of DNMT1 in MDA-MB-231 cells, however, knockdown of miR-148a blocked this phenomenon (Fig. 7D and E). So we further hypothesized that there might be a feedback loop between miR-148a and DNMT1 in CaA-treated human cancer cells, that was reduced DNMT1 would further reduce the demethylation on miR-148a promoter.
Conflict of interest
There have been no involvements that might raise the question of bias in the work reported or in the conclusions, implications, or opinions stated.
Author contributions
Planned experiments: Yuan Li, Zhong Li, Lei Li; Performed experiments: Yuan Li, Fei Jiang, Lijun Chen, Ye Yang, Shuyuan Cao; Analyzed data: Lijun Chen, Yuting Ye, Xingxing Wang, Juan Mu; Contributed reagents/materials/analysis tools: Lei Li; Wrote the paper: Yuan Li, Fei Jiang.
Acknowledgements
This work was supported by National Natural Science Foundation of China (81473020 and 81402667), a Research Fund for the Doctoral Program of Higher Education of China (20133234110007), a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), a Natural Science Foundation of Jiangsu Higher Education Institutions (14KJB330003), and a Technology Development Fund of Nanjing Medical University (2013NJMU021).
Appendix A. Supplementary data
References
- 1.Eaves C.J. Cancer stem cells: Here, there, everywhere? Nature. 2008;456:581–582. doi: 10.1038/456581a. [DOI] [PubMed] [Google Scholar]
- 2.Hutz K., Mejias-Luque R., Farkasova K. The stem cell factor SOX2 regulates the tumorigenic potential in human gastric cancer cells. Carcinogenesis. 2014;35:942–950. doi: 10.1093/carcin/bgt410. [DOI] [PubMed] [Google Scholar]
- 3.Philip B., Ito K., Moreno-Sanchez R. HIF expression and the role of hypoxic microenvironments within primary tumours as protective sites driving cancer stem cell renewal and metastatic progression. Carcinogenesis. 2013;34:1699–1707. doi: 10.1093/carcin/bgt209. [DOI] [PubMed] [Google Scholar]
- 4.Jiang R., Li Y., Xu Y. EMT and CSC-like properties mediated by the IKKbeta/IkappaBalpha/RelA signal pathway via the transcriptional regulator, Snail, are involved in the arsenite-induced neoplastic transformation of human keratinocytes. Arch. Toxicol. 2013;87:991–1000. doi: 10.1007/s00204-012-0933-0. [DOI] [PubMed] [Google Scholar]
- 5.Bhola N.E., Balko J.M., Dugger T.C. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013;123:1348–1358. doi: 10.1172/JCI65416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vidal S.J., Rodriguez-Bravo V., Galsky M. Targeting cancer stem cells to suppress acquired chemotherapy resistance. Oncogene. 2014;33:4451–4463. doi: 10.1038/onc.2013.411. [DOI] [PubMed] [Google Scholar]
- 7.Abubaker K., Latifi A., Luwor R. Short-term single treatment of chemotherapy results in the enrichment of ovarian cancer stem cell-like cells leading to an increased tumor burden. Mol. Cancer. 2013;12:24. doi: 10.1186/1476-4598-12-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Onori P., DeMorrow S., Gaudio E. Caffeic acid phenethyl ester decreases cholangiocarcinoma growth by inhibition of NF-kappaB and induction of apoptosis. Int. J. Cancer J. Int. du Cancer. 2009;125:565–576. doi: 10.1002/ijc.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Touaibia M., Jean-Francois J., Doiron J. Caffeic acid, a versatile pharmacophore: an overview. Mini Rev. Med. Chem. 2011;11:695–713. doi: 10.2174/138955711796268750. [DOI] [PubMed] [Google Scholar]
- 10.Guerriero E., Sorice A., Capone F. Effects of lipoic acid, caffeic acid and a synthesized lipoyl-caffeic conjugate on human hepatoma cell lines. Molecules. 2011;16:6365–6377. doi: 10.3390/molecules16086365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee W.J., Zhu B.T. Inhibition of DNA methylation by caffeic acid and chlorogenic acid, two common catechol-containing coffee polyphenols. Carcinogenesis. 2006;27:269–277. doi: 10.1093/carcin/bgi206. [DOI] [PubMed] [Google Scholar]
- 12.Park W.H., Kim S.H., Kim C.H. A new matrix metalloproteinase-9 inhibitor 3,4-dihydroxycinnamic acid (caffeic acid) from methanol extract of Euonymus alatus: isolation and structure determination. Toxicology. 2005;207:383–390. doi: 10.1016/j.tox.2004.10.008. [DOI] [PubMed] [Google Scholar]
- 13.Jung J.E., Kim H.S., Lee C.S. Caffeic acid and its synthetic derivative CADPE suppress tumor angiogenesis by blocking STAT3-mediated VEGF expression in human renal carcinoma cells. Carcinogenesis. 2007;28:1780–1787. doi: 10.1093/carcin/bgm130. [DOI] [PubMed] [Google Scholar]
- 14.Omene C.O., Wu J., Frenkel K. Caffeic Acid Phenethyl Ester (CAPE) derived from propolis, a honeybee product, inhibits growth of breast cancer stem cells. Invest. New Drugs. 2012;30:1279–1288. doi: 10.1007/s10637-011-9667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang F., Mu J., Wang X. The repressive effect of miR-148a on TGF beta-SMADs signal pathway is involved in the Glabridin-induced inhibition of the cancer stem cells-like properties in hepatocellular carcinoma cells. PLoS ONE. 2014;9:e96698. doi: 10.1371/journal.pone.0096698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding S.J., Li Y., Shao X.X. Proteome analysis of hepatocellular carcinoma cell strains, MHCC97-H and MHCC97-L, with different metastasis potentials. Proteomics. 2004;4:982–994. doi: 10.1002/pmic.200300653. [DOI] [PubMed] [Google Scholar]
- 17.Lu L., Katsaros D., de la Longrais I.A. Hypermethylation of let-7a-3 in epithelial ovarian cancer is associated with low insulin-like growth factor-II expression and favorable prognosis. Cancer Res. 2007;67:10117–10122. doi: 10.1158/0008-5472.CAN-07-2544. [DOI] [PubMed] [Google Scholar]
- 18.Yamashita T., Honda M., Nakamoto Y. Discrete nature of EpCAM+ and CD90+ cancer stem cells in human hepatocellular carcinoma. Hepatology. 2013;57:1484–1497. doi: 10.1002/hep.26168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirsch H.A., Iliopoulos D., Struhl K. Metformin inhibits the inflammatory response associated with cellular transformation and cancer stem cell growth. Proc. Natl. Acad. Sci. U.S.A. 2013;110:972–977. doi: 10.1073/pnas.1221055110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu L., Katsaros D., Mayne S.T. Functional study of risk loci of stem cell-associated gene lin-28B and associations with disease survival outcomes in epithelial ovarian cancer. Carcinogenesis. 2012;33:2119–2125. doi: 10.1093/carcin/bgs243. [DOI] [PubMed] [Google Scholar]
- 21.Lu L., Katsaros D., Shaverdashvili K. Pluripotent factor lin-28 and its homologue lin-28b in epithelial ovarian cancer and their associations with disease outcomes and expression of let-7a and IGF-II. Eur. J. Cancer. 2009;45:2212–2218. doi: 10.1016/j.ejca.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 22.Ji H., Li Y., Jiang F. Inhibition of transforming growth factor beta/SMAD signal by MiR-155 is involved in arsenic trioxide-induced anti-angiogenesis in prostate cancer. Cancer Sci. 2014;105:1541–1549. doi: 10.1111/cas.12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Q., Jiang Y., Yin Y. A regulatory circuit of miR-148a/152 and DNMT1 in modulating cell transformation and tumor angiogenesis through IGF-IR and IRS1. J. Mol. Cell Biol. 2013;5:3–13. doi: 10.1093/jmcb/mjs049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanoun N., Delpu Y., Suriawinata A.A. The silencing of microRNA 148a production by DNA hypermethylation is an early event in pancreatic carcinogenesis. Clin. Chem. 2010;56:1107–1118. doi: 10.1373/clinchem.2010.144709. [DOI] [PubMed] [Google Scholar]
- 25.Wang P., Heber D., Henning S.M. Quercetin increased bioavailability and decreased methylation of green tea polyphenols in vitro and in vivo. Food Funct. 2012;3:635–642. doi: 10.1039/c2fo10254d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li C.L., Nie H., Wang M. MicroRNA-155 is downregulated in gastric cancer cells and involved in cell metastasis. Oncol. Rep. 2012;27:1960–1966. doi: 10.3892/or.2012.1719. [DOI] [PubMed] [Google Scholar]
- 27.Yang Y., Li Y., Wang K. P38/NF-kappaB/snail pathway is involved in caffeic acid-induced inhibition of cancer stem cells-like properties and migratory capacity in malignant human keratinocyte. PLoS ONE. 2013;8:e58915. doi: 10.1371/journal.pone.0058915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michels K.B., Willett W.C., Fuchs C.S. Coffee, tea, and caffeine consumption and incidence of colon and rectal cancer. J. Natl Cancer Inst. 2005;97:282–292. doi: 10.1093/jnci/dji039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tavani A., Pregnolato A., La Vecchia C. Coffee and tea intake and risk of cancers of the colon and rectum: a study of 3530 cases and 7057 controls. Int. J. Cancer J. Int. du Cancer. 1997;73:193–197. doi: 10.1002/(sici)1097-0215(19971009)73:2<193::aid-ijc5>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 30.Olthof M.R., Hollman P.C., Katan M.B. Chlorogenic acid and caffeic acid are absorbed in humans. J. Nutr. 2001;131:66–71. doi: 10.1093/jn/131.1.66. [DOI] [PubMed] [Google Scholar]
- 31.Zavadil J., Bitzer M., Liang D. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc. Natl. Acad. Sci. U.S.A. 2001;98:6686–6691. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu K., Ding J., Chen C. Hepatic transforming growth factor beta gives rise to tumor-initiating cells and promotes liver cancer development. Hepatology. 2012;56:2255–2267. doi: 10.1002/hep.26007. [DOI] [PubMed] [Google Scholar]
- 33.Zhong Z., Carroll K.D., Policarpio D. Anti-transforming growth factor beta receptor II antibody has therapeutic efficacy against primary tumor growth and metastasis through multi effects on cancer, stroma, and immune cells. Clin. Cancer Res. 2010;16:1191–1205. doi: 10.1158/1078-0432.CCR-09-1634. [DOI] [PubMed] [Google Scholar]
- 34.Liu Z., Bandyopadhyay A., Nichols R.W. Blockade of autocrine TGF-beta signaling inhibits stem cell phenotype, survival, and metastasis of murine breast cancer cells. J. Stem Cell Res. Therap. 2012;2:1–8. doi: 10.4172/2157-7633.1000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.West N.R., Murray J.I., Watson P.H. Oncostatin-M promotes phenotypic changes associated with mesenchymal and stem cell-like differentiation in breast cancer. Oncogene. 2014;33:1485–1494. doi: 10.1038/onc.2013.105. [DOI] [PubMed] [Google Scholar]
- 36.Bueno L., de Alwis D.P., Pitou C. Semi-mechanistic modelling of the tumour growth inhibitory effects of LY2157299, a new type I receptor TGF-beta kinase antagonist, in mice. Eur. J. Cancer. 2008;44:142–150. doi: 10.1016/j.ejca.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 37.Yan H., Dong X., Zhong X. Inhibitions of epithelial to mesenchymal transition and cancer stem cells-like properties are involved in miR-148a-mediated anti-metastasis of hepatocellular carcinoma. Mol. Carcinog. 2014;53:960–969. doi: 10.1002/mc.22064. [DOI] [PubMed] [Google Scholar]
- 38.Winbanks C.E., Wang B., Beyer C. TGF-beta regulates miR-206 and miR-29 to control myogenic differentiation through regulation of HDAC4. J. Biol. Chem. 2011;286:13805–13814. doi: 10.1074/jbc.M110.192625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takagi S., Nakajima M., Mohri T. Post-transcriptional regulation of human pregnane X receptor by micro-RNA affects the expression of cytochrome P450 3A4. J. Biol. Chem. 2008;283:9674–9680. doi: 10.1074/jbc.M709382200. [DOI] [PubMed] [Google Scholar]
- 40.Zhang H., Li Y., Huang Q. MiR-148a promotes apoptosis by targeting Bcl-2 in colorectal cancer. Cell Death Differ. 2011;18:1702–1710. doi: 10.1038/cdd.2011.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Budhu A., Jia H.L., Forgues M. Identification of metastasis-related microRNAs in hepatocellular carcinoma. Hepatology. 2008;47:897–907. doi: 10.1002/hep.22160. [DOI] [PubMed] [Google Scholar]
- 42.Yu J., Li Q., Xu Q. MiR-148a inhibits angiogenesis by targeting ERBB3. J. Biomed. Res. 2011;25:170–177. doi: 10.1016/S1674-8301(11)60022-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.